
Chemotherapy-Induced Changes in the Lung Microenvironment: The Role of MMP-2 in Facilitating Intravascular Arrest of Breast Cancer Cells


Abstract
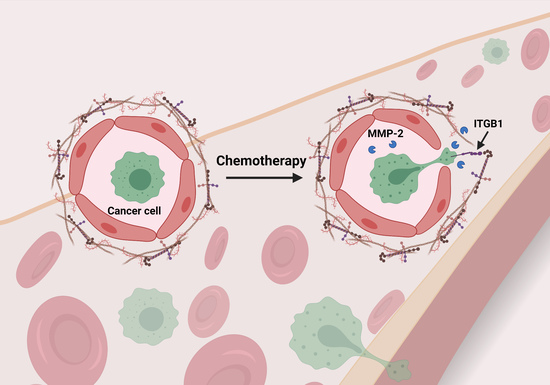
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
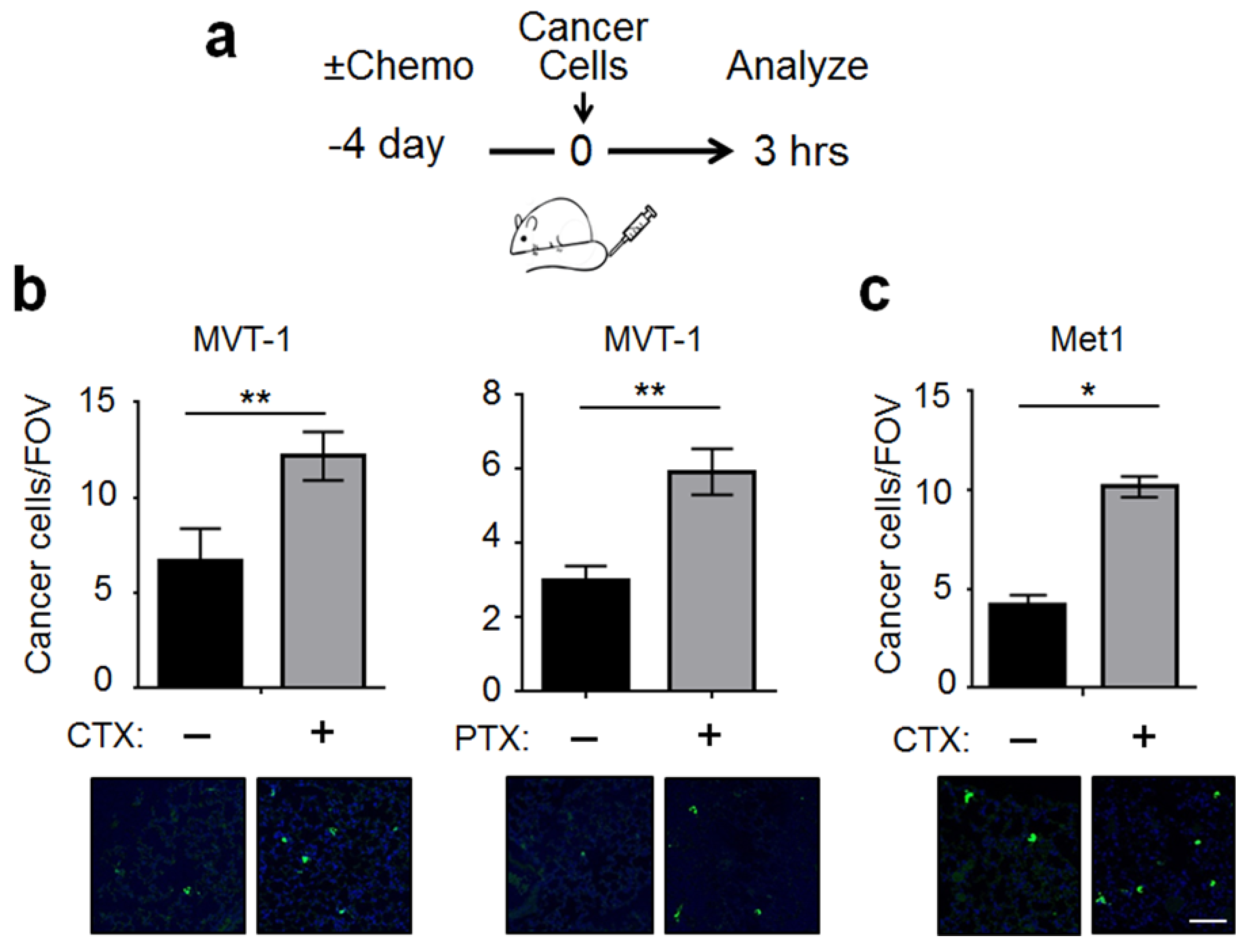
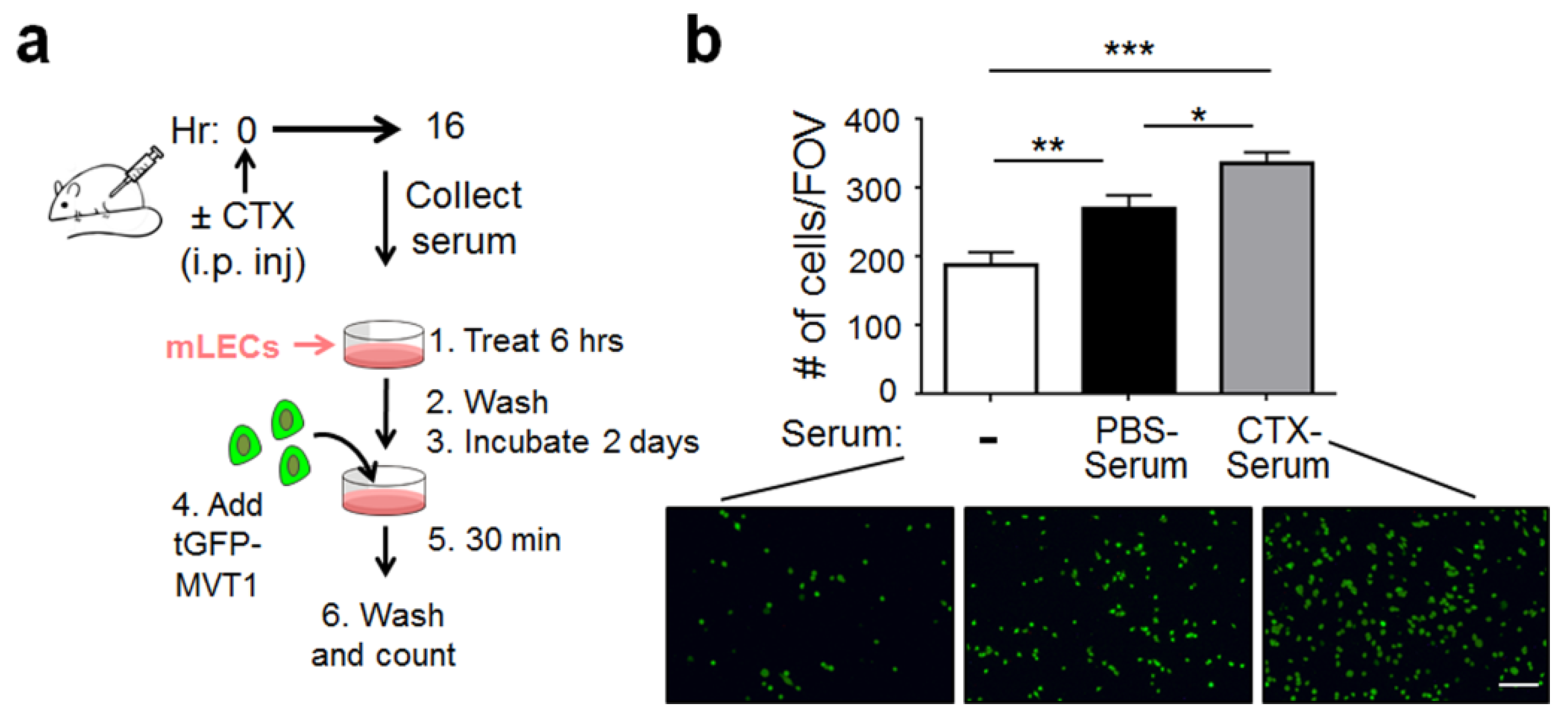
2.1. CTX Modified the Lung Endothelium to Allow More Cancer Cells Adhesion—In a Manner Dependent on Host-Derived Serum Factors
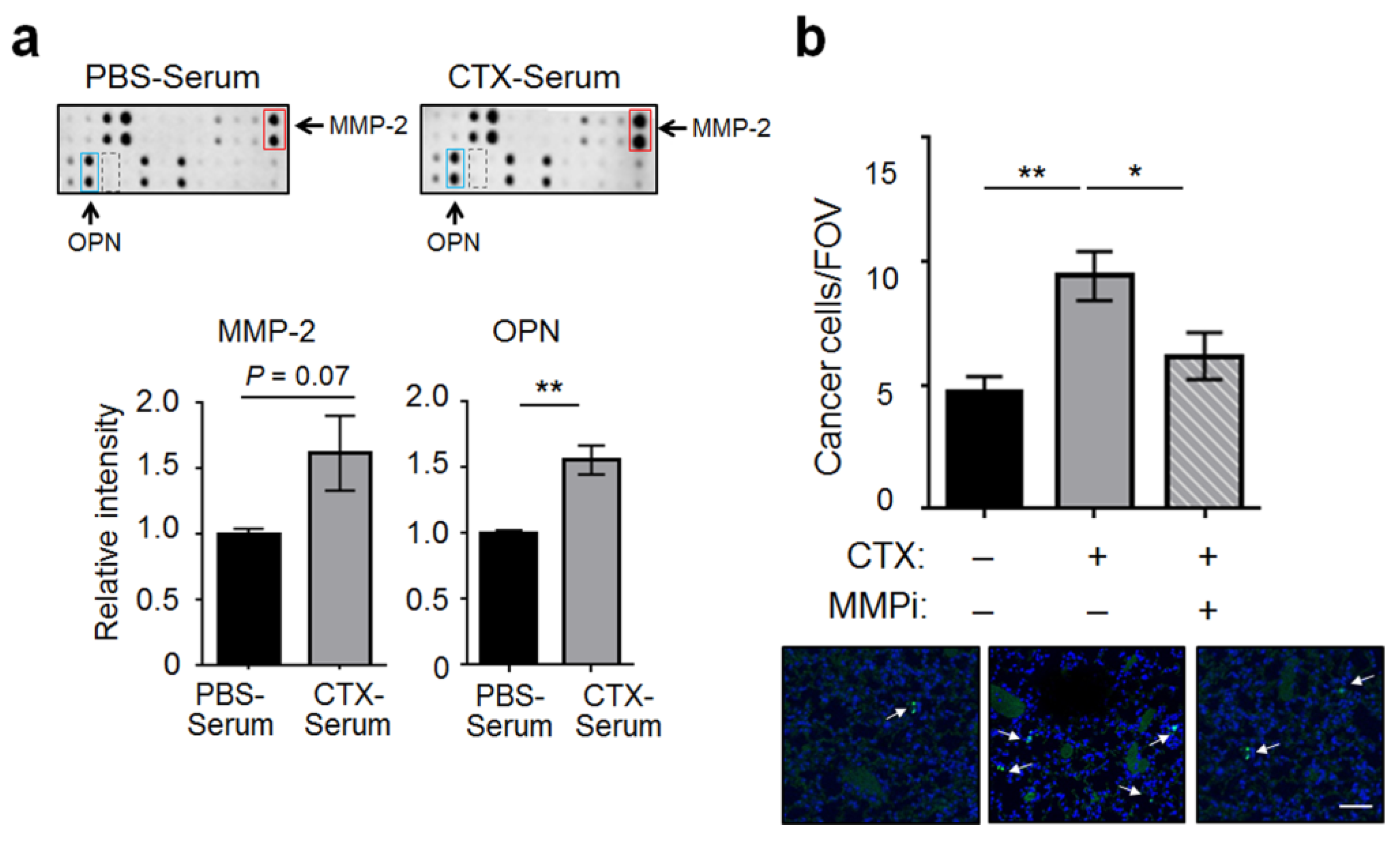
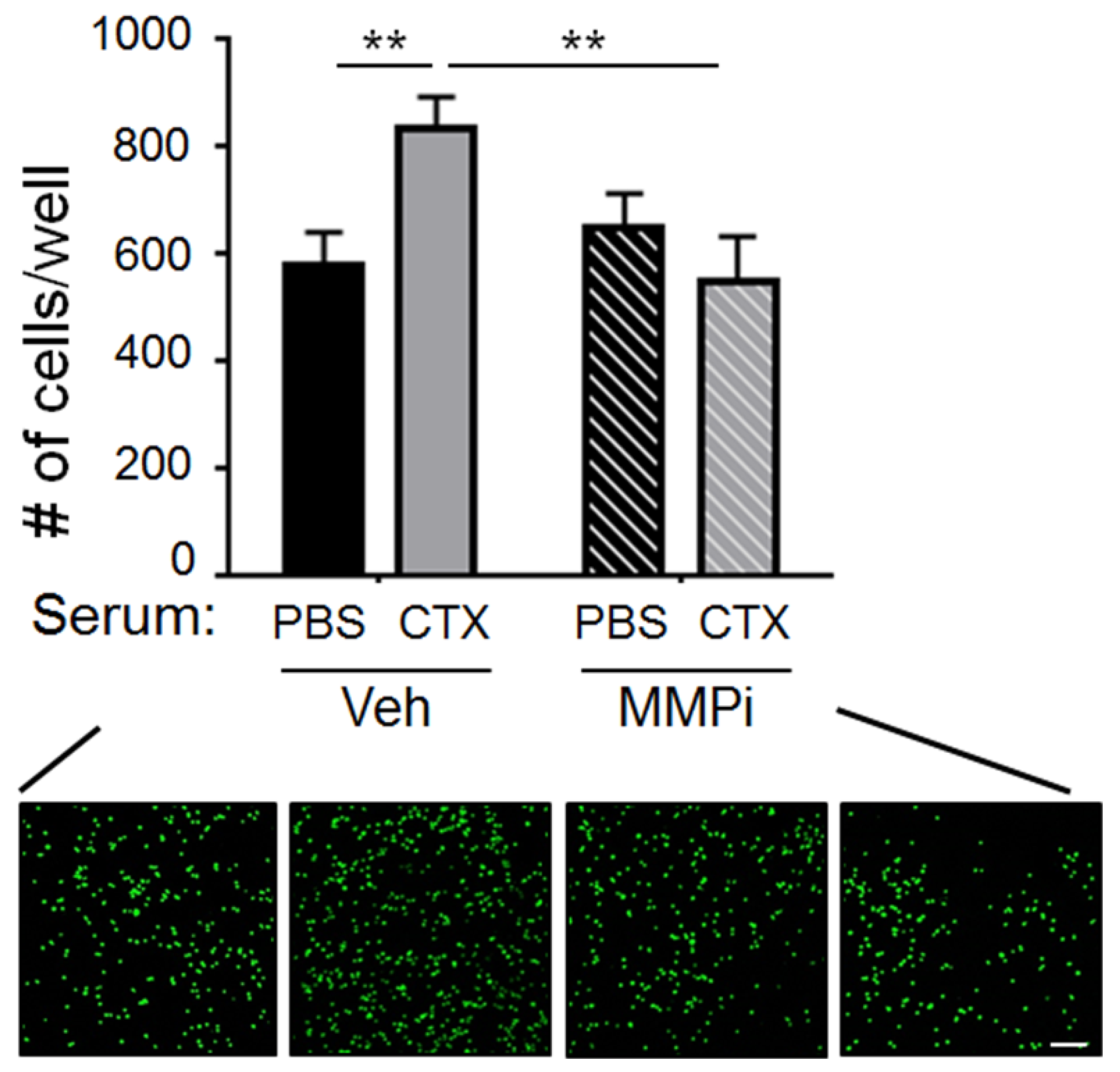
2.2. Matrix Metalloproteinase-2 (MMP-2) Was a Functionally Important Serum Factor for CTX to Increase the Vascular Adhesiveness
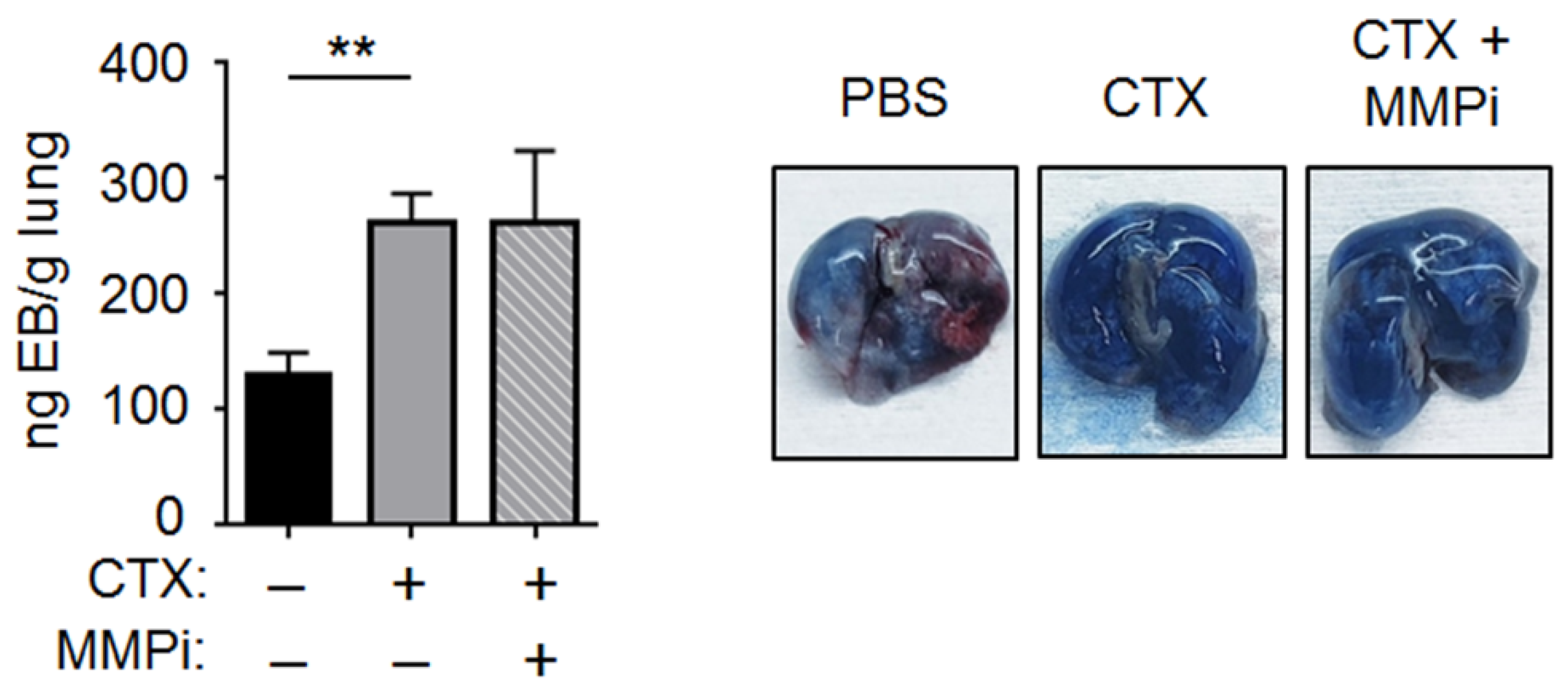
2.3. CTX Increased Vascular Permeability in the Lung
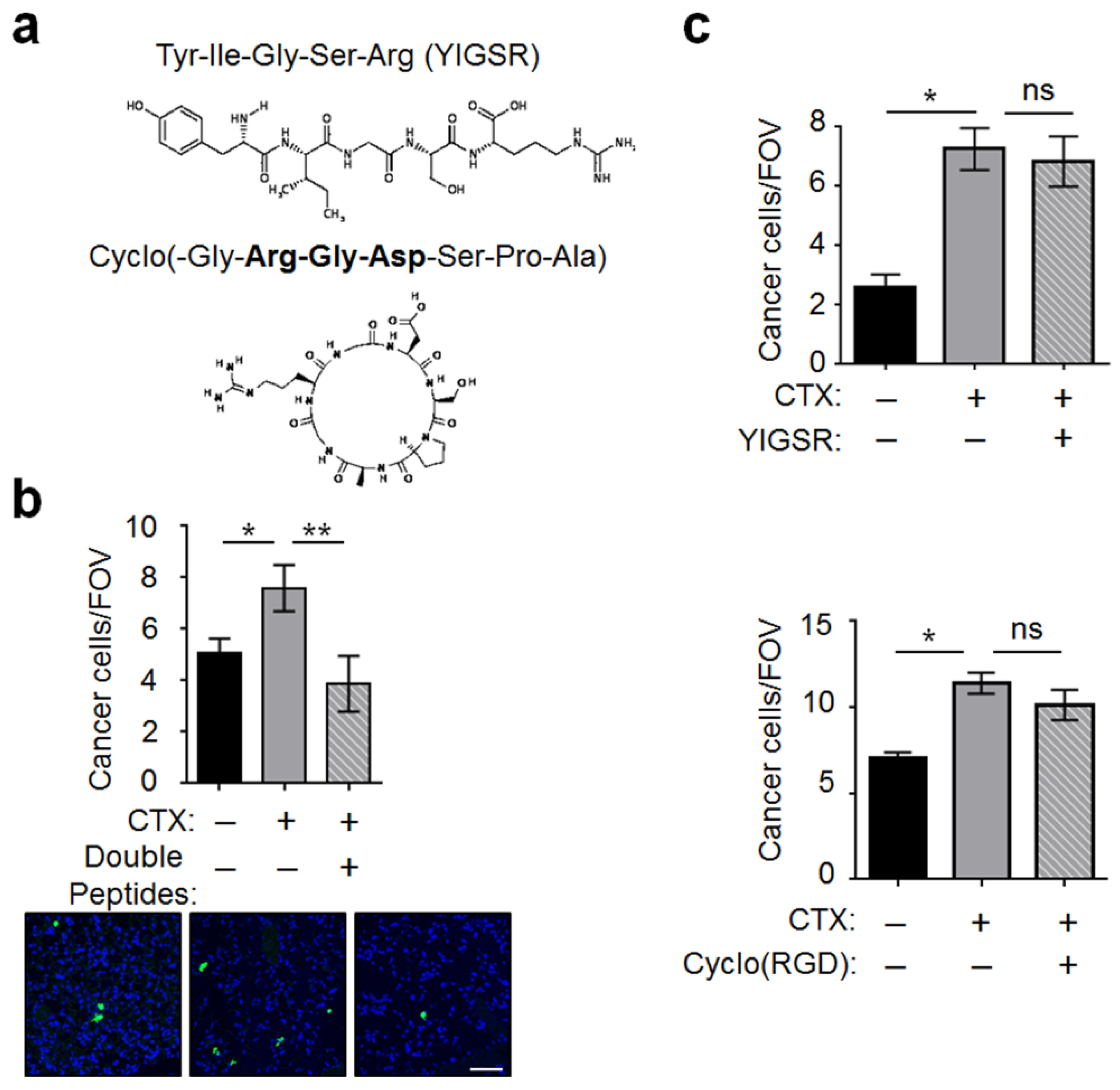
2.4. MMP-2 Remodels BM and Two ECM Protein Domains Were Required for CTX to Increase Intravascular Cancer Cell Arrest
2.5. Integrin β1 on Cancer Cells Played an Important Role for Cancer Cells to Interact with the Host Vascular Walls
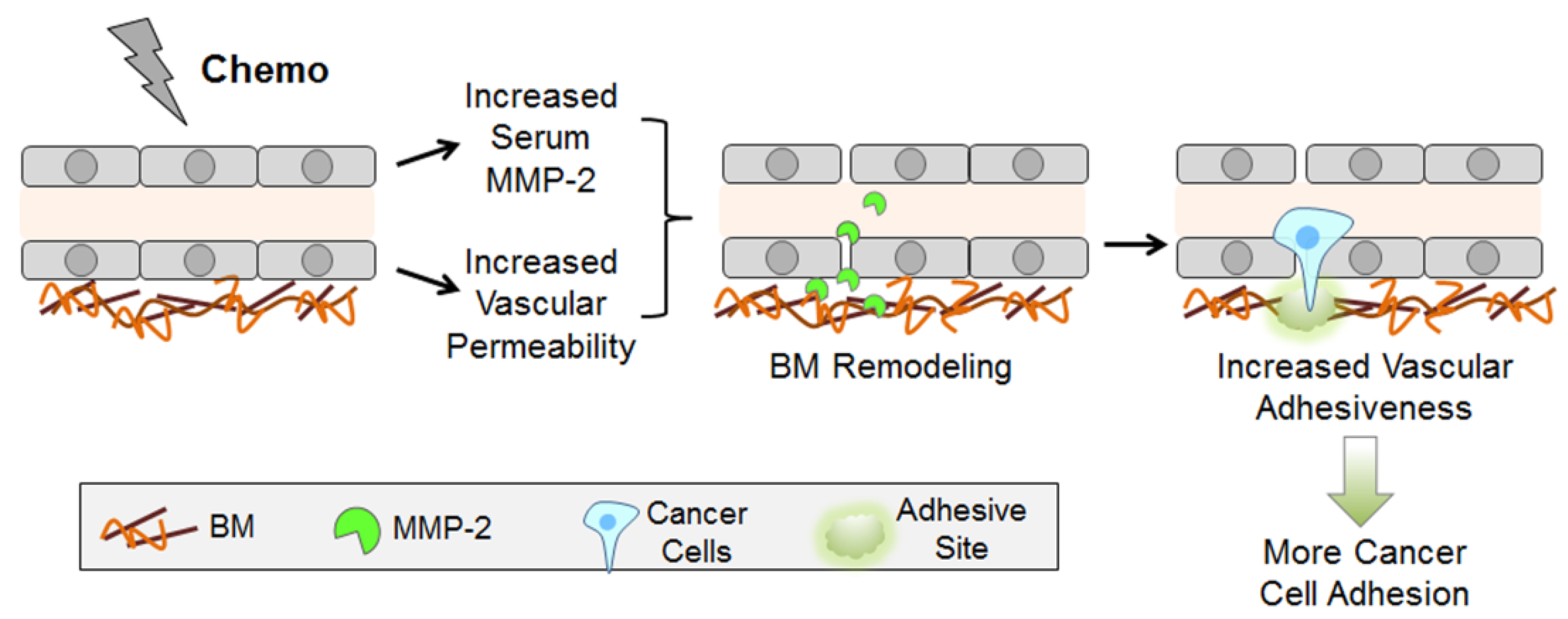
3. Discussion
4. Materials and Methods
4.1. Animal Studies
4.2. Cell Culture, Treatment, and siRNA Knockdown
4.3. Cell Isolation from Mouse Tissues
4.4. Generation of CTX-Serum and PBS-Serum
4.5. In Vitro EC-Cancer Cell Adhesion Assay
4.6. Protein Array
4.7. Image Analysis
4.8. RNA Isolation and RT-qPCR
4.9. Immunofluorescence Analysis
4.10. Evans Blue Vascular Permeability Assay
4.11. Analytical Flow Cytometry and Fluorescence Activated Cell Sorting (FACS)
4.12. In Vitro Matrigel (ECM)-Cancer Cell Adhesion Assay
4.13. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Kitamura, T.; Qian, B.Z.; Pollard, J.W. Immune cell promotion of metastasis. Nat. Rev. Immunol. 2015, 15, 73–86. [Google Scholar] [CrossRef]
- Valastyan, S.; Weinberg, R.A. Tumor metastasis: Molecular insights and evolving paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef] [Green Version]
- Alishekevitz, D.; Gingis-Velitski, S.; Kaidar-Person, O.; Gutter-Kapon, L.; Scherer, S.D.; Raviv, Z.; Merquiol, E.; Ben-Nun, Y.; Miller, V.; Rachman-Tzemah, C.; et al. Macrophage-Induced Lymphangiogenesis and Metastasis following Paclitaxel Chemotherapy Is Regulated by VEGFR3. Cell Rep. 2016, 17, 1344–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.S.; Jalgaonkar, S.P.; Middleton, J.D.; Hai, T. Stress-inducible gene Atf3 in the noncancer host cells contributes to chemotherapy-exacerbated breast cancer metastasis. Proc. Natl. Acad. Sci. USA 2017, 114, E7159–E7168. [Google Scholar] [CrossRef] [Green Version]
