
Impact of Dietary Fat on the Progression of Liver Fibrosis: Lessons from Animal and Cell Studies



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
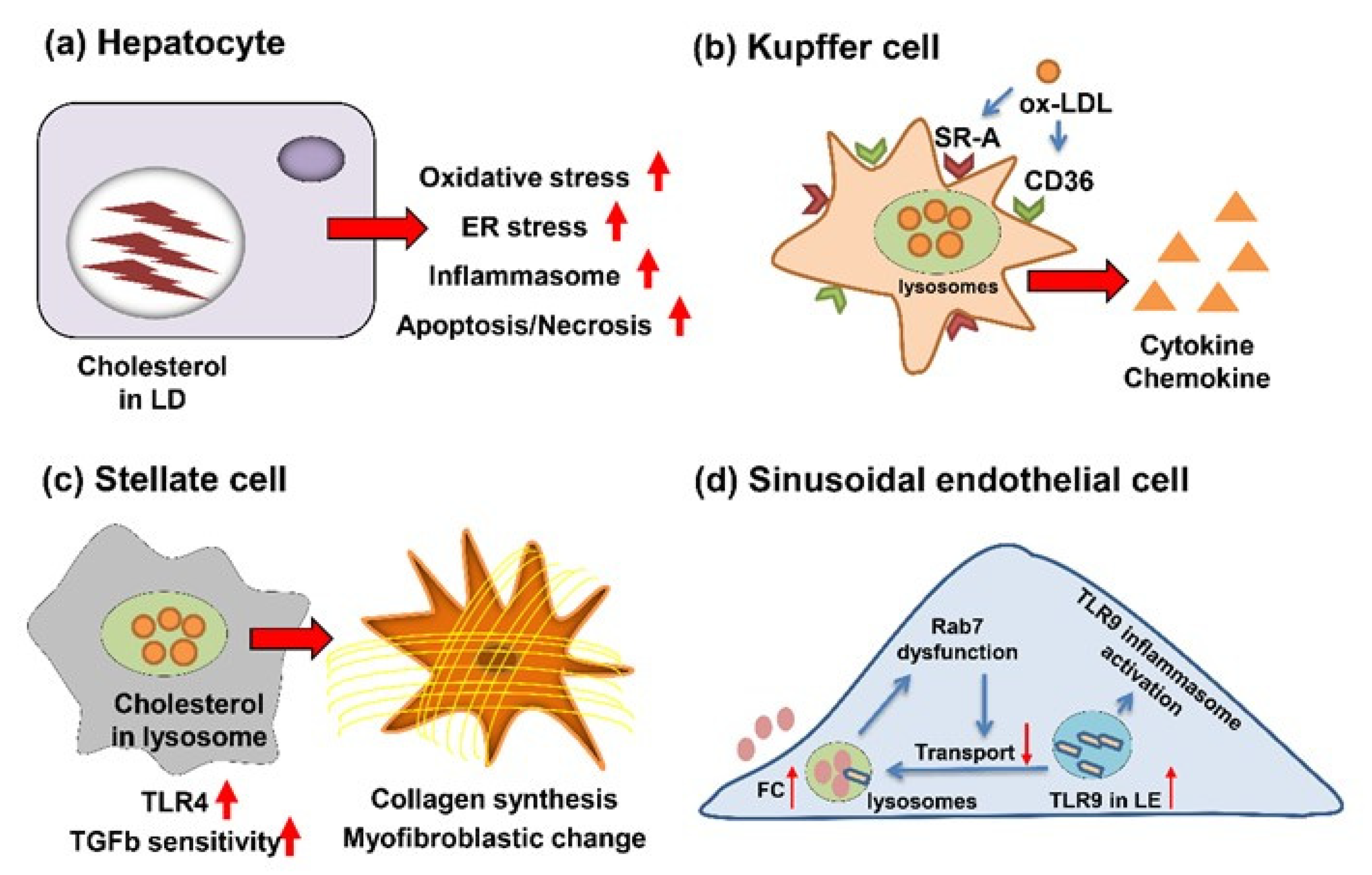
2. Role of Cholesterol in the Progression of Liver Fibrosis
2.1. Cholesterol’s Action for Hepatocytes
2.2. Cholesterol’s Action for KC
2.3. Cholesterol’s Action for HSC
2.4. Cholesterol’s Action for Cholangiocytes
2.5. Cholesterol’s Action for LSECs
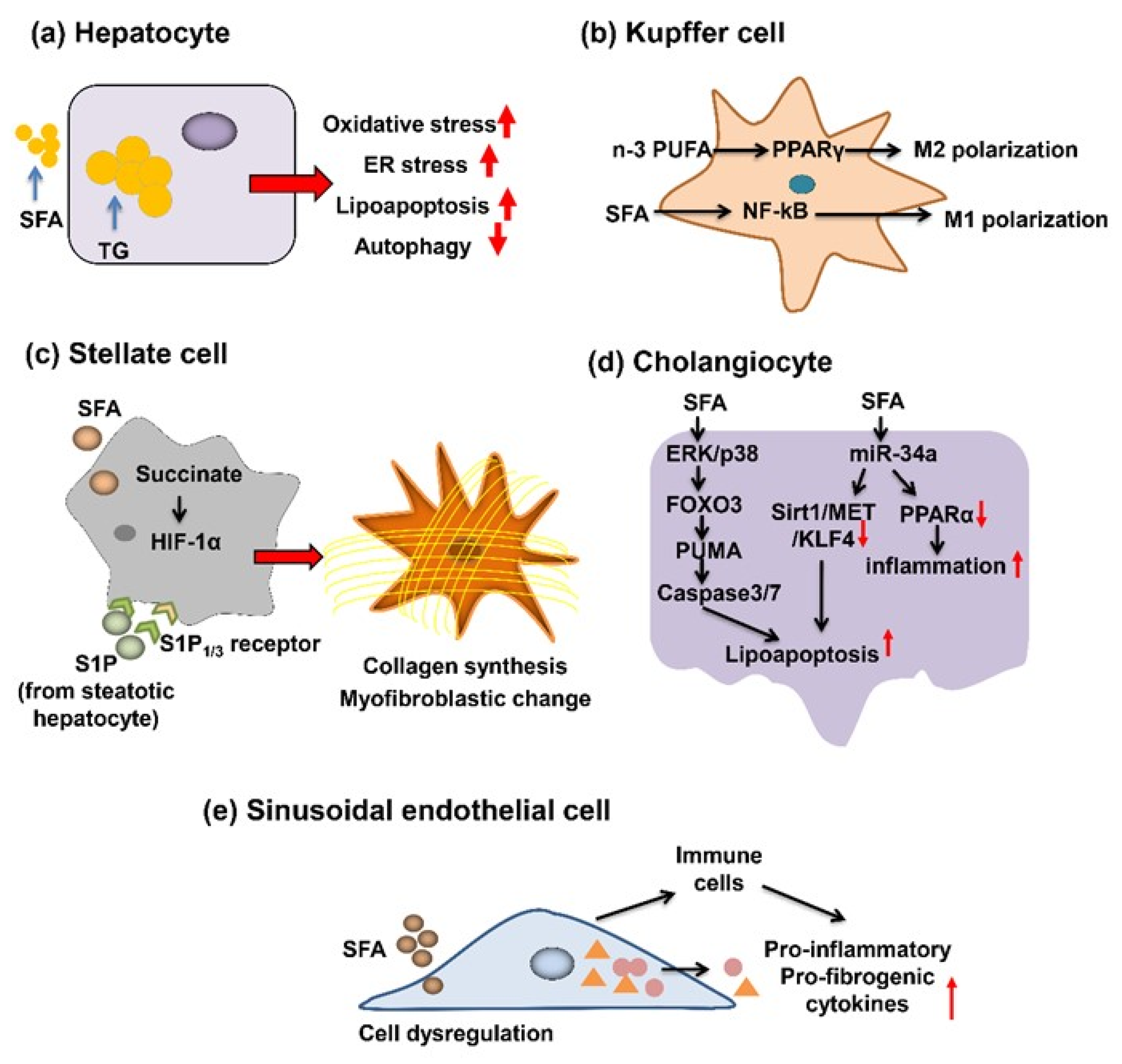
3. Role of SFA in the Progression of Liver Fibrosis
3.1. SFA’s Action for Hepatocytes
3.2. SFA’s Action for KC
3.3. SFA’s Action for HSC
3.4. SFA’s Action for Cholangiocytes
3.5. SFA’s Action for LSECs
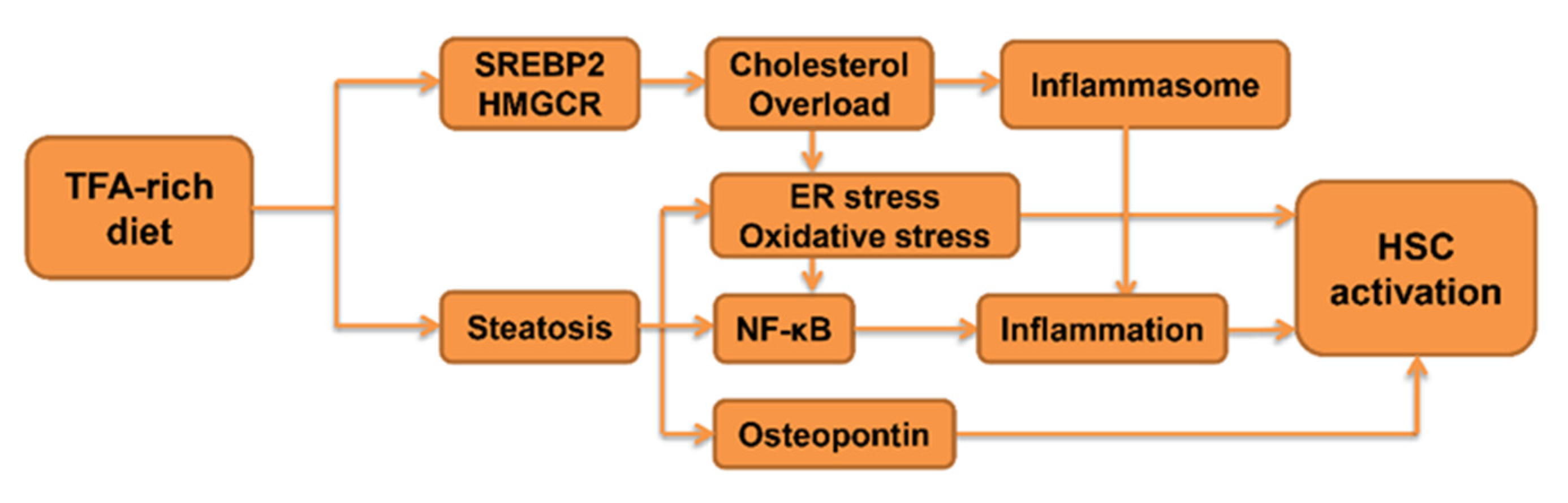
4. Role of TFA in the Progression of Fibrosis
4.1. The Impact of TFA on Inflammatory Signaling
4.2. The Impact of TFA on Lipid Metabolism
5. Future Perspectives
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACAT | acyltransferase |
| α-SMA | α-smooth muscle actin |
| CD36 | cluster differentiation 36 |
| CM | conditioned media |
| COL1A1 | collagen type 1A1 |
| COX | cyclooxygenase |
| CTGF | connective tissue growth factor |
| DGAT2 | diacylglycerol acyltransferase 2 |
| DR | ductular reaction |
| EA | elaidic acid |
| ER | endoplasmic reticulum |
| ERK | extracellular signal-regulated kinase |
| FFA | free fatty acid |
| FOXO3 | forkhead family transcription factors 3 |
| HCVcpTg | hepatitis C virus core gene transgenic |
| HIF | hypoxia-inducible transcription factor |
| HSC | hepatic stellate cells |
| IL | interleukin |
| JNK | jun-N-terminal kinase |
| KC | Kupffer cell |
| LDL | low-density-lipoprotein |
| LD | lipid droplet |
| LOX-1 | lectin-like oxidized LDL receptor-1 |
| LSEC | liver sinusoidal endothelial cell |
| NAFL | non-alcoholic fatty liver |
| NAFLD | non-alcoholic fatty liver disease |
| NASH | non-alcoholic steatohepatitis |
| NF-κB | nuclear factor-kappa B |
| NKT | natural killer T |
| OPN | osteopontin |
| oxLDL | oxidized LDL |
| PA | palmitic acid |
| PPAR | peroxisome proliferator-activated receptor |
| PUFA | polyunsaturated fatty acid |
| PUMA | p53-up-regulated modulator of apoptosis |
| ROS | reactive oxygen species |
| S1P | sphingosine 1-phosphate |
| SFA | saturated fatty acid |
| SR-A | scavenger receptor A |
| SREBP 2 | sterol regulatory element-binding protein 2 |
| TFA | trans fatty acid |
| TG | triglyceride |
| TGF | transforming growth factor |
| TIMP-1 | tissue inhibitor of metallo-proteinase-1 |
| TLR | Toll-like receptor |
| TNF | tumor necrosis factor |
References
- Michelotti, G.A.; Machado, M.V.; Diehl, A.M. NAFLD, NASH and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Kimura, T.; Fujimori, N.; Nagaya, T.; Komatsu, M.; Tanaka, E. Current status, problems, and perspectives of non-alcoholic fatty liver disease research. World J. Gastroenterol. 2019, 25, 163–177. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Kobayashi, A.; Tanaka, N.; Sano, K.; Komatsu, M.; Fujimori, N.; Yamazaki, T.; Shibata, S.; Ichikawa, Y.; Joshita, S.; et al. Clinicopathological characteristics of non-B non-C hepatocellular carcinoma without past hepatitis B virus infection. Hepatol. Res. 2017, 47, 405–418. [Google Scholar] [CrossRef]
- Nagaya, T.; Tanaka, N.; Suzuki, T.; Sano, K.; Horiuchi, A.; Komatsu, M.; Nakajima, T.; Nishizawa, T.; Joshita, S.; Umemura, T.; et al. Down-regulation of SREBP-1c is associated with the development of burned-out NASH. J. Hepatol. 2010, 53, 724–731. [Google Scholar] [CrossRef] [Green Version]
- Nagaya, T.; Tanaka, N.; Komatsu, M.; Ichijo, T.; Sano, K.; Horiuchi, A.; Joshita, S.; Umemura, T.; Matsumoto, A.; Yoshizawa, K.; et al. Development from simple steatosis to liver cirrhosis and hepatocellular carcinoma: A 27-year follow-up case. Clin. J. Gastroenterol. 2008, 1, 116–121. [Google Scholar] [CrossRef]
- Tanaka, N.; Aoyama, T.; Kimura, S.; Gonzalez, F.J. Targeting nuclear receptors for the treatment of fatty liver disease. Pharmacol. Ther. 2017, 179, 142–157. [Google Scholar] [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metab. Clin. Exp. 2016, 65, 1038–1048. [Google Scholar] [CrossRef]
- Ioannou, G.N. The Role of Cholesterol in the Pathogenesis of NASH. Trends Endocrinol. Metab. 2016, 27, 84–95. [Google Scholar] [CrossRef]
