
The PPARα and PPARγ Epigenetic Landscape in Cancer and Immune and Metabolic Disorders


Abstract
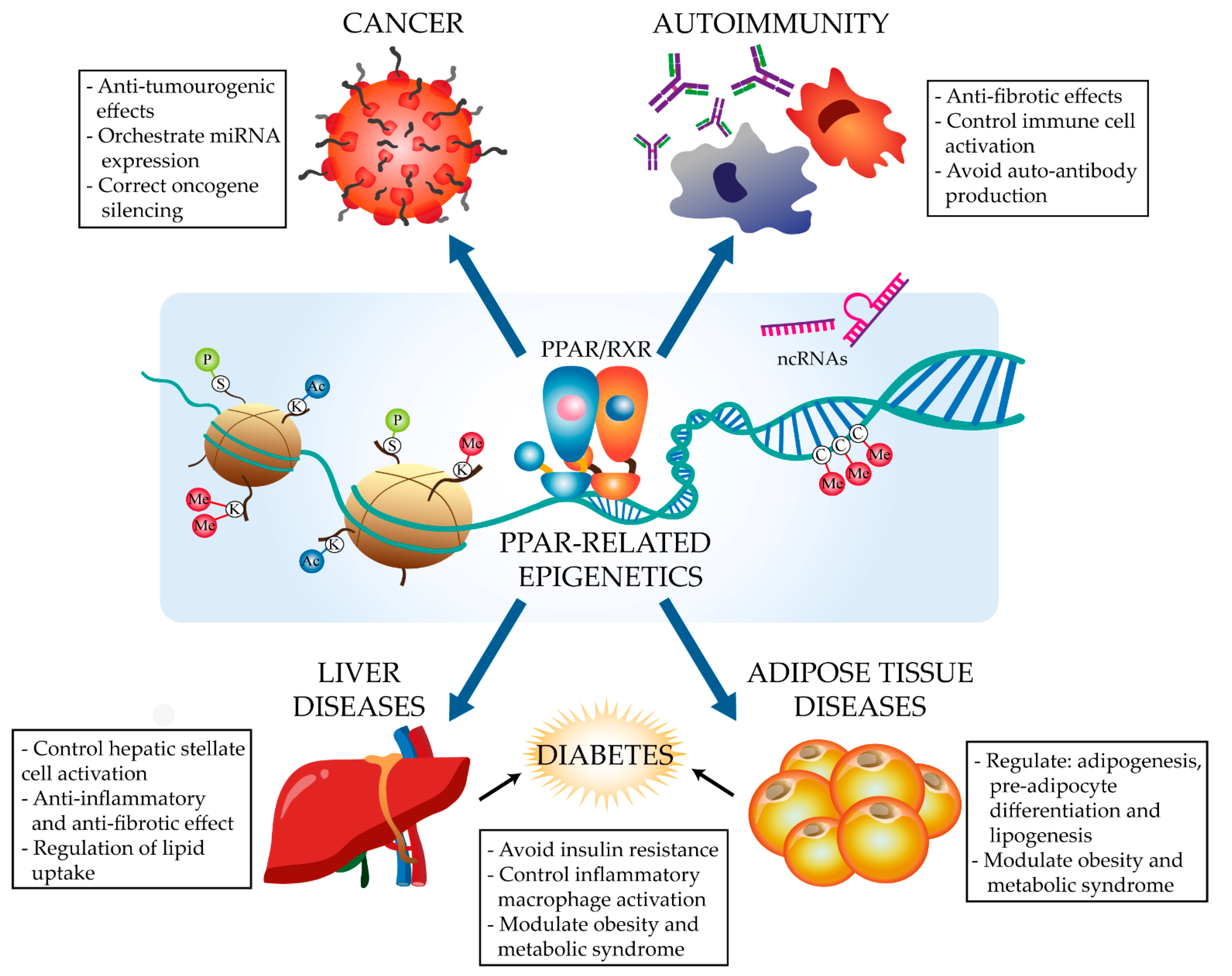
:1. Introduction
1.1. Peroxisome Proliferator Activated Receptors
1.2. Epigenetics
1.2.1. Major Epigenetic Modifications
DNA Methylation
Histone Modification
Non-Coding RNAs
2. The PPARα and PPARγ Epigenetic Landscape in Disease
2.1. Cancer
2.1.1. Colorectal Cancer
2.1.2. Liver Cancer
2.1.3. Other Cancers

{kind=link}
| Condition | PPAR Isoform | Epigenetic Player | Effect | References |
|---|---|---|---|---|
| Colorectal cancer | PPARα | miR-506 | PPARα expression inhibition in a hydroxicamptothecin resistant colon cancer cell line. | [62] |
| DNMT1 | Absence of PPARα caused P21 and P27 methylation by DNMT1. | [68] | ||
| PPARγ | miR-27b, miR-130b and miR-138 | Potential downregulation of PPARγ. | [53] | |
| UHRF1 | Epigenetic PPARγ inactivation in human-derived CRC cell lines. | [64] | ||
| Promoter hypermethylation | Hypermethylation of Pparg promoter caused PPARγ suppression. | [53] | ||
| Hepatocellular carcinoma | PPARα | miR-9 | Putative biding sites to PPARα 3’ UTR. | [75] |
| PPARγ | miR-30, miR-29c and miR-338 | Antifibrotic miRNAs regulated by PPARγ during HCC-related liver fibrosis. | [71] | |
| miR-27a | PPARγ inhibition in hepatocarcinoma cells. | [72] | ||
| Thyroid cancer | PPARγ | miR-27a | no relation obsrved yet. | [77] |
| Lung cancer | PPARγ | Promoter methylation | Significantly loss of 5′-methylation. | [78] |
| Gingivo-buccal oral squamous cell carcinoma | PPARγ | DNMTs | DNA methyltransferase inhibitors could renew PPARγ transcription. | [79] |
| Prostate cancer | PPARα | miR-17/92 | Possible direct PPARα targetting and dowregulation. | [80] |
2.2. Immune Disorders
2.2.1. Asthma
2.2.2. Systemic Lupus Erythematosus
2.2.3. Systemic Sclerosis (Scleroderma)
| Condition | PPAR Isoform | Epigenetic Player | Effect | References |
|---|---|---|---|---|
| Asthma | PPARα | DNA methylation | Human white blood cells showed DNA methylation in several PPAR pathway. | [84] |
| PPARγ | miR-21 | The profibroti Smad-TGFβ1-miR-21c axis was supress upon PPARγ pioglitazone activation. | [87] | |
| miR-98 | This profibrotic miRNA was downregulated upon PPARγ rosiglitazone activation. | [80] | ||
| Not specified | set of lncRNAs | Modulation of PPAR signalling pathway in sputa from eosinophilic asthma patients. | [90] | |
| Systemic Lupus Erythematosus | PPARγ | H4K20me1 and HDAC9 | Decreased H3K9ac and H3K18ac in the Pparg promoter leading to pro-inflammatory T cell cytokines and B cell auto-antibodies. | [93,94] |
| PPARγ | Sirt1 | Reduced PPARγ expression due to H3 deacetylation, avoiding M2 monocytic transition. | [85] | |
| Systemic sclerosis | PPARγ | p300 | Ligand-activated PPARγ blocks histone acetylatransferase p300 avoiding Smad3 pathway activation and Col1a2 locus histone 4 hyperacetylation. | [99,100,101] |
2.3. Metabolism-Related Diseases
2.3.1. Liver Diseases
2.3.2. Adipose Tissue Diseases
| Condition | PPAR Isoform | Epigenetic Player | Effect | References |
|---|---|---|---|---|
| Adipose tissue diseases | PPARα | Lsd1 | Targets PPARα to control beige adipocyte numbers | [153] |
| Bta-miR-199a-3p, -154c, -320a and -432 | Control lipid metabolism through PPARα | [154] | ||
| miR-519d | Suppresses PPARα protein translation in obese patients | [155] |
| Condition | PPAR Isoform | Epigenetic Player | Effect | References |
|---|---|---|---|---|
| Adipose tissue diseases | PPARγ | U90926 | Inhibition of Pparg transcription activity | [180] |
| NEAT1 | Regulation of Pparg splicing | [178] | ||
| HOTAIR | Increased expression of PPARγ | [182] | ||
| miR-155, miR-221 and miR-122 | Decreased expression of PPARγ in human bone-marrow-derived stromal cells | [184] | ||
| miR-540 | Decreased expression of PPARγ in adipose tissue-derived stromal cells | [185] | ||
| miR-27a/b, miR-31, miR-130/b, miR301a, miR-302a and miR-548d5p | Negative regulation of PPARγ and adipogenesis | [186,187] | ||
| miR-103, miR-143, miR-200a, miR-335 and miR-375 | Upregulation of Pparg | [187,188] | ||
| p400/Brd8 complex | Incorporation of the histone variant H2A.Z, which facilitates the expression of PPARγ target genes | [156] | ||
| MLL3 and MLL4 | Complex with ASC-2. Migration to the Pparg locus and methylation of H3K4, promoting enhanced Pparg expression | [159] | ||
| EZH2 | H3K27 methylation in the Hdac9c promoter. Enhanced adipogenesis | [162] | ||
| SETD8 (KMT5A) | Enhanced H4K20me marks in PPARγ target genes. | [93] | ||
| JMJD2C | Downregulation of PPARγ transcriptional activation | [166] | ||
| JHDM2A (JMJD1A) | Decreased H3K9me2 marks and facilitated recruitment of PPARγ, RXRα and PGC1α | [167,168] | ||
| Cyclin D1 | Interaction with p300 and HDACs to inhibit Pparg expression | [172] | ||
| SIRT1 | Blocked PPARγ mechanism of action | [173,174] | ||
| LncRNA TUG1 and miR-294 | Control fatty acid accumulation through GLUT4/PPARγ/AKT axis | [183] |
2.3.3. Insulin Sensitivity and Resistance: Type 2 Diabetes
| Condition | PPAR Isoform | Epigenetic Player | Effect | References |
|---|---|---|---|---|
| Insulin sensitivity and resistance: Type 2 Diabetes | PPARγ | miR27-a | Target of Pparg transcripts, promoting insulin resistance. Induction of inflammatory ATM activation in obesity | [213,214] |
| HDAC3 | Decreased expression of PPARγ in E3 rat livers. Correlated with inflammation and insulin resistance | [196,197,198] | ||
| SIRT1 | Control of the PPARγ acetylation status and its activity | [175] | ||
| DNMT3b | Pparg promoter methylation. Increased inflammatory macrophage activation and insulin resistance | [209,210] | ||
| DNMT3a | Fgf21 hypermethylation in human adipocytes, insulin resistance | [211] | ||
| DNMT1 | Adiponectin promoter methylation in obese mice. Glucose intolerance | [212] |
3. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Mirza, A.Z.; AlThagafi, I.I.; Shamshad, H. Role of PPAR receptor in different diseases and their ligands: Physiological importance and clinical implications. Eur. J. Med. Chem. 2019, 166, 502–513. [Google Scholar] [CrossRef] [PubMed]
- Amber-Vitos, O.; Chaturvedi, N.; Nachliel, E.; Gutman, M.; Tsfadia, Y. The effect of regulating molecules on the structure of the PPAR-RXR complex. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2016, 1861, 1852–1863. [Google Scholar] [CrossRef] [PubMed]
- Feige, J.N.; Gelman, L.; Michalik, L.; Desvergne, B.; Wahli, W. From molecular action to physiological outputs: Peroxisome proliferator-activated receptors are nuclear receptors at the crossroads of key cellular functions. Prog. Lipid Res. 2006, 45, 120–159. [Google Scholar] [CrossRef] [PubMed]
- Hong, F.; Pan, S.; Guo, Y.; Xu, P.; Zhai, Y. PPARs as Nuclear Receptors for Nutrient and Energy Metabolism. Molecules 2019, 24, 2545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grygiel-Gorniak, B. Peroxisome proliferator-activated receptors and their ligands: Nutritional and clinical implications—A review. Nutr. J. 2014, 13, 17. [Google Scholar] [CrossRef] [Green Version]
- Hall, J.A.; Rusten, M.; AbuGhazaleh, R.D.; Wuertz, B.; Souksavong, V.; Escher, P.; Ondrey, F. Effects of PPAR-gamma agonists on oral cancer cell lines: Potential horizons for chemopreventives and adjunctive therapies. Head Neck 2020, 42, 2542–2554. [Google Scholar] [CrossRef] [PubMed]
- Zoete, V.; Grosdidier, A.; Michielin, O. Peroxisome proliferator-activated receptor structures: Ligand specificity, molecular switch and interactions with regulators. Biochim. Biophys. Acta 2007, 1771, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Font-Diaz, J.; Jimenez-Panizo, A.; Caelles, C.; Vivanco, M.D.; Perez, P.; Aranda, A.; Estébanez-Perpina, E.; Castrillo, A.; Ricote, M.; Valledor, A.F. Nuclear receptors: Lipid and hormone sensors with essential roles in the control of cancer development. Semin. Cancer Biol. 2021, 73, 58–75. [Google Scholar] [CrossRef]