- Karagiannis, G.S.; Pastoriza, J.M.; Wang, Y.; Harney, A.S.; Entenberg, D.; Pignatelli, J.; Sharma, V.P.; Xue, E.A.; Cheng, E.; D’Alfonso, T.M.; et al. Neoadjuvant chemotherapy induces breast cancer metastasis through a TMEM-mediated mechanism. Sci. Transl. Med. 2017, 9, 397–411. [Google Scholar] [CrossRef] [Green Version]
- Robinson, B.D.; Sica, G.L.; Liu, Y.F.; Rohan, T.E.; Gertler, F.B.; Condeelis, J.S.; Jones, J.G. Tumor microenvironment of metastasis in human breast carcinoma: A potential prognostic marker linked to hematogenous dissemination. Clin. Cancer Res. 2009, 15, 2433–2441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harney, A.S.; Arwert, E.N.; Entenberg, D.; Wang, Y.; Guo, P.; Qian, B.Z.; Oktay, M.H.; Pollard, J.W.; Jones, J.G.; Condeelis, J.S. Real-Time Imaging Reveals Local, Transient Vascular Permeability, and Tumor Cell Intravasation Stimulated by TIE2hi Macrophage-Derived VEGFA. Cancer Discov. 2015, 5, 932–943. [Google Scholar] [CrossRef] [Green Version]
- Massague, J.; Obenauf, A.C. Metastatic colonization by circulating tumour cells. Nature 2016, 529, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Daenen, L.G.; Roodhart, J.M.; van Amersfoort, M.; Dehnad, M.; Roessingh, W.; Ulfman, L.H.; Derksen, P.W.; Voest, E.E. Chemotherapy enhances metastasis formation via VEGFR-1-expressing endothelial cells. Cancer Res. 2011, 71, 6976–6985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gingis-Velitski, S.; Loven, D.; Benayoun, L.; Munster, M.; Bril, R.; Voloshin, T.; Alishekevitz, D.; Bertolini, F.; Shaked, Y. Host response to short-term, single-agent chemotherapy induces matrix metalloproteinase-9 expression and accelerates metastasis in mice. Cancer Res. 2011, 71, 6986–6996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.I.; Liao, J.; Berry, J.E.; Li, X.; Koh, A.J.; Michalski, M.E.; Eber, M.R.; Soki, F.N.; Sadler, D.; Sud, S.; et al. Cyclophosphamide creates a receptive microenvironment for prostate cancer skeletal metastasis. Cancer Res. 2012, 72, 2522–2532. [Google Scholar] [CrossRef] [Green Version]
- Middleton, J.D.; Fehlman, J.; Sivakumar, S.; Stover, D.G.; Hai, T. Stress-Inducible Gene Atf3 Dictates a Dichotomous Macrophage Activity in Chemotherapy-Enhanced Lung Colonization. Int. J. Mol. Sci. 2021, 22, 7356. [Google Scholar] [CrossRef] [PubMed]
