
Achievements and Challenges of Genomics-Assisted Breeding in Forest Trees: From Marker-Assisted Selection to Genome Editing




Abstract
:1. Introduction
2. Conventional Forest Tree Breeding and Marker-Assisted Selection (MAS)
3. High-Throughput Genotyping Techniques Enable Different Fields of Studies on Plants
4. Genomic Selection/Prediction, an Extension of BLUP Methods to Maximize the Predictive Power of Traits of Interest
5. Factors That Determine the Accuracy of Genomic Prediction Models in Forest Trees
6. The Detection of Alleles in Narrow LD Allows the Optimization of the Accuracy of Genomic Prediction Models
7. Genome-Wide Association Studies (GWAS)
8. GRF-GIF Chimeras Could Be Gamer Changer Tools in Forest Editing to Boost Tree Regeneration

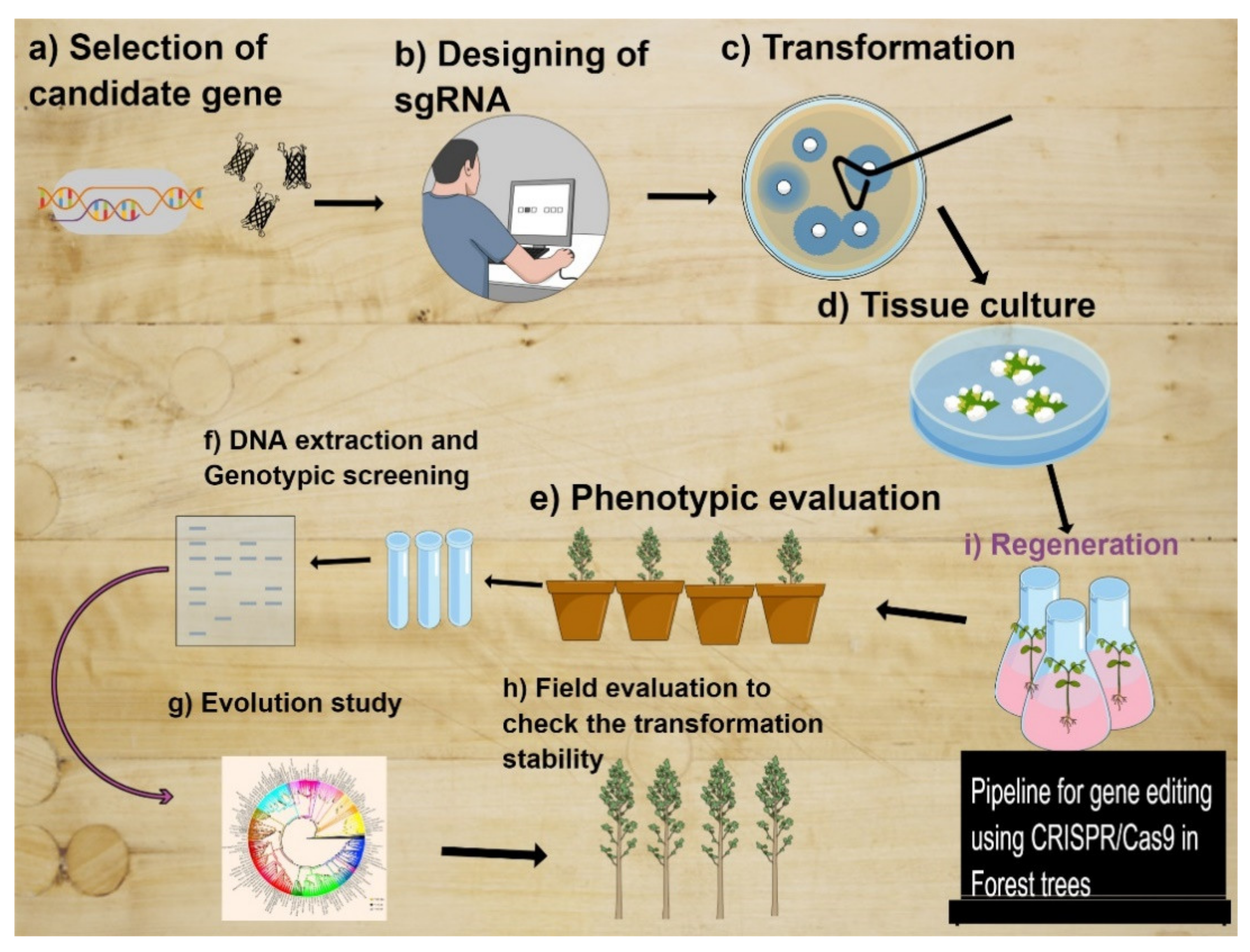{kind=link}
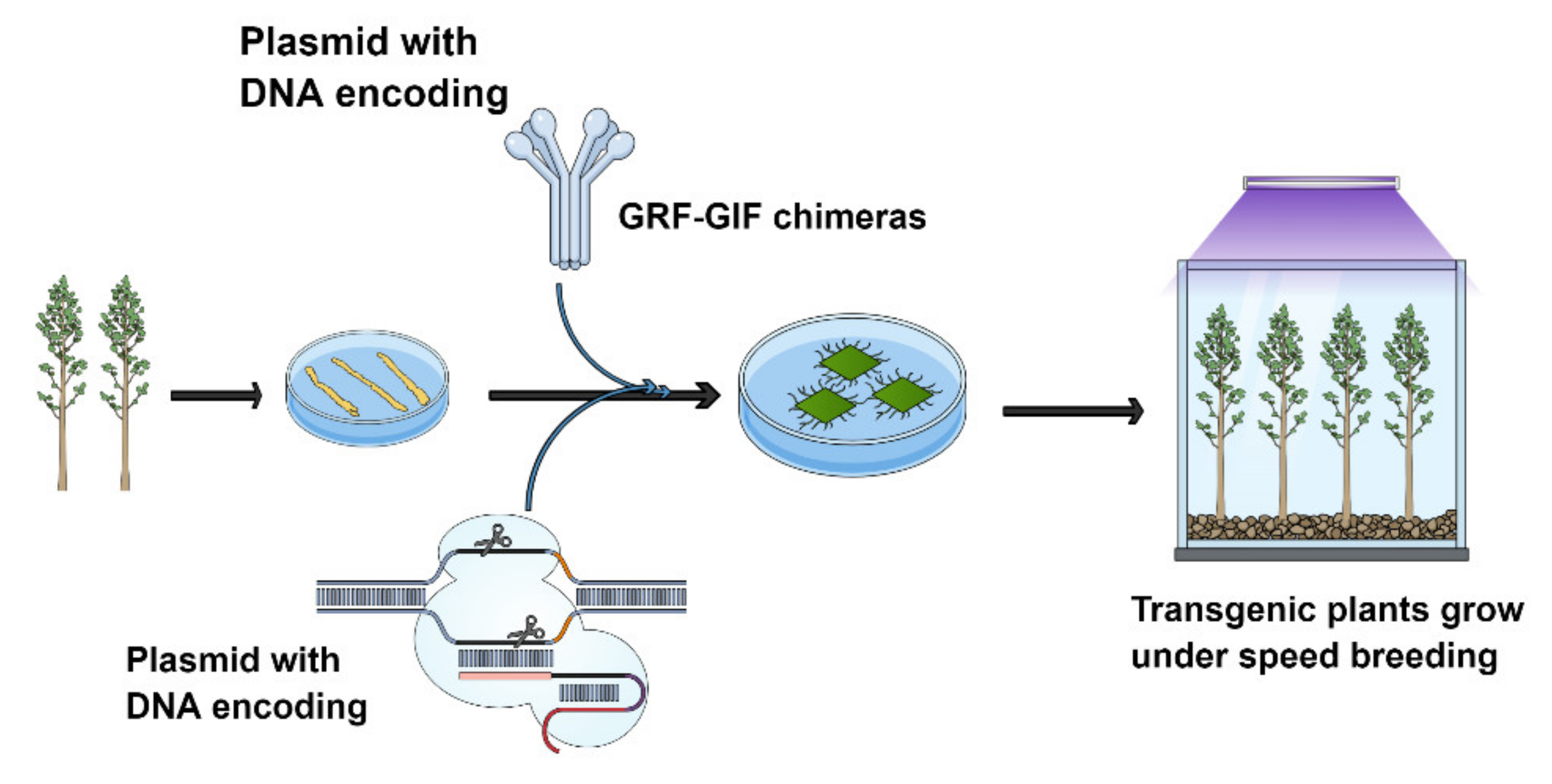{kind=link}
| Tree Species | Method | Targeted Gene | Transformation Method | Findings | References |
|---|---|---|---|---|---|
| Populus | CRISPR/Cas9 | 4CL1, 4CL2, 4CL5 | AT | Role in lignin production. The lignin content of all edited transgenic plants was decreased by about 23%, with a corresponding reduction in the S/G lignin ratio of around 30%. | [259] |
| Populus tomentosa Carr | CRISPR/Cas9 | PtoPDS | AT | Chlorophyll biosynthesis, albino phenotype | [248] |
| Populus tomentosa Carr | CRISPR/Cas9 | MYB57, MYB115, MYB156, MYB170 | AT | Ectopic deposition of lignin, xylan and cellulose during secondary cell wall formation | [261] |
| Populus tomentosa Carr | CRISPR/Cas9 | BRC1-1, BRC2- | AT | Secondary wall synthesis, which is responsible the involvement of brassinosteroids in the development of wood | [263] |
| Populus tremula × P. alba | CRISPR/Cas9 | AG1, AG2, LFY | AT | A distinct mutation spectrum was observed LFY and AG in sgRNA-gene combinations | [241] |
| Parasponia andersonii Planch (tropical tree) | CRISPR/Cas9 | EIN2, HK4, NSP1, NSP2 | AT | Regulate cytokinin, ethylene, or strigolactone hormonal pathways and, in legumes, perform important symbiotic activities | [256] |
| Populus tremula L. | CRISPR/Cas9 | SOC1, FUL, NFP TOZ19 | AT | GC content, purine residues in the final four nucleotides of the gRNA and an at least partially unpaired seed region all affected the gRNAs effectiveness for target cleavage | [267] |
| Pinus radiata D. Don | CRISPR/Cas9 | GUX1 | AT | biallelic and monoallelic INDELs can be generated in the coniferous tree P. radiata using DNA and RNPs | [282] |
| Populus davidiana × Populus bolleana | CRISPR/Cas9 | PdbPDS1 | AT | Second, regeneration could produce homozygous mutant shoots at a high frequency and that kanamycin selection could increase the frequency of homozygous mutant shoots. | [266] |
| Eucalyptus grandis x urophylla | CRISPR/Cas9 | LFY, FT | AT | The absence of male and female gametes and indeterminacy in floral development due to floral alteration because of disruption of ELFY function | [264] |
| Populus alba × Populus glandulosa | CRISPR/Cas12 | PDS | AT | AsCas12a system is the most efficient and optimization of the co-cultivation temperature after Agrobacterium-mediated transformation from 22 to 28 °C to increase the Cas12a nuclease editing efficiency in poplar | [272] |
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Maldonado, C.; Mora-Poblete, F.; Contreras-Soto, R.I.; Ahmar, S.; Chen, J.-T.; Júnior, A.T.D.A.; Scapim, C.A. Genome-Wide Prediction of Complex Traits in Two Outcrossing Plant Species Through Deep Learning and Bayesian Regularized Neural Network. Front. Plant Sci. 2020, 11, 1734. [Google Scholar] [CrossRef]
- Gray, L.K.; Rweyongeza, D.; Hamann, A.; John, S.; Thomas, B.R. Developing management strategies for tree improvement programs under climate change: Insights gained from long-term field trials with lodgepole pine. For. Ecol. Manag. 2016, 377, 128–138. [Google Scholar] [CrossRef]
- Mora-Poblete, F.; Ballesta, P.; Lobos, G.A.; Molina-Montenegro, M.; Gleadow, R.; Ahmar, S.; Jiménez-Aspee, F. Genome-wide association study of cyanogenic glycosides, proline, sugars, and pigments in Eucalyptus cladocalyx after 18 consecutive dry summers. Physiol. Plant. 2021. [Google Scholar] [CrossRef]