- Leamy, A.K.; Egnatchik, R.A.; Young, J.D. Molecular mechanisms and the role of saturated fatty acids in the progression of non-alcoholic fatty liver disease. Prog. Lipid Res. 2013, 52, 165–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obara, N.; Fukushima, K.; Ueno, Y.; Wakui, Y.; Kimura, O.; Tamai, K.; Kakazu, E.; Inoue, J.; Kondo, Y.; Ogawa, N.; et al. Possible involvement and the mechanisms of excess trans-fatty acid consumption in severe NAFLD in mice. J. Hepatol. 2010, 53, 326–334. [Google Scholar] [CrossRef]
- Ioannou, G.N.; Morrow, O.B.; Connole, M.L.; Lee, S.P. Association between dietary nutrient composition and the incidence of cirrhosis or liver cancer in the united states population. Hepatology 2009, 50, 175–184. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Morishima, C.; Ioannou, G.N. Dietary Cholesterol Intake Is Associated with Progression of Liver Disease in Patients with Chronic Hepatitis C: Analysis of the Hepatitis C Antiviral Long-term Treatment Against Cirrhosis Trial. Clin. Gastroenterol. Hepatol. 2013, 11, 1661–1666.e3. [Google Scholar] [CrossRef] [PubMed]
- Diao, P.; Jia, F.; Wang, X.; Hu, X.; Kimura, T.; Nakajima, T.; Aoyama, T.; Moriya, K.; Koike, K.; Tanaka, N. Mechanisms of steatosis-derived hepatocarcinogenesis: Lessons from HCV core gene transgenic mice. Engineering 2021, in press. [Google Scholar]
- Moriya, K.; Matsuura, Y.; Miyamura, T.; Koike, K.; Shintani, Y.; Ishibashi, K.; Fujie, H.; Yotsuyanagi, H. Hepatitis C virus core protein induces hepatic steatosis in transgenic mice. J. Gen. Virol. 1997, 78, 1527–1531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriya, K.; Fujie, H.; Shintani, Y.; Yotsuyanagi, H.; Tsutsumi, T.; Ishibashi, K.; Matsuura, Y.; Kimura, S.; Miyamura, T.; Koike, K. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat. Med. 1998, 4, 1065–1067. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Moriya, K.; Kiyosawa, K.; Koike, K.; Aoyama, T. Hepatitis C virus core protein induces spontaneous and persistent activation of peroxisome proliferator-activated receptor alpha in transgenic mice: Implications for HCV-associated hepatocarcinogenesis. Int. J. Cancer 2008, 122, 124–131. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, N.; Moriya, K.; Kiyosawa, K.; Koike, K.; Gonzalez, F.J.; Aoyama, T. PPARalpha activation is essential for HCV core protein-induced hepatic steatosis and hepatocellular carcinoma in mice. J. Clin. Investig. 2008, 118, 683–694. [Google Scholar]
- Wang, X.; Tanaka, N.; Hu, X.; Kimura, T.; Lu, Y.; Jia, F.; Sato, Y.; Nakayama, J.; Moriya, K.; Koike, K.; et al. A high-cholesterol diet promotes steatohepatitis and liver tumorigenesis in HCV core gene transgenic mice. Arch. Toxicol. 2019, 93, 1713–1725. [Google Scholar] [CrossRef]
- Diao, P.; Wang, X.; Jia, F.; Kimura, T.; Hu, X.; Shirotori, S.; Nakamura, I.; Sato, Y.; Nakayama, J.; Moriya, K.; et al. A saturated fatty acid-rich diet enhances hepatic lipogenesis and tumorigenesis in HCV core gene transgenic mice. J. Nutr. Biochem. 2020, 85, 108460. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Wang, X.; Jia, F.; Tanaka, N.; Kimura, T.; Nakajima, T.; Sato, Y.; Moriya, K.; Koike, K.; Gonzalez, F.J.; et al. A trans-fatty acid-rich diet promotes liver tumorigenesis in HCV core gene transgenic mice. Carcinogenesis 2020, 41, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.-S.; Shin, E.-J.; Yeom, J.-W.; Park, Y.-H.; Kim, S.-K. Association between Nutrient Intake and Metabolic Syndrome in Patients with Colorectal Cancer. Clin. Nutr. Res. 2017, 6, 38–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cascio, G.; Schiera, G.; Di Liegro, I. Dietary fatty acids in metabolic syndrome, diabetes and cardiovascular diseases. Curr. Diabetes Rev. 2012, 8, 2–17. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.D.; Li, Y.; Chiuve, S.; Stampfer, M.J.; Manson, J.E.; Rimm, E.B.; Willett, W.C.; Hu, F.B. Association of Specific Dietary Fats with Total and Cause-Specific Mortality. JAMA Intern. Med. 2016, 176, 1134–1145. [Google Scholar] [CrossRef]
- Ioannou, G.N.; Subramanian, S.; Chait, A.; Haigh, W.G.; Yeh, M.M.; Farrell, G.C.; Lee, S.P.; Savard, C. Cholesterol crystallization within hepatocyte lipid droplets and its role in murine NASH. J. Lipid Res. 2017, 58, 1067–1079. [Google Scholar] [CrossRef] [Green Version]