- Wolford, C.C.; McConoughey, S.J.; Jalgaonkar, S.P.; Leon, M.; Merchant, A.S.; Dominick, J.L.; Yin, X.; Chang, Y.; Zmuda, E.J.; Toole, S.A.; et al. Transcription factor ATF3 links host adaptive response to breast cancer metastasis. J. Clin. Investig. 2013, 123, 2893–2906. [Google Scholar] [CrossRef] [Green Version]
- Kanekal, S.; Fraiser, L.; Kehrer, J.P. Pharmacokinetics, metabolic activation, and lung toxicity of cyclophosphamide in C57/B16 and ICR mice. Toxicol. Appl. Pharmacol. 1992, 114, 1–8. [Google Scholar] [CrossRef]
- Innocenti, F.; Danesi, R.; Di Paolo, A.; Agen, C.; Nardini, D.; Bocci, G.; Del Tacca, M. Plasma and tissue disposition of paclitaxel (taxol) after intraperitoneal administration in mice. Drug Metab. Dispos. 1995, 23, 713–717. [Google Scholar]
- Labelle, M.; Begum, S.; Hynes, R.O. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 2011, 20, 576–590. [Google Scholar] [CrossRef] [Green Version]
- Mook, O.R.; Van Marle, J.; Vreeling-Sindelarova, H.; Jonges, R.; Frederiks, W.M.; Van Noorden, C.J. Visualization of early events in tumor formation of eGFP-transfected rat colon cancer cells in liver. Hepatology 2003, 38, 295–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kienast, Y.; von Baumgarten, L.; Fuhrmann, M.; Klinkert, W.E.; Goldbrunner, R.; Herms, J.; Winkler, F. Real-time imaging reveals the single steps of brain metastasis formation. Nat. Med. 2010, 16, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Glinskii, O.V.; Huxley, V.H.; Glinsky, G.V.; Pienta, K.J.; Raz, A.; Glinsky, V.V. Mechanical entrapment is insufficient and intercellular adhesion is essential for metastatic cell arrest in distant organs. Neoplasia 2005, 7, 522–527. [Google Scholar] [CrossRef] [Green Version]
- Gassmann, P.; Hemping-Bovenkerk, A.; Mees, S.T.; Haier, J. Metastatic tumor cell arrest in the liver-lumen occlusion and specific adhesion are not exclusive. Int. J. Colorectal Dis. 2009, 24, 851–858. [Google Scholar] [CrossRef]
- Enns, A.; Gassmann, P.; Schluter, K.; Korb, T.; Spiegel, H.U.; Senninger, N.; Haier, J. Integrins can directly mediate metastatic tumor cell adhesion within the liver sinusoids. J. Gastrointest. Surg. 2004, 8, 1049–1059, discussion 1060. [Google Scholar] [CrossRef]