- Ding, C.; Hamann, A.; Yang, R.C.; Brouard, J.S. Genetic parameters of growth and adaptive traits in aspen (Populus tremuloides): Implications for tree breeding in a warming world. PLoS ONE 2020, 15, e0229225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isaac-Renton, M.; Stoehr, M.; Statland, C.B.; Woods, J. Tree breeding and silviculture: Douglas-fir volume gains with minimal wood quality loss under variable planting densities. For. Ecol. Manag. 2020, 465, 118094. [Google Scholar] [CrossRef]
- Cortés, A.J.; Restrepo-Montoya, M.; Bedoya-Canas, L.E. Modern Strategies to Assess and Breed Forest Tree Adaptation to Changing Climate. Front. Plant Sci. 2020, 11, 583323. [Google Scholar] [CrossRef] [PubMed]
- Bouvet, J.-M.; Ekomono, C.G.M.; Brendel, O.; Laclau, J.-P.; Bouillet, J.-P.; Epron, D. Selecting for water use efficiency, wood chemical traits and biomass with genomic selection in a Eucalyptus breeding program. For. Ecol. Manag. 2020, 465, 118092. [Google Scholar] [CrossRef]
- Alves, R.S.; Rocha, J.R.D.A.S.D.C.; Teodoro, P.E.; De Resende, M.D.V.; Henriques, E.P.; Silva, L.A.; Carneiro, P.C.S.; Bhering, L.L. Multiple-trait BLUP: A suitable strategy for genetic selection of Eucalyptus. Tree Genet. Genomes 2018, 14, 77. [Google Scholar] [CrossRef]
- Pastorino, M.J.; Marchelli, P. Low Intensity Breeding of Native Forest Trees in Argentina; Springer: Berlin/Heidelberg, Germany, 2021. [Google Scholar]
- Lebedev, V.G.; Lebedeva, T.N.; Chernodubov, A.I.; Shestibratov, K.A. Genomic Selection for Forest Tree Improvement: Methods, Achievements and Perspectives. Forests 2020, 11, 1190. [Google Scholar] [CrossRef]
- Crossa, J.; Pérez-Rodríguez, P.; Cuevas, J.; Montesinos-Lopez, O.A.; Jarquín, D.; Campos, G.D.L.; Burgueño, J.; González-Camacho, J.M.; Pérez-Elizalde, S.; Beyene, Y.; et al. Genomic Selection in Plant Breeding: Methods, Models, and Perspectives. Trends Plant Sci. 2017, 22, 961–975. [Google Scholar] [CrossRef]
- Beaulieu, J.; Doerksen, T.; Clément, S.; Mackay, J.; Bousquet, J. Accuracy of genomic selection models in a large population of open-pollinated families in white spruce. Heredity 2014, 113, 343–352. [Google Scholar] [CrossRef] [Green Version]
- Ballesta, P.; Serra, N.; Guerra, F.P.; Hasbún, R.; Mora, F. Genomic Prediction of Growth and Stem Quality Traits in Eucalyptus globulus Labill. at Its Southernmost Distribution Limit in Chile. Forests 2018, 9, 779. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Song, Q.; Cregan, P.B.; Jiang, G.L. Genome-wide association study, genomic prediction and marker-assisted selec-tion for seed weight in soybean (Glycine max). Theor. Appl. Genet. 2016, 129, 117–130. [Google Scholar] [CrossRef] [Green Version]
- Goddard, M.E.; Hayes, B.J.; Meuwissen, T.H.E. Using the genomic relationship matrix to predict the accuracy of genomic selection. J. Anim. Breed. Genet. 2011, 128, 409–421. [Google Scholar] [CrossRef]
- Scutari, M.; Mackay, I.; Balding, D. Using Genetic Distance to Infer the Accuracy of Genomic Prediction. PLoS Genet. 2016, 12, e1006288. [Google Scholar] [CrossRef]
- Lee, S.H.; Clark, S.; Van Der Werf, J.H.J. Estimation of genomic prediction accuracy from reference populations with vary-ing degrees of relationship. PLoS ONE 2017, 12, e0189775. [Google Scholar] [CrossRef] [Green Version]
- Ly, D.; Hamblin, M.; Rabbi, I.Y.; Gedil, M.; Bakare, M.A.; Gauch Jr, H.; Okechukwu, R.U.; Dixon, A.; Kulakow, P.A.; Jan-nink, J.-L. Relatedness and genotype x environment interaction affect prediction accuracies in genomic selection: A study in cassava. Crop Sci. 2013, 53, 1312–1325. [Google Scholar] [CrossRef] [Green Version]
- Thistlethwaite, F.R.; El-Dien, O.G.; Ratcliffe, B.; Klápště, J.; Porth, I.; Chen, C.; Stoehr, M.U.; Ingvarsson, P.K.; El-Kassaby, Y.A. Linkage disequilibrium vs. pedigree: Genomic selection prediction accuracy in conifer species. PLoS ONE 2020, 15, e0232201. [Google Scholar] [CrossRef] [PubMed]