- Fougerat, A.; Montagner, A.; Loiseau, N.; Guillou, H.; Wahli, W. Peroxisome Proliferator-Activated Receptors and Their Novel Ligands as Candidates for the Treatment of Non-Alcoholic Fatty Liver Disease. Cells 2020, 9, 1638. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, J.; Luo, S.; Zhan, Y.; Lu, Q. The roles of PPARgamma and its agonists in autoimmune diseases: A comprehensive review. J. Autoimmun. 2020, 113, 102510. [Google Scholar] [CrossRef]
- Nobs, S.P.; Natali, S.; Pohlmeier, L.; Okreglicka, K.; Schneider, C.; Kurrer, M.; Sallusto, F.; Kopf, M. PPARgamma in dendritic cells and T cells drives pathogenic type-2 effector responses in lung inflammation. J. Exp. Med. 2017, 214, 3015–3035. [Google Scholar] [CrossRef] [PubMed]
- Housley, W.J.; Adams, C.O.; Vang, A.G.; Brocke, S.; Nichols, F.C.; Lacombe, M.; Rajan, T.V.; Clark, R.B. Peroxisome Proliferator-Activated Receptor gamma Is Required for CD4+ T Cell-Mediated Lymphopenia-Associated Autoimmunity. J. Immunol. 2011, 187, 4161–4169. [Google Scholar] [CrossRef] [Green Version]
- Porcuna, J.; Menendez-Gutierrez, M.P.; Ricote, M. Molecular control of tissue-resident macrophage identity by nuclear receptors. Curr. Opin. Pharmacol. 2020, 53, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Tronick, E.; Hunter, R.G. Waddington, Dynamic Systems, and Epigenetics. Front. Behav. Neurosci. 2016, 10, 107. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [Green Version]
- Rajabi, H.; Tagde, A.; Alam, M.; Bouillez, A.; Pitroda, S.; Suzuki, Y.; Kufe, D. DNA methylation by DNMT1 and DNMT3b methyltransferases is driven by the MUC1-C oncoprotein in human carcinoma cells. Oncogene 2016, 35, 6439–6445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, Y.M.; Mahar, M.; Ewan, E.E.; Leahy, K.M.; Zhao, G.; Cavalli, V. Epigenetic regulator UHRF1 inactivates REST and growth suppressor gene expression via DNA methylation to promote axon regeneration. Proc. Natl. Acad. Sci. USA 2018, 115, E12417–E12426. [Google Scholar] [CrossRef] [Green Version]
- Ginno, P.A.; Gaidatzis, D.; Feldmann, A.; Hoerner, L.; Imanci, D.; Burger, L.; Zilbermann, F.; Peters, A.; Edenhofer, F.; Smallwood, S.A.; et al. A genome-scale map of DNA methylation turnover identifies site-specific dependencies of DNMT and TET activity. Nat. Commun. 2020, 11, 2680. [Google Scholar] [CrossRef]
- Giaimo, B.D.; Ferrante, F.; Herchenrother, A.; Hake, S.B.; Borggrefe, T. The histone variant H2A.Z in gene regulation. Epigenetics Chromatin 2019, 12, 37. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.L.; Grant, P.A. The Role of DNA Methylation and Histone Modifications in Transcriptional Regulation in Humans. Subcell Biochem. 2013, 61, 289–317. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, D.; Avvakumov, N.; Cote, J. Histone phosphorylation: A chromatin modification involved in diverse nuclear events. Epigenetics 2012, 7, 1098–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Blythe, S.A.; Cha, S.W.; Tadjuidje, E.; Heasman, J.; Klein, P.S. beta-Catenin Primes Organizer Gene Expression by Recruiting a Histone H3 Arginine 8 Methyltransferase, Prmt2. Dev. Cell 2010, 19, 220–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svobodova Kovarikova, A.; Legartova, S.; Krejci, J.; Bartova, E. H3K9me3 and H4K20me3 represent the epigenetic landscape for 53BP1 binding to DNA lesions. Aging (Albany NY) 2018, 10, 2585–2605. [Google Scholar] [CrossRef]
- Hervouet, E.; Peixoto, P.; Delage-Mourroux, R.; Boyer-Guittaut, M.; Cartron, P.-F. Specific or not specific recruitment of DNMTs for DNA methylation, an epigenetic dilemma. Clin. Epigenet. 2018, 10, 17. [Google Scholar] [CrossRef]
- Di Croce, L.; Raker, V.; Corsaro, M.; Fazi, F.; Fanelli, M.; Faretta, M.; Fuks, F.; Lo Coco, F.; Kouzarides, T.; Nervi, C.; et al. Methyltransferase Recruitment and DNA Hyermethylation of Target Promoters by an Oncogenic Transcription Factor. Science 2002, 295, 1079–1082. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, K.J.; Scelfo, A.; Jammula, S.; Cuomo, A.; Barozzi, I.; Stutzer, A.; Fischle, W.; Bonaldi, T.; Pasini, D. Polycomb-Dependent H3K27me1 and H3K27me2 Regulate Active Transcription and Enhancer Fidelity. Mol. Cell 2014, 53, 49–62. [Google Scholar] [CrossRef] [Green Version]
- Kaikkonen, M.U.; Spann, N.J.; Heinz, S.; Romanoski, C.E.; Allison, K.A.; Stender, J.D.; Chun, H.B.; Tough, D.F.; Prinjha, R.K.; Benner, C.; et al. Remodeling of the Enhancer Landscape during Macrophage Activation Is Coupled to Enhancer Transcription. Mol. Cell 2013, 51, 310–325. [Google Scholar] [CrossRef] [Green Version]
- Lara-Astiaso, D.; Weiner, A.; Lorenzo-Vivas, E.; Zaretsky, I.; Jaitin, D.A.; David, E.; Keren-Shaul, H.; Mildner, A.; Winter, D.; Jung, S.; et al. Immunogenetics. Chromatin state dynamics during blood formation. Science 2014, 345, 943–949. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Wu, W.; Chen, Q.; Chen, M. Non-Coding RNAs and their Integrated Networks. J. Integr. Bioinform. 2019, 16. [Google Scholar] [CrossRef]
- Frias-Lasserre, D.; Villagra, C.A. The Importance of ncRNAs as Epigenetic Mechanisms in Phenotypic Variation and Organic Evolution. Front. Microbiol. 2017, 8, 2483. [Google Scholar] [CrossRef]
- Ratti, M.; Lampis, A.; Ghidini, M.; Salati, M.; Mirchev, M.B.; Valeri, N.; Hahne, J.C. MicroRNAs (miRNAs) and Long Non-Coding RNAs (lncRNAs) as New Tools for Cancer Therapy: First Steps from Bench to Bedside. Target. Oncol. 2020, 15, 261–278. [Google Scholar] [CrossRef]
- Zhang, W.; Qu, J.; Liu, G.-H.; Belmonte, J.C.I. The ageing epigenome and its rejuvenation. Nat. Rev. Mol. Cell Biol. 2020, 21, 137–150. [Google Scholar] [CrossRef]
- Zhao, Y.; Zheng, D.; Cvekl, A. Profiling of chromatin accessibility and identification of general cis-regulatory mechanisms that control two ocular lens differentiation pathways. Epigenet. Chromatin 2019, 12, 27. [Google Scholar] [CrossRef]
- Chen, S.; Yang, J.; Wei, Y.; Wei, X. Epigenetic regulation of macrophages: From homeostasis maintenance to host defense. Cell. Mol. Immunol. 2020, 17, 36–49. [Google Scholar] [CrossRef] [Green Version]
- Elnady, H.G.; Sherif, L.S.; Kholoussi, N.M.; Ali Azzam, M.; Foda, A.R.; Helwa, I.; Sabry, R.N.; Eissa, E.; Fahmy, R.F. Aberrant Expression of Immune-related MicroRNAs in Pediatric Patients with Asthma. Int. J. Mol. Cell Med. 2020, 9, 246–255. [Google Scholar]
- Neganova, M.E.; Klochkov, S.G.; Aleksandrova, Y.R.; Aliev, G. Histone modifications in epigenetic regulation of cancer: Perspectives and achieved progress. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2020. [Google Scholar] [CrossRef]
- Islam, A.; Mohammad, E.; Khan, M.A. Aberration of the modulatory functions of intronic microRNA hsa-miR-933 on its host gene ATF2 results in type II diabetes mellitus and neurodegenerative disease development. Hum. Genom. 2020, 14, 34. [Google Scholar] [CrossRef]
- Yang, Y.; Wicks, J.; Haitchi, H.M.; Powell, R.M.; Manuyakorn, W.; Howarth, P.H.; Holgate, S.T.; Davies, D.E. Regulation of A Disintegrin and Metalloprotease-33Expression by Transforming Growth Factor-β. Am. J. Respir. Cell Mol. Biol. 2012, 46, 633–640. [Google Scholar] [CrossRef] [Green Version]
- Harb, H.; Raedler, D.; Ballenberger, N.; Bock, A.; Kesper, D.A.; Renz, H.; Schaub, B. Childhood allergic asthma is associated with increased IL-13 and FOXP3 histone acetylation. J. Allergy Clin. Immunol. 2015, 136, 200–202. [Google Scholar] [CrossRef]
- Le, T.N.; Williams, S.R.; Alaimo, J.T.; Elsea, S.H. Genotype and phenotype correlation in 103 individuals with 2q37 deletion syndrome reveals incomplete penetrance and supports HDAC4 as the primary genetic contributor. Am. J. Med Genet. Part A 2019, 179, 782–791. [Google Scholar] [CrossRef]
- Wu, Y.; Cui, W.; Zhang, D.; Wu, W.; Yang, Z. The shortening of leukocyte telomere length relates to DNA hypermethylation of LINE-1 in type 2 diabetes mellitus. Oncotarget 2017, 8, 73964–73973. [Google Scholar] [CrossRef]