- Azevedo, A.S.; Follain, G.; Patthabhiraman, S.; Harlepp, S.; Goetz, J.G. Metastasis of circulating tumor cells: Favorable soil or suitable biomechanics, or both? Cell Adhes. Migr. 2015, 9, 345–356. [Google Scholar] [CrossRef]
- Perea Paizal, J.; Au, S.H.; Bakal, C. Squeezing through the microcirculation: Survival adaptations of circulating tumour cells to seed metastasis. Br. J. Cancer 2021, 124, 58–65. [Google Scholar] [CrossRef]
- Wirtz, D.; Konstantopoulos, K.; Searson, P.C. The physics of cancer: The role of physical interactions and mechanical forces in metastasis. Nat. Rev. Cancer 2011, 11, 512–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fehrenbach, M.L.; Cao, G.; Williams, J.T.; Finklestein, J.M.; Delisser, H.M. Isolation of murine lung endothelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 296, L1096–L1103. [Google Scholar] [CrossRef] [PubMed]
- Sobczak, M.; Dargatz, J.; Chrzanowska-Wodnicka, M. Isolation and culture of pulmonary endothelial cells from neonatal mice. J. Vis. Exp. 2010, 46, 2316. [Google Scholar] [CrossRef]
- Koivunen, E.; Arap, W.; Valtanen, H.; Rainisalo, A.; Medina, O.P.; Heikkila, P.; Kantor, C.; Gahmberg, C.G.; Salo, T.; Konttinen, Y.T.; et al. Tumor targeting with a selective gelatinase inhibitor. Nat. Biotechnol. 1999, 17, 768–774. [Google Scholar] [CrossRef]
- Wang, H.; Fu, W.; Im, J.H.; Zhou, Z.; Santoro, S.A.; Iyer, V.; DiPersio, C.M.; Yu, Q.C.; Quaranta, V.; Al-Mehdi, A.; et al. Tumor cell alpha3beta1 integrin and vascular laminin-5 mediate pulmonary arrest and metastasis. J. Cell Biol. 2004, 164, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Gibson-Corley, K.N.; Herndon, M.E.; Sun, Y.; Gustafson-Wagner, E.; Teoh-Fitzgerald, M.; Domann, F.E.; Henry, M.D.; Stipp, C.S. Integrin alpha3beta1 can function to promote spontaneous metastasis and lung colonization of invasive breast carcinoma. Mol. Cancer Res. 2014, 12, 143–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Jin, X.; Liu, K.J.; Liu, W. Matrix metalloproteinase-2-mediated occludin degradation and caveolin-1-mediated claudin-5 redistribution contribute to blood-brain barrier damage in early ischemic stroke stage. J. Neurosci. 2012, 32, 3044–3057. [Google Scholar] [CrossRef]
- Giannelli, G.; Falk-Marzillier, J.; Schiraldi, O.; Stetler-Stevenson, W.G.; Quaranta, V. Induction of cell migration by matrix metalloprotease-2 cleavage of laminin-5. Science 1997, 277, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Rodriguez, D.; Petitclerc, E.; Kim, J.J.; Hangai, M.; Moon, Y.S.; Davis, G.E.; Brooks, P.C. Proteolytic exposure of a cryptic site within collagen type IV is required for angiogenesis and tumor growth in vivo. J. Cell Biol. 2001, 154, 1069–1079. [Google Scholar] [CrossRef] [PubMed]