- Norman, A.; Taylor, J.; Edwards, J.; Kuchel, H. Optimising Genomic Selection in Wheat: Effect of Marker Density, Population Size and Population Structure on Prediction Accuracy. G3 Genes Genomes Genet. 2018, 8, 2889–2899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wientjes, Y.C.J.; Veerkamp, R.F.; Calus, M.P.L. The Effect of Linkage Disequilibrium and Family Relationships on the Reliability of Genomic Prediction. Genetics 2013, 193, 621–631. [Google Scholar] [CrossRef] [Green Version]
- Wolc, A.; Zhao, H.H.; Arango, J.; Settar, P.; Fulton, J.E.; O’Sullivan, N.P.; Preisinger, R.; Stricker, C.; Habier, D.; Fernando, R.L.; et al. Response and inbreeding from a genomic selection experiment in layer chickens. Genet. Sel. Evol. 2015, 47, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Fernando, R.; Dekkers, J. Contributions of linkage disequilibrium and co-segregation information to the accuracy of genomic prediction. Genet. Sel. Evol. 2016, 48, 77. [Google Scholar] [CrossRef] [Green Version]
- Schopp, P.; Müller, D.; Technow, F.; Melchinger, A.E. Accuracy of Genomic Prediction in Synthetic Populations Depending on the Number of Parents, Relatedness, and Ancestral Linkage Disequilibrium. Genetics 2017, 205, 441–454. [Google Scholar] [CrossRef] [Green Version]
- Velazco, J.G.; Malosetti, M.; Hunt, C.H.; Mace, E.S.; Jordan, D.R.; van Eeuwijk, F.A. Combining pedigree and genomic in-formation to improve prediction quality: An example in sorghum. Theor. Appl. Genet. 2019, 132, 2055–2067. [Google Scholar] [CrossRef] [Green Version]
- Klápště, J.; Suontama, M.; Telfer, E.; Graham, N.; Low, C.; Stovold, T.; McKinley, R.; Dungey, H. Exploration of genetic ar-chitecture through sib-ship reconstruction in advanced breeding population of Eucalyptus nitens. PLoS ONE 2017, 12, e0185137. [Google Scholar] [CrossRef] [Green Version]
- Edwards, D. Two molecular measures of relatedness based on haplotype sharing. BMC Bioinform. 2015, 16, 383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howard, N.P.; Van De Weg, E.; Bedford, D.S.; Peace, C.P.; Vanderzande, S.; Clark, M.D.; Teh, S.L.; Cai, L.; Luby, J.J. Eluci-dation of the Honeycrisp’ pedigree through haplotype analysis with a multi-family integrated SNP linkage map and a large apple (Malus × domestica) pedigree-connected SNP data set. Horticult. Res. 2017, 4, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Mathew, B.; Léon, J.; Sillanpää, M.J. A novel linkage-disequilibrium corrected genomic relationship matrix for SNP-heritability estimation and genomic prediction. Heredity 2018, 120, 356–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calus, M.P.L.; Meuwissen, T.H.E.; de Roos, A.P.W.; Veerkamp, R.F. Accuracy of Genomic Selection Using Different Methods to Define Haplotypes. Genetics 2008, 178, 553–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matias, F.I.; Galli, G.; Granato, I.S.C.; Fritsche-Neto, R. Genomic Prediction of Autogamous and Allogamous Plants by SNPs and Haplotypes. Crop Sci. 2017, 57, 2951–2958. [Google Scholar] [CrossRef]
- He, S.; Thistlethwaite, R.; Forrest, K.; Shi, F.; Hayden, M.; Trethowan, R.; Daetwyler, H.D. Extension of a haplotype-based genomic prediction model to manage multi-environment wheat data using environmental covariates. Theor. Appl. Genet. 2019, 132, 3143–3154. [Google Scholar] [CrossRef]
- Sallam, A.H.; Conley, E.; Prakapenka, D.; Da, Y.; Anderson, J.A. Improving prediction accuracy using multi-allelic haplo-type prediction and training population optimization in wheat. G3 Genes Genomes Genet. 2020, 10, 2265–2273. [Google Scholar]
- Lan, S.; Zheng, C.; Hauck, K.; McCausland, M.; Duguid, S.D.; Booker, H.M.; Cloutier, S.; You, F.M. Genomic Prediction Accuracy of Seven Breeding Selection Traits Improved by QTL Identification in Flax. Int. J. Mol. Sci. 2020, 21, 1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villumsen, T.M.; Janss, L. Bayesian genomic selection: The effect of haplotype length and priors. BMC Proc. 2009, 3, S11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballesta, P.; Maldonado, C.; Pérez-Rodríguez, P.; Mora, F. SNP and Haplotype-Based Genomic Selection of Quantitative Traits in Eucalyptus globulus. Plants 2019, 8, 331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmar, S.; Saeed, S.; Khan, M.; Khan, S.U.; Mora-Poblete, F.; Kamran, M.; Faheem, A.; Maqsood, A.; Rauf, M.; Saleem, S.; et al. A Revolution toward Gene-Editing Technology and Its Application to Crop Improvement. Int. J. Mol. Sci. 2020, 21, 5665. [Google Scholar] [CrossRef] [PubMed]
