
The Role of PPAR Alpha in the Modulation of Innate Immunity




Abstract
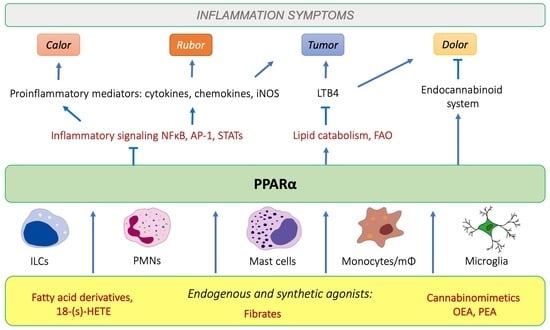
:
1. Introduction
2. The New Perspective on Innate Immunity
3. The Main Populations of Innate Immune Cells
4. Peroxisome Proliferator-Activated Receptor alpha (PPARα) and Its Role in Inflammation
4.1. PPARα as a Nuclear Receptor Present in Peripheral Tissues and Immunocompetent Cells
4.2. PPARα-Mediated Transrepression of Main Inflammatory Transcription Factors
4.3. PPARα and Inflammatory Lipid Mediators
4.4. PPARα Crosstalk with Pattern Recognition Receptors
4.5. PPARα and the Regulation of Inflammasomes
5. PPARα’s Role in the Innate Immunity Effector Processes: ROS/RNS Production
6. PPARα as an Immunomodulator during Infections
7. Interplay between PPARα and the Endocannabinoid System: Implications for Inflamma-Tion, Neuroprotection, and Analgesia
7.1. Analgesic Lipid Mediators as PPARα Agonists
7.2. PPARα Involvement in Resolution of Neuroinflammation
7.3. PPARα-Mediated Regulation of Microglia and Macrophage Functions
7.4. PPARα’s Role in Restoration of Neural Function after Injury or Infection
7.5. PPARα and Endocannabinoid Involvement in the Regulation of Mast-Cell Functions
8. Evolutionary Aspects of PPARα-Mediated Immunomodulation
9. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Boraschi, D.; Italiani, P. Innate immune memory: Time for adopting a correct terminology. Front. Immunol. 2018, 9, 799. [Google Scholar] [CrossRef]
- Netea, M.G.; Quintin, J.; van der Meer, J.W. Trained immunity: A memory for innate host defense. Cell Host Microbe 2011, 9, 355–361. [Google Scholar] [CrossRef] [Green Version]
- Moret, Y.; Siva-Jothy, M.T. Adaptive innate immunity? Responsive-mode prophylaxis in the mealworm beetle, Tenebrio molitor. Proc. Biol. Sci. 2003, 270, 2475–2480. [Google Scholar] [CrossRef] [Green Version]
- Torre, C.; Abnave, P.; Tsoumtsa, L.L.; Mottola, G.; Lepolard, C.; Trouplin, V.; Gimenez, G.; Desrousseaux, J.; Gempp, S.; Levasseur, A.; et al. Staphylococcus aureus promotes Smed-PGRP-2/Smed-setd8-1 methyltransferase signalling in planarian neoblasts to sensitize anti-bacterial gene responses during re-infection. EBioMedicine 2017, 20, 150–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Qiu, L.; Sun, Z.; Wang, L.; Zhou, Z.; Liu, R.; Yue, F.; Sun, R.; Song, L. The specifically enhanced cellular immune responses in Pacific oyster (Crassostrea gigas) against secondary challenge with Vibrio splendidus. Dev. Comp. Immunol. 2014, 45, 141–150. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, Q.; Christensen, B.M. Identification and characterization of the fibrinogen-like domain of fibrinogen-related proteins in the mosquito, Anopheles gambiae, and the fruitfly, Drosophila melanogaster, genomes. BMC Genom. 2005, 6, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melillo, D.; Marino, R.; Italiani, P.; Boraschi, D. Innate immune memory in invertebrate metazoans: A Critical appraisal. Front. Immunol. 2018, 9, 1915. [Google Scholar] [CrossRef]
- Cerenius, L.; Soderhall, K. Immune properties of invertebrate phenoloxidases. Dev. Comp. Immunol. 2021, 122, 104098. [Google Scholar] [CrossRef]
- Coates, C.J.; Nairn, J. Diverse immune functions of hemocyanins. Dev. Comp. Immunol. 2014, 45, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Han, Z.; Oppenheim, J.J. Alarmins and immunity. Immunol. Rev. 2017, 280, 41–56. [Google Scholar] [CrossRef]
- Amarante-Mendes, G.P.; Adjemian, S.; Branco, L.M.; Zanetti, L.C.; Weinlich, R.; Bortoluci, K.R. Pattern Recognition Receptors and the Host Cell Death Molecular Machinery. Front. Immunol. 2018, 9, 2379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olive, C. Pattern recognition receptors: Sentinels in innate immunity and targets of new vaccine adjuvants. Expert Rev. Vaccines 2012, 11, 237–256. [Google Scholar] [CrossRef]
- Rabinovitch, M. Professional and non-professional phagocytes: An introduction. Trends Cell Biol. 1995, 5, 85–87. [Google Scholar] [CrossRef]
- Lim, J.J.; Grinstein, S.; Roth, Z. Diversity and versatility of phagocytosis: Roles in innate immunity, tissue remodeling, and homeostasis. Front. Cell Infect. Microbiol. 2017, 7, 191. [Google Scholar] [CrossRef] [Green Version]
- Witko-Sarsat, V.; Descamps-Latscha, B. Neutrophil-derived Oxidants and proteinases as immunomodulatory mediators in inflammation. Mediat. Inflamm. 1994, 3, 257–273. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, G.T.; Green, E.R.; Mecsas, J. Neutrophils to the ROScue: Mechanisms of NADPH oxidase activation and bacterial resistance. Front. Cell Infect. Microbiol. 2017, 7, 373. [Google Scholar] [CrossRef] [PubMed]
- Kettle, A.J.; Winterbourn, C.C. Myeloperoxidase: A key regulator of neutrophil oxidant production. Redox Rep. 1997, 3, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Prolo, C.; Alvarez, M.N.; Radi, R. Peroxynitrite, a potent macrophage-derived oxidizing cytotoxin to combat invading pathogens. Biofactors 2014, 40, 215–225. [Google Scholar] [CrossRef] [Green Version]
- Yamasaki, R.; Lu, H.; Butovsky, O.; Ohno, N.; Rietsch, A.M.; Cialic, R.; Wu, P.M.; Doykan, C.E.; Lin, J.; Cotleur, A.C.; et al. Differential roles of microglia and monocytes in the inflamed central nervous system. J. Exp. Med. 2014, 211, 1533–1549. [Google Scholar] [CrossRef] [Green Version]
- Schulz, D.; Severin, Y.; Zanotelli, V.R.T.; Bodenmiller, B. In-Depth characterization of monocyte-derived macrophages using a mass cytometry-based phagocytosis assay. Sci. Rep. 2019, 9, 1925. [Google Scholar] [CrossRef]
- Gerner, E.W.; Meyskens, F.L., Jr. Polyamines and cancer: Old molecules, new understanding. Nat. Rev. Cancer 2004, 4, 781–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luk, G.D. Essential role of polyamine metabolism in hepatic regeneration. Inhibition of deoxyribonucleic acid and protein synthesis and tissue regeneration by difluoromethylornithine in the rat. Gastroenterology 1986, 90, 1261–1267. [Google Scholar] [CrossRef]
- Murray, P.J. Macrophage polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef]
