
Design, Synthesis and Anticancer Profile of New 4-(1H-benzo[d]imidazol-1-yl)pyrimidin-2-amine-Linked Sulfonamide Derivatives with V600EBRAF Inhibitory Effect

 , , ,
, , ,  ,
, 
Abstract
:1. Introduction
2. Results and Discussion
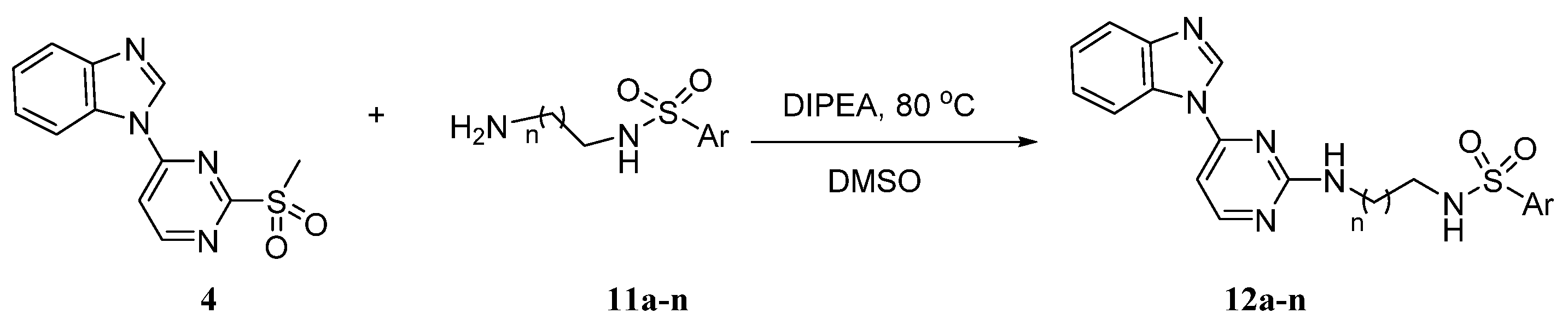
2.1. Chemistry
2.2. Biology
2.2.1. Enzyme Assay
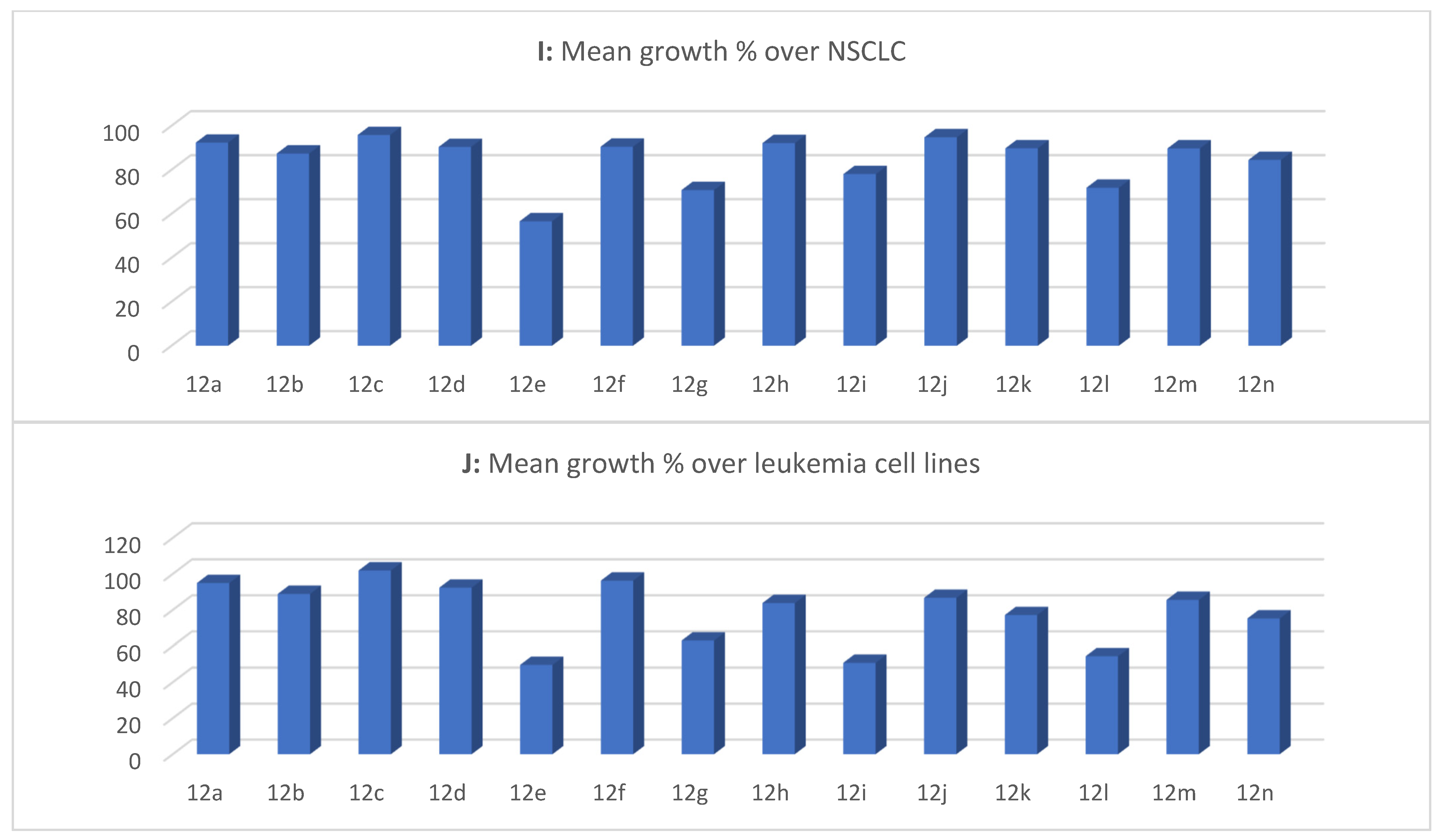
2.2.2. In Vitro Antiproliferative Activity
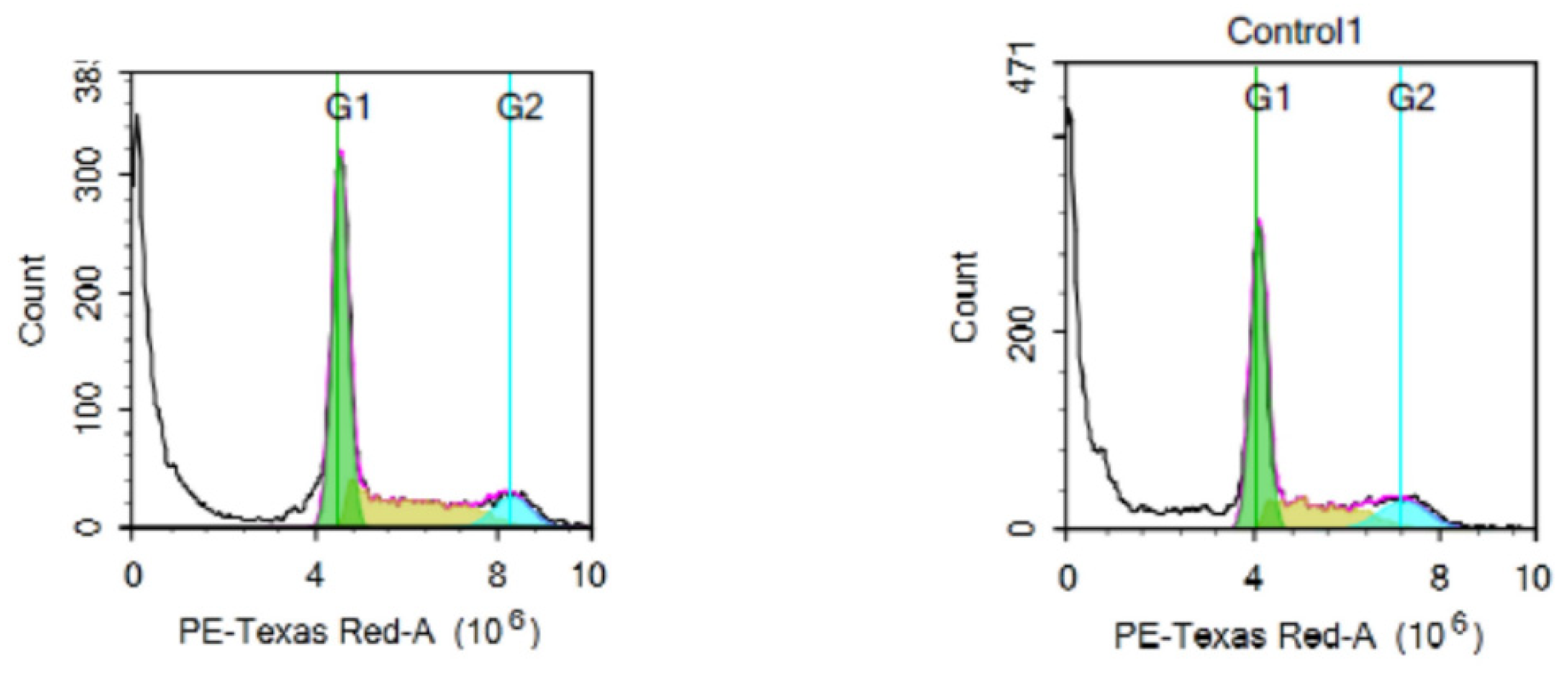
2.2.3. Cell Cycle Analysis
2.3. Molecular Docking
3. Conclusions
4. Materials and Methods
4.1. Chemistry
4.1.1. Synthesis of 1H-benzo[d]imidazole (2)
4.1.2. Synthesis of 1-(2-(methylthio)pyrimidin-4-yl)-1H-benzo[d]imidazole (3)
4.1.3. Synthesis of 1-(2-(methyl sulfonyl) pyrimidin-4-yl)-1H-benzo[d]imidazole (4)
4.1.4. Synthesis of Sustituted Ethanediamine and Propanediamine (11a–n)
4.1.5. Synthesis of Target Compounds (12a–n)
4.2. In Vitro Anticancer Activity
4.2.1. Anticancer Screening over 60 Cell Lines
4.2.2. Antiproliferative Effect over A375 and SK-MEL-5
4.3. Enzyme Assay
4.4. Cell Cycle Analysis
4.5. Molecular Docking
4.6. Molecular Dynamics Simulations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Trivedi, P.; Adhikari, N.; Amin, S.A.; Jha, T.; Ghosh, B. Design, synthesis and biological screening of 2-aminobenzamides as selective hdac3 inhibitors with promising anticancer effects. Eur. J. Pharm. Sci. 2018, 124, 165–181. [Google Scholar] [CrossRef]
- Hassan, R.M.; Abd-Allah, W.H.; Salman, A.M.; El-Azzouny, A.A.; Aboul-Enein, M.N. Design, synthesis and anticancer evaluation of novel 1,3-benzodioxoles and 1,4-benzodioxines. Eur. J. Pharm. Sci. 2019, 139, 105045. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.P.; Liu, Z.Y.; Xie, C.; Zhou, L.; Sun, X. Novel fluorinated docetaxel analog for anti-hepatoma: Molecular docking and biological evaluation. Eur. J. Pharm. Sci. 2016, 88, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Al-Sanea, M.M.; Gotina, L.; Mohamed, M.F.; Grace Thomas Parambi, D.; Gomaa, H.A.M.; Mathew, B.; Youssif, B.G.M.; Alharbi, K.S.; Elsayed, Z.M.; Abdelgawad, M.A.; et al. Design, synthesis and biological evaluation of new hdac1 and hdac2 inhibitors endowed with ligustrazine as a novel cap moiety. Drug Des. Devel. Ther. 2020, 14, 497–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumimoto, H.; Imabayashi, F.; Iwata, T.; Kawakami, Y. The braf-mapk signaling pathway is essential for cancer-immune evasion in human melanoma cells. J. Exp. Med. 2006, 203, 1651–1656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fecher, L.A.; Amaravadi, R.K.; Flaherty, K.T. The mapk pathway in melanoma. Curr. Opin. Oncol. 2008, 20, 183–189. [Google Scholar] [CrossRef]