- Ahmar, S.; Gill, R.A.; Jung, K.-H.; Faheem, A.; Qasim, M.U.; Mubeen, M.; Zhou, W. Conventional and Molecular Techniques from Simple Breeding to Speed Breeding in Crop Plants: Recent Advances and Future Outlook. Int. J. Mol. Sci. 2020, 21, 2590. [Google Scholar] [CrossRef] [Green Version]
- Ran, Y.; Patron, N.; Kay, P.; Wong, D.; Buchanan, M.; Cao, Y.Y.; Sawbridge, T.; Davies, J.P.; Mason, J.; Webb, S.R.; et al. Zinc finger nuclease-mediated precision genome editing of an endogenous gene in hexaploid bread wheat (Triticum aes-tivum) using a DNA repair template. Plant Biotechnol. J. 2018, 16, 2088–2101. [Google Scholar] [CrossRef] [Green Version]
- Luo, M.; Li, H.; Chakraborty, S.; Morbitzer, R.; Rinaldo, A.; Upadhyaya, N.; Bhatt, D.; Louis, S.; Richardson, T.; Lahaye, T.; et al. Efficient TALEN -mediated gene editing in wheat. Plant Biotechnol. J. 2019, 17, 2026–2028. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhang, S.; Zhang, R.; Gao, J.; Qi, Y.; Song, G.; Li, W.; Li, Y.; Li, G. Efficient multiplex genome editing by CRISPR/Cas9 in common wheat. Plant Biotechnol. J. 2020, 19, 427–429. [Google Scholar] [CrossRef]
- Kieu, N.P.; Lenman, M.; Wang, E.S.; Petersen, B.L.; Andreasson, E. Mutations introduced in susceptibility genes through CRISPR/Cas9 genome editing confer increased late blight resistance in potatoes. Sci. Rep. 2021, 11, 1–12. [Google Scholar] [CrossRef]
- Lee, J.E.; Neumann, M.; Duro, D.I.; Schmid, M. CRISPR-based tools for targeted transcriptional and epigenetic regulation in plants. PLoS ONE 2019, 14, e0222778. [Google Scholar] [CrossRef]
- Moradpour, M.; Abdulah, S.N.A. CRISPR/dCas9 platforms in plants: Strategies and applications beyond genome editing. Plant Biotechnol. J. 2019, 18, 32–44. [Google Scholar] [CrossRef] [Green Version]
- De Almeida Filho, J.E.; Guimarães, J.F.R.; Fonsceca, E.; Silva, F.; De Resende, M.D.V.; Muñoz, P.; Kirst, M.; De Resende, M.F.R. Genomic prediction of additive and non-additive effects using genetic markers and pedigrees. G3 Genes Genomes Genet. 2019, 9, 2739–2748. [Google Scholar] [CrossRef] [Green Version]
- Zapata-Valenzuela, J.; Whetten, R.W.; Neale, D.; McKeand, S.; Isik, F. Genomic Estimated Breeding Values Using Genomic Relationship Matrices in a Cloned Population of Loblolly Pine. G3 Genes Genomes Genet. 2013, 3, 909–916. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Molloy, C.; Muñoz, P.; Daetwyler, H.; Chagné, D.; Volz, R. Genome-enabled estimates of additive and nonaddi-tive genetic variances and prediction of apple phenotypes across environments. G3 Genes Genomes Genet. 2015, 5, 2711–2718. [Google Scholar]
- Perron, M.; DeBlois, J.; Desponts, M. Use of resampling to assess optimal subgroup composition for estimating genetic pa-rameters from progeny trials. Tree Genet. Genomes 2013, 9, 129–143. [Google Scholar] [CrossRef]
- Tambarussi, E.V.; Pereira, F.B.; Da Silva, P.H.M.; Lee, D.; Bush, D. Are tree breeders properly predicting genetic gain? A case study involving Corymbia species. Euphytica 2018, 214, 150. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.X. Benefits and risks of using clones in forestry—A review. Scand. J. For. Res. 2019, 34, 352–359. [Google Scholar] [CrossRef]
- Mora, F.; Serra, N. Bayesian estimation of genetic parameters for growth, stem straightness, and survival in Eucalyptus globulus on an Andean Foothill site. Tree Genet. Genomes 2014, 10, 711–719. [Google Scholar] [CrossRef]