- Locati, M.; Curtale, G.; Mantovani, A. Diversity, Mechanisms, and significance of macrophage plasticity. Annu. Rev. Pathol. 2020, 15, 123–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasteiger, G.; D’Osualdo, A.; Schubert, D.A.; Weber, A.; Bruscia, E.M.; Hartl, D. Cellular innate immunity: An old game with new players. J. Innate. Immun. 2017, 9, 111–125. [Google Scholar] [CrossRef]
- Erb, K.J.; Holloway, J.W.; Le Gros, G. Mast cells in the front line. Innate immunity. Curr. Biol. 1996, 6, 941–942. [Google Scholar] [CrossRef] [Green Version]
- Sendo, S.; Saegusa, J.; Morinobu, A. Myeloid-derived suppressor cells in non-neoplastic inflamed organs. Inflamm. Regen. 2018, 38, 19. [Google Scholar] [CrossRef]
- Killig, M.; Glatzer, T.; Romagnani, C. Recognition strategies of group 3 innate lymphoid cells. Front. Immunol. 2014, 5, 142. [Google Scholar] [CrossRef] [Green Version]
- Trabanelli, S.; Gomez-Cadena, A.; Salome, B.; Michaud, K.; Mavilio, D.; Landis, B.N.; Jandus, P.; Jandus, C. Human innate lymphoid cells (ILCs): Toward a uniform immune-phenotyping. Cytom. B Clin. Cytom. 2018, 94, 392–399. [Google Scholar] [CrossRef]
- Manoharan, I.; Suryawanshi, A.; Hong, Y.; Ranganathan, P.; Shanmugam, A.; Ahmad, S.; Swafford, D.; Manicassamy, B.; Ramesh, G.; Koni, P.A.; et al. Homeostatic PPARalpha signaling limits inflammatory responses to commensal microbiota in the intestine. J. Immunol. 2016, 196, 4739–4749. [Google Scholar] [CrossRef]
- Sagebiel, A.F.; Steinert, F.; Lunemann, S.; Korner, C.; Schreurs, R.; Altfeld, M.; Perez, D.; Reinshagen, K.; Bunders, M.J. Tissue-resident Eomes(+) NK cells are the major innate lymphoid cell population in human infant intestine. Nat. Commun. 2019, 10, 975. [Google Scholar] [CrossRef] [Green Version]
- Gordon, S.M.; Chaix, J.; Rupp, L.J.; Wu, J.; Madera, S.; Sun, J.C.; Lindsten, T.; Reiner, S.L. The transcription factors T-bet and Eomes control key checkpoints of natural killer cell maturation. Immunity 2012, 36, 55–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbert, D.R.; Douglas, B.; Zullo, K. Group 2 innate lymphoid cells (ILC2): Type 2 immunity and helminth immunity. Int. J. Mol. Sci. 2019, 20, 2276. [Google Scholar] [CrossRef] [Green Version]
- Hepworth, M.R.; Fung, T.C.; Masur, S.H.; Kelsen, J.R.; McConnell, F.M.; Dubrot, J.; Withers, D.R.; Hugues, S.; Farrar, M.A.; Reith, W.; et al. Immune tolerance. Group 3 innate lymphoid cells mediate intestinal selection of commensal bacteria-specific CD4(+) T cells. Science 2015, 348, 1031–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Withers, D.R.; Hepworth, M.R. Group 3 innate lymphoid cells: Communications hubs of the intestinal immune system. Front. Immunol. 2017, 8, 1298. [Google Scholar] [CrossRef] [Green Version]
- Celsus, A.C. De Medicina, 1st ed.; Nicolaus Laurentii: Florence, Italy, 1478. [Google Scholar]
- Frias, B.; Merighi, A. Capsaicin, nociception and pain. Molecules 2016, 21, 797. [Google Scholar] [CrossRef] [Green Version]
- Nilius, B.; Owsianik, G. The transient receptor potential family of ion channels. Genome Biol. 2011, 12, 218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desvergne, B.; Wahli, W. Peroxisome proliferator-activated receptors: Nuclear control of metabolism. Endocrinol. Rev. 1999, 20, 649–688. [Google Scholar] [CrossRef] [Green Version]
- Sertznig, P.; Seifert, M.; Tilgen, W.; Reichrath, J. Present concepts and future outlook: Function of peroxisome proliferator-activated receptors (PPARs) for pathogenesis, progression, and therapy of cancer. J. Cell Physiol. 2007, 212, 1–12. [Google Scholar] [CrossRef]
- Kamata, S.; Oyama, T.; Saito, K.; Honda, A.; Yamamoto, Y.; Suda, K.; Ishikawa, R.; Itoh, T.; Watanabe, Y.; Shibata, T.; et al. PPARalpha ligand-binding domain structures with endogenous fatty acids and fibrates. iScience 2020, 23, 101727. [Google Scholar] [CrossRef]
- Wahli, W.; Michalik, L. PPARs at the crossroads of lipid signaling and inflammation. Trends Endocrinol. Metab. 2012, 23, 351–363. [Google Scholar] [CrossRef]
- Lo Verme, J.; Fu, J.; Astarita, G.; La Rana, G.; Russo, R.; Calignano, A.; Piomelli, D. The nuclear receptor peroxisome proliferator-activated receptor-alpha mediates the anti-inflammatory actions of palmitoylethanolamide. Mol. Pharmacol. 2005, 67, 15–19. [Google Scholar] [CrossRef]
- LoVerme, J.; La Rana, G.; Russo, R.; Calignano, A.; Piomelli, D. The search for the palmitoylethanolamide receptor. Life Sci. 2005, 77, 1685–1698. [Google Scholar] [CrossRef] [Green Version]
- de Lera, A.R.; Krezel, W.; Ruhl, R. An endogenous mammalian retinoid X receptor ligand, at last! ChemMedChem 2016, 11, 1027–1037. [Google Scholar] [CrossRef]
- Ruhl, R.; Krzyzosiak, A.; Niewiadomska-Cimicka, A.; Rochel, N.; Szeles, L.; Vaz, B.; Wietrzych-Schindler, M.; Alvarez, S.; Szklenar, M.; Nagy, L.; et al. 9-cis-13,14-dihydroretinoic acid is an endogenous retinoid acting as RXR ligand in mice. PLoS Genet. 2015, 11, e1005213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najib, J. Fenofibrate in the treatment of dyslipidemia: A review of the data as they relate to the new suprabioavailable tablet formulation. Clin. Ther. 2002, 24, 2022–2050. [Google Scholar] [CrossRef]
- Blais, J.E.; Tong, G.K.Y.; Pathadka, S.; Mok, M.; Wong, I.C.K.; Chan, E.W. Comparative efficacy and safety of statin and fibrate monotherapy: A systematic review and meta-analysis of head-to-head randomized controlled trials. PLoS ONE 2021, 16, e0246480. [Google Scholar] [CrossRef] [PubMed]
- Alsheikh-Ali, A.A.; Kuvin, J.T.; Karas, R.H. Risk of adverse events with fibrates. Am. J. Cardiol. 2004, 94, 935–938. [Google Scholar] [CrossRef] [PubMed]
- Kaipainen, A.; Kieran, M.W.; Huang, S.; Butterfield, C.; Bielenberg, D.; Mostoslavsky, G.; Mulligan, R.; Folkman, J.; Panigrahy, D. PPARalpha deficiency in inflammatory cells suppresses tumor growth. PLoS ONE 2007, 2, e260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimura, Y.; Tachibana, H.; Yamada, K. Peroxisome proliferator-activated receptor ligands negatively regulate the expression of the high-affinity IgE receptor Fc epsilon RI in human basophilic KU812 cells. Biochem. Biophys. Res. Commun. 2002, 297, 193–201. [Google Scholar] [CrossRef]