- Sun, Z.H.; Liu, Y.H.; Liu, J.D.; Xu, D.D.; Li, X.F.; Meng, X.M.; Ma, T.T.; Huang, C.; Li, J. MeCP2 Regulates PTCH1 Expression Through DNA Methylation in Rheumatoid Arthritis. Inflammation 2017, 40, 1497–1508. [Google Scholar] [CrossRef]
- Raveche, E.S.; Salerno, E.; Scaglione, B.J.; Manohar, V.; Abbasi, F.; Lin, Y.C.; Fredrickson, T.; Landgraf, P.; Ramachandra, S.; Huppi, K.; et al. Abnormal microRNA-16 locus with synteny to human 13q14 linked to CLL in NZB mice. Blood 2007, 109, 5079–5086. [Google Scholar] [CrossRef] [Green Version]
- Zernecke, A.; Bidzhekov, K.; Noels, H.; Shagdarsuren, E.; Gan, L.; Denecke, B.; Hristov, M.; Koppel, T.; Jahantigh, M.N.; Lutgens, E.; et al. Delivery of MicroRNA-126 by Apoptotic Bodies Induces CXCL12-Dependent Vascular Protection. Sci. Signal. 2009, 2, ra81. [Google Scholar] [CrossRef]
- Rana, Z.; Diermeier, S.; Hanif, M.; Rosengren, R.J. Understanding Failure and Improving Treatment Using HDAC Inhibitors for Prostate Cancer. Biomedicines 2020, 8, 22. [Google Scholar] [CrossRef] [Green Version]
- Dovey, O.M.; Foster, C.T.; Conte, N.; Edwards, S.A.; Edwards, J.M.; Singh, R.; Vassiliou, G.; Bradley, A.; Cowley, S.M. Histone deacetylase 1 and 2 are essential for normal T-cell development and genomic stability in mice. Blood 2013, 121, 1335–1344. [Google Scholar] [CrossRef] [Green Version]
- Tian, J.; Hu, L.; Li, X.; Geng, J.; Dai, M.; Bai, X. MicroRNA-130b promotes lung cancer progression via PPARγ/VEGF-A/BCL-2-mediated suppression of apoptosis. J. Exp. Clin. Cancer Res. 2016, 35, 105. [Google Scholar] [CrossRef] [Green Version]
- Koga, H.; Sakisaka, S.; Harada, M.; Takagi, T.; Hanada, S.; Taniguchi, E.; Kawaguchi, T.; Sasatomi, K.; Kimura, R.; Hashimoto, O.; et al. Involvement of p21(WAF1/Cip1), p27(Kip1), and p18(INK4c) in troglitazone-induced cell-cycle arrest in human hepatoma cell lines. Hepatology 2001, 33, 1087–1097. [Google Scholar] [CrossRef]
- Rawla, P.; Sunkara, T.; Barsouk, A. Epidemiology of colorectal cancer: Incidence, mortality, survival, and risk factors. Prz. Gastroenterol. 2019, 14, 89–103. [Google Scholar] [CrossRef]
- Park, J.-I.; Kwak, J.-Y. The Role of Peroxisome Proliferator-Activated Receptors in Colorectal Cancer. PPAR Res. 2012, 2012, 876418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tait, S.; Baldassarre, A.; Masotti, A.; Calura, E.; Martini, P.; Vari, R.; Scazzocchio, B.; Gessani, S.; Del Corno, M. Integrated Transcriptome Analysis of Human Visceral Adipocytes Unravels Dysregulated microRNA-Long Non-coding RNA-mRNA Networks in Obesity and Colorectal Cancer. Front. Oncol. 2020, 10, 1089. [Google Scholar] [CrossRef]
- Motawi, T.K.; Shaker, O.G.; Ismail, M.F.; Sayed, N.H. Peroxisome Proliferator-Activated Receptor Gamma in Obesity and Colorectal Cancer: The Role of Epigenetics. Sci. Rep. 2017, 7, 10714. [Google Scholar] [CrossRef]
- Baffa, R.; Fassan, M.; Volinia, S.; O’Hara, B.; Liu, C.G.; Palazzo, J.P.; Gardiman, M.; Rugge, M.; Gomella, L.G.; Croce, C.M.; et al. MicroRNA expression profiling of human metastatic cancers identifies cancer gene targets. J. Pathol. 2009, 219, 214–221. [Google Scholar] [CrossRef]
- Colangelo, T.; Fucci, A.; Votino, C.; Sabatino, L.; Pancione, M.; Laudanna, C.; Binaschi, M.; Bigioni, M.; Maggi, C.A.; Parente, D.; et al. MicroRNA-130b Promotes Tumor Development and Is Associated with Poor Prognosis in Colorectal Cancer. Neoplasia 2013, 15, 1086–1099. [Google Scholar] [CrossRef]
- Karbiener, M.; Fischer, C.; Nowitsch, S.; Opriessnig, P.; Papak, C.; Ailhaud, G.; Dani, C.; Amri, E.Z.; Scheideler, M. microRNA miR-27b impairs human adipocyte differentiation and targets PPARγ. Biochem. Biophys. Res. Commun. 2009, 390, 247–251. [Google Scholar] [CrossRef]
- Lin, Q.; Gao, Z.; Alarcon, R.M.; Ye, J.; Yun, Z. A role of miR-27 in the regulation of adipogenesis. FEBS J. 2009, 276, 2348–2358. [Google Scholar] [CrossRef]
- Yi, R.; Li, Y.; Wang, F.; Gu, J.; Isaji, T.; Li, J.; Qi, R.; Zhu, X.; Zhao, Y. Transforming growth factor (TGF) β1 acted through miR-130b to increase integrin α5 to promote migration of colorectal cancer cells. Tumor Biol. 2016, 37, 10763–10773. [Google Scholar] [CrossRef]
- Matsuyama, R.; Okuzaki, D.; Okada, M.; Oneyama, C. Micro RNA -27b suppresses tumor progression by regulating ARFGEF 1 and focal adhesion signaling. Cancer Sci. 2016, 107, 28–35. [Google Scholar] [CrossRef]
- Qin, Y.Z.; Xie, X.C.; Liu, H.Z.; Lai, H.; Qiu, H.; Ge, L.Y. Screening and preliminary validation of miRNAs with the regulation of hTERT in colorectal cancer. Oncol. Rep. 2015, 33, 2728–2736. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Yu, H.; Yi, S.; Peng, X.; Su, P.; Xiao, Z.; Liu, R.; Tang, A.; Li, X.; Liu, F.; et al. The tumor suppressor miR-138-5p targets PD-L1 in colorectal cancer. Oncotarget 2016, 7, 45370–45384. [Google Scholar] [CrossRef] [Green Version]
- Tong, J.L.; Zhang, C.P.; Nie, F.; Xu, X.T.; Zhu, M.M.; Xiao, S.D.; Ran, Z.H. MicroRNA 506 regulates expression of PPAR alpha in hydroxycamptothecin-resistant human colon cancer cells. FEBS Lett. 2011, 585, 3560–3568. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Li, H.; Sun, L.; Shen, S.; Zhou, Q.; Yuan, Y.; Xing, C. Epigenetic Alternations of MicroRNAs and DNA Methylation Contribute to Liver Metastasis of Colorectal Cancer. Dig. Dis. Sci. 2019, 64, 1523–1534. [Google Scholar] [CrossRef] [PubMed]
- Sabatino, L.; Fucci, A.; Pancione, M.; Carafa, V.; Nebbioso, A.; Pistore, C.; Babbio, F.; Votino, C.; Laudanna, C.; Ceccarelli, M.; et al. UHRF1 coordinates peroxisome proliferator activated receptor gamma (PPARG) epigenetic silencing and mediates colorectal cancer progression. Oncogene 2012, 31, 5061–5072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, B.B.; Lee, E.J.; Jung, E.H.; Chun, H.K.; Chang, D.K.; Song, S.Y.; Park, J.; Kim, D.H. Aberrant Methylation of APC, MGMT, RASSF2A, and Wif-1 Genes in Plasma as a Biomarker for Early Detection of Colorectal Cancer. Clin. Cancer Res. 2009, 15, 6185–6191. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Chu, F.H.; Xu, W.R.; Sun, J.Q.; Sun, X.; Ma, X.M.; Yu, M.W.; Yang, G.W.; Wang, X.M. Identification of regulatory role of DNA methylation in colon cancer gene expression via systematic bioinformatics analysis. Medicine 2017, 96, e8487. [Google Scholar] [CrossRef] [PubMed]
- Pancione, M.; Sabatino, L.; Fucci, A.; Carafa, V.; Nebbioso, A.; Forte, N.; Febbraro, A.; Parente, D.; Ambrosino, C.; Normanno, N.; et al. Epigenetic Silencing of Peroxisome Proliferator-Activated Receptor γ Is a Biomarker for Colorectal Cancer Progression and Adverse Patients’ Outcome. PLoS ONE 2010, 5, e14229. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Xie, C.; Brocker, C.N.; Fan, J.; Wu, X.; Feng, L.; Wang, Q.; Zhao, J.; Lu, D.; Tandon, M.; et al. Intestinal PPARα Protects Against Colon Carcinogenesis via Regulation of Methyltransferases DNMT1 and PRMT6. Gastroenterology 2019, 157, 744–759.e4. [Google Scholar] [CrossRef] [PubMed]
- Yaghoubizadeh, M.; Pishkar, L.; Basati, G. Aberrant Expression of Peroxisome Proliferator-Activated Receptors in Colorectal Cancer and Their Association with Cancer Progression and Prognosis. Gastrointest. Tumors 2020, 7, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Nagano, T.; Shah, Y.; Cheung, C.; Ito, S.; Gonzalez, F.J. The PPARα-Humanized Mouse: A Model to Investigate Species Differences in Liver Toxicity Mediated by PPARα. Toxicol. Sci. 2008, 101, 132–139. [Google Scholar] [CrossRef] [Green Version]
- Winkler, I.; Bitter, C.; Winkler, S.; Weichenhan, D.; Thavamani, A.; Hengstler, J.G.; Borkham-Kamphorst, E.; Kohlbacher, O.; Plass, C.; Geffers, R.; et al. Identification of Pparγ-modulated miRNA hubs that target the fibrotic tumor microenvironment. Proc. Natl. Acad. Sci. USA 2020, 117, 454–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Li, J.; Fei, B.-Y.; Shao, D.; Pan, Y.; Mo, Z.-H.; Sun, B.-Z.; Zhang, D.; Zheng, X.; Zhang, M.; et al. MiR-27a Promotes Hepatocellular Carcinoma Cell Proliferation Through Suppression of its Target Gene Peroxisome Proliferator-activated Receptor γ. Chin. Med. J. 2015, 128, 941–947. [Google Scholar] [CrossRef] [PubMed]