- Mutgan, A.C.; Jandl, K.; Kwapiszewska, G. Endothelial Basement Membrane Components and Their Products, Matrikines: Active Drivers of Pulmonary Hypertension? Cells 2020, 9, 2029. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Li, Y.; Shen, Y.; Wang, A.; Wang, S.; Xie, T. The functions and applications of RGD in tumor therapy and tissue engineering. Int. J. Mol. Sci. 2013, 14, 13447–13462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieberler, M.; Reuning, U.; Reichart, F.; Notni, J.; Wester, H.J.; Schwaiger, M.; Weinmuller, M.; Rader, A.; Steiger, K.; Kessler, H. Exploring the Role of RGD-Recognizing Integrins in Cancer. Cancers 2017, 9, 116. [Google Scholar] [CrossRef] [PubMed]
- Graf, J.; Ogle, R.C.; Robey, F.A.; Sasaki, M.; Martin, G.R.; Yamada, Y.; Kleinman, H.K. A pentapeptide from the laminin B1 chain mediates cell adhesion and binds the 67,000 laminin receptor. Biochemistry 1987, 26, 6896–6900. [Google Scholar] [CrossRef]
- Iwamoto, Y.; Robey, F.A.; Graf, J.; Sasaki, M.; Kleinman, H.K.; Yamada, Y.; Martin, G.R. YIGSR, a synthetic laminin pentapeptide, inhibits experimental metastasis formation. Science 1987, 238, 1132–1134. [Google Scholar] [CrossRef]
- Kumagai, H.; Tajima, M.; Ueno, Y.; Giga-Hama, Y.; Ohba, M. Effect of cyclic RGD peptide on cell adhesion and tumor metastasis. Biochem. Biophys. Res. Commun. 1991, 177, 74–82. [Google Scholar] [CrossRef]
- Chen, M.B.; Lamar, J.M.; Li, R.; Hynes, R.O.; Kamm, R.D. Elucidation of the Roles of Tumor Integrin beta1 in the Extravasation Stage of the Metastasis Cascade. Cancer Res. 2016, 76, 2513–2524. [Google Scholar] [CrossRef] [Green Version]
- Ardini, E.; Pesole, G.; Tagliabue, E.; Magnifico, A.; Castronovo, V.; Sobel, M.E.; Colnaghi, M.I.; Menard, S. The 67-kDa laminin receptor originated from a ribosomal protein that acquired a dual function during evolution. Mol. Biol. Evol. 1998, 15, 1017–1025. [Google Scholar] [CrossRef] [Green Version]
- Menard, S.; Tagliabue, E.; Colnaghi, M.I. The 67 kDa laminin receptor as a prognostic factor in human cancer. Breast Cancer Res. Treat. 1998, 52, 137–145. [Google Scholar] [CrossRef]