- Abdelazem, A.Z.; Al-Sanea, M.M.; Park, B.S.; Park, H.M.; Yoo, K.H.; Sim, T.; Park, J.B.; Lee, S.H.; Lee, S.H. Synthesis and biological evaluation of new pyrazol-4-ylpyrimidine derivatives as potential ros1 kinase inhibitors. Eur. J. Med. Chem. 2015, 90, 195–208. [Google Scholar] [CrossRef]
- Al-Sanea, M.M.; Elkamhawy, A.; Paik, S.; Lee, K.; El Kerdawy, A.M.; Syed Nasir Abbas, B.; Joo Roh, E.; Eldehna, W.M.; Elshemy, H.A.H.; Bakr, R.B.; et al. Sulfonamide-based 4-anilinoquinoline derivatives as novel dual aurora kinase (aurka/b) inhibitors: Synthesis, biological evaluation and in silico insights. Bioorg. Med. Chem. 2020, 28, 115525. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the braf gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Pollock, P.M.; Meltzer, P.S. A genome-based strategy uncovers frequent braf mutations in melanoma. Cancer Cell 2002, 2, 5–7. [Google Scholar] [CrossRef]
- Di Nicolantonio, F.; Martini, M.; Molinari, F.; Sartore Bianchi, A.; Arena, S.; Saletti, P.; De Dosso, S.; Mazzucchelli, L.; Frattini, M.; Siena, S. Wild-type braf is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J. Clin. Oncol. 2008, 26, 5705–5712. [Google Scholar] [CrossRef]
- Nikiforova, M.N.; Kimura, E.T.; Gandhi, M.; Biddinger, P.W.; Knauf, J.A.; Basolo, F.; Zhu, Z.; Giannini, R.; Salvatore, G.; Fusco, A.; et al. Braf mutations in thyroid tumors are restricted to papillary carcinomas and anaplastic or poorly differentiated carcinomas arising from papillary carcinomas. J. Clin. Endocrinol. Metab. 2003, 88, 5399–5404. [Google Scholar] [CrossRef] [PubMed]
- Kimura, E.T.; Nikiforova, M.N.; Zhu, Z.; Knauf, J.A.; Nikiforov, Y.E.; Fagin, J.A. High prevalence of braf mutations in thyroid cancer: Genetic evidence for constitutive activation of the ret/ptc-ras-braf signaling pathway in papillary thyroid carcinoma. Cancer Res. 2003, 63, 1454–1457. [Google Scholar] [PubMed]