- Tombolan, L.; Zampini, M.; Casara, S.; Boldrin, E.; Zin, A.; Bisogno, G.; Rosolen, A.; De Pitta, C.; Lanfranchi, G. MicroRNA-27a Contributes to Rhabdomyosarcoma Cell Proliferation by Suppressing RARA and RXRA. PLoS ONE 2015, 10, e0125171. [Google Scholar] [CrossRef]
- Oda, Y.; Nakajima, M.; Tsuneyama, K.; Takamiya, M.; Aoki, Y.; Fukami, T.; Yokoi, T. Retinoid X receptor α in human liver is regulated by miR-34a. Biochem. Pharmacol. 2014, 90, 179–187. [Google Scholar] [CrossRef] [Green Version]
- Cui, M.; Xiao, Z.; Wang, Y.; Zheng, M.; Song, T.; Cai, X.; Sun, B.; Ye, L.; Zhang, X. Long Noncoding RNA HULC Modulates Abnormal Lipid Metabolism in Hepatoma Cells through an miR-9–Mediated RXRA Signaling Pathway. Cancer Res. 2015, 75, 846–857. [Google Scholar] [CrossRef] [Green Version]
- Catalano, M.G.; Poli, R.; Pugliese, M.; Fortunati, N.; Boccuzzi, G. Emerging molecular therapies of advanced thyroid cancer. Mol. Asp. Med. 2010, 31, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Toraih, E.A.; Fawzy, M.S.; Abushouk, A.I.; Shaheen, S.; Hobani, Y.H.; Alruwetei, A.M.; Mansouri, O.A.; Kandil, E.; Badran, D.I. Prognostic value of the miRNA-27a and PPAR/RXRα signaling axis in patients with thyroid carcinoma. Epigenomics 2020, 12, 1825–1843. [Google Scholar] [CrossRef]
- Herrera, C.L.; Kim, D.Y.; Kumar, S.R.; Bryan, J.N. Peroxisome proliferator activated receptor γ protein expression is asymmetrically distributed in primary lung tumor and metastatic to lung osteosarcoma samples and does not correlate with gene methylation. BMC Veter-Res. 2015, 11, 230. [Google Scholar] [CrossRef] [Green Version]
- Das, D.; Ghosh, S.; Maitra, A.; Biswas, N.K.; Panda, C.K.; Roy, B.; Sarin, R.; Majumder, P.P. Epigenomic dysregulation-mediated alterations of key biological pathways and tumor immune evasion are hallmarks of gingivo-buccal oral cancer. Clin. Epigenet. 2019, 11, 178. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.-L.W.; Welsh, J.; Tenniswood, M. 1,25-Dihydroxyvitamin D3 modulates lipid metabolism in prostate cancer cells through miRNA mediated regulation of PPARA. J. Steroid Biochem. Mol. Biol. 2013, 136, 247–251. [Google Scholar] [CrossRef]
- Mohan, M.; Okeoma, C.M.; Sestak, K. Dietary Gluten and Neurodegeneration: A Case for Preclinical Studies. Int. J. Mol. Sci. 2020, 21, 5407. [Google Scholar] [CrossRef] [PubMed]
- Aprahamian, T.; Bonegio, R.G.; Weitzner, Z.; Gharakhanian, R.; Rifkin, I.R. Peroxisome proliferator-activated receptor gamma agonists in the prevention and treatment of murine systemic lupus erythematosus. Immunology 2014, 142, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Luo, G.; Li, X.; Chen, J.; Wu, J.; Peng, Y. PPARγ inhibits HMGB1 expression through upregulation of miR-142-3p in vitro and in vivo. Cell. Signal. 2016, 28, 158–164. [Google Scholar] [CrossRef]
- Jeong, A.; Imboden, M.; Ghantous, A.; Novoloaca, A.; Carsin, A.-E.; Kogevinas, M.; Schindler, C.; Lovison, G.; Herceg, Z.; Cuenin, C.; et al. DNA Methylation in Inflammatory Pathways Modifies the Association between BMI and Adult-Onset Non-Atopic Asthma. Int. J. Environ. Res. Public Health 2019, 16, 600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Luo, S.; Zhan, Y.; Wang, J.; Zhao, R.; Li, Y.; Zeng, J.; Lu, Q. Increased Expression of PPAR-γ Modulates Monocytes Into a M2-Like Phenotype in SLE Patients: An Implicative Protective Mechanism and Potential Therapeutic Strategy of Systemic Lupus Erythematosus. Front. Immunol. 2021, 11, 579372. [Google Scholar] [CrossRef]
- Banno, A.; Reddy, A.; Lakshmi, S.; Reddy, A.R.C. PPARs: Key Regulators of Airway Inflammation and Potential Therapeutic Targets in Asthma. Nucl. Recept. Res. 2018, 5, 101306. [Google Scholar] [CrossRef] [Green Version]
- Wongtrakool, C.; Ko, J.; Jang, A.J.; Grooms, K.; Chang, S.; Sylber, C.; Kosmider, B.; Bahmed, K.; Blackburn, M.R.; Sutliff, R.L.; et al. MicroRNA-98 reduces nerve growth factor expression in nicotine-induced airway remodeling. J. Biol. Chem. 2020, 295, 18051–18064. [Google Scholar] [CrossRef]
- Liu, L.; Pan, Y.; Zhai, C.; Zhu, Y.; Ke, R.; Shi, W.; Wang, J.; Yan, X.; Su, X.; Song, Y.; et al. Activation of peroxisome proliferation–activated receptor-γ inhibits transforming growth factor-β1-induced airway smooth muscle cell proliferation by suppressing Smad–miR-21 signaling. J. Cell. Physiol. 2018, 234, 669–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, L.; Wang, X.; Sun, Q.; Papakonstantinou, E.; S’Ng, C.; Tamm, M.; Stolz, D.; Roth, M. IgE Downregulates PTEN through MicroRNA-21-5p and Stimulates Airway Smooth Muscle Cell Remodeling. Int. J. Mol. Sci. 2019, 20, 875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.-J.; Mao, D.; Gao, W.; Hu, H. Peripheral whole blood lncRNA expression analysis in patients with eosinophilic asthma. Medicine 2018, 97, e9817. [Google Scholar] [CrossRef]
- Moulton, V.R.; Suárez-Fueyo, A.; Meidan, E.; Li, H.; Mizui, M.; Tsokos, G.C. Pathogenesis of Human Systemic Lupus Erythematosus: A Cellular Perspective. Trends Mol. Med. 2017, 23, 615–635. [Google Scholar] [CrossRef]
- Rőszer, T.; Menendez-Gutierrez, M.P.; Lefterova, M.I.; Alameda, D.; Nunez, V.; Lazar, M.A.; Fischer, T.; Ricote, M. Autoimmune kidney disease and impaired engulfment of apoptotic cells in mice with macrophage peroxisome proliferator-activated receptor gamma or retinoid X receptor alpha deficiency. J. Immunol. 2011, 186, 621–631. [Google Scholar] [CrossRef]
- Wakabayashi, K.-I.; Okamura, M.; Tsutsumi, S.; Nishikawa, N.S.; Tanaka, T.; Sakakibara, I.; Kitakami, J.-I.; Ihara, S.; Hashimoto, Y.; Hamakubo, T.; et al. The Peroxisome Proliferator-Activated Receptor γ/Retinoid X Receptor α Heterodimer Targets the Histone Modification Enzyme PR-Set7/Setd8 Gene and Regulates Adipogenesis through a Positive Feedback Loop. Mol. Cell. Biol. 2009, 29, 3544–3555. [Google Scholar] [CrossRef] [Green Version]
- Yan, K.; Cao, Q.; Reilly, C.; Young, N.; Garcia, B.A.; Mishra, N. Histone Deacetylase 9 Deficiency Protects against Effector T Cell-mediated Systemic Autoimmunity. J. Biol. Chem. 2011, 286, 28833–28843. [Google Scholar] [CrossRef] [Green Version]
- Hu, N.; Qiu, X.; Luo, Y.; Yuan, J.; Li, Y.; Lei, W.; Zhang, G.; Zhou, Y.; Su, Y.; Lu, Q. Abnormal histone modification patterns in lupus CD4+ T cells. J. Rheumatol. 2008, 35, 804–810. [Google Scholar]
- Xu, X.; Ramanujam, M.; Visvanathan, S.; Assassi, S.; Liu, Z.; Li, L. Transcriptional insights into pathogenesis of cutaneous systemic sclerosis using pathway driven meta-analysis assisted by machine learning methods. PLoS ONE 2020, 15, e0242863. [Google Scholar] [CrossRef]
- Wei, J.; Ghosh, A.K.; Sargent, J.L.; Komura, K.; Wu, M.; Huang, Q.-Q.; Jain, M.; Whitfield, M.L.; Feghali-Bostwick, C.; Varga, J. PPARγ Downregulation by TGFß in Fibroblast and Impaired Expression and Function in Systemic Sclerosis: A Novel Mechanism for Progressive Fibrogenesis. PLoS ONE 2010, 5, e13778. [Google Scholar] [CrossRef] [Green Version]
- Kohno, S.; Endo, H.; Hashimoto, A.; Hayashi, I.; Murakami, Y.; Kitasato, H.; Kojima, F.; Kawai, S.; Kondo, H. Inhibition of skin sclerosis by 15deoxy Δ12,14-prostaglandin J2 and retrovirally transfected prostaglandin D synthase in a mouse model of bleomycin-induced scleroderma. Biomed. Pharmacother. 2006, 60, 18–25. [Google Scholar] [CrossRef]
- Nuwormegbe, S.A.; Sohn, J.H.; Kim, S.W. A PPAR-Gamma Agonist Rosiglitazone Suppresses Fibrotic Response in Human Pterygium Fibroblasts by Modulating the p38 MAPK Pathway. Investig. Opthalmol. Vis. Sci. 2017, 58, 5217–5226. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.K.; Bhattacharyya, S.; Wei, J.; Kim, S.; Barak, Y.; Mori, Y.; Varga, J. Peroxisome proliferator-activated receptor-γ abrogates Smad-dependent collagen stimulation by targeting the p300 transcriptional coactivator. FASEB J. 2009, 23, 2968–2977. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Xia, Y.; Lin, X.; Feng, X.-H.; Wang, Y. Smad3 signaling activates bone marrow-derived fibroblasts in renal fibrosis. Lab. Investig. 2014, 94, 545–556. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.K.; Bhattacharyya, S.; Lakos, G.; Chen, S.-J.; Mori, Y.; Varga, J. Disruption of transforming growth factor ? Signaling and profibrotic responses in normal skin fibroblasts by peroxisome proliferator-activated receptor? Arthritis Rheum. 2004, 50, 1305–1318. [Google Scholar] [CrossRef]