- Woerly, G.; Honda, K.; Loyens, M.; Papin, J.P.; Auwerx, J.; Staels, B.; Capron, M.; Dombrowicz, D. Peroxisome proliferator-activated receptors alpha and gamma down-regulate allergic inflammation and eosinophil activation. J. Exp. Med. 2003, 198, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Chinetti, G.; Fruchart, J.C.; Staels, B. Peroxisome proliferator-activated receptors: New targets for the pharmacological modulation of macrophage gene expression and function. Curr. Opin. Lipidol. 2003, 14, 459–468. [Google Scholar] [CrossRef]
- Babaev, V.R.; Ishiguro, H.; Ding, L.; Yancey, P.G.; Dove, D.E.; Kovacs, W.J.; Semenkovich, C.F.; Fazio, S.; Linton, M.F. Macrophage expression of peroxisome proliferator-activated receptor-alpha reduces atherosclerosis in low-density lipoprotein receptor-deficient mice. Circulation 2007, 116, 1404–1412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Zhang, X.; Zheng, L.; Zhao, H.; Yan, G.; Zhang, Q.; Zhou, Y.; Lei, J.; Zhang, J.; Wang, J.; et al. RIPK3 orchestrates fatty acid metabolism in tumor-associated macrophages and hepatocarcinogenesis. Cancer Immunol. Res. 2020, 8, 710–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brocker, C.N.; Yue, J.; Kim, D.; Qu, A.; Bonzo, J.A.; Gonzalez, F.J. Hepatocyte-specific PPARA expression exclusively promotes agonist-induced cell proliferation without influence from nonparenchymal cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G283–G299. [Google Scholar] [CrossRef]
- Dubrac, S.; Stoitzner, P.; Pirkebner, D.; Elentner, A.; Schoonjans, K.; Auwerx, J.; Saeland, S.; Hengster, P.; Fritsch, P.; Romani, N.; et al. Peroxisome proliferator-activated receptor-alpha activation inhibits Langerhans cell function. J. Immunol. 2007, 178, 4362–4372. [Google Scholar] [CrossRef] [Green Version]
- Poulsen, R.C.; Moughan, P.J.; Kruger, M.C. Long-chain polyunsaturated fatty acids and the regulation of bone metabolism. Exp. Biol. Med. 2007, 232, 1275–1288. [Google Scholar] [CrossRef]
- Warden, A.; Truitt, J.; Merriman, M.; Ponomareva, O.; Jameson, K.; Ferguson, L.B.; Mayfield, R.D.; Harris, R.A. Localization of PPAR isotypes in the adult mouse and human brain. Sci. Rep. 2016, 6, 27618. [Google Scholar] [CrossRef]
- Kroetz, D.L.; Yook, P.; Costet, P.; Bianchi, P.; Pineau, T. Peroxisome proliferator-activated receptor alpha controls the hepatic CYP4A induction adaptive response to starvation and diabetes. J. Biol. Chem. 1998, 273, 31581–31589. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, T.; Fujita, T.; Usuda, N.; Cook, W.; Qi, C.; Peters, J.M.; Gonzalez, F.J.; Yeldandi, A.V.; Rao, M.S.; Reddy, J.K. Peroxisomal and mitochondrial fatty acid beta-oxidation in mice nullizygous for both peroxisome proliferator-activated receptor alpha and peroxisomal fatty acyl-CoA oxidase. Genotype correlation with fatty liver phenotype. J. Biol. Chem. 1999, 274, 19228–19236. [Google Scholar] [CrossRef] [Green Version]
- Vila-Brau, A.; De Sousa-Coelho, A.L.; Mayordomo, C.; Haro, D.; Marrero, P.F. Human HMGCS2 regulates mitochondrial fatty acid oxidation and FGF21 expression in HepG2 cell line. J. Biol. Chem. 2011, 286, 20423–20430. [Google Scholar] [CrossRef] [Green Version]
- Daynes, R.A.; Jones, D.C. Emerging roles of PPARs in inflammation and immunity. Nat. Rev. Immunol. 2002, 2, 748–759. [Google Scholar] [CrossRef]
- Ammazzalorso, A.; Bruno, I.; Florio, R.; De Lellis, L.; Laghezza, A.; Cerchia, C.; De Filippis, B.; Fantacuzzi, M.; Giampietro, L.; Maccallini, C.; et al. Sulfonimide and amide derivatives as novel PPARalpha antagonists: Synthesis, antiproliferative activity, and docking studies. ACS Med. Chem. Lett. 2020, 11, 624–632. [Google Scholar] [CrossRef] [Green Version]
- Delerive, P.; De Bosscher, K.; Besnard, S.; Vanden Berghe, W.; Peters, J.M.; Gonzalez, F.J.; Fruchart, J.C.; Tedgui, A.; Haegeman, G.; Staels, B. Peroxisome proliferator-activated receptor alpha negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-kappaB and AP-1. J. Biol. Chem. 1999, 274, 32048–32054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Planavila, A.; Iglesias, R.; Giralt, M.; Villarroya, F. Sirt1 acts in association with PPARalpha to protect the heart from hypertrophy, metabolic dysregulation, and inflammation. Cardiovasc. Res. 2011, 90, 276–284. [Google Scholar] [CrossRef] [Green Version]
- Rothgiesser, K.M.; Fey, M.; Hottiger, M.O. Acetylation of p65 at lysine 314 is important for late NF-kappaB-dependent gene expression. BMC Genom. 2010, 11, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, N.; Chu, E.S.; Zhang, J.; Li, X.; Liang, Q.; Chen, J.; Chen, M.; Teoh, N.; Farrell, G.; Sung, J.J.; et al. Peroxisome proliferator activated receptor alpha inhibits hepatocarcinogenesis through mediating NF-kappaB signaling pathway. Oncotarget 2014, 5, 8330–8340. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, M.D.; Harrison, S.C. Structure of an IkappaBalpha/NF-kappaB complex. Cell 1998, 95, 749–758. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.F.; Greene, W.C. Shaping the nuclear action of NF-kappaB. Nat. Rev. Mol. Cell Biol. 2004, 5, 392–401. [Google Scholar] [CrossRef]
- Murakami, K.; Bujo, H.; Unoki, H.; Saito, Y. Effect of PPARalpha activation of macrophages on the secretion of inflammatory cytokines in cultured adipocytes. Eur. J. Pharmacol. 2007, 561, 206–213. [Google Scholar] [CrossRef]
- Marx, N.; Mackman, N.; Schonbeck, U.; Yilmaz, N.; Hombach, V.; Libby, P.; Plutzky, J. PPARalpha activators inhibit tissue factor expression and activity in human monocytes. Circulation 2001, 103, 213–219. [Google Scholar] [CrossRef] [Green Version]
- Neve, B.P.; Corseaux, D.; Chinetti, G.; Zawadzki, C.; Fruchart, J.C.; Duriez, P.; Staels, B.; Jude, B. PPARalpha agonists inhibit tissue factor expression in human monocytes and macrophages. Circulation 2001, 103, 207–212. [Google Scholar] [CrossRef] [Green Version]
- Haque, S.J.; Sharma, P. Interleukins and STAT signaling. Vitam. Horm. 2006, 74, 165–206. [Google Scholar] [CrossRef]