- Chen, Z.-Q.; Karlsson, B.; Lundqvist, S.-O.; Gil, M.R.G.; Olsson, L.; Wu, H.X. Estimating solid wood properties using Pilodyn and acoustic velocity on standing trees of Norway spruce. Ann. For. Sci. 2015, 72, 499–508. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Zhu, Y.; Xu, J.; Lu, Z.; Chen, G.; Song, P.; Guo, W. Genetic variation and genetic gain for energy production, growth traits and wood properties in Eucalyptus hybrid clones in China. Aust. For. 2017, 80, 57–65. [Google Scholar] [CrossRef]
- Isik, F.; McKeand, S.E. Fourth cycle breeding and testing strategy for Pinus taeda in the NC State University Cooperative Tree Improvement Program. Tree Genet. Genomes 2019, 15, 1–2. [Google Scholar] [CrossRef]
- Hiraoka, Y.; Miura, M.; Fukatsu, E.; Iki, T.; Yamanobe, T.; Kurita, M.; Isoda, K.; Kubota, M.; Takahashi, M. Time trends of genetic parameters and genetic gains and optimum selection age for growth traits in sugi (Cryptomeria japonica) based on progeny tests conducted throughout Japan. J. For. Res. 2019, 24, 303–312. [Google Scholar] [CrossRef]
- Gamal El-Dien, O.; Ratcliffe, B.; Klápště, J.; Chen, C.; Porth, I.; El-Kassaby, Y.A. Prediction accuracies for growth and wood attributes of interior spruce in space using genotyping-by-sequencing. BMC Genom. 2015, 16, 370. [Google Scholar] [CrossRef] [Green Version]
- Klápště, J.; Suontama, M.; Dungey, H.S.; Telfer, E.J.; Graham, N.J.; Low, C.B.; Stovold, G.T. Effect of Hidden Relatedness on Single-Step Genetic Evaluation in an Advanced Open-Pollinated Breeding Program. J. Hered. 2018, 109, 802–810. [Google Scholar] [CrossRef] [Green Version]
- Bush, D.; Kain, D.; Kanowski, P.; Matheson, C. Genetic parameter estimates informed by a marker-based pedigree: A case study with Eucalyptus cladocalyx in southern Australia. Tree Genet. Genomes 2014, 11, 798. [Google Scholar] [CrossRef]
- Henderson, C.R. Applications of Linear Models in Animal Breeding; Cabi: Wallingford, UK, 1984. [Google Scholar]
- Henderson, C.R. Selection index and expected genetic advance. Stat. Genet. Plant Breed. 1963, 141–153. [Google Scholar]
- Henderson, C.R. Sire Evaluation and Genetic Trends. J. Anim. Sci. 1973, 1973, 10–41. [Google Scholar] [CrossRef]
- White, T.L.; Hodge, G.R. Best linear prediction of breeding values in a forest tree improvement program. Theor. Appl. Genet. 1988, 76, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Viana, J.M.S.; Sobreira, F.M.; De Resende, M.D.V.; Faria, V.R. Multi-trait BLUP in half-sib selection of annual crops. Plant Breed. 2010, 129, 599–604. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Chen, Z.; Olsson, L.; Grahn, T.; Karlsson, B.; Wu, H.X.; Lundqvist, S.O.; García-Gil, M.R. Effect of number of an-nual rings and tree ages on genomic predictive ability for solid wood properties of Norway spruce. BMC Genom. 2020, 21, 323. [Google Scholar] [CrossRef] [Green Version]
- Mphahlele, M.M.; Isik, F.; Mostert-O’Neill, M.M.; Reynolds, S.M.; Hodge, G.R.; Myburg, A.A. Expected benefits of genomic selection for growth and wood quality traits in Eucalyptus grandis. Tree Genet. Genomes 2020, 16, 1–12. [Google Scholar] [CrossRef]
- Sverrisdóttir, E.; Byrne, S.; Sundmark, E.H.R.; Johnsen, H.; Kirk, H.G.; Asp, T.; Janss, L.; Nielsen, E.H.R. Genomic prediction of starch content and chipping quality in tetraploid potato using genotyping-by-sequencing. Theor. Appl. Genet. 2017, 130, 2091–2108. [Google Scholar] [CrossRef]
- Kumar, S.; Banks, T.W.; Cloutier, S. SNP discovery through next-generation sequencing and its applications. Int. J. Plant Genom. 2012, 2012, 503–511. [Google Scholar] [CrossRef]
- Hayward, A.C.; Tollenaere, R.; Dalton-Morgan, J.; Batley, J. Molecular marker applications in plants. Methods Mol. Biol. 2015, 13–27. [Google Scholar]
- Nadeem, M.A.; Nawaz, M.A.; Shahid, M.Q.; Doğan, Y.; Comertpay, G.; Yıldız, M.; Hatipoğlu, R.; Ahmad, F.; Alsaleh, A.; Labhane, N.; et al. DNA molecular markers in plant breeding: Current status and recent advancements in genomic selection and genome editing. Biotechnol. Biotechnol. Equip. 2017, 32, 261–285. [Google Scholar] [CrossRef] [Green Version]
- Cobb, J.N.; Biswas, P.S.; Platten, J.D. Back to the future: Revisiting MAS as a tool for modern plant breeding. Theor. Appl. Genet. 2018, 132, 647–667. [Google Scholar] [CrossRef] [Green Version]