- Trovisco, V.; de Castro, I.V.; Soares, P.; Maximo, V.; Silva, P.; Magalhaes, J.; Abrosimov, A.; Guiu, X.M.; Sobrinho-Simoes, M. Braf mutations are associated with some histological types of papillary thyroid carcinoma. J. Pathol. 2004, 202, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Hoeflich, K.P.; Herter, S.; Tien, J.; Wong, L.; Berry, L.; Chan, J.; O’Brien, C.; Modrusan, Z.; Seshagiri, S.; Lackner, M. Antitumor efficacy of the novel raf inhibitor gdc-0879 is predicted by brafv600e mutational status and sustained extracellular signal-regulated kinase/mitogen-activated protein kinase pathway suppression. Cancer Res. 2009, 69, 3042–3051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDermott, U.; Sharma, S.V.; Dowell, L.; Greninger, P.; Montagut, C.; Lamb, J.; Archibald, H.; Raudales, R.; Tam, A.; Lee, D.; et al. Identification of genotype-correlated sensitivity to selective kinase inhibitors by using high-throughput tumor cell line profiling. Proc. Natl. Acad. Sci. USA 2007, 104, 19936–19941. [Google Scholar] [CrossRef] [Green Version]
- Solit, D.B.; Garraway, L.A.; Pratilas, C.A.; Sawai, A.; Getz, G.; Basso, A.; Ye, Q.; Lobo, J.M.; She, Y.; Osman, I.; et al. Braf mutation predicts sensitivity to mek inhibition. Nature 2006, 439, 358–362. [Google Scholar] [CrossRef] [Green Version]
- Wan, P.T.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; Barford, D.; et al. Mechanism of activation of the raf-erk signaling pathway by oncogenic mutations of b-raf. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef] [Green Version]
- Wellbrock, C.; Ogilvie, L.; Hedley, D.; Karasarides, M.; Martin, J.; Niculescu-Duvaz, D.; Springer, C.J.; Marais, R. V599eb-raf is an oncogene in melanocytes. Cancer Res. 2004, 64, 2338–2342. [Google Scholar] [CrossRef] [Green Version]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with braf v600e mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bucheit, A.D.; Davies, M.A. Emerging insights into resistance to braf inhibitors in melanoma. Biochem. Pharmacol. 2014, 87, 381–389. [Google Scholar] [CrossRef]
- Al-Sanea, M.M.; Abdelazem, A.Z.; Park, B.S.; Yoo, K.H.; Sim, T.; Kwon, Y.J.; Lee, S.H. Ros1 kinase inhibitors for molecular-targeted therapies. Curr. Med. Chem. 2016, 23, 142–160. [Google Scholar] [CrossRef]