- Shipley, J.M.; Waxman, D.J. Down-regulation of STAT5b transcriptional activity by ligand-activated peroxisome proliferator-activated receptor (PPAR) alpha and PPARgamma. Mol. Pharmacol. 2003, 64, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Shipley, J.M.; Waxman, D.J. Simultaneous, bidirectional inhibitory crosstalk between PPAR and STAT5b. Toxicol. Appl. Pharmacol. 2004, 199, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.C.; Waxman, D.J. STAT5b down-regulates peroxisome proliferator-activated receptor alpha transcription by inhibition of ligand-independent activation function region-1 trans-activation domain. J. Biol. Chem. 1999, 274, 29874–29882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.X.; Leonard, W.J. The role of Stat5a and Stat5b in signaling by IL-2 family cytokines. Oncogene 2000, 19, 2566–2576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bendickova, K.; Fric, J. Roles of IL-2 in bridging adaptive and innate immunity, and as a tool for cellular immunotherapy. J. Leukoc. Biol. 2020, 108, 427–437. [Google Scholar] [CrossRef]
- Devchand, P.R.; Keller, H.; Peters, J.M.; Vazquez, M.; Gonzalez, F.J.; Wahli, W. The PPARalpha-leukotriene B4 pathway to inflammation control. Nature 1996, 384, 39–43. [Google Scholar] [CrossRef]
- Dahlen, S.E.; Bjork, J.; Hedqvist, P.; Arfors, K.E.; Hammarstrom, S.; Lindgren, J.A.; Samuelsson, B. Leukotrienes promote plasma leakage and leukocyte adhesion in postcapillary venules: In vivo effects with relevance to the acute inflammatory response. Proc. Natl. Acad. Sci. USA 1981, 78, 3887–3891. [Google Scholar] [CrossRef] [Green Version]
- Lindbom, L.; Hedqvist, P.; Dahlen, S.E.; Lindgren, J.A.; Arfors, K.E. Leukotriene B4 induces extravasation and migration of polymorphonuclear leukocytes in vivo. Acta Physiol. Scand. 1982, 116, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Marleau, S.; Fruteau de Laclos, B.; Sanchez, A.B.; Poubelle, P.E.; Borgeat, P. Role of 5-lipoxygenase products in the local accumulation of neutrophils in dermal inflammation in the rabbit. J. Immunol. 1999, 163, 3449–3458. [Google Scholar]
- Yu, K.; Bayona, W.; Kallen, C.B.; Harding, H.P.; Ravera, C.P.; McMahon, G.; Brown, M.; Lazar, M.A. Differential activation of peroxisome proliferator-activated receptors by eicosanoids. J. Biol. Chem. 1995, 270, 23975–23983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christofides, A.; Konstantinidou, E.; Jani, C.; Boussiotis, V.A. The role of peroxisome proliferator-activated receptors (PPAR) in immune responses. Metabolism 2021, 114, 154338. [Google Scholar] [CrossRef]
- Wolf, A.J.; Reyes, C.N.; Liang, W.; Becker, C.; Shimada, K.; Wheeler, M.L.; Cho, H.C.; Popescu, N.I.; Coggeshall, K.M.; Arditi, M.; et al. Hexokinase Is an innate immune receptor for the detection of bacterial peptidoglycan. Cell 2016, 166, 624–636. [Google Scholar] [CrossRef] [Green Version]
- Frosali, S.; Pagliari, D.; Gambassi, G.; Landolfi, R.; Pandolfi, F.; Cianci, R. How the intricate interaction among toll-like receptors, microbiota, and intestinal immunity can influence gastrointestinal pathology. J. Immunol. Res. 2015, 2015, 489821. [Google Scholar] [CrossRef]
- Wang, Y.; Yin, Y.; Chen, X.; Zhao, Y.; Wu, Y.; Li, Y.; Wang, X.; Chen, H.; Xiang, C. Induction of intestinal Th17 cells by flagellins from segmented filamentous bacteria. Front. Immunol. 2019, 10, 2750. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Jeong, S.Y.; Choi, A.J.; Kim, S.J. Lipopolysaccharide directly stimulates Th17 differentiation in vitro modulating phosphorylation of RelB and NF-kappaB1. Immunol. Lett. 2015, 165, 10–19. [Google Scholar] [CrossRef]
- Silveira, L.S.; Pimentel, G.D.; Souza, C.O.; Biondo, L.A.; Teixeira, A.A.S.; Lima, E.A.; Batatinha, H.A.P.; Rosa Neto, J.C.; Lira, F.S. Effect of an acute moderate-exercise session on metabolic and inflammatory profile of PPAR-alpha knockout mice. Cell Biochem. Funct. 2017, 35, 510–517. [Google Scholar] [CrossRef]
- Becker, J.; Delayre-Orthez, C.; Frossard, N.; Pons, F. Regulation of peroxisome proliferator-activated receptor-alpha expression during lung inflammation. Pulm. Pharmacol. Ther. 2008, 21, 324–330. [Google Scholar] [CrossRef]
- Chistyakov, D.V.; Aleshin, S.E.; Astakhova, A.A.; Sergeeva, M.G.; Reiser, G. Regulation of peroxisome proliferator-activated receptors (PPAR) alpha and -gamma of rat brain astrocytes in the course of activation by toll-like receptor agonists. J. Neurochem. 2015, 134, 113–124. [Google Scholar] [CrossRef]
- Dana, N.; Javanmard, H.S.; Vaseghi, G. The effect of fenofibrate, a PPARalpha activator on toll-like receptor-4 signal transduction in melanoma both in vitro and in vivo. Clin. Transl. Oncol. 2020, 22, 486–494. [Google Scholar] [CrossRef]
- Shen, W.; Gao, Y.; Lu, B.; Zhang, Q.; Hu, Y.; Chen, Y. Negatively regulating TLR4/NF-kappaB signaling via PPARalpha in endotoxin-induced uveitis. Biochim. Biophys. Acta 2014, 1842, 1109–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamkanfi, M.; Dixit, V.M. Mechanisms and functions of inflammasomes. Cell 2014, 157, 1013–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, D.; Liwinski, T.; Elinav, E. Inflammasome activation and regulation: Toward a better understanding of complex mechanisms. Cell Discov. 2020, 6, 36. [Google Scholar] [CrossRef] [PubMed]
- Seok, J.K.; Kang, H.C.; Cho, Y.Y.; Lee, H.S.; Lee, J.Y. Regulation of the NLRP3 inflammasome by post-translational modifications and small molecules. Front. Immunol. 2020, 11, 618231. [Google Scholar] [CrossRef]
- Gugliandolo, E.; Fusco, R.; Ginestra, G.; D’Amico, R.; Bisignano, C.; Mandalari, G.; Cuzzocrea, S.; Di Paola, R. Involvement of TLR4 and PPAR-alpha receptors in host response and NLRP3 Inflammasome activation, against pulmonary infection with Pseudomonas aeruginosa. Shock 2019, 51, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Brocker, C.N.; Kim, D.; Melia, T.; Karri, K.; Velenosi, T.J.; Takahashi, S.; Aibara, D.; Bonzo, J.A.; Levi, M.; Waxman, D.J.; et al. Long non-coding RNA Gm15441 attenuates hepatic inflammasome activation in response to PPARA agonism and fasting. Nat. Commun. 2020, 11, 5847. [Google Scholar] [CrossRef]
- Hu, J.; Zhu, Z.; Ying, H.; Yao, J.; Ma, H.; Li, L.; Zhao, Y. Oleoylethanolamide protects against acute liver injury by regulating Nrf-2/HO-1 and NLRP3 pathways in mice. Front. Pharmacol. 2020, 11, 605065. [Google Scholar] [CrossRef]