- Ioannou, G.N.; Landis, C.S.; Jin, G.; Haigh, W.G.; Farrell, G.C.; Kuver, R.; Lee, S.P.; Savard, C. Cholesterol Crystals in Hepatocyte Lipid Droplets Are Strongly Associated with Human Nonalcoholic Steatohepatitis. Hepatol. Commun. 2019, 3, 776–791. [Google Scholar] [CrossRef] [PubMed]
- Makino, A.; Hullin-Matsuda, F.; Murate, M.; Abe, M.; Tomishige, N.; Fukuda, M.; Yamashita, S.; Fujimoto, T.; Vidal, H.; Lagarde, M.; et al. Acute accumulation of free cholesterol induces the degradation of perilipin 2 and Rab18-dependent fusion of ER and lipid droplets in cultured human hepatocytes. Mol. Biol. Cell 2016, 27, 3293–3304. [Google Scholar] [CrossRef]
- Arguello, G.; Balboa, E.; Arrese, M.; Zanlungo, S. Recent insights on the role of cholesterol in non-alcoholic fatty liver disease. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 1765–1778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Rooyen, D.M.; Larter, C.Z.; Haigh, W.G.; Yeh, M.M.; Ioannou, G.; Kuver, R.; Lee, S.P.; Teoh, N.C.; Farrell, G.C. Hepatic Free Cholesterol Accumulates in Obese, Diabetic Mice and Causes Nonalcoholic Steatohepatitis. Gastroenterology 2011, 141, 1393–1403.e5. [Google Scholar] [CrossRef] [Green Version]
- Ioannou, G.N.; Haigh, W.G.; Thorning, D.; Savard, C. Hepatic cholesterol crystals and crown-like structures distinguish NASH from simple steatosis. J. Lipid Res. 2013, 54, 1326–1334. [Google Scholar] [CrossRef] [Green Version]
- Abreu, I.C.M.E.D.; Guerra, J.F.D.C.; Pereira, R.R.; Silva, M.; Lima, W.G.D.; Silva, M.E.; Pedrosa, M.L. Hypercholesterolemic diet induces hepatic steatosis and alterations in mRNA expression of NADPH oxidase in rat livers. Arq. Bras. Endocrinol. Metabol. 2014, 58, 251–259. [Google Scholar] [CrossRef] [Green Version]
- Hager, L.; Li, L.; Pun, H.; Liu, L.; Hossain, M.A.; Maguire, G.F.; Naples, M.; Baker, C.; Magomedova, L.; Tam, J.; et al. Lecithin: Cholesterol acyltransferase deficiency protects against cholesterol-induced hepatic endoplasmic reticulum stress in mice. J. Biol. Chem. 2012, 87, 20755–20768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Röhrl, C.; Eigner, K.; Winter, K.; Korbelius, M.; Obrowsky, S.; Kratky, D.; Kovacs, W.; Stangl, H. Endoplasmic reticulum stress impairs cholesterol efflux and synthesis in hepatic cells. J. Lipid Res. 2014, 55, 94–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Liu, Z.; Guo, J.; Chen, J.; Yang, P.; Tian, J.; Sun, J.; Zong, Y.; Qu, S. Cholesterol overloading leads to hepatic L02 cell damage through activation of the unfolded protein response. Int. J. Mol. Med. 2009, 24, 459–464. [Google Scholar]
- Zhu, C.; Xie, P.; Zhao, F.; Zhang, L.; An, W.; Zhan, Y. Mechanism of the promotion of steatotic HepG2 cell apoptosis by cholesterol. Int. J. Clin. Exp. Pathol. 2014, 7, 6807–6813. [Google Scholar] [PubMed]
- Dixon, L.J.; Barnes, M.; Tang, H.; Pritchard, M.T.; Nagy, L.E. Kupffer Cells in the Liver. Compr. Physiol. 2013, 3, 785–797. [Google Scholar]
- Hendrikx, T.; Walenbergh, S.M.A.; Hofker, M.H.; Shiri-Sverdlov, R. Lysosomal cholesterol accumulation: Driver on the road to inflammation during atherosclerosis and non-alcoholic steatohepatitis. Obes. Rev. 2014, 15, 424–433. [Google Scholar] [CrossRef]
- Wenfeng, Z.; Yakun, W.; Di, M.; Jianping, G.; Chuanxin, W.; Chun, H. Kupffer cells: Increasingly significant role in nonalcoholic fatty liver disease. Ann. Hepatol. 2014, 13, 489–495. [Google Scholar] [CrossRef]
- Funke, A.; Schreurs, M.; Aparicio-Vergara, M.; Sheedfar, F.; Gruben, N.; Kloosterhuis, N.J.; Shiri-Sverdlov, R.; Groen, A.K.; Van De Sluis, B.; Hofker, M.H.; et al. Cholesterol-induced hepatic inflammation does not contribute to the development of insulin resistance in male LDL receptor knockout mice. Atherosclerosis 2014, 232, 390–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamps, J.A.A.M.; Kruijt, J.K.; Kuiper, J.; Van Berkel, T.J.C. Uptake and degradation of human low-density lipoprotein by human liver parenchymal and Kupffer cells in culture. Biochem. J. 1991, 276, 135–140. [Google Scholar] [CrossRef] [Green Version]
- Nenseter, M.S.; Gudmundsen, O.; Roos, N.; Maelandsmo, G.; Drevon, C.; Berg, T. Role of liver endothelial and Kupffer cells in clearing low density lipoprotein from blood in hypercholesterolemic rabbits. J. Lipid Res. 1992, 33, 867–877. [Google Scholar] [CrossRef]
- Levitan, I.; Volkov, S.; Subbaiah, P.V. Oxidized LDL: Diversity, Patterns of Recognition, and Pathophysiology. Antioxid. Redox Signal. 2010, 13, 39–75. [Google Scholar] [CrossRef] [Green Version]