- Belkin, A.M.; Stepp, M.A. Integrins as receptors for laminins. Microsc. Res. Tech. 2000, 51, 280–301. [Google Scholar] [CrossRef]
- Stipp, C.S. Laminin-binding integrins and their tetraspanin partners as potential antimetastatic targets. Expert Rev. Mol. Med. 2010, 12, e3. [Google Scholar] [CrossRef] [Green Version]
- Romanov, V.; Sobel, M.E.; Pinto da Silva, P.; Menard, S.; Castronovo, V. Cell localization and redistribution of the 67 kD laminin receptor and alpha 6 beta 1 integrin subunits in response to laminin stimulation: An immunogold electron microscopy study. Cell Adhes. Commun. 1994, 2, 201–209. [Google Scholar] [CrossRef]
- DiGiacomo, V.; Meruelo, D. Looking into laminin receptor: Critical discussion regarding the non-integrin 37/67-kDa laminin receptor/RPSA protein. Biol. Rev. Camb. Philos. Soc. 2016, 91, 288–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mammoto, A.; Mammoto, T.; Kanapathipillai, M.; Wing Yung, C.; Jiang, E.; Jiang, A.; Lofgren, K.; Gee, E.P.; Ingber, D.E. Control of lung vascular permeability and endotoxin-induced pulmonary oedema by changes in extracellular matrix mechanics. Nat. Commun. 2013, 4, 1759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.J.; Zhong, Z.J.; Cao, L.H.; Li, H.T.; Zhang, T.H.; Lin, W.Q. Paclitaxel-induced lung injury and its amelioration by parecoxib sodium. Sci. Rep. 2015, 5, 12977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpenter, T.C.; Schroeder, W.; Stenmark, K.R.; Schmidt, E.P. Eph-A2 promotes permeability and inflammatory responses to bleomycin-induced lung injury. Am. J. Respir. Cell Mol. Biol. 2012, 46, 40–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Ning, Z.; Cui, J.; Yu, F.; Sioutas, C.; Hsiai, T. Diesel exhaust particles modulate vascular endothelial cell permeability: Implication of ZO-1 expression. Toxicol. Lett. 2010, 197, 163–168. [Google Scholar] [CrossRef] [Green Version]
- Goetzl, E.J.; Banda, M.J.; Leppert, D. Matrix metalloproteinases in immunity. J. Immunol. 1996, 156, 1–4. [Google Scholar]
- Hassona, Y.; Cirillo, N.; Heesom, K.; Parkinson, E.K.; Prime, S.S. Senescent cancer-associated fibroblasts secrete active MMP-2 that promotes keratinocyte dis-cohesion and invasion. Br. J. Cancer 2014, 111, 1230–1237. [Google Scholar] [CrossRef] [Green Version]
- Unemori, E.N.; Bouhana, K.S.; Werb, Z. Vectorial secretion of extracellular matrix proteins, matrix-degrading proteinases, and tissue inhibitor of metalloproteinases by endothelial cells. J. Biol. Chem. 1990, 265, 445–451. [Google Scholar] [CrossRef]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [Green Version]