- Wilhelm, S.; Carter, C.; Lynch, M.; Lowinger, T.; Dumas, J.; Smith, R.A.; Schwartz, B.; Simantov, R.; Kelley, S. Discovery and development of sorafenib: A multikinase inhibitor for treating cancer. Nat. Rev. Drug. Discov. 2006, 5, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.M.; Adnane, L.; Newell, P.; Villanueva, A.; Llovet, J.M.; Lynch, M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both raf and vegf and pdgf receptor tyrosine kinase signaling. Mol. Cancer Ther. 2008, 7, 3129–3140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bollag, G.; Tsai, J.; Zhang, J.; Zhang, C.; Ibrahim, P.; Nolop, K.; Hirth, P. Vemurafenib: The first drug approved for braf-mutant cancer. Nat. Rev. Drug. Discov. 2012, 11, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Qu, M.; Liu, Z.; Zhao, D.; Wang, C.; Zhang, J.; Tang, Z.; Liu, K.; Shu, X.; Yuan, H.; Ma, X. Design, synthesis and biological evaluation of sulfonamide-substituted diphenylpyrimidine derivatives (sul-dppys) as potent focal adhesion kinase (fak) inhibitors with antitumor activity. Bioorg. Med. Chem. 2017, 25, 3989–3996. [Google Scholar] [CrossRef]
- Liu, H.; Qu, M.; Xu, L.; Han, X.; Wang, C.; Shu, X.; Yao, J.; Liu, K.; Peng, J.; Li, Y.; et al. Design and synthesis of sulfonamide-substituted diphenylpyrimidines (sfa-dppys) as potent bruton’s tyrosine kinase (btk) inhibitors with improved activity toward b-cell lymphoblastic leukemia. Eur. J. Med. Chem. 2017, 135, 60–69. [Google Scholar] [CrossRef]
- Gibney, G.T.; Zager, J.S. Clinical development of dabrafenib in braf mutant melanoma and other malignancies. Expert. Opin. Drug Metab. Toxicol. 2013, 9, 893–899. [Google Scholar] [CrossRef]
- Bryan, M.C.; Falsey, J.R.; Frohn, M.; Reichelt, A.; Yao, G.; Bartberger, M.D.; Bailis, J.M.; Zalameda, L.; Miguel, T.S.; Doherty, E.M.; et al. N-substituted azaindoles as potent inhibitors of cdc7 kinase. Bioorg. Med. Chem. Lett. 2013, 23, 2056–2060. [Google Scholar] [CrossRef]
- Nofal, Z.M.; Soliman, E.A.; Abd El-Karim, S.S.; El-Zahar, M.I.; Srour, A.M.; Sethumadhavan, S.; Maher, T.J. Novel benzimidazole derivatives as expected anticancer agents. Acta Pol. Pharm. 2011, 68, 519–534. [Google Scholar]
- Shaharyar, M.; Abdullah, M.M.; Bakht, M.A.; Majeed, J. Pyrazoline bearing benzimidazoles: Search for anticancer agent. Eur. J. Med. Chem. 2010, 45, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Refaat, H.M. Synthesis and anticancer activity of some novel 2-substituted benzimidazole derivatives. Eur. J. Med. Chem. 2010, 45, 2949–2956. [Google Scholar] [CrossRef] [PubMed]