- Crucet, M.; Wüst, S.J.A.; Spielmann, P.; Lüscher, T.F.; Wenger, R.H.; Matter, C.M. Hypoxia enhances lipid uptake in macro-phages: Role of the scavenger receptors Lox1, SRA, and CD36. Atherosclerosis 2013, 229, 110–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bieghs, V.; van Gorp, P.J.; Walenbergh, S.M.; Gijbels, M.J.; Verheyen, F.; Buurman, W.A.; Briles, D.E.; Hofker, M.H.; Binder, C.J.; Shiri-Sverdlov, R. Specific immunization strategies against oxidized low-density lipoprotein: A novel way to reduce non-alcoholic steatohepatitis in mice. Hepatology 2012, 56, 894–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, M.J.; Malik, T.H.; Ehrenstein, M.R.; Boyle, J.J.; Botto, M.; Haskard, D.O. Immunoglobulin M Is Required for Protection Against Atherosclerosis in Low-Density Lipoprotein Receptor–Deficient Mice. Circulation 2009, 120, 417–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeurissen, M.L.; Walenbergh, S.M.; Houben, T.; Gijbels, M.J.; Li, J.; Hendrikx, T.; Oligschlaeger, Y.; van Gorp, P.J.; Binder, C.J.; Donners, M.M.; et al. Prevention of oxLDL uptake leads to decreased atherosclerosis in hematopoietic NPC1-deficient Ldlr−/− mice. Atherosclerosis 2016, 255, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, Y.-T.; Wang, N.; Tan, H.Y.; Li, S.; Feng, Y. Targeting Hepatic Stellate Cells for the Treatment of Liver Fibrosis by Natural Products: Is It the Dawning of a New Era? Front. Pharmacol. 2020, 11, 548. [Google Scholar] [CrossRef]
- Li, J.; Zhao, Y.-R.; Tian, Z. Roles of hepatic stellate cells in acute liver failure: From the perspective of inflammation and fibrosis. World J. Hepatol. 2019, 11, 412–420. [Google Scholar] [CrossRef]
- Ho, C.-M.; Ho, S.-L.; Jeng, Y.-M.; Lai, Y.-S.; Chen, Y.-H.; Lu, S.-C.; Chen, H.-L.; Chang, P.-Y.; Hu, R.-H.; Lee, P.-H. Accumulation of free cholesterol and oxidized low-density lipoprotein is associated with portal inflammation and fibrosis in nonalcoholic fatty liver disease. J. Inflamm. 2019, 16, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Ozturk, O.; Colak, Y.; Senates, E.; Yilmaz, Y.; Ulasoglu, C.; Doganay, L.; Ozkanli, S.; Oltulu, Y.M.; Coskunpinar, E.; Tuncer, I. Increased serum soluble lectin-like oxidized low-density lipoprotein receptor-1 levels in patients with biopsy-proven non-alcoholic fatty liver disease. World J. Gastroenterol. 2015, 21, 8096–8102. [Google Scholar] [CrossRef]
- Musso, G.; Cassader, M.; De Michieli, F.; Saba, F.; Bo, S.; Gambino, R. Effect of lectin-like oxidized LDL receptor-1 polymorphism on liver disease, glucose homeostasis, and postprandial lipoprotein metabolism in nonalcoholic steatohepatitis. Am. J. Clin. Nutr. 2011, 94, 1033–1042. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Liu, J.; Liu, J.; Huang, W.; Tian, L.; Quan, J.; Wang, Y.; Niu, R. oxLDL induces injury and defenestration of human liver sinusoidal endothelial cells via LOX1. J. Mol. Endocrinol. 2014, 53, 281–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, Q.; Chen, A. Curcumin eliminates oxidized LDL roles in activating hepatic stellate cells by suppressing gene expression of lectin-like oxidized LDL receptor-1. Lab. Invest. 2009, 89, 1275–1290. [Google Scholar] [CrossRef] [Green Version]
- Tomita, K.; Teratani, T.; Suzuki, T.; Shimizu, M.; Sato, H.; Narimatsu, K.; Okada, Y.; Kurihara, C.; Irie, R.; Yokoyama, H.; et al. Free cholesterol accumulation in hepatic stellate cells: Mechanism of liver fibrosis aggravation in nonalcoholic steatohepatitis in mice. Hepatology 2014, 59, 154–169. [Google Scholar] [CrossRef] [PubMed]
- Teratani, T.; Tomita, K.; Suzuki, T.; Oshikawa, T.; Yokoyama, H.; Shimamura, K.; Tominaga, S.; Hiroi, S.; Irie, R.; Okada, Y.; et al. A High-Cholesterol Diet Exacerbates Liver Fibrosis in Mice via Accumulation of Free Cholesterol in Hepatic Stellate Cells. Gastroenterology 2012, 142, 152–164.e10. [Google Scholar] [CrossRef]
- Tomita, K.; Teratani, T.; Suzuki, T.; Shimizu, M.; Sato, H.; Narimatsu, K.; Usui, S.; Furuhashi, H.; Kimura, A.; Nishiyama, K.; et al. Acyl-CoA: Cholesterol acyltransferase 1 mediates liver fibrosis by regulating free cholesterol accumulation in hepatic stellate cells. J. Hepatol. 2014, 61, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Dewidar, B.; Meyer, C.; Dooley, S.; Meindl-Beinker, A.N. TGF-β in Hepatic Stellate Cell Activation and Liver Fibrogenesis—Updated 2019. Cells 2019, 8, 1419. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.M.; Seki, E. TNFα in liver fibrosis. Curr. Pathobiol. Rep. 2015, 3, 253–261. [Google Scholar] [CrossRef] [Green Version]