- Connolly, M.K. Systemic sclerosis (scleroderma): Remaining challenges. Ann. Transl. Med. 2021, 9, 438. [Google Scholar] [CrossRef]
- Ding, W.; Pu, W.; Wang, L.; Jiang, S.; Zhou, X.; Tu, W.; Yu, L.; Zhang, J.; Guo, S.; Liu, Q.; et al. Genome-Wide DNA Methylation Analysis in Systemic Sclerosis Reveals Hypomethylation of IFN-Associated Genes in CD4+ and CD8+ T Cells. J. Investig. Dermatol. 2018, 138, 1069–1077. [Google Scholar] [CrossRef] [Green Version]
- Hemmatazad, H.; Rodrigues, H.M.; Maurer, B.; Brentano, F.; Pileckyte, M.; Distler, J.H.W.; Gay, R.E.; Michel, B.A.; Gay, S.; Huber, L.C.; et al. Histone deacetylase 7, a potential target for the antifibrotic treatment of systemic sclerosis. Arthritis Rheum. 2009, 60, 1519–1529. [Google Scholar] [CrossRef] [Green Version]
- Huber, L.C.; Distler, J.H.W.; Moritz, F.; Hemmatazad, H.; Hauser, T.; Michel, B.A.; Gay, R.E.; Matucci-Cerinic, M.; Gay, S.; Distler, O.; et al. Trichostatin A prevents the accumulation of extracellular matrix in a mouse model of bleomycin-induced skin fibrosis. Arthritis Rheum. 2007, 56, 2755–2764. [Google Scholar] [CrossRef]
- Saklayen, M.G. The Global Epidemic of the Metabolic Syndrome. Curr. Hypertens. Rep. 2018, 20, 12. [Google Scholar] [CrossRef] [Green Version]
- Sherling, D.H.; Perumareddi, P.; Hennekens, C.H. Metabolic Syndrome. J. Cardiovasc. Pharmacol. Ther. 2017, 22, 365–367. [Google Scholar] [CrossRef]
- McCracken, E.; Monaghan, M.; Sreenivasan, S. Pathophysiology of the metabolic syndrome. Clin. Dermatol. 2018, 36, 14–20. [Google Scholar] [CrossRef]
- Hong, F.; Xu, P.; Zhai, Y. The Opportunities and Challenges of Peroxisome Proliferator-Activated Receptors Ligands in Clinical Drug Discovery and Development. Int. J. Mol. Sci. 2018, 19, 2189. [Google Scholar] [CrossRef] [Green Version]
- Bansal, G.; Thanikachalam, P.V.; Maurya, R.K.; Chawla, P.; Ramamurthy, S. An overview on medicinal perspective of thiazolidine-2,4-dione: A remarkable scaffold in the treatment of type 2 diabetes. J. Adv. Res. 2020, 23, 163–205. [Google Scholar] [CrossRef] [PubMed]
- Hevener, A.L.; Olefsky, J.M.; Reichart, D.; Nguyen, M.T.; Bandyopadyhay, G.; Leung, H.Y.; Watt, M.J.; Benner, C.; Febbraio, M.A.; Nguyen, A.K.; et al. Macrophage PPAR gamma is required for normal skeletal muscle and hepatic insulin sensitivity and full antidiabetic effects of thiazolidinediones. J. Clin. Investig. 2007, 117, 1658–1669. [Google Scholar] [CrossRef]
- Brunt, E.M.; Wong, V.W.-S.; Nobili, V.; Day, C.P.; Sookoian, S.; Maher, J.J.; Bugianesi, E.; Sirlin, C.B.; Neuschwander-Tetri, B.A.; Rinella, M.E. Nonalcoholic fatty liver disease. Nat. Rev. Dis. Prim. 2015, 1, 15080. [Google Scholar] [CrossRef]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Hajri, T.; Zaiou, M.; Fungwe, T.; Ouguerram, K.; Besong, S. Epigenetic Regulation of Peroxisome Proliferator-Activated Receptor Gamma Mediates High-Fat Diet-Induced Non-Alcoholic Fatty Liver Disease. Cells 2021, 10, 1355. [Google Scholar] [CrossRef]
- Moran-Salvador, E.; Mann, J. Epigenetics and Liver Fibrosis. Cell. Mol. Gastroenterol. Hepatol. 2017, 4, 125–134. [Google Scholar] [CrossRef] [Green Version]
- Claveria-Cabello, A.; Colyn, L.; Arechederra, M.; Urman, J.M.; Berasain, C.; Avila, M.A.; Fernandez-Barrena, M.G. Epigenetics in Liver Fibrosis: Could HDACs Be a Therapeutic Target? Cells 2020, 9, 2321. [Google Scholar] [CrossRef]
- Sodum, N.; Kumar, G.; Bojja, S.L.; Kumar, N.; Rao, C.M. Epigenetics in NAFLD/NASH: Targets and therapy. Pharmacol. Res. 2021, 167, 105484. [Google Scholar] [CrossRef]
- Rodrigues, P.M.; Rodrigues, C.; Castro, R.E. Modulation of liver steatosis by miR-21/PPARα. Cell Death Discov. 2018, 4, 9. [Google Scholar] [CrossRef]
- Moody, L.; Xu, G.B.; Chen, H.; Pan, Y.-X. Epigenetic regulation of carnitine palmitoyltransferase 1 (Cpt1a) by high fat diet. Biochim. Biophys. Acta (BBA)-Bioenerg. 2018, 1862, 141–152. [Google Scholar] [CrossRef]
- Wang, L.; Chen, L.; Tan, Y.; Wei, J.; Chang, Y.; Jin, T.; Zhu, H. Betaine supplement alleviates hepatic triglyceride accumulation of apolipoprotein E deficient mice via reducing methylation of peroxisomal proliferator-activated receptor alpha promoter. Lipids Health Dis. 2013, 12, 34. [Google Scholar] [CrossRef] [Green Version]
- Ehara, T.; Kamei, Y.; Yuan, X.; Takahashi, M.; Kanai, S.; Tamura, E.; Tsujimoto, K.; Tamiya, T.; Nakagawa, Y.; Shimano, H.; et al. Ligand-Activated PPARα-Dependent DNA Demethylation Regulates the Fatty Acid β-Oxidation Genes in the Postnatal Liver. Diabetes 2014, 64, 775–784. [Google Scholar] [CrossRef] [Green Version]
- Byun, S.; Seok, S.; Kim, Y.-C.; Zhang, Y.; Yau, P.; Iwamori, N.; Xu, H.E.; Ma, J.; Kemper, B.; Kemper, J.K. Fasting-induced FGF21 signaling activates hepatic autophagy and lipid degradation via JMJD3 histone demethylase. Nat. Commun. 2020, 11, 807. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Liu, J.; Zhang, X.-O.; Sibley, K.; Najjar, S.M.; Lee, M.M.; Wu, Q. Inhibition of protein arginine methyltransferase 5 enhances hepatic mitochondrial biogenesis. J. Biol. Chem. 2018, 293, 10884–10894. [Google Scholar] [CrossRef] [Green Version]
- Mann, J.; Chu, D.C.; Maxwell, A.; Oakley, F.; Zhu, N.; Tsukamoto, H.; Mann, D.A. MeCP2 Controls an Epigenetic Pathway That Promotes Myofibroblast Transdifferentiation and Fibrosis. Gastroenterology 2010, 138, 705–714.e4. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Tang, T.; Wang, G.-D.; Liu, B. LncRNA-H19 promotes hepatic lipogenesis by directly regulating miR-130a/PPARγ axis in non-alcoholic fatty liver disease. Biosci. Rep. 2019, 39, BSR20181722. [Google Scholar] [CrossRef] [Green Version]
- Hardy, T.; Zeybel, M.; Day, C.P.; Dipper, C.; Masson, S.; McPherson, S.; Henderson, E.; Tiniakos, D.; White, S.; French, J.; et al. Plasma DNA methylation: A potential biomarker for stratification of liver fibrosis in non-alcoholic fatty liver disease. Gut 2016, 66, 1321–1328. [Google Scholar] [CrossRef]
- Hwang, J.W.; So, Y.; Bae, G.; Kim, S.; Kim, Y.K. Protein arginine methyltransferase 6 suppresses adipogenic differentiation by repressing peroxisome proliferator-activated receptor γ activity. Int. J. Mol. Med. 2019, 43, 2462–2470. [Google Scholar] [CrossRef]
- Jiang, Y.; Wang, S.; Zhao, Y.; Lin, C.; Zhong, F.; Jin, L.; He, F.; Wang, H. Histone H3K9 demethylase JMJD1A modulates hepatic stellate cells activation and liver fibrosis by epigenetically regulating peroxisome proliferator-activated receptor γ. FASEB J. 2015, 29, 1830–1841. [Google Scholar] [CrossRef]
- Kim, J.-H.; Jung, D.Y.; Nagappan, A.; Jung, M.H. Histone H3K9 demethylase JMJD2B induces hepatic steatosis through upregulation of PPARγ2. Sci. Rep. 2018, 8, 13734. [Google Scholar] [CrossRef]
- Pang, H.; Ling, D.; Cheng, Y.; Akbar, R.; Jin, L.; Ren, J.; Wu, H.; Chen, B.; Zhou, Y.; Zhu, H.; et al. Gestational high-fat diet impaired demethylation of Ppar α and induced obesity of offspring. J. Cell. Mol. Med. 2021, 25, 5404–5416. [Google Scholar] [CrossRef] [PubMed]
- Kawahori, K.; Kondo, Y.; Yuan, X.; Kawasaki, Y.; Hanzawa, N.; Tsujimoto, K.; Wada, F.; Kohda, T.; Ishigami, A.; Yamada, T.; et al. Ascorbic acid during the suckling period is required for proper DNA demethylation in the liver. Sci. Rep. 2020, 10, 21228. [Google Scholar] [CrossRef]
- Kharitonenkov, A.; Shiyanova, T.L.; Koester, A.; Ford, A.M.; Micanovic, R.; Galbreath, E.; Sandusky, G.E.; Hammond, L.J.; Moyers, J.S.; Owens, R.A.; et al. FGF-21 as a novel metabolic regulator. J. Clin. Investig. 2005, 115, 1627–1635. [Google Scholar] [CrossRef] [Green Version]