- Paul, K.; Sharma, A.; Luxami, V. Synthesis and in vitro antitumor evaluation of primary amine substituted quinazoline linked benzimidazole. Bioorg. Med. Chem. Lett. 2014, 24, 624–629. [Google Scholar] [CrossRef]
- Tonelli, M.; Simone, M.; Tasso, B.; Novelli, F.; Boido, V.; Sparatore, F.; Paglietti, G.; Pricl, S.; Giliberti, G.; Blois, S.; et al. Antiviral activity of benzimidazole derivatives. Ii. Antiviral activity of 2-phenylbenzimidazole derivatives. Bioorg. Med. Chem. 2010, 18, 2937–2953. [Google Scholar] [CrossRef] [PubMed]
- Kharitonova, M.I.; Konstantinova, I.D.; Miroshnikov, A.I. Benzimidazole nucleosides: Antiviral and antitumour activities and methods of synthesis. Russ. Chem. Rev. 2018, 87, 1111–1138. [Google Scholar] [CrossRef]
- Li, Y.F.; Wang, G.F.; He, P.L.; Huang, W.G.; Zhu, F.H.; Gao, H.Y.; Tang, W.; Luo, Y.; Feng, C.L.; Shi, L.P.; et al. Synthesis and anti-hepatitis b virus activity of novel benzimidazole derivatives. J. Med. Chem. 2006, 49, 4790–4794. [Google Scholar] [CrossRef]
- Elnima, E.I.; Zubair, M.U.; Al-Badr, A.A. Antibacterial and antifungal activities of benzimidazole and benzoxazole derivatives. Antimicrob. Agents Chemother. 1981, 19, 29–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singla, P.; Luxami, V.; Paul, K. Benzimidazole-biologically attractive scaffold for protein kinase inhibitors. Rsc. Adv. 2014, 4, 12422–12440. [Google Scholar] [CrossRef]
- Waller, P.J. Anthelmintic resistance. Vet. Parasitol. 1997, 72, 391–405. [Google Scholar] [CrossRef]
- Martin, R.J. Modes of action of anthelmintic drugs. Vet. J. 1997, 154, 11–34. [Google Scholar] [CrossRef]
- Brown, H.; Matzuk, A.; Ilves, I.; Peterson, L.; Harris, S.; Sarett, L.; Egerton, J.; Yakstis, J.; Campbell, W.; Cuckler, A.C. Antiparasitic drugs. Iv. 2-(4′-thiazolyl)-benzimidazole, a new anthelmintic. J. Am. Chem. Soc. 1961, 83, 1764–1765. [Google Scholar] [CrossRef]
- Abdel-Maksoud, M.S.; El-Gamal, M.I.; Lee, B.S.; Gamal El-Din, M.M.; Jeon, H.R.; Kwon, D.; Ammar, U.M.; Mersal, K.I.; Ali, E.M.H.; Lee, K.T.; et al. Discovery of new imidazo[2,1-b]thiazole derivatives as potent pan-raf inhibitors with promising in vitro and in vivo anti-melanoma activity. J. Med. Chem. 2021, 64, 6877–6901. [Google Scholar]