- Kiagiadaki, F.; Kampa, M.; Voumvouraki, A.; Castanas, E.; Kouroumalis, E.; Notas, G. Activin-A causes Hepatic stellate cell activation via the induction of TNFα and TGFβ in Kupffer cells. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 891–899. [Google Scholar] [CrossRef]
- Williams, M.J.; Clouston, A.; Forbes, S.J. Links between Hepatic Fibrosis, Ductular Reaction, and Progenitor Cell Expansion. Gastroenterology 2014, 146, 349–356. [Google Scholar] [CrossRef]
- Clouston, A.D.; Powell, E.; Walsh, M.J.; Richardson, M.M.; Demetris, A.J.; Jonsson, J.R. Fibrosis correlates with a ductular reaction in hepatitis C: Roles of impaired replication, progenitor cells and steatosis. Hepatology 2005, 41, 809–818. [Google Scholar] [CrossRef]
- Rókusz, A.; Veres, D.; Szücs, A.; Bugyik, E.; Mózes, M.; Paku, S.; Nagy, P.; Dezső, K. Ductular reaction correlates with fibrogenesis but does not contribute to liver regeneration in experimental fibrosis models. PLoS ONE 2017, 12, e0176518. [Google Scholar] [CrossRef]
- Hammoutene, A.; Rautou, P.-E. Role of liver sinusoidal endothelial cells in non-alcoholic fatty liver disease. J. Hepatol. 2019, 70, 1278–1291. [Google Scholar] [CrossRef] [Green Version]
- Poisson, J.; Lemoinne, S.; Boulanger, C.M.; Durand, F.; Moreau, R.; Valla, D.; Rautou, P.-E. Liver sinusoidal endothelial cells: Physiology and role in liver diseases. J. Hepatol. 2017, 66, 212–227. [Google Scholar] [CrossRef] [Green Version]
- Teratani, T.; Tomita, K.; Suzuki, T.; Furuhashi, H.; Irie, R.; Hida, S.; Okada, Y.; Kurihara, C.; Ebinuma, H.; Nakamoto, N.; et al. Free cholesterol accumulation in liver sinusoidal endothelial cells exacerbates acetaminophen hepatotoxicity via TLR9 signaling. J. Hepatol. 2017, 67, 780–790. [Google Scholar] [CrossRef]
- Wei, Y.; Wang, D.; Topczewski, F.; Pagliassotti, M.J. Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am. J. Physiol. Metab. 2006, 291, E275–E281. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Kaufman, R.J. The role of ER stress in lipid metabolism and lipotoxicity. J. Lipid Res. 2016, 57, 1329–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhanasekaran, D.N.; Reddy, E.P. JNK signaling in apoptosis. Oncogene 2008, 27, 6245–6251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, M.; Strudwick, N.; Suwara, M.; Sutcliffe, L.K.; Mihai, A.D.; Ali, A.A.; Watson, J.N.; Schröder, M. An initial phase of JNK activation inhibits cell death early in the endoplasmic reticulum stress response. J. Cell Sci. 2016, 129, 2317–2328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schattenberg, J.M.; Singh, R.; Wang, Y.; Lefkowitch, J.H.; Rigoli, R.M.; Scherer, P.E.; Czaja, M.J. Jnk1 but not jnk2 promotes the development of steatohepatitis in mice. Hepatology 2006, 43, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Nemcová-Fürstová, V.; Balu Íková, K.; Rámek, J.; James, R.F.; Kovár, J. Caspase-2 and JNK Activated by Saturated Fatty Acids are Not Involved in Apoptosis Induction but Modulate ER Stress in Human Pancreatic β-cells. Cell. Physiol. Biochem. 2013, 31, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Yang, L.; McCall, S.; Huang, J.; Yu, X.X.; Pandey, S.K.; Bhanot, S.; Monia, B.P.; Li, Y.-X.; Diehl, A.M. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 2007, 45, 1366–1374. [Google Scholar] [CrossRef]
- Liu, J.; Han, L.; Zhu, L.; Yu, Y. Free fatty acids, not triglycerides, are associated with non-alcoholic liver injury progression in high fat diet induced obese rats. Lipids Health Dis. 2016, 15, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mei, S.; Ni, H.-M.; Manley, S.; Bockus, A.; Kassel, K.M.; Luyendyk, J.P.; Copple, B.L.; Ding, W.-X. Differential Roles of Unsaturated and Saturated Fatty Acids on Autophagy and Apoptosis in Hepatocytes. J. Pharmacol. Exp. Ther. 2011, 339, 487–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.-Y.; Li, X.-F.; Meng, X.-M.; Huang, C.; Zhang, L.; Li, J. Macrophage Phenotype in Liver Injury and Repair. Scand. J. Immunol. 2016, 85, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Liu, J.; Xu, Y.; Cao, H. Role of macrophages in experimental liver injury and repair in mice. Exp. Ther. Med. 2019, 17, 3835–3847. [Google Scholar] [CrossRef] [Green Version]