- Jimenez, V.; Jambrina, C.; Casana, E.; Sacristan, V.; Muñoz, S.; Darriba, S.; Rodó, J.; Mallol, C.; Garcia, M.; León, X.; et al. FGF21 gene therapy as treatment for obesity and insulin resistance. EMBO Mol. Med. 2018, 10, e8791. [Google Scholar] [CrossRef]
- Longo, R.; Peri, C.; Cricrì, D.; Coppi, L.; Caruso, D.; Mitro, N.; De Fabiani, E.; Crestani, M. Ketogenic Diet: A New Light Shining on Old but Gold Biochemistry. Nutrients 2019, 11, 2497. [Google Scholar] [CrossRef] [Green Version]
- Seok, S.; Kim, Y.-C.; Byun, S.; Choi, S.; Xiao, Z.; Iwamori, N.; Zhang, Y.; Wang, C.; Ma, J.; Ge, K.; et al. Fasting-induced JMJD3 histone demethylase epigenetically activates mitochondrial fatty acid β-oxidation. J. Clin. Investig. 2018, 128, 3144–3159. [Google Scholar] [CrossRef]
- Yuan, X.; Tsujimoto, K.; Hashimoto, K.; Kawahori, K.; Hanzawa, N.; Hamaguchi, M.; Seki, T.; Nawa, M.; Ehara, T.; Kitamura, Y.; et al. Epigenetic modulation of Fgf21 in the perinatal mouse liver ameliorates diet-induced obesity in adulthood. Nat. Commun. 2018, 9, 636. [Google Scholar] [CrossRef]
- Hazra, S.; Miyahara, T.; Rippe, R.A.; Tsukamoto, H. PPAR Gamma and Hepatic Stellate Cells. Comp. Hepatol. 2004, 3 (Suppl. 1), S7. [Google Scholar] [CrossRef] [Green Version]
- Perugorria, M.J.; Wilson, C.L.; Zeybel, M.; Walsh, M.; Amin, S.; Robinson, S.; White, S.A.; Burt, A.D.; Oakley, F.; Tsukamoto, H.; et al. Histone methyltransferase ASH1 orchestrates fibrogenic gene transcription during myofibroblast transdifferentiation. Hepatology 2012, 56, 1129–1139. [Google Scholar] [CrossRef]
- Byrd, K.N.; Shearn, A. ASH1, a Drosophila trithorax group protein, is required for methylation of lysine 4 residues on histone H3. Proc. Natl. Acad. Sci. USA 2003, 100, 11535–11540. [Google Scholar] [CrossRef] [Green Version]
- Zong, Y.; Yan, J.; Jin, L.; Xu, B.; He, Z.; Zhang, R.; Hu, C.; Jia, W. Relationship between circulating miR-132 and non-alcoholic fatty liver disease in a Chinese population. Hereditas 2020, 157, 22. [Google Scholar] [CrossRef] [PubMed]
- Lau-Corona, D.; Bae, W.K.; Hennighausen, L.; Waxman, D.J. Sex-biased genetic programs in liver metabolism and liver fibrosis are controlled by EZH1 and EZH2. PLoS Genet. 2020, 16, e1008796. [Google Scholar] [CrossRef]
- Moran-Salvador, E.; Garcia-Macia, M.; Sivaharan, A.; Sabater, L.; Zaki, M.Y.; Oakley, F.; Knox, A.; Page, A.; Luli, S.; Mann, J.; et al. Fibrogenic Activity of MECP2 Is Regulated by Phosphorylation in Hepatic Stellate Cells. Gastroenterology 2019, 157, 1398–1412.e9. [Google Scholar] [CrossRef] [Green Version]
- Jing, F.; Geng, Y.; Xu, X.-Y.; Xu, H.-Y.; Shi, J.-S.; Xu, Z.-H. MicroRNA29a Reverts the Activated Hepatic Stellate Cells in the Regression of Hepatic Fibrosis through Regulation of ATPase H+ Transporting V1 Subunit C1. Int. J. Mol. Sci. 2019, 20, 796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xuan, J.; Guo, S.-L.; Huang, A.; Xu, H.-B.; Shao, M.; Yang, Y.; Wen, W. MiR-29a and miR-652 Attenuate Liver Fibrosis by Inhibiting the Differentiation of CD4+ T Cells. Cell Struct. Funct. 2017, 42, 95–103. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, Y.; Itami, S.; Kuroda, M.; Yoshizato, K.; Kawada, N.; Murakami, Y. MiR-29a Assists in Preventing the Activation of Human Stellate Cells and Promotes Recovery From Liver Fibrosis in Mice. Mol. Ther. 2016, 24, 1848–1859. [Google Scholar] [CrossRef] [Green Version]
- Galic, S.; Oakhill, J.; Steinberg, G.R. Adipose tissue as an endocrine organ. Mol. Cell. Endocrinol. 2010, 316, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Khandekar, M.J.; Cohen, P.; Spiegelman, B.M. Molecular mechanisms of cancer development in obesity. Nat. Rev. Cancer 2011, 11, 886–895. [Google Scholar] [CrossRef] [PubMed]
- Heydari, H.; Ghiasi, R.; Ghaderpour, S.; Keyhanmanesh, R. The Mechanisms Involved in Obesity-Induced Male Infertility. Curr. Diabetes Rev. 2021, 17, 259–267. [Google Scholar] [CrossRef]
- Siersbaek, R.; Nielsen, R.; Mandrup, S. PPARγ in adipocyte differentiation and metabolism—Novel insights from genome-wide studies. FEBS Lett. 2010, 584, 3242–3249. [Google Scholar] [CrossRef] [Green Version]
- Lefterova, M.I.; Zhang, Y.; Steger, D.J.; Schupp, M.; Schug, J.; Cristancho, A.; Feng, D.; Zhuo, D.; Stoeckert, J.C.J.; Liu, X.S.; et al. PPAR and C/EBP factors orchestrate adipocyte biology via adjacent binding on a genome-wide scale. Genes Dev. 2008, 22, 2941–2952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiki, K.; Kano, F.; Shiota, K.; Murata, M. Expression of the peroxisome proliferator activated receptor γ gene is repressed by DNA methylation in visceral adipose tissue of mouse models of diabetes. BMC Biol. 2009, 7, 38. [Google Scholar] [CrossRef] [Green Version]
- Duteil, D.; Tosic, M.; Willmann, D.; Georgiadi, A.; Kanouni, T.; Schüle, R. Lsd1 prevents age-programed loss of beige adipocytes. Proc. Natl. Acad. Sci. USA 2017, 114, 5265–5270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Zhang, B.; Lan, X.; Zhang, C.; Lei, C.; Chen, H. Comparative Transcriptome Analysis Reveals Significant Differences in MicroRNA Expression and Their Target Genes between Adipose and Muscular Tissues in Cattle. PLoS ONE 2014, 9, e102142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinelli, R.; Nardelli, C.; Pilone, V.; Buonomo, T.; Liguori, R.; Castanò, I.; Buono, P.; Masone, S.; Persico, G.; Forestieri, P.; et al. miR-519d Overexpression Is Associated With Human Obesity. Obesity 2010, 18, 2170–2176. [Google Scholar] [CrossRef] [PubMed]
- Couture, J.-P.; Nolet, G.; Beaulieu, E.; Blouin, R.; Gévry, N. The p400/Brd8 Chromatin Remodeling Complex Promotes Adipogenesis by Incorporating Histone Variant H2A.Z at PPARγ Target Genes. Endocrinology 2012, 153, 5796–5808. [Google Scholar] [CrossRef] [Green Version]
- Mikkelsen, T.S.; Xu, Z.; Zhang, X.; Wang, L.; Gimble, J.M.; Lander, E.S.; Rosen, E.D. Comparative Epigenomic Analysis of Murine and Human Adipogenesis. Cell 2010, 143, 156–169. [Google Scholar] [CrossRef] [Green Version]
- Park, U.; Hwang, J.; Youn, H.; Kim, E.; Um, S. Piperine inhibits adipocyte differentiation via dynamic regulation of histone modifications. Phytother. Res. 2019, 33, 2429–2439. [Google Scholar] [CrossRef]
- Lee, J.-E.; Wang, C.; Xu, S.; Cho, Y.-W.; Wang, L.; Feng, X.; Baldridge, A.; Sartorelli, V.; Zhuang, L.; Peng, W.; et al. H3K4 mono- and di-methyltransferase MLL4 is required for enhancer activation during cell differentiation. Elife 2013, 2, e01503. [Google Scholar] [CrossRef]
- Cho, Y.-W.; Hong, S.; Jin, Q.; Wang, L.; Lee, J.-E.; Gavrilova, O.; Ge, K. Histone Methylation Regulator PTIP Is Required for PPARγ and C/EBPα Expression and Adipogenesis. Cell Metab. 2009, 10, 27–39. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Lee, H.H.; Ye, B.J.; Lee-Kwon, W.; Choi, S.Y.; Kwon, H.M. TonEBP suppresses adipogenesis and insulin sensitivity by blocking epigenetic transition of PPARγ2. Sci. Rep. 2015, 5, 10937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.-H.; Chung, C.-C.; Liu, Y.-C.; Yeh, S.-P.; Hsu, J.L.; Hung, M.-C.; Su, H.-L.; Li, L.-Y. Enhancer of Zeste Homolog 2 and Histone Deacetylase 9c Regulate Age-Dependent Mesenchymal Stem Cell Differentiation into Osteoblasts and Adipocytes. Stem Cells 2016, 34, 2183–2193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.-H.; Yeh, F.-L.; Yeh, S.-P.; Ma, H.-T.; Hung, S.-C.; Hung, M.-C.; Li, L.-Y. Myocyte Enhancer Factor-2 Interacting Transcriptional Repressor (MITR) Is a Switch That Promotes Osteogenesis and Inhibits Adipogenesis of Mesenchymal Stem Cells by Inactivating Peroxisome Proliferator-activated Receptor γ-2. J. Biol. Chem. 2011, 286, 10671–10680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, L.; Jang, Y.; Park, Y.-K.; Lee, J.-E.; Jain, S.; Froimchuk, E.; Broun, A.; Liu, C.; Gavrilova, O.; Ge, K. Depletion of Nsd2-mediated histone H3K36 methylation impairs adipose tissue development and function. Nat. Commun. 2018, 9, 1796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nic-Can, G.I.; Rodas-Junco, B.A.; Carrillo-Cocom, L.M.; Zepeda-Pedreguera, A.; Peñaloza-Cuevas, R.; Aguilar-Ayala, F.J.; Rojas-Herrera, R.A. Epigenetic Regulation of Adipogenic Differentiation by Histone Lysine Demethylation. Int. J. Mol. Sci. 2019, 20, 3918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lizcano, F.; Romero, C.; Vargas, D. Regulation of adipogenesis by nucelar receptor PPARγ is modulated by the histone demethylase JMJD2C. Genet. Mol. Biol. 2010, 34, 19–24. [Google Scholar] [CrossRef]