- El-Gamal, M.I.; Khan, M.A.; Tarazi, H.; Abdel-Maksoud, M.S.; Gamal El-Din, M.M.; Yoo, K.H.; Oh, C.H. Design and synthesis of new raf kinase-inhibiting antiproliferative quinoline derivatives. Part 2: Diarylurea derivatives. Eur. J. Med. Chem. 2017, 127, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Maksoud, M.S.; El-Gamal, M.I.; El-Din, M.M.G.; Kwak, S.S.; Kim, H.I.; Oh, C.H. Broad-spectrum antiproliferative activity of a series of 6-(4-fluorophenyl)-5-(2-substituted pyrimidin-4-yl)imidazo[2,1-b]thiazole derivatives. Med. Chem. Res. 2016, 25, 824–833. [Google Scholar] [CrossRef]
- Abdel-Maksoud, M.S.; Ammar, U.M.; El-Gamal, M.I.; Gamal El-Din, M.M.; Mersal, K.I.; Ali, E.M.H.; Yoo, K.H.; Lee, K.T.; Oh, C.H. Design, synthesis, and anticancer activity of imidazo[2,1-b]oxazole-based raf kinase inhibitors. Bioorg. Chem. 2019, 93, 103349. [Google Scholar] [CrossRef] [PubMed]
- Ali, E.M.; El-Telbany, R.F.A.; Abdel-Maksoud, M.S.; Ammar, U.M.; Mersal, K.I.; Zaraei, S.-O.; El-Gamal, M.I.; Choi, S.-I.; Lee, K.-T.; Kim, H.-K. Design, synthesis, biological evaluation, and docking studies of novel (imidazol-5-yl) pyrimidine-based derivatives as dual brafv600e/p38α inhibitors. Eur. J. Med. Chem. 2021, 215, 113277. [Google Scholar] [PubMed]
- Abdel-Maksoud, M.S.; Ali, E.M.H.; Ammar, U.M.; Mersal, K.I.; Yoo, K.H.; Oh, C.H. Design and synthesis of novel pyrrolo[2,3-b]pyridine derivatives targeting (v600e)braf. Bioorg. Med. Chem. 2020, 28, 115493. [Google Scholar] [CrossRef]
- Eldehna, W.M.; Nocentini, A.; Elsayed, Z.M.; Al-Warhi, T.; Aljaeed, N.; Alotaibi, O.J.; Al-Sanea, M.M.; Abdel-Aziz, H.A.; Supuran, C.T. Benzofuran-based carboxylic acids as carbonic anhydrase inhibitors and antiproliferative agents against breast cancer. ACS Med. Chem. Lett. 2020, 11, 1022–1027. [Google Scholar] [CrossRef]
- Vogel, A.I.; Furniss, B.S.; Hannaford, A.J.; Smith, P.W.; Tatchell, A.R. Vogel’s Textbook of Practical Organic Chemistry; Longman Scientific & Technical: London, UK, 1989; Volume 5. [Google Scholar]
- Al-Warhi, T.; Abo-Ashour, M.F.; Almahli, H.; Alotaibi, O.J.; Al-Sanea, M.M.; Al-Ansary, G.H.; Ahmed, H.Y.; Elaasser, M.M.; Eldehna, W.M.; Abdel-Aziz, H.A. Novel [(n-alkyl-3-indolylmethylene)hydrazono]oxindoles arrest cell cycle and induce cell apoptosis by inhibiting cdk2 and bcl-2: Synthesis, biological evaluation and in silico studies. J. Enzyme Inhib. Med. Chem. 2020, 35, 1300–1309. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. Charmm-gui: A web-based graphical user interface for charmm. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Abagyan, R.; Totrov, M.; Kuznetsov, D. Icm—A new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem. 1994, 15, 488–506. [Google Scholar] [CrossRef]