- Pootakham, W.; Jomchai, N.; Ruang-Areerate, P.; Shearman, J.R.; Sonthirod, C.; Sangsrakru, D.; Tragoonrung, S.; Tangphatsornruang, S. Genome-wide SNP discovery and identification of QTL associated with agronomic traits in oil palm using genotyping-by-sequencing (GBS). Genomics 2015, 105, 288–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenner, C.; Weir, B. Issues and strategies in the DNA identification of World Trade Center victims. Theor. Popul. Biol. 2003, 63, 173–178. [Google Scholar] [CrossRef]
- Seddon, J.M.; Parker, H.G.; Ostrander, E.A.; Ellegren, H. SNPs in ecological and conservation studies: A test in the Scandi-navian wolf population. Mol. Ecol. 2005, 14, 503–511. [Google Scholar] [CrossRef]
- Yu, H.; Xie, W.; Wang, J.; Xing, Y.; Xu, C.; Li, X.; Xiao, J.; Zhang, Q. Gains in QTL detection using an ultra-high density SNP map based on population sequencing relative to traditional RFLP/SSR markers. PLoS ONE 2011, 6, e17595. [Google Scholar]
- Adhikari, S.; Saha, S.; Biswas, A.; Rana, T.S.; Bandyopadhyay, T.K.; Ghosh, P. Application of molecular markers in plant genome analysis: A review. Nucleus 2017, 60, 283–297. [Google Scholar] [CrossRef]
- Garrido-Cardenas, J.A.; Valle, C.M.; Manzano-Agugliaro, F. Trends in plant research using molecular markers. Planta 2017, 247, 543–557. [Google Scholar] [CrossRef]
- Harismendy, O.; Ng, P.C.; Strausberg, R.L.; Wang, X.; Stockwell, T.B.; Beeson, K.Y.; Schork, N.J.; Murray, S.S.; Topol, E.J.; Levy, S.; et al. Evaluation of next generation sequencing platforms for population targeted sequencing studies. Genome Biol. 2009, 10, R32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paszkiewicz, K.; Studholme, D.J. High-throughput sequencing data analysis software: Current state and future developments. In Bioinformatics for High Throughput Sequencing; Springer: Berlin/Heidelberg, Germany, 2012; pp. 231–248. [Google Scholar]
- Kharabian-Masouleh, A.; Waters, D.L.E.; Reinke, R.F.; Henry, R.J. Discovery of polymorphisms in starch-related genes in rice germplasm by amplification of pooled DNA and deeply parallel sequencing. Plant Biotechnol. J. 2011, 9, 1074–1085. [Google Scholar] [CrossRef]
- Chandra, S.; Singh, D.; Pathak, J.; Kumari, S.; Kumar, M.; Poddar, R.; Balyan, H.S.; Prabhu, K.V.; Gupta, P.K.; Mukhopadhyay, K. SNP discovery from next-generation transcriptome sequencing data and their validation using KASP assay in wheat (Triticum aestivum L.). Mol. Breed. 2017, 37. [Google Scholar] [CrossRef]
- Silva-Junior, O.B.; Faria, D.A.; Grattapaglia, D. A flexible multi-species genome-wide 60K SNP chip developed from pooled resequencing of 240 Eucalyptus tree genomes across 12 species. New Phytol. 2015, 206, 1527–1540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulaszewski, B.; Meger, J.; Burczyk, J. Comparative analysis of SNP discovery and genotyping in Fagus sylvatica L. and Quercus robur L. using RADseq, GBS, and ddRAD methods. Forests 2021, 12, 222. [Google Scholar] [CrossRef]
- Livingstone, D.; Royaert, S.; Stack, C.; Mockaitis, K.; May, G.; Farmer, A.; Saski, C.; Schnell, R.; Kuhn, D.; Motamayor, J.C. Making a chocolate chip: Development and evaluation of a 6K SNP array forTheobroma cacao. DNA Res. 2015, 22, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Peace, C.; Bassil, N.; Main, R.; Ficklin, S.; Rosyara, U.R.; Stegmeir, T.; Sebolt, A.; Gilmore, B.; Lawley, C.; Mockler, T.C.; et al. Development and Evaluation of a Genome-Wide 6K SNP Array for Diploid Sweet Cherry and Tetraploid Sour Cherry. PLoS ONE 2012, 7, e48305. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Dong, Z.; Zhao, L.; Ren, Y.; Zhang, N.; Chen, F. The Wheat 660K SNP array demonstrates great potential for marker-assisted selection in polyploid wheat. Plant Biotechnol. J. 2020, 18, 1354–1360. [Google Scholar] [CrossRef]
- Singh, N.; Jayaswal, P.K.; Panda, K.; Mandal, P.; Kumar, V.; Singh, B.; Mishra, S.; Singh, Y.; Singh, R.; Rai, V.; et al. Sin-gle-copy gene based 50 K SNP chip for genetic studies and molecular breeding in rice. Sci. Rep. 2015, 5, 1–9. [Google Scholar]