- Lama, A.; Provensi, G.; Amoriello, R.; Pirozzi, C.; Rani, B.; Mollica, M.P.; Raso, G.M.; Ballerini, C.; Meli, R.; Passani, M.B. The anti-inflammatory and immune-modulatory effects of OEA limit DSS-induced colitis in mice. Biomed. Pharmacother. 2020, 129, 110368. [Google Scholar] [CrossRef]
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric oxide synthases: Structure, function and inhibition. Biochem. J. 2001, 357, 593–615. [Google Scholar] [CrossRef]
- Kleinert, H.; Schwarz, P.M.; Forstermann, U. Regulation of the expression of inducible nitric oxide synthase. Biol. Chem. 2003, 384, 1343–1364. [Google Scholar] [CrossRef] [PubMed]
- Forstermann, U.; Boissel, J.P.; Kleinert, H. Expressional control of the ‘constitutive’ isoforms of nitric oxide synthase (NOS I and NOS III). FASEB J. 1998, 12, 773–790. [Google Scholar] [CrossRef] [PubMed]
- Pou, S.; Keaton, L.; Surichamorn, W.; Rosen, G.M. Mechanism of superoxide generation by neuronal nitric-oxide synthase. J. Biol. Chem. 1999, 274, 9573–9580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paukkeri, E.L.; Leppanen, T.; Sareila, O.; Vuolteenaho, K.; Kankaanranta, H.; Moilanen, E. PPARalpha agonists inhibit nitric oxide production by enhancing iNOS degradation in LPS-treated macrophages. Br. J. Pharmacol. 2007, 152, 1081–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Ginderachter, J.A.; Movahedi, K.; Van den Bossche, J.; De Baetselier, P. Macrophages, PPARs, and cancer. PPAR Res. 2008, 2008, 169414. [Google Scholar] [CrossRef] [Green Version]
- Maccallini, C.; Mollica, A.; Amoroso, R. The positive regulation of eNOS signaling by PPAR agonists in cardiovascular diseases. Am. J. Cardiovasc. Drugs 2017, 17, 273–281. [Google Scholar] [CrossRef]
- Tanaka, S.; Hosogi, S.; Sawabe, Y.; Shimamoto, C.; Matsumura, H.; Inui, T.; Marunaka, Y.; Nakahari, T. PPARalpha induced NOS1 phosphorylation via PI3K/Akt in guinea pig antral mucous cells: NO-enhancement in Ca(2+)-regulated exocytosis. Biomed. Res. 2016, 37, 167–178. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, S.; Sugiyama, N.; Takahashi, Y.; Mantoku, D.; Sawabe, Y.; Kuwabara, H.; Nakano, T.; Shimamoto, C.; Matsumura, H.; Marunaka, Y.; et al. PPARalpha autocrine regulation of Ca(2)(+)-regulated exocytosis in guinea pig antral mucous cells: NO and cGMP accumulation. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, G1169–G1179. [Google Scholar] [CrossRef] [Green Version]
- Bune, A.J.; Shergill, J.K.; Cammack, R.; Cook, H.T. L-arginine depletion by arginase reduces nitric oxide production in endotoxic shock: An electron paramagnetic resonance study. FEBS Lett. 1995, 366, 127–130. [Google Scholar] [CrossRef] [Green Version]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 macrophages and the Th1/Th2 paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabacka, M.; Placha, W.; Plonka, P.M.; Pajak, S.; Urbanska, K.; Laidler, P.; Slominski, A. Inhibition of melanoma metastases by fenofibrate. Arch. Dermatol. Res. 2004, 296, 54–58. [Google Scholar] [CrossRef]
- Gallorini, M.; Rapino, M.; Schweikl, H.; Cataldi, A.; Amoroso, R.; Maccallini, C. Selective inhibitors of the inducible nitric oxide synthase as modulators of cell responses in LPS-stimulated human monocytes. Molecules 2021, 26, 4419. [Google Scholar] [CrossRef]
- Teissier, E.; Nohara, A.; Chinetti, G.; Paumelle, R.; Cariou, B.; Fruchart, J.C.; Brandes, R.P.; Shah, A.; Staels, B. Peroxisome proliferator-activated receptor alpha induces NADPH oxidase activity in macrophages, leading to the generation of LDL with PPAR-alpha activation properties. Circ. Res. 2004, 95, 1174–1182. [Google Scholar] [CrossRef] [Green Version]
- Deininger, P. Alu elements: Know the SINEs. Genome Biol. 2011, 12, 236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynolds, W.F.; Kumar, A.P.; Piedrafita, F.J. The human myeloperoxidase gene is regulated by LXR and PPARalpha ligands. Biochem. Biophys. Res. Commun. 2006, 349, 846–854. [Google Scholar] [CrossRef] [Green Version]
- Penas, F.; Mirkin, G.A.; Vera, M.; Cevey, A.; Gonzalez, C.D.; Gomez, M.I.; Sales, M.E.; Goren, N.B. Treatment in vitro with PPARalpha and PPARgamma ligands drives M1-to-M2 polarization of macrophages from T. cruzi-infected mice. Biochim. Biophys. Acta 2015, 1852, 893–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redlich, S.; Ribes, S.; Schutze, S.; Nau, R. Palmitoylethanolamide stimulates phagocytosis of Escherichia coli K1 by macrophages and increases the resistance of mice against infections. J. Neuroinflamm. 2014, 11, 108. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.S.; Lee, H.M.; Kim, J.K.; Yang, C.S.; Kim, T.S.; Jung, M.; Jin, H.S.; Kim, S.; Jang, J.; Oh, G.T.; et al. PPAR-alpha activation mediates innate host defense through induction of TFEB and lipid catabolism. J. Immunol. 2017, 198, 3283–3295. [Google Scholar] [CrossRef] [Green Version]
- Peyron, P.; Vaubourgeix, J.; Poquet, Y.; Levillain, F.; Botanch, C.; Bardou, F.; Daffe, M.; Emile, J.F.; Marchou, B.; Cardona, P.J.; et al. Foamy macrophages from tuberculous patients’ granulomas constitute a nutrient-rich reservoir for M. tuberculosis persistence. PLoS Pathog. 2008, 4, e1000204. [Google Scholar] [CrossRef]
- Tam, V.C.; Suen, R.; Treuting, P.M.; Armando, A.; Lucarelli, R.; Gorrochotegui-Escalante, N.; Diercks, A.H.; Quehenberger, O.; Dennis, E.A.; Aderem, A.; et al. PPARalpha exacerbates necroptosis, leading to increased mortality in postinfluenza bacterial superinfection. Proc. Natl. Acad. Sci. USA 2020, 117, 15789–15798. [Google Scholar] [CrossRef] [PubMed]
- Ng, V.Y.; Huang, Y.; Reddy, L.M.; Falck, J.R.; Lin, E.T.; Kroetz, D.L. Cytochrome P450 eicosanoids are activators of peroxisome proliferator-activated receptor alpha. Drug Metab. Dispos. 2007, 35, 1126–1134. [Google Scholar] [CrossRef]
- Yang, M.; Wang, Y.; Chen, J.; Wang, Q.; Wei, S.; Wang, S.; Qin, Q. Functional analysis of Epinephelus coioides peroxisome proliferative-activated receptor alpha (PPARalpha): Involvement in response to viral infection. Fish. Shellfish Immunol. 2020, 102, 257–266. [Google Scholar] [CrossRef]
- Ehrlich, A.; Uhl, S.; Ioannidis, K.; Hofree, M.; tenOever, B.R.; Nahmias, Y. The SARS-CoV-2 Transcriptional Metabolic Signature in Lung Epithelium. Available online: https://ssrn.com/abstract=3650499 (accessed on 28 September 2021).