- Sahakyan, H. Improving virtual screening results with mm/gbsa and mm/pbsa rescoring. J. Comput. Aided Mol. Des. 2021, 35, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Oliver, D.S.; Cunha, L.B.; Reynolds, A.C. Markov chain monte carlo methods for conditioning a permeability field to pressure data. Math. Geol. 1997, 29, 61–91. [Google Scholar] [CrossRef]
- Wu, X.W.; Brooks, B.R. Self-guided langevin dynamics simulation method. Chem. Phys. Lett. 2003, 381, 512–518. [Google Scholar] [CrossRef]
- Miller, B.R., III; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. Mmpbsa. Py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||
|---|---|---|---|---|---|---|---|
| Comp. | n | R | Yield 1 | Comp. | n | R | Yield 1 |
| 12a | 1 | H | 71% | 12h | 2 | H | 64% |
| 12b | 1 | 4-F | 74% | 12i | 2 | 4-F | 59% |
| 12c | 1 | 4-Cl | 77% | 12j | 2 | 4-Cl | 59% |
| 12d | 1 | 4-Br | 70% | 12k | 2 | 4-Br | 64% |
| 12e | 1 | 4-CF3 | 62% | 12l | 2 | 4-CF3 | 69% |
| 12f | 1 | 4-OCH3 | 81% | 12m | 2 | 4-OCH3 | 70% |
| 12g | 1 | 4-CH3 | 52% | 12n | 2 | Fused benzene | 72% |
| Comp. | % Inhibition at 1 µM 1 | Comp. | % Inhibition at 1 µM 1 |
|---|---|---|---|
| 12a | 30.35 ± 0.41 | 12h | 49.54 ± 0.61 |
| 12b | 75.17 ± 0.27 | 12i | 97.81 ± 0.82 |
| 12c | 70.55 ± 0.71 | 12j | 88.14 ± 0.19 |
| 12d | 69.28 ± 0.51 | 12k | 73.81 ± 0.75 |
| 12e | 98.24 ± 0.44 | 12l | 98.75 ± 0.58 |
| 12f | 55.21 ± 0.52 | 12m | 51.26 ± 0.29 |
| 12g | 82.11 ± 0.32 | 12n | 41.27 ± 0.46 |
| Sorafenib | 84.57 ± 0.77 |
| Comp. | BRAF (Wild Type) | V600EBRAF | CRAF |
|---|---|---|---|
| 12a | 3.250 ± 0.450 | 1.910 ± 0.140 | 2.980 ± 0.210 |
| 12b | 1.510 ± 0.220 | 0.740 ± 0.051 | 1.340 ± 0.190 |
| 12c | 1.380 ± 0.170 | 0.820 ± 0.024 | 1.490 ± 0.230 |
| 12d | 1.790 ± 0.150 | 0.910 ± 0.019 | 2.10 ± 0.180 |
| 12e | 1.250 ± 0.095 | 0.620 ± 0.031 | 1.140 ± 0.088 |
| 12f | 1.980 ± 0.120 | 0.980 ± 0.020 | 1.670 ± 0.210 |
| 12g | 1.530 ± 0.260 | 0.790 ± 0.042 | 1.480 ± 0.099 |
| 12h | 2.320 ± 0.110 | 1.120 ± 0.098 | 2.140 ± 0.078 |
| 12i | 1.330 ± 0.081 | 0.530 ± 0.022 | 0.980 ± 0.044 |
| 12j | 1.130 ± 0.099 | 0.790 ± 0.051 | 1.020 ± 0.077 |
| 12k | 1.530 ± 0.110 | 0.810 ± 0.056 | 0.990 ± 0.085 |
| 12l | 0.940 ± 0.065 | 0.490 ± 0.061 | 0.840 ± 0.076 |
| 12m | 1.460 ± 0.086 | 0.820 ± 0.048 | 0.960 ± 0.079 |