- Tateishi, K.; Okada, Y.; Kallin, E.M.; Zhang, Y. Role of Jhdm2a in regulating metabolic gene expression and obesity resistance. Nature 2009, 458, 757–761. [Google Scholar] [CrossRef] [Green Version]
- Inagaki, T.; Tachibana, M.; Magoori, K.; Kudo, H.; Tanaka, T.; Okamura, M.; Naito, M.; Kodama, T.; Shinkai, Y.; Sakai, J. Obesity and metabolic syndrome in histone demethylase JHDM2a-deficient mice. Genes Cells 2009, 14, 991–1001. [Google Scholar] [CrossRef]
- Lee, J.-E.; Ge, K. Transcriptional and epigenetic regulation of PPARγ expression during adipogenesis. Cell Biosci. 2014, 4, 29. [Google Scholar] [CrossRef] [Green Version]
- Steger, D.J.; Grant, G.; Schupp, M.; Tomaru, T.; Lefterova, M.I.; Schug, J.; Manduchi, E.; Stoeckert, C.J.; Lazar, M.A. Propagation of adipogenic signals through an epigenomic transition state. Genes Dev. 2010, 24, 1035–1044. [Google Scholar] [CrossRef] [Green Version]
- Erener, S.; Hesse, M.; Kostadinova, R.; Hottiger, M.O. Poly(ADP-Ribose)Polymerase-1 (PARP1) Controls Adipogenic Gene Expression and Adipocyte Function. Mol. Endocrinol. 2012, 26, 79–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, M.; Rao, M.; Bouras, T.; Wang, C.; Wu, K.; Zhang, X.; Li, Z.; Yao, T.-P.; Pestell, R.G. Cyclin D1 Inhibits Peroxisome Proliferator-activated Receptor γ-mediated Adipogenesis through Histone Deacetylase Recruitment. J. Biol. Chem. 2005, 280, 16934–16941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Q.; Zhang, F.; Yan, T.; Liu, Z.; Wang, C.; Ge, X.; Zhai, Q. C/EBPα regulates SIRT1 expression during adipogenesis. Cell Res. 2010, 20, 470–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picard, F.; Kurtev, M.; Chung, N.; Topark-Ngarm, A.; Senawong, T.; Machado De Oliveira, R.; Leid, M.; McBurney, M.W.; Guarente, L. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature 2004, 429, 771–776. [Google Scholar] [CrossRef]
- Mayoral, R.; Osborn, O.; McNelis, J.; Johnson, A.M.; Oh, D.Y.; Izquierdo, C.L.; Chung, H.; Li, P.; Traves, P.G.; Bandyopadhyay, G.; et al. Adipocyte SIRT1 knockout promotes PPARγ activity, adipogenesis and insulin sensitivity in chronic-HFD and obesity. Mol. Metab. 2015, 4, 378–391. [Google Scholar] [CrossRef]
- Ferrari, A.; Fiorino, E.; Longo, R.; Barilla, S.; Mitro, N.; Cermenati, G.; Giudici, M.; Caruso, D.; Mai, A.; Guerrini, U.; et al. Attenuation of diet-induced obesity and induction of white fat browning with a chemical inhibitor of histone deacetylases. Int. J. Obes. 2016, 41, 289–298. [Google Scholar] [CrossRef] [Green Version]
- Aguilar, E.C.; Silva, J.; Navia-Pelaez, J.M.; Leonel, A.J.; Lopes, L.G.; Menezes-Garcia, Z.; Ferreira, A.; Capettini, L.; Teixeira, L.G.; Lemos, V.S.; et al. Sodium butyrate modulates adipocyte expansion, adipogenesis, and insulin receptor signaling by upregulation of PPAR-γ in obese Apo E knockout mice. Nutrition 2017, 47, 75–82. [Google Scholar] [CrossRef]
- Yoon, G.-E.; Jung, J.K.; Lee, Y.-H.; Jang, B.-C.; Kim, J.I. Histone deacetylase inhibitor CG200745 ameliorates high-fat diet-induced hypertension via inhibition of angiotensin II production. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2019, 393, 491–500. [Google Scholar] [CrossRef] [Green Version]
- Bian, F.; Ma, X.; Villivalam, S.D.; You, D.; Choy, L.R.; Paladugu, A.; Fung, S.; Kang, S. TET2 facilitates PPARγ agonist–mediated gene regulation and insulin sensitization in adipocytes. Metabolism 2018, 89, 39–47. [Google Scholar] [CrossRef]
- Chen, J.; Liu, Y.; Lu, S.; Yin, L.; Zong, C.; Cui, S.; Qin, D.; Yang, Y.; Guan, Q.; Li, X.; et al. The role and possible mechanism of lncRNA U90926 in modulating 3T3-L1 preadipocyte differentiation. Int. J. Obes. 2016, 41, 299–308. [Google Scholar] [CrossRef] [Green Version]
- Cooper, D.R.; Carter, G.; Li, P.; Patel, R.; Watson, J.E.; Patel, N.A. Long Non-Coding RNA NEAT1 Associates with SRp40 to Temporally Regulate PPARγ2 Splicing during Adipogenesis in 3T3-L1 Cells. Genes 2014, 5, 1050–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Divoux, A.; Karastergiou, K.; Xie, H.; Guo, W.; Perera, R.J.; Fried, S.K.; Smith, S.R. Identification of a novel lncRNA in gluteal adipose tissue and evidence for its positive effect on preadipocyte differentiation. Obesity 2014, 22, 1781–1785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Gu, M.; Ma, Y.; Peng, Y. LncRNA TUG1 reduces inflammation and enhances insulin sensitivity in white adipose tissue by regulating miR-204/SIRT1 axis in obesity mice. Mol. Cell. Biochem. 2020, 475, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Skårn, M.; Namløs, H.M.; Noordhuis, P.; Wang, M.-Y.; Meza-Zepeda, L.A.; Myklebost, O. Adipocyte Differentiation of Human Bone Marrow-Derived Stromal Cells Is Modulated by MicroRNA-155, MicroRNA-221, and MicroRNA-222. Stem Cells Dev. 2012, 21, 873–883. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chen, Y.; Zhang, S.; Ye, L.; Cui, J.; Sun, Q.; Li, K.; Wu, H.; Liu, L. MiR-540 as a Novel Adipogenic Inhibitor Impairs Adipogenesis Via Suppression of PPARγ. J. Cell. Biochem. 2015, 116, 969–976. [Google Scholar] [CrossRef]
- Li, H.; Xue, M.; Xu, J.; Qin, X. MiR-301a is involved in adipocyte dysfunction during obesity-related inflammation via suppression of PPARgamma. Pharmazie 2016, 71, 84–88. [Google Scholar]
- Huang, Q.; Ma, C.; Chen, L.; Luo, D.; Chen, R.; Liang, F. Mechanistic Insights into the Interaction between Transcription Factors and Epigenetic Modifications and the Contribution to the Development of Obesity. Front. Endocrinol. 2018, 9, 370. [Google Scholar] [CrossRef] [Green Version]
- McGregor, R.A.; Choi, M.S. microRNAs in the Regulation of Adipogenesis and Obesity. Curr. Mol. Med. 2011, 11, 304–316. [Google Scholar] [CrossRef]
- Xie, H.; Lim, B.; Lodish, H.F. MicroRNAs Induced During Adipogenesis that Accelerate Fat Cell Development Are Downregulated in Obesity. Diabetes 2009, 58, 1050–1057. [Google Scholar] [CrossRef] [Green Version]
- Kolb, H.; Martin, S. Environmental/lifestyle factors in the pathogenesis and prevention of type 2 diabetes. BMC Med. 2017, 15, 131. [Google Scholar] [CrossRef]
- Ojo, O. Dietary Intake and Type 2 Diabetes. Nutrients 2019, 11, 2177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henning, R.J. Type-2 diabetes mellitus and cardiovascular disease. Future Cardiol. 2018, 14, 491–509. [Google Scholar] [CrossRef]
- Ogurtsova, K.; da Rocha Fernandes, J.D.; Huang, Y.; Linnenkamp, U.; Guariguata, L.; Cho, N.; Cavan, D.; Shaw, J.; Makaroff, L. IDF Diabetes Atlas: Global estimates for the prevalence of diabetes for 2015 and 2040. Diabetes Res. Clin. Pract. 2017, 128, 40–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naidoo, V.; Naidoo, M.; Ghai, M. Cell- and tissue-specific epigenetic changes associated with chronic inflammation in insulin resistance and type 2 diabetes mellitus. Scand. J. Immunol. 2018, 88, e12723. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Tsai, L.; Zhou, Y.; Evertts, A.G.; Xu, S.; Griffin, M.; Issner, R.; Whitton, H.J.; Garcia, B.A.; Epstein, C.B.; et al. Identification of nuclear hormone receptor pathways causing insulin resistance by transcriptional and epigenomic analysis. Nat. Cell. Biol. 2015, 17, 44–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.; Ye, X.; Guo, W.; Lu, H.; Gao, Z. Inhibition of HDAC3 promotes ligand-independent PPARγ activation by protein acetylation. J. Mol. Endocrinol. 2014, 53, 191–200. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Wang, X.; Ren, W.; Ren, J.; Lan, X.; Wang, F.; Zhang, F.; Han, Y.; Song, T.; Holmdahl, R.; et al. High expression of liver histone deacetylase 3 contributes to high-fat-diet-induced metabolic syndrome by suppressing the PPAR-γ and LXR-α-pathways in E3 rats. Mol. Cell. Endocrinol. 2011, 344, 69–80. [Google Scholar] [CrossRef]