- Heffernan, K.S.; Ranadive, S.M.; Jae, S.Y. Exercise as medicine for COVID-19: On PPAR with emerging pharmacotherapy. Med. Hypotheses 2020, 143, 110197. [Google Scholar] [CrossRef]
- Piomelli, D.; Hohmann, A.G.; Seybold, V.; Hammock, B.D. A lipid gate for the peripheral control of pain. J. Neurosci. 2014, 34, 15184–15191. [Google Scholar] [CrossRef]
- Chiurchiu, V.; Battistini, L.; Maccarrone, M. Endocannabinoid signalling in innate and adaptive immunity. Immunology 2015, 144, 352–364. [Google Scholar] [CrossRef]
- Guindon, J.; Hohmann, A.G. The endocannabinoid system and pain. CNS Neurol. Disord. Drug Targets 2009, 8, 403–421. [Google Scholar] [CrossRef] [PubMed]
- Bisogno, T.; Ventriglia, M.; Milone, A.; Mosca, M.; Cimino, G.; Di Marzo, V. Occurrence and metabolism of anandamide and related acyl-ethanolamides in ovaries of the sea urchin Paracentrotus lividus. Biochim. Biophys. Acta 1997, 1345, 338–348. [Google Scholar] [CrossRef]
- Sepe, N.; De Petrocellis, L.; Montanaro, F.; Cimino, G.; Di Marzo, V. Bioactive long chain N-acylethanolamines in five species of edible bivalve molluscs. Possible implications for mollusc physiology and sea food industry. Biochim. Biophys. Acta 1998, 1389, 101–111. [Google Scholar] [CrossRef]
- Matias, I.; Bisogno, T.; Melck, D.; Vandenbulcke, F.; Verger-Bocquet, M.; De Petrocellis, L.; Sergheraert, C.; Breton, C.; Di Marzo, V.; Salzet, M. Evidence for an endocannabinoid system in the central nervous system of the leech Hirudo medicinalis. Brain Res. Mol. Brain Res. 2001, 87, 145–159. [Google Scholar] [CrossRef]
- Fu, J.; Gaetani, S.; Oveisi, F.; Lo Verme, J.; Serrano, A.; Rodriguez De Fonseca, F.; Rosengarth, A.; Luecke, H.; Di Giacomo, B.; Tarzia, G.; et al. Oleylethanolamide regulates feeding and body weight through activation of the nuclear receptor PPAR-alpha. Nature 2003, 425, 90–93. [Google Scholar] [CrossRef]
- Bradshaw, H.B.; Walker, J.M. The expanding field of cannabimimetic and related lipid mediators. Br. J. Pharmacol. 2005, 144, 459–465. [Google Scholar] [CrossRef] [Green Version]
- Oddi, S.; Fezza, F.; Pasquariello, N.; De Simone, C.; Rapino, C.; Dainese, E.; Finazzi-Agro, A.; Maccarrone, M. Evidence for the intracellular accumulation of anandamide in adiposomes. Cell Mol. Life Sci. 2008, 65, 840–850. [Google Scholar] [CrossRef]
- Kaczocha, M.; Glaser, S.T.; Chae, J.; Brown, D.A.; Deutsch, D.G. Lipid droplets are novel sites of N-acylethanolamine inactivation by fatty acid amide hydrolase-2. J. Biol. Chem. 2010, 285, 2796–2806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bisogno, T.; Maurelli, S.; Melck, D.; De Petrocellis, L.; Di Marzo, V. Biosynthesis, uptake, and degradation of anandamide and palmitoylethanolamide in leukocytes. J. Biol. Chem. 1997, 272, 3315–3323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuboi, K.; Ikematsu, N.; Uyama, T.; Deutsch, D.G.; Tokumura, A.; Ueda, N. Biosynthetic pathways of bioactive N-acylethanolamines in brain. CNS Neurol. Disord. Drug Targets 2013, 12, 7–16. [Google Scholar] [CrossRef]
- Fegley, D.; Gaetani, S.; Duranti, A.; Tontini, A.; Mor, M.; Tarzia, G.; Piomelli, D. Characterization of the fatty acid amide hydrolase inhibitor cyclohexyl carbamic acid 3′-carbamoyl-biphenyl-3-yl ester (URB597): Effects on anandamide and oleoylethanolamide deactivation. J. Pharmacol. Exp. Ther. 2005, 313, 352–358. [Google Scholar] [CrossRef] [Green Version]
- Ogura, Y.; Parsons, W.H.; Kamat, S.S.; Cravatt, B.F. A calcium-dependent acyltransferase that produces N-acyl phosphatidylethanolamines. Nat. Chem. Biol. 2016, 12, 669–671. [Google Scholar] [CrossRef] [Green Version]
- Ueda, N.; Tsuboi, K.; Uyama, T. Metabolism of endocannabinoids and related N-acylethanolamines: Canonical and alternative pathways. FEBS J. 2013, 280, 1874–1894. [Google Scholar] [CrossRef] [PubMed]
- LoVerme, J.; Russo, R.; La Rana, G.; Fu, J.; Farthing, J.; Mattace-Raso, G.; Meli, R.; Hohmann, A.; Calignano, A.; Piomelli, D. Rapid broad-spectrum analgesia through activation of peroxisome proliferator-activated receptor-alpha. J. Pharmacol. Exp. Ther. 2006, 319, 1051–1061. [Google Scholar] [CrossRef]
- Suardiaz, M.; Estivill-Torrus, G.; Goicoechea, C.; Bilbao, A.; de Fonseca, R.F. Analgesic properties of oleoylethanolamide (OEA) in visceral and inflammatory pain. Pain 2007, 133, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.K.; Dadia, N.; Yang, C.B.; Krishnan, S.; Badr, M. Peroxisome proliferator-activated receptor agonists inhibit inflammatory edema and hyperalgesia. Inflammation 2002, 26, 121–127. [Google Scholar] [CrossRef]
- Russo, R.; LoVerme, J.; La Rana, G.; D’Agostino, G.; Sasso, O.; Calignano, A.; Piomelli, D. Synergistic antinociception by the cannabinoid receptor agonist anandamide and the PPAR-alpha receptor agonist GW7647. Eur. J. Pharmacol. 2007, 566, 117–119. [Google Scholar] [CrossRef] [Green Version]
- D’Agostino, G.; La Rana, G.; Russo, R.; Sasso, O.; Iacono, A.; Esposito, E.; Raso, G.M.; Cuzzocrea, S.; Lo Verme, J.; Piomelli, D.; et al. Acute intracerebroventricular administration of palmitoylethanolamide, an endogenous peroxisome proliferator-activated receptor-alpha agonist, modulates carrageenan-induced paw edema in mice. J. Pharmacol. Exp. Ther. 2007, 322, 1137–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, R.; Loverme, J.; La Rana, G.; Compton, T.R.; Parrott, J.; Duranti, A.; Tontini, A.; Mor, M.; Tarzia, G.; Calignano, A.; et al. The fatty acid amide hydrolase inhibitor URB597 (cyclohexylcarbamic acid 3′-carbamoylbiphenyl-3-yl ester) reduces neuropathic pain after oral administration in mice. J. Pharmacol. Exp. Ther. 2007, 322, 236–242. [Google Scholar] [CrossRef]