| 12n | 3.540 ± 0.175 | 1.280 ± 0.144 | 1.780 ± 0.156 |
| Sorafenib | ND | 0.814 ± 0.071 | 0.910 ± 0.094 |
| Comp. | SK-MEL-5 | A375 |
|---|---|---|
| 12a | 20.45 ± 0.86 | 12.78 ± 0.41 |
| 12b | 15.63 ± 0.37 | 10.54 ± 0.22 |
| 12c | 17.58 ±0.49 | 10.95 ± 0.36 |
| 12d | 19.67 ± 0.51 | 11.78 ± 0.71 |
| 12e | 6.54 ± 0.18 | 2.89 ± 0.14 |
| 12f | 18.74 ± 0.69 | 12.59 ± 0.44 |
| 12g | 8.25 ± 0.12 | 7.22 ± 0.23 |
| 12h | 11.28 ± 0.29 | 9.26 ± 0.16 |
| 12i | 2.36 ± 0.21 | 1.95 ± 0.13 |
| 12j | 12.88 ± 0.72 | 10.39 ± 0.24 |
| 12k | 13.46 ± 0.39 | 11.98 ± 0.47 |
| 12l | 2.02 ± 0.09 | 1.85 ± 0.11 |
| 12m | 14.56 ± 0.71 | 13.13 ± 0.27 |
| 12n | 17.85 ± 0.62 | 16.87 ± 0.81 |
| Sorafenib | 9.22 ± 0.81 | 5.25 ± 0.74 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdel-Maksoud, M.S.; Mohamed, A.A.B.; Hassan, R.M.; Abdelgawad, M.A.; Chilingaryan, G.; Selim, S.; Abdel-Bakky, M.S.; Al-Sanea, M.M. Design, Synthesis and Anticancer Profile of New 4-(1H-benzo[d]imidazol-1-yl)pyrimidin-2-amine-Linked Sulfonamide Derivatives with V600EBRAF Inhibitory Effect. Int. J. Mol. Sci. 2021, 22, 10491. https://doi.org/10.3390/ijms221910491
Abdel-Maksoud MS, Mohamed AAB, Hassan RM, Abdelgawad MA, Chilingaryan G, Selim S, Abdel-Bakky MS, Al-Sanea MM. Design, Synthesis and Anticancer Profile of New 4-(1H-benzo[d]imidazol-1-yl)pyrimidin-2-amine-Linked Sulfonamide Derivatives with V600EBRAF Inhibitory Effect. International Journal of Molecular Sciences. 2021; 22(19):10491. https://doi.org/10.3390/ijms221910491
Chicago/Turabian StyleAbdel-Maksoud, Mohammed S., Ahmed A. B. Mohamed, Rasha M. Hassan, Mohamed A. Abdelgawad, Garri Chilingaryan, Samy Selim, Mohamed S. Abdel-Bakky, and Mohammad M. Al-Sanea. 2021. "Design, Synthesis and Anticancer Profile of New 4-(1H-benzo[d]imidazol-1-yl)pyrimidin-2-amine-Linked Sulfonamide Derivatives with V600EBRAF Inhibitory Effect" International Journal of Molecular Sciences 22, no. 19: 10491. https://doi.org/10.3390/ijms221910491
APA StyleAbdel-Maksoud, M. S., Mohamed, A. A. B., Hassan, R. M., Abdelgawad, M. A., Chilingaryan, G., Selim, S., Abdel-Bakky, M. S., & Al-Sanea, M. M. (2021). Design, Synthesis and Anticancer Profile of New 4-(1H-benzo[d]imidazol-1-yl)pyrimidin-2-amine-Linked Sulfonamide Derivatives with V600EBRAF Inhibitory Effect. International Journal of Molecular Sciences, 22(19), 10491. https://doi.org/10.3390/ijms221910491