- Sathishkumar, C.; Prabu, P.; Balakumar, M.; Lenin, R.; Prabhu, D.; Anjana, R.M.; Mohan, V.; Balasubramanyam, M. Augmentation of histone deacetylase 3 (HDAC3) epigenetic signature at the interface of proinflammation and insulin resistance in patients with type 2 diabetes. Clin. Epigenet. 2016, 8, 125. [Google Scholar] [CrossRef] [Green Version]
- Ahmadian, M.; Suh, J.M.; Hah, N.; Liddle, C.; Atkins, A.R.; Downes, M.; Evans, R.M. PPARγ signaling and metabolism: The good, the bad and the future. Nat. Med. 2013, 19, 557–566. [Google Scholar] [CrossRef] [Green Version]
- Makkar, R.; Behl, T.; Arora, S. Role of HDAC inhibitors in diabetes mellitus. Curr. Res. Transl. Med. 2020, 68, 45–50. [Google Scholar] [CrossRef]
- Hu, Y.; Liu, J.; Yuan, Y.; Chen, J.; Cheng, S.; Wang, H.; Xu, Y. Sodium butyrate mitigates type 2 diabetes by inhibiting PERK-CHOP pathway of endoplasmic reticulum stress. Environ. Toxicol. Pharmacol. 2018, 64, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Rafehi, H.; Kaspi, A.; Ziemann, M.; Okabe, J.; Karagiannis, T.C.; El-Osta, A. Systems approach to the pharmacological actions of HDAC inhibitors reveals EP300 activities and convergent mechanisms of regulation in diabetes. Epigenetics 2017, 12, 991–1003. [Google Scholar] [CrossRef] [PubMed]
- Fujisaka, S. The role of adipose tissue M1/M2 macrophages in type 2 diabetes mellitus. Diabetol. Int. 2020, 12, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Zhao, J.; Meng, H.; Zhang, X. Adipose Tissue-Resident Immune Cells in Obesity and Type 2 Diabetes. Front. Immunol. 2019, 10, 1173. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; de Winther, M.P.; Bossche, J. Epigenetic mechanisms of macrophage activation in type 2 diabetes. Immunobiology 2017, 222, 937–943. [Google Scholar] [CrossRef]
- Bouhlel, M.A.; Derudas, B.; Rigamonti, E.; Dievart, R.; Brozek, J.; Haulon, S.; Zawadzki, C.; Jude, B.; Torpier, G.; Marx, N.; et al. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 2007, 6, 137–143. [Google Scholar] [CrossRef] [Green Version]
- Bouhlel, M.A.; Brozek, J.; Derudas, B.; Zawadzki, C.; Jude, B.; Staels, B.; Chinetti-Gbaguidi, G. Unlike PPARgamma, PPARalpha or PPARbeta/delta activation does not promote human monocyte differentiation toward alternative macrophages. Biochem. Biophys. Res. Commun. 2009, 386, 459–462. [Google Scholar] [CrossRef]
- Charo, I.F. Macrophage Polarization and Insulin Resistance: PPARγ in Control. Cell Metab. 2007, 6, 96–98. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Cao, Q.; Yu, L.; Shi, H.; Xue, B.; Shi, H. Epigenetic regulation of macrophage polarization and inflammation by DNA methylation in obesity. JCI Insight 2016, 1, e87748. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Wang, X.; Liu, D.; Yu, L.; Xue, B.; Shi, H. Epigenetic Regulation of Macrophage Polarization by DNA Methyltransferase 3b. Mol. Endocrinol. 2014, 28, 565–574. [Google Scholar] [CrossRef] [Green Version]
- You, D.; Nilsson, E.; Tenen, D.E.; Lyubetskaya, A.; Lo, J.C.; Jiang, R.; Deng, J.; Dawes, B.A.; Vaag, A.; Ling, C.; et al. Dnmt3a is an epigenetic mediator of adipose insulin resistance. Elife 2017, 6, e30766. [Google Scholar] [CrossRef]
- Kim, A.Y.; Park, Y.J.; Pan, X.; Shin, K.C.; Kwak, S.-H.; Bassas, A.F.; Sallam, R.M.; Park, K.S.; Alfadda, A.; Xu, A.; et al. Obesity-induced DNA hypermethylation of the adiponectin gene mediates insulin resistance. Nat. Commun. 2015, 6, 7585. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Zhang, Y.; Liu, Y.; Zhu, D.; Yu, J.; Li, G.; Sun, Z.; Wang, W.; Jiang, H.; Hong, Z. MiR-27a promotes insulin resistance and mediates glucose metabolism by targeting PPAR-γ-mediated PI3K/AKT signaling. Aging (Albany NY) 2019, 11, 7510–7524. [Google Scholar] [CrossRef]
- Yao, F.; Yu, Y.; Feng, L.; Li, J.; Zhang, M.; Lan, X.; Yan, X.; Liu, Y.; Guan, F.; Zhang, M.; et al. Adipogenic miR-27a in adipose tissue upregulates macrophage activation via inhibiting PPARγ of insulin resistance induced by high-fat diet-associated obesity. Exp. Cell Res. 2017, 355, 105–112. [Google Scholar] [CrossRef]
- Liang, H.; Ward, W.F. PGC-1α: A key regulator of energy metabolism. Adv. Physiol. Educ. 2006, 30, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Marcos, P.J.; Auwerx, J. Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr. 2011, 93, 884S–890S. [Google Scholar] [CrossRef] [Green Version]
- Jeninga, E.H.; Schoonjans, K.; Auwerx, J. Reversible acetylation of PGC-1: Connecting energy sensors and effectors to guarantee metabolic flexibility. Oncogene 2010, 29, 4617–4624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodgers, J.; Lerin, C.; Gerhart-Hines, Z.; Puigserver, P. Metabolic adaptations through the PGC-1α and SIRT1 pathways. FEBS Lett. 2007, 582, 46–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerhart-Hines, Z.; Rodgers, J.; Bare, O.; Lerin, C.; Kim, S.-H.; Mostoslavsky, R.; Alt, F.W.; Wu, Z.; Puigserver, P. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1α. EMBO J. 2007, 26, 1913–1923. [Google Scholar] [CrossRef] [PubMed]
- Sharabi, K.; Lin, H.; Tavares, C.D.; Dominy, J.E.; Camporez, J.P.; Perry, R.J.; Schilling, R.; Rines, A.K.; Lee, J.; Hickey, M.; et al. Selective Chemical Inhibition of PGC-1α Gluconeogenic Activity Ameliorates Type 2 Diabetes. Cell 2017, 169, 148–160.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]

| Condition | PPAR Isoform | Epigenetic Player | Effect | References |
|---|---|---|---|---|
| NASH | PPARα | miR-21 | Diminished PPARα expression and activation of HSCs in obesogenic models | [119] |
| TET1 and TET2 | Downregulated enzymes under high fat diet conditions, promoting Ppara hypermethylation | [121] | ||
| Ascorbic acid | Cofactor of TET enzymes. Its lack promotes PPARα target genes hypermethylation | [122] | ||
| JMJD3 | Phosphorylated upon fasting-induced FGF21 signaling. Direct interaction with PPARα for the upregulation of autophagy-related genes | [123] | ||
| PRMT5 | Downregulation of Ppara expression | [124] | ||
| PPARγ | miR-132 | miR-132 downregulation induces the expression of MeCP2 in HSCs | [125] | |
| miR-29a | Expressed upon Rosiglitazone-mediated PPARγ activation. Repression of profibrotic genes | [126] | ||
| MeCP2 | H3K9 and H3K27 methylation and HP1α repressor recruitment in Pparg locus of HSCs. MeCP2 also induces the expression of EZH2 and ASH1 in HSCs. | [125] | ||
| Pparg promoter CpG methylation | Downregulation of PPARγ. Potential non-invasive fibrosis marker in cell-free DNA in plasma. | [127] | ||
| PRMT6 | Repression of PPARγ activity | [128] | ||
| JMJD1A and JMJD2B | Upregulation of Pparg and increased lipid uptake | [129,130] | ||
| LncRNA-H19 | Control of hepatic lipogenesis through mi-130A/PPARγ axis | [126] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Porcuna, J.; Mínguez-Martínez, J.; Ricote, M. The PPARα and PPARγ Epigenetic Landscape in Cancer and Immune and Metabolic Disorders. Int. J. Mol. Sci. 2021, 22, 10573. https://doi.org/10.3390/ijms221910573
Porcuna J, Mínguez-Martínez J, Ricote M. The PPARα and PPARγ Epigenetic Landscape in Cancer and Immune and Metabolic Disorders. International Journal of Molecular Sciences. 2021; 22(19):10573. https://doi.org/10.3390/ijms221910573
Chicago/Turabian StylePorcuna, Jesús, Jorge Mínguez-Martínez, and Mercedes Ricote. 2021. "The PPARα and PPARγ Epigenetic Landscape in Cancer and Immune and Metabolic Disorders" International Journal of Molecular Sciences 22, no. 19: 10573. https://doi.org/10.3390/ijms221910573
APA StylePorcuna, J., Mínguez-Martínez, J., & Ricote, M. (2021). The PPARα and PPARγ Epigenetic Landscape in Cancer and Immune and Metabolic Disorders. International Journal of Molecular Sciences, 22(19), 10573. https://doi.org/10.3390/ijms221910573