- Ben-Shabat, S.; Fride, E.; Sheskin, T.; Tamiri, T.; Rhee, M.H.; Vogel, Z.; Bisogno, T.; De Petrocellis, L.; Di Marzo, V.; Mechoulam, R. An entourage effect: Inactive endogenous fatty acid glycerol esters enhance 2-arachidonoyl-glycerol cannabinoid activity. Eur. J. Pharmacol. 1998, 353, 23–31. [Google Scholar] [CrossRef]
- Ho, W.S.; Barrett, D.A.; Randall, M.D. ‘Entourage’ effects of N-palmitoylethanolamide and N-oleoylethanolamide on vasorelaxation to anandamide occur through TRPV1 receptors. Br. J. Pharmacol. 2008, 155, 837–846. [Google Scholar] [CrossRef] [Green Version]
- Steardo, L., Jr.; Bronzuoli, M.R.; Iacomino, A.; Esposito, G.; Steardo, L.; Scuderi, C. Does neuroinflammation turn on the flame in Alzheimer’s disease? Focus on astrocytes. Front. Neurosci. 2015, 9, 259. [Google Scholar] [CrossRef] [Green Version]
- Skaper, S.D.; Facci, L.; Giusti, P. Glia and mast cells as targets for palmitoylethanolamide, an anti-inflammatory and neuroprotective lipid mediator. Mol. Neurobiol. 2013, 48, 340–352. [Google Scholar] [CrossRef] [PubMed]
- Amor, S.; Puentes, F.; Baker, D.; van der Valk, P. Inflammation in neurodegenerative diseases. Immunology 2010, 129, 154–169. [Google Scholar] [CrossRef] [PubMed]
- Colombo, E.; Farina, C. Astrocytes: Key regulators of neuroinflammation. Trends Immunol. 2016, 37, 608–620. [Google Scholar] [CrossRef] [PubMed]
- Aloe, L.; Leon, A.; Levi-Montalcini, R. A proposed autacoid mechanism controlling mastocyte behaviour. Agents Actions 1993, 39, C145–C147. [Google Scholar] [CrossRef]
- Benito, C.; Tolon, R.M.; Castillo, A.I.; Ruiz-Valdepenas, L.; Martinez-Orgado, J.A.; Fernandez-Sanchez, F.J.; Vazquez, C.; Cravatt, B.F.; Romero, J. beta-Amyloid exacerbates inflammation in astrocytes lacking fatty acid amide hydrolase through a mechanism involving PPAR-alpha, PPAR-gamma and TRPV1, but not CB(1) or CB(2) receptors. Br. J. Pharmacol. 2012, 166, 1474–1489. [Google Scholar] [CrossRef] [Green Version]
- Hohmann, U.; Pelzer, M.; Kleine, J.; Hohmann, T.; Ghadban, C.; Dehghani, F. Opposite effects of neuroprotective cannabinoids, palmitoylethanolamide, and 2-arachidonoylglycerol on function and morphology of microglia. Front. Neurosci. 2019, 13, 1180. [Google Scholar] [CrossRef] [PubMed]
- Bronzuoli, M.R.; Facchinetti, R.; Steardo, L., Jr.; Romano, A.; Stecca, C.; Passarella, S.; Steardo, L.; Cassano, T.; Scuderi, C. Palmitoylethanolamide dampens reactive astrogliosis and improves neuronal trophic support in a triple transgenic model of Alzheimer’s disease: In vitro and in vivo evidence. Oxid. Med. Cell. Longev. 2018, 2018, 4720532. [Google Scholar] [CrossRef] [Green Version]
- Scuderi, C.; Stecca, C.; Valenza, M.; Ratano, P.; Bronzuoli, M.R.; Bartoli, S.; Steardo, L.; Pompili, E.; Fumagalli, L.; Campolongo, P.; et al. Palmitoylethanolamide controls reactive gliosis and exerts neuroprotective functions in a rat model of Alzheimer’s disease. Cell Death Dis. 2014, 5, e1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scuderi, C.; Valenza, M.; Stecca, C.; Esposito, G.; Carratu, M.R.; Steardo, L. Palmitoylethanolamide exerts neuroprotective effects in mixed neuroglial cultures and organotypic hippocampal slices via peroxisome proliferator-activated receptor-alpha. J. Neuroinflamm. 2012, 9, 49. [Google Scholar] [CrossRef] [Green Version]
- Guida, F.; Luongo, L.; Boccella, S.; Giordano, M.E.; Romano, R.; Bellini, G.; Manzo, I.; Furiano, A.; Rizzo, A.; Imperatore, R.; et al. Palmitoylethanolamide induces microglia changes associated with increased migration and phagocytic activity: Involvement of the CB2 receptor. Sci. Rep. 2017, 7, 375. [Google Scholar] [CrossRef]
- Rinne, P.; Guillamat-Prats, R.; Rami, M.; Bindila, L.; Ring, L.; Lyytikainen, L.P.; Raitoharju, E.; Oksala, N.; Lehtimaki, T.; Weber, C.; et al. Palmitoylethanolamide promotes a proresolving macrophage phenotype and attenuates atherosclerotic plaque formation. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2562–2575. [Google Scholar] [CrossRef]
- Luo, D.; Zhang, Y.; Yuan, X.; Pan, Y.; Yang, L.; Zhao, Y.; Zhuo, R.; Chen, C.; Peng, L.; Li, W.; et al. Oleoylethanolamide inhibits glial activation via moudulating PPARalpha and promotes motor function recovery after brain ischemia. Pharmacol. Res. 2019, 141, 530–540. [Google Scholar] [CrossRef]
- Xu, X.; Guo, H.; Jing, Z.; Yang, L.; Chen, C.; Peng, L.; Wang, X.; Yan, L.; Ye, R.; Jin, X.; et al. N-oleoylethanolamine reduces inflammatory cytokines and adhesion molecules in TNF-alpha-induced human umbilical vein endothelial cells by activating CB2 and PPAR-alpha. J. Cardiovasc. Pharmacol. 2016, 68, 280–291. [Google Scholar] [CrossRef]
- Holubiec, M.I.; Romero, J.I.; Suarez, J.; Portavella, M.; Fernandez-Espejo, E.; Blanco, E.; Galeano, P.; de Fonseca, F.R. Palmitoylethanolamide prevents neuroinflammation, reduces astrogliosis and preserves recognition and spatial memory following induction of neonatal anoxia-ischemia. Psychopharmacology 2018, 235, 2929–2945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.C.; Guo, H.; Zhou, H.; Suo, D.Q.; Li, W.J.; Zhou, Y.; Zhao, Y.; Yang, W.S.; Jin, X. Chronic oleoylethanolamide treatment improves spatial cognitive deficits through enhancing hippocampal neurogenesis after transient focal cerebral ischemia. Biochem. Pharmacol. 2015, 94, 270–281. [Google Scholar] [CrossRef]