- Kato, H.; Liao, Z.; Mitsios, J.V.; Wang, H.Y.; Deryugina, E.I.; Varner, J.A.; Quigley, J.P.; Shattil, S.J. The primacy of beta1 integrin activation in the metastatic cascade. PLoS ONE 2012, 7, e46576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamidi, H.; Ivaska, J. Every step of the way: Integrins in cancer progression and metastasis. Nat. Rev. Cancer 2018, 18, 533–548. [Google Scholar] [CrossRef] [Green Version]
- Gloe, T.; Pohl, U. Laminin binding conveys mechanosensing in endothelial cells. News Physiol. Sci. 2002, 17, 166–169. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, H.; Boelte, K.C.; Lin, P.C. Endothelial cell adhesion molecules and cancer progression. Curr. Med. Chem. 2007, 14, 377–386. [Google Scholar] [CrossRef]
- Bendas, G.; Borsig, L. Cancer cell adhesion and metastasis: Selectins, integrins, and the inhibitory potential of heparins. Int. J. Cell Biol. 2012, 2012, 676731. [Google Scholar] [CrossRef] [PubMed]
- Reymond, N.; d’Agua, B.B.; Ridley, A.J. Crossing the endothelial barrier during metastasis. Nat. Rev. Cancer 2013, 13, 858–870. [Google Scholar] [CrossRef] [PubMed]
- Gay, L.J.; Felding-Habermann, B. Contribution of platelets to tumour metastasis. Nat. Rev. Cancer 2011, 11, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Ivetic, A.; Hoskins Green, H.L.; Hart, S.J. L-selectin: A Major Regulator of Leukocyte Adhesion, Migration and Signaling. Front. Immunol. 2019, 10, 1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savi, P.; Nurden, P.; Nurden, A.T.; Levy-Toledano, S.; Herbert, J.M. Clopidogrel: A review of its mechanism of action. Platelets 1998, 9, 251–255. [Google Scholar] [CrossRef]
- Esumi, N.; Fan, D.; Fidler, I.J. Inhibition of murine melanoma experimental metastasis by recombinant desulfatohirudin, a highly specific thrombin inhibitor. Cancer Res. 1991, 51, 4549–4556. [Google Scholar]
- Barkan, D.; Chambers, A.F. beta1-integrin: A potential therapeutic target in the battle against cancer recurrence. Clin. Cancer Res. 2011, 17, 7219–7223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fields, G.B. The rebirth of matrix metalloproteinase inhibitors: Moving beyond the dogma. Cells 2019, 8, 984. [Google Scholar] [CrossRef] [Green Version]