- Geraldes, A.; Difazio, S.P.; Slavov, G.T.; Ranjan, P.; Muchero, W.; Hannemann, J.; Gunter, L.E.; Wymore, A.M.; Grassa, C.J.; Farzaneh, N.; et al. A 34K SNP genotyping array for Populus trichocarpa: Design, application to the study of natural popula-tions and transferability to other Populus species. Mol. Ecol. Res. 2013, 13, 306–323. [Google Scholar] [CrossRef]
- Pavy, N.; Gagnon, F.; Rigault, P.; Blais, S.; Deschênes, A.; Boyle, B.; Pelgas, B.; Deslauriers, M.; Clément, S.; Lavigne, P.; et al. Development of high-density SNP genotyping arrays for white spruce (Picea glauca) and transferability to subtropical and nordic congeners. Mol. Ecol. Res. 2013, 13, 324–336. [Google Scholar] [CrossRef]
- Lepoittevin, C.; Bodénès, C.; Chancerel, E.; Villate, L.; Lang, T.; Lesur, I.; Boury, C.; Ehrenmann, F.; Zelenika, D.; Boland, A.; et al. Single-nucleotide polymorphism discovery and validation in high-density SNP array for genetic analysis in European white oaks. Mol. Ecol. Res. 2015, 15, 1446–1459. [Google Scholar] [CrossRef] [PubMed]
- Silva, P.I.T.; Silva-Junior, O.B.; Resende, L.V.; Sousa, V.A.; Aguiar, A.V.; Grattapaglia, D. A 3K Axiom SNP array from a transcriptome-wide SNP resource sheds new light on the genetic diversity and structure of the iconic subtropical conifer tree Araucaria angustifolia (Bert.) Kuntze. PLoS ONE 2020, 15, e0230404. [Google Scholar] [CrossRef]
- Perry, A.; Wachowiak, W.; Downing, A.; Talbot, R.; Cavers, S. Development of a single nucleotide polymorphism array for population genomic studies in four European pine species. Mol. Ecol. Resour. 2020, 20, 1697–1705. [Google Scholar] [CrossRef]
- Howe, G.T.; Jayawickrama, K.; Kolpak, S.E.; Kling, J.; Trappe, M.; Hipkins, V.; Ye, T.; Guida, S.; Cronn, R.; Cushman, S.A.; et al. An Axiom SNP genotyping array for Douglas-fir. BMC Genom. 2020, 21, 9. [Google Scholar] [CrossRef] [Green Version]
- Bernhardsson, C.; Zan, Y.; Chen, Z.; Ingvarsson, P.K.; Wu, H.X. Development of a highly efficient 50K single nucleotide polymorphism genotyping array for the large and complex genome of Norway spruce (Picea abies L. Karst) by whole ge-nome resequencing and its transferability to other spruce species. Mol. Ecol. Res. 2021, 21, 880–896. [Google Scholar] [CrossRef]
- Pellicer, J.; Hidalgo, O.; Dodsworth, S.; Leitch, I.J. Genome Size Diversity and Its Impact on the Evolution of Land Plants. Genes 2018, 9, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faivre-Rampant, P.; Zaina, G.; Jorge, V.; Giacomello, S.; Segura, V.; Scalabrin, S.; Guérin, V.; De Paoli, E.; Aluome, C.; Viger, M.; et al. New resources for genetic studies in Populus nigra: Genome-wide SNP discovery and development of a 12k Infin-ium array. Mol. Ecol. Resour. 2016, 16, 1023–1036. [Google Scholar] [CrossRef] [PubMed]
- Telfer, E.; Graham, N.; Macdonald, L.; Li, Y.; Klápště, J.; Jr, M.R.; Neves, L.G.; Dungey, H.; Wilcox, P. A high-density exome capture genotype-by-sequencing panel for forestry breeding in Pinus radiata. PLoS ONE 2019, 14, e0222640. [Google Scholar] [CrossRef] [Green Version]
- Ramos, A.; Usié, A.; Barbosa, P.; Barros, P.M.; Capote, T.; Chaves, I.; Simões, F.; Abreu, I.; Carrasquinho, I.; Faro, C.; et al. The draft genome sequence of cork oak. Sci. Data 2018, 5, 180069. [Google Scholar] [CrossRef]
- Zoldoš, V.; Papeš, D.; Brown, S.C.; Panaud, O.; Šiljak-Yakovlev, S. Genome size and base composition of seven Quercus species: Inter- and intra-population variation. Genome 1998, 41, 162–168. [Google Scholar] [CrossRef]
- Tuskan, G.; DiFazio, S.; Jansson, S.; Bohlmann, J.; Grigoriev, I.; Hellsten, U.; Putnam, N.; Ralph, S.; Rombauts, S.; Salamov, A.; et al. The Genome of Black Cottonwood, Populus trichocarpa (Torr. & Gray). Science 2006, 313, 1596–1604. [Google Scholar] [CrossRef] [Green Version]
- Kovach, A.; Wegrzyn, J.L.; Parra, G.; Holt, C.; Bruening, G.E.; Loopstra, C.A.; Hartigan, J.; Yandell, M.; Langley, C.H.; Korf, I.; et al. The Pinus taeda genome is characterized by diverse and highly diverged repetitive sequences. BMC Genom. 2010, 11, 420. [Google Scholar] [CrossRef] [Green Version]