- Braga, T.T.; Agudelo, J.S.H.; Camara, N. Macrophages During the Fibrotic Process: M2 as Friend and Foe. Front. Immunol. 2015, 6, 602. [Google Scholar] [CrossRef] [Green Version]
- Luo, W.; Xu, Q.; Wang, Q.; Wu, H.; Hua, J. Effect of modulation of PPAR-γ activity on Kupffer cells M1/M2 polarization in the development of non-alcoholic fatty liver disease. Sci. Rep. 2017, 7, srep44612. [Google Scholar] [CrossRef] [Green Version]
- Leroux, A.; Ferrere, G.; Godie, V.; Cailleux, F.; Renoud, M.-L.; Gaudin, F.; Naveau, S.; Prévot, S.; Makhzami, S.; Perlemuter, G.; et al. Toxic lipids stored by Kupffer cells correlates with their pro-inflammatory phenotype at an early stage of steatohepatitis. J. Hepatol. 2012, 57, 141–149. [Google Scholar] [CrossRef]
- Wu, H.M.; Ni, X.X.; Xu, Q.Y.; Wang, Q.; Li, X.Y.; Hua, J. Regulation of lipid-induced macrophage polarization through modulating peroxisome proliferator-activated receptor-gamma activity affects hepatic lipid metabolism via a Toll-like receptor 4/NF-κB signaling pathway. J. Gastroenterol. Hepatol. 2020, 35, 1998–2008. [Google Scholar] [CrossRef]
- Villanueva, C.J.; Tontonoz, P. Licensing PPARγ to work in macrophages. Immunity 2010, 33, 647–649. [Google Scholar] [CrossRef] [Green Version]
- Chawla, A. Control of Macrophage Activation and Function by PPARs. Circ. Res. 2010, 106, 1559–1569. [Google Scholar] [CrossRef]
- Wan, J.; Benkdane, M.; Teixeira-Clerc, F.; Bonnafous, S.; Louvet, A.; Lafdil, F.; Pecker, F.; Tran, A.; Gual, P.; Mallat, A.; et al. M2 Kupffer cells promote M1 Kupffer cell apoptosis: A protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology 2014, 59, 130–142. [Google Scholar] [CrossRef]
- Liu, B.; Qu, L.; Yan, S. Cancer Cell Int Cyclooxygenase-2 promotes tumor growth and suppresses tumor immunity. Cancer Cell Int. 2015, 15, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inaba, T.; Sano, H.; Kawahito, Y.; Hla, T.; Akita, K.; Toda, M.; Yamashina, I.; Inoue, M.; Nakada, H. Induction of cyclooxy-genase-2 in monocyte/macrophage by mucins secreted from colon cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 2736–2741. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Sohn, K.H.; Rhee, S.H.; Hwang, D. Saturated Fatty Acids, but Not Unsaturated Fatty Acids, Induce the Expression of Cyclooxygenase-2 Mediated through Toll-like Receptor 4. J. Biol. Chem. 2001, 276, 16683–16689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Xuefeng, Y.; Shandong, W.; Jianhua, X. COX-2 in liver fibrosis. Clin. Chim. Acta 2020, 506, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Wobser, H.; Dorn, C.; Weiss, T.; Amann, T.; Bollheimer, C.; Büttner, R.; Schölmerich, J.; Hellerbrand, C. Lipid accumulation in hepatocytes induces fibrogenic activation of hepatic stellate cells. Cell Res. 2009, 19, 996–1005. [Google Scholar] [CrossRef] [Green Version]
- Al Fadel, F.; Fayyaz, S.; Japtok, L.; Kleuser, B. Involvement of Sphingosine 1-Phosphate in Palmitate-Induced Non-Alcoholic Fatty Liver Disease. Cell. Physiol. Biochem. 2016, 40, 1637–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- She, L.; Xu, D.; Wang, Z.; Zhang, Y.; Wei, Q.; Aa, J.; Wang, G.; Liu, B.; Xie, Y. Curcumin inhibits hepatic stellate cell activation via suppression of succinate-associated HIF-1α induction. Mol. Cell. Endocrinol. 2018, 476, 129–138. [Google Scholar] [CrossRef]
- Hu, S.; Bae, M.; Park, Y.-K.; Lee, J.-Y. n-3 PUFAs inhibit TGFβ1-induced profibrogenic gene expression by ameliorating the repression of PPARγ in hepatic stellate cells. J. Nutr. Biochem. 2020, 85, 108452. [Google Scholar] [CrossRef]
- Natarajan, S.K.; Ingham, S.A.; Mohr, A.M.; Wehrkamp, C.J.; Ray, A.; Roy, S.; Cazanave, S.C.; Phillippi, M.A.; Mott, J.L. Saturated free fatty acids induce cholangiocyte lipoapoptosis. Hepatology 2014, 60, 1942–1956. [Google Scholar] [CrossRef] [Green Version]