- Friedrichs, B.; Siegel, S.; Kloess, M.; Barsoum, A.; Coggin, J.; Rohrer, J.; Jakob, I.; Tiemann, M.; Heidorn, K.; Schulte, C.; et al. Humoral immune responses against the immature laminin receptor protein show prognostic significance in patients with chronic lymphocytic leukemia. J. Immunol. 2008, 180, 6374–6384. [Google Scholar] [CrossRef]
- Hartman, M.G.; Lu, D.; Kim, M.L.; Kociba, G.J.; Shukri, T.; Buteau, J.; Wang, X.; Frankel, W.L.; Guttridge, D.; Prentki, M.; et al. Role for activating transcription factor 3 in stress-induced beta-cell apoptosis. Mol. Cell. Biol. 2004, 24, 5721–5732. [Google Scholar] [CrossRef] [Green Version]
- Lim, Y.C.; Luscinskas, F.W. Isolation and culture of murine heart and lung endothelial cells for in vitro model systems. Methods Mol. Biol. 2006, 341, 141–154. [Google Scholar] [CrossRef]
- Yin, X.; DeWille, J.; Hai, T. A potential dichotomous role of ATF3, an adaptive-response gene, in cancer development. Oncogene 2008, 27, 2118–2127. [Google Scholar] [CrossRef] [Green Version]
- Hai, T.; Jalgaonkar, S.; Wolford, C.C.; Yin, X. Immunohistochemical detection of Activating Transcription Factor 3, a hub of the cellular adaptive-response network. Methods Enzymol. 2011, 490, 175–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Middleton, J.D.; Sivakumar, S.; Hai, T. Chemotherapy-Induced Changes in the Lung Microenvironment: The Role of MMP-2 in Facilitating Intravascular Arrest of Breast Cancer Cells. Int. J. Mol. Sci. 2021, 22, 10280. https://doi.org/10.3390/ijms221910280
Middleton JD, Sivakumar S, Hai T. Chemotherapy-Induced Changes in the Lung Microenvironment: The Role of MMP-2 in Facilitating Intravascular Arrest of Breast Cancer Cells. International Journal of Molecular Sciences. 2021; 22(19):10280. https://doi.org/10.3390/ijms221910280
Chicago/Turabian StyleMiddleton, Justin D., Subhakeertana Sivakumar, and Tsonwin Hai. 2021. "Chemotherapy-Induced Changes in the Lung Microenvironment: The Role of MMP-2 in Facilitating Intravascular Arrest of Breast Cancer Cells" International Journal of Molecular Sciences 22, no. 19: 10280. https://doi.org/10.3390/ijms221910280
APA StyleMiddleton, J. D., Sivakumar, S., & Hai, T. (2021). Chemotherapy-Induced Changes in the Lung Microenvironment: The Role of MMP-2 in Facilitating Intravascular Arrest of Breast Cancer Cells. International Journal of Molecular Sciences, 22(19), 10280. https://doi.org/10.3390/ijms221910280