- Flannery, L.E.; Kerr, D.M.; Hughes, E.M.; Kelly, C.; Costello, J.; Thornton, A.M.; Humphrey, R.M.; Finn, D.P.; Roche, M. N-acylethanolamine regulation of TLR3-induced hyperthermia and neuroinflammatory gene expression: A role for PPARalpha. J. Neuroimmunol. 2021, 358, 577654. [Google Scholar] [CrossRef] [PubMed]
- Taquet, M.; Geddes, J.R.; Husain, M.; Luciano, S.; Harrison, P.J. 6-month neurological and psychiatric outcomes in 236 379 survivors of COVID-19: A retrospective cohort study using electronic health records. Lancet Psychiatry 2021, 8, 416–427. [Google Scholar] [CrossRef]
- Vaia, M.; Petrosino, S.; De Filippis, D.; Negro, L.; Guarino, A.; Carnuccio, R.; Di Marzo, V.; Iuvone, T. Palmitoylethanolamide reduces inflammation and itch in a mouse model of contact allergic dermatitis. Eur. J. Pharmacol. 2016, 791, 669–674. [Google Scholar] [CrossRef] [PubMed]
- Skaper, S.D.; Facci, L.; Romanello, S.; Leon, A. Mast cell activation causes delayed neurodegeneration in mixed hippocampal cultures via the nitric oxide pathway. J. Neurochem. 1996, 66, 1157–1166. [Google Scholar] [CrossRef]
- Facci, L.; Dal Toso, R.; Romanello, S.; Buriani, A.; Skaper, S.D.; Leon, A. Mast cells express a peripheral cannabinoid receptor with differential sensitivity to anandamide and palmitoylethanolamide. Proc. Natl. Acad. Sci. USA 1995, 92, 3376–3380. [Google Scholar] [CrossRef] [Green Version]
- Di Paola, R.; Impellizzeri, D.; Torre, A.; Mazzon, E.; Cappellani, A.; Faggio, C.; Esposito, E.; Trischitta, F.; Cuzzocrea, S. Effects of palmitoylethanolamide on intestinal injury and inflammation caused by ischemia-reperfusion in mice. J. Leukoc. Biol. 2012, 91, 911–920. [Google Scholar] [CrossRef] [PubMed]
- Borrelli, F.; Romano, B.; Petrosino, S.; Pagano, E.; Capasso, R.; Coppola, D.; Battista, G.; Orlando, P.; Di Marzo, V.; Izzo, A.A. Palmitoylethanolamide, a naturally occurring lipid, is an orally effective intestinal anti-inflammatory agent. Br. J. Pharmacol. 2015, 172, 142–158. [Google Scholar] [CrossRef] [Green Version]
- Misto, A.; Provensi, G.; Vozella, V.; Passani, M.B.; Piomelli, D. Mast cell-derived histamine regulates liver ketogenesis via oleoylethanolamide signaling. Cell Metab. 2019, 29, 91–102e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, J.; Kim, J.; Oveisi, F.; Astarita, G.; Piomelli, D. Targeted enhancement of oleoylethanolamide production in proximal small intestine induces across-meal satiety in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 295, R45–R50. [Google Scholar] [CrossRef] [Green Version]
- Golderer, G.; Werner, E.R.; Leitner, S.; Grobner, P.; Werner-Felmayer, G. Nitric oxide synthase is induced in sporulation of Physarum polycephalum. Genes Dev. 2001, 15, 1299–1309. [Google Scholar] [CrossRef] [Green Version]
- Hall, D.A.; Vander Kooi, C.W.; Stasik, C.N.; Stevens, S.Y.; Zuiderweg, E.R.; Matthews, R.G. Mapping the interactions between flavodoxin and its physiological partners flavodoxin reductase and cobalamin-dependent methionine synthase. Proc. Natl. Acad. Sci. USA 2001, 98, 9521–9526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radomski, M.W.; Palmer, R.M.; Moncada, S. Modulation of platelet aggregation by an L-arginine-nitric oxide pathway. Trends Pharmacol. Sci. 1991, 12, 87–88. [Google Scholar] [CrossRef]
- Escriva, H.; Safi, R.; Hanni, C.; Langlois, M.C.; Saumitou-Laprade, P.; Stehelin, D.; Capron, A.; Pierce, R.; Laudet, V. Ligand binding was acquired during evolution of nuclear receptors. Proc. Natl. Acad. Sci. USA 1997, 94, 6803–6808. [Google Scholar] [CrossRef] [Green Version]
- Zhou, T.; Yan, X.; Wang, G.; Liu, H.; Gan, X.; Zhang, T.; Wang, J.; Li, L. Evolutionary pattern and regulation analysis to support why diversity functions existed within PPAR gene family members. Biomed. Res. Int. 2015, 2015, 613910. [Google Scholar] [CrossRef] [Green Version]
- Crawford, N.M. Mechanisms for nitric oxide synthesis in plants. J. Exp. Bot. 2006, 57, 471–478. [Google Scholar] [CrossRef] [Green Version]
- Dohrmann, M.; Worheide, G. Dating early animal evolution using phylogenomic data. Sci. Rep. 2017, 7, 3599. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PPARα Agonists and Antagonists | |
|---|---|
| Natural agonists |  |
| Synthetic agonists |  |
| Agonists applied in clinic: fibrate derivatives |  |
| Synthetic antagonists |  |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grabacka, M.; Pierzchalska, M.; Płonka, P.M.; Pierzchalski, P. The Role of PPAR Alpha in the Modulation of Innate Immunity. Int. J. Mol. Sci. 2021, 22, 10545. https://doi.org/10.3390/ijms221910545
Grabacka M, Pierzchalska M, Płonka PM, Pierzchalski P. The Role of PPAR Alpha in the Modulation of Innate Immunity. International Journal of Molecular Sciences. 2021; 22(19):10545. https://doi.org/10.3390/ijms221910545
Chicago/Turabian StyleGrabacka, Maja, Małgorzata Pierzchalska, Przemysław M. Płonka, and Piotr Pierzchalski. 2021. "The Role of PPAR Alpha in the Modulation of Innate Immunity" International Journal of Molecular Sciences 22, no. 19: 10545. https://doi.org/10.3390/ijms221910545
APA StyleGrabacka, M., Pierzchalska, M., Płonka, P. M., & Pierzchalski, P. (2021). The Role of PPAR Alpha in the Modulation of Innate Immunity. International Journal of Molecular Sciences, 22(19), 10545. https://doi.org/10.3390/ijms221910545