- Natarajan, S.K.; Stringham, B.A.; Mohr, A.M.; Wehrkamp, C.J.; Lu, S.; Phillippi, M.A.; Harrison-Findik, D.; Mott, J.L. FoxO3 increases miR-34a to cause palmitate-induced cholangiocyte lipoapoptosis. J. Lipid Res. 2017, 58, 866–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pizarro, M.; Balasubramaniyan, N.; Solís, N.; Solar, A.; Duarte, I.; Miquel, J.F.; Suchy, F.J.; Trauner, M.; Accatino, L.; Ananthanarayanan, M.; et al. Bile secretory function in the obese Zucker rat: Evidence of cholestasis and altered canalicular transport function. Gut 2004, 53, 1837–1843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wehr, A.; Baeck, C.; Heymann, F.; Niemietz, P.M.; Hammerich, L.; Martin, C.; Zimmermann, H.W.; Pack, O.; Gassler, N.; Hittatiya, K.; et al. Chemokine Receptor CXCR6-Dependent Hepatic NK T Cell Accumulation Promotes Inflammation and Liver Fibrosis. J. Immunol. 2013, 190, 5226–5236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novobrantseva, T.I.; Majeau, G.R.; Amatucci, A.; Kogan, S.; Brenner, I.; Casola, S.; Shlomchik, M.J.; Koteliansky, V.; Hochman, P.S.; Ibraghimov, A. Attenuated liver fibrosis in the absence of B cells. J. Clin. Investig. 2005, 115, 3072–3082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neubauer, K.; Krüger, M.; Quondamatteo, F.; Knittel, T.; Saile, B.; Ramadori, G. Transforming growth factor-beta1 stimulates the synthesis of basement membrane proteins laminin, collagen type IV and entactin in rat liver sinusoidal endothelial cells. J. Hepatol. 1999, 31, 692–702. [Google Scholar] [CrossRef]
- Mozaffarian, D.; Katan, M.B.; Ascherio, A.; Stampfer, M.J.; Willett, W.C. Trans Fatty Acids and Cardiovascular Disease. N. Engl. J. Med. 2006, 354, 1601–1613. [Google Scholar] [CrossRef] [Green Version]
- Micha, R.; Mozaffarian, D. Trans fatty acids: Effects on metabolic syndrome, heart disease and diabetes. Nat. Rev. Endocrinol. 2009, 5, 335–344. [Google Scholar] [CrossRef]
- Makino, Y.; Hikita, H.; Kodama, T.; Shigekawa, M.; Yamada, R.; Sakamori, R.; Eguchi, H.; Morii, E.; Yokoi, H.; Mukoyama, M.; et al. CTGF Mediates Tumor-Stroma Interactions between Hepatoma Cells and Hepatic Stellate Cells to Accelerate HCC Progression. Cancer Res. 2018, 78, 4902–4914. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Allen, A.M.; Wang, Z.; Prokop, L.J.; Murad, M.H.; Loomba, R. Fibrosis Progression in Nonalcoholic Fatty Liver vs Nonalcoholic Steatohepatitis: A Systematic Review and Meta-analysis of Paired-Biopsy Studies. Clin. Gastroenterol. Hepatol. 2015, 13, 643–654.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, S.E.; Poolman, T.; Arvaniti, A.; Cox, R.D.; Gathercole, L.L.; Tomlinson, J.W. The American lifestyle-induced obesity syndrome diet in male and female rodents recapitulates the clinical and transcriptomic features of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Am. J. Physiol. Liver Physiol. 2020, 319, G345–G360. [Google Scholar] [CrossRef]
- Kohli, R.; Kirby, M.; Xanthakos, S.A.; Softic, S.; Feldstein, A.E.; Saxena, V.; Tang, P.H.; Miles, L.; Miles, M.V.; Balistreri, W.F.; et al. High-fructose, medium chain trans fat diet induces liver fibrosis and elevates plasma coenzyme Q9 in a novel murine model of obesity and nonalcoholic steatohepatitis. Hepatology 2010, 52, 934–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeyapal, S.; Putcha, U.K.; Mullapudi, V.S.; Ghosh, S.; Sakamuri, A.; Kona, S.R.; Vadakattu, S.S.; Madakasira, C.; Ibrahim, A. Chronic consumption of fructose in combination with trans fatty acids but not with saturated fatty acids induces nonalcoholic steatohepatitis with fibrosis in rats. Eur. J. Nutr. 2017, 57, 2171–2187. [Google Scholar] [CrossRef]
- Biswas, S.K. Does the Interdependence between Oxidative Stress and Inflammation Explain the Antioxidant Paradox? Oxid. Med. Cell. Longev. 2016, 2016, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urtasun, R.; de la Rosa, L.C.; Nieto, N. Oxidative and Nitrosative Stress and Fibrogenic Response. Clin. Liver Dis. 2008, 12, 769–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syn, W.-K.; Agboola, K.M.; Swiderska, M.; Michelotti, G.; Liaskou, E.; Pang, H.; Xie, G.; Philips, G.; Chan, I.; Karaca, G.F.; et al. NKT-associated hedgehog and osteopontin drive fibrogenesis in non-alcoholic fatty liver disease. Gut 2012, 61, 1323–1329. [Google Scholar] [CrossRef]
- Wan, Y.; Wu, Y.L.; Lian, L.H.; Xie, W.X.; Li, X.; Ouyang, B.Q.; Bai, T.; Li, Q.; Yang, N.; Nan, J.X. The anti-fibrotic effect of betulinic acid is mediated through the inhibition of NF-κB nuclear protein translocation. Chem. Biol. Interact. 2012, 195, 215–223. [Google Scholar] [CrossRef]
- Saile, B.; Matthes, N.; El Armouche, H.; Neubauer, K.; Ramadori, G. The bcl, NFκB and p53/p21WAF1 systems are involved in spontaneous apoptosis and in the anti-apoptotic effect of TGF-β or TNF-α on activated hepatic stellate cells. Eur. J. Cell Biol. 2001, 80, 554–561. [Google Scholar] [CrossRef]
- Santos, J.D.; Mendonça, A.A.; Sousa, R.C.; Silva, T.G.; Bigonha, S.M.; Santos, E.C.; Gonçalves, R.V.; Novaes, R.D. Food-drug interaction: Anabolic steroids aggravate hepatic lipotoxicity and nonalcoholic fatty liver disease induced by trans fatty acids. Food Chem. Toxicol. 2018, 116, 360–368. [Google Scholar] [CrossRef]
- Dhibi, M.; Brahmi, F.; Mnari, A.; Houas, Z.; Chargui, I.; Bchir, L.; Gazzah, N.; Alsaif, M.; Hammami, M. The intake of high fat diet with different trans fatty acid levels differentially induces oxidative stress and non alcoholic fatty liver disease (NAFLD) in rats. Nutr. Metab. 2011, 8, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Tanaka, N.; Guo, R.; Lu, Y.; Nakajima, T.; Gonzalez, F.J.; Aoyama, T. PPARα protects against trans -fatty-acid-containing diet-induced steatohepatitis. J. Nutr. Biochem. 2017, 39, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Tetri, L.H.; Basaranoglu, M.; Brunt, E.M.; Yerian, L.M.; Neuschwander-Tetri, B.A. Severe NAFLD with hepatic necroinflam-matory changes in mice fed trans fats and a high-fructose corn syrup equivalent. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, G987–G995. [Google Scholar] [CrossRef] [PubMed]
- Larner, D.P.; Morgan, S.A.; Gathercole, L.L.; Doig, C.L.; Guest, P.; Weston, C.; Hazeldine, J.; Tomlinson, J.W.; Stewart, P.M.; Lavery, G.G. Male 11β-HSD1 Knockout Mice Fed Trans-Fats and Fructose Are Not Protected from Metabolic Syndrome or Nonalcoholic Fatty Liver Disease. Endocrinology 2016, 157, 3493–3504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhnt, K.; Baehr, M.; Rohrer, C.; Jahreis, G. Trans fatty acid isomers and the trans-9/trans-11 index in fat containing foods. Eur. J. Lipid Sci. Technol. 2011, 113, 1281–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginter, E.; Simko, V. New data on harmful effects of trans-fatty acids. Bratisl. Med. J. 2016, 117, 251–253. [Google Scholar] [CrossRef]
- Oteng, A.; Loregger, A.; van Weeghel, M.; Zelcer, N.; Kersten, S. Industrial Trans Fatty Acids Stimulate SREBP2-Mediated Cholesterogenesis and Promote Non-Alcoholic Fatty Liver Disease. Mol. Nutr. Food Res. 2019, 63, 1900385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krogager, T.P.; Nielsen, L.V.; Kahveci, D.; Dyrlund, T.F.; Scavenius, C.; Sanggaard, K.W.; Enghild, J.J. Hepatocytes respond differently to major dietary trans fatty acid isomers, elaidic acid and trans-vaccenic acid. Proteome Sci. 2015, 13, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, L.V.; Krogager, T.P.; Young, C.; Ferreri, C.; Chatgilialoglu, C.; Jensen, O.N.; Enghild, J.J. Effects of Elaidic Acid on Lipid Metabolism in HepG2 Cells, Investigated by an Integrated Approach of Lipidomics, Transcriptomics and Proteomics. PLoS ONE 2013, 8, e74283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, F.; Diao, P.; Wang, X.; Hu, X.; Kimura, T.; Nakamuta, M.; Nakamura, I.; Shirotori, S.; Sato, Y.; Moriya, K.; et al. Dietary Restriction Suppresses Steatosis-Associated Hepatic Tumorigenesis in Hepatitis C Virus Core Gene Transgenic Mice. Liver Cancer 2020, 9, 529–548. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jia, F.; Hu, X.; Kimura, T.; Tanaka, N. Impact of Dietary Fat on the Progression of Liver Fibrosis: Lessons from Animal and Cell Studies. Int. J. Mol. Sci. 2021, 22, 10303. https://doi.org/10.3390/ijms221910303
Jia F, Hu X, Kimura T, Tanaka N. Impact of Dietary Fat on the Progression of Liver Fibrosis: Lessons from Animal and Cell Studies. International Journal of Molecular Sciences. 2021; 22(19):10303. https://doi.org/10.3390/ijms221910303
Chicago/Turabian StyleJia, Fangping, Xiao Hu, Takefumi Kimura, and Naoki Tanaka. 2021. "Impact of Dietary Fat on the Progression of Liver Fibrosis: Lessons from Animal and Cell Studies" International Journal of Molecular Sciences 22, no. 19: 10303. https://doi.org/10.3390/ijms221910303
APA StyleJia, F., Hu, X., Kimura, T., & Tanaka, N. (2021). Impact of Dietary Fat on the Progression of Liver Fibrosis: Lessons from Animal and Cell Studies. International Journal of Molecular Sciences, 22(19), 10303. https://doi.org/10.3390/ijms221910303