- Zimin, A.; Stevens, K.A.; Crepeau, M.; Holtz-Morris, A.; Koriabine, M.; Marçais, G.; Puiu, D.; Roberts, M.; Wegrzyn, J.; de Jong, P.J.; et al. Sequencing and Assembly of the 22-Gb Loblolly Pine Genome. Genetics 2014, 196, 875–890. [Google Scholar] [CrossRef] [Green Version]
- Zimin, A.V.; Stevens, K.A.; Crepeau, M.; Puiu, D.; Wegrzyn, J.; Yorke, J.A.; Langley, C.H.; Neale, D.B.; Salzberg, S.L. An improved assembly of the loblolly pine mega-genome using long-read single-molecule sequencing. GigaScience 2017, 6, 1–4. [Google Scholar] [CrossRef]
- De La Torre, A.R.; Puiu, D.; Crepeau, M.W.; Stevens, K.; Salzberg, S.L.; Langley, C.H.; Neale, D.B. Genomic architecture of complex traits in loblolly pine. New Phytol. 2018, 221, 1789–1801. [Google Scholar] [CrossRef]
- Myburg, A.A.; Grattapaglia, D.; Tuskan, G.; Hellsten, U.; Hayes, R.; Grimwood, J.; Jenkins, J.; Lindquist, E.; Tice, H.; Bauer, D.; et al. The genome of Eucalyptus grandis. Nature 2014, 510, 356–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meuwissen, E.T.H.; Hayes, B.; Goddard, M. Prediction of Total Genetic Value Using Genome-Wide Dense Marker Maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [CrossRef]
- Crossa, J.; De Los Campos, G.; Pérez, P.; Gianola, D.; Burgueño, J.; Araus, J.L.; Makumbi, D.; Singh, R.P.; Dreisigacker, S.; Yan, J.; et al. Prediction of genetic values of quantitative traits in plant breeding using pedigree and molecular markers. Genetics 2010, 186, 713–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; Zhao, Y.; Gowda, M.; Longin, C.F.H.; Reif, J.C.; Mette, M.F. Predicting Hybrid Performances for Quality Traits through Genomic-Assisted Approaches in Central European Wheat. PLoS ONE 2016, 11, e0158635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Q.; Gong, C.; Wang, Q.; Zhou, D.; Yang, H.; Pan, W.; Li, B.; Zhang, D. Genetic architecture of growth traits in Populus revealed by integrated quantitative trait locus (QTL) analysis and association studies. New Phytol. 2015, 209, 1067–1082. [Google Scholar] [CrossRef] [PubMed]
- Bartholomé, J.; Bink, M.C.; Van Heerwaarden, J.; Chancerel, E.; Boury, C.; Lesur, I.; Isik, F.; Bouffier, L.; Plomion, C. Linkage and Association Mapping for Two Major Traits Used in the Maritime Pine Breeding Program: Height Growth and Stem Straightness. PLoS ONE 2016, 11, e0165323. [Google Scholar] [CrossRef] [PubMed]
- Mori, H.; Ueno, S.; Ujino-Ihara, T.; Fujiwara, T.; Yamashita, K.; Kanetani, S.; Endo, R.; Matsumoto, A.; Uchiyama, K.; Matsui, Y.; et al. Mapping quantitative trait loci for growth and wood property traits in Cryptomeria japonica across multiple environments. Tree Genet. Genomes 2019, 15, 43. [Google Scholar] [CrossRef]
- Valenzuela, C.E.; Ballesta, P.; Ahmar, S.; Fiaz, S.; Heidari, P.; Maldonado, C.; Mora-Poblete, F. Haplotype- and SNP-Based GWAS for Growth and Wood Quality Traits in Eucalyptus cladocalyx Trees under Arid Conditions. Plants 2021, 10, 148. [Google Scholar] [CrossRef]
- Missiaggia, A.A.; Piacezzi, A.L.; Grattapaglia, D. Genetic mapping of Eef1, a major effect QTL for early flowering in Eucalyptus grandis. Tree Genet. Genomes 2005, 1, 79–84. [Google Scholar] [CrossRef] [Green Version]
- Bundock, P.C.; Potts, B.M.; Vaillancourt, R.E. Detection and stability of quantitative trait loci (QTL) in Eucalyptus globulus. Tree Genet. Genomes 2007, 4, 85–95. [Google Scholar] [CrossRef]
- Arriagada, O.; Júnior, A.T.D.A.; Mora, F. Thirteen years under arid conditions: Exploring marker-trait associations in Eucalyptus cladocalyx for complex traits related to flowering, stem form and growth. Breed. Sci. 2018, 68, 367–374. [Google Scholar] [CrossRef] [Green Version]
- Thumma, B.R.; Baltunis, B.S.; Bell, J.C.; Emebiri, L.C.; Moran, G.F.; Southerton, S.G. Quantitative trait locus (QTL) analysis of growth and vegetative propagation traits in Eucalyptus nitens full-sib families. Tree Genet. Genomes 2010, 6, 877–889. [Google Scholar] [CrossRef]
- Thavamanikumar, S.; McManus, L.J.; Ades, P.K.; Bossinger, G.; Stackpole, D.J.; Kerr, R.; Hadjigol, S.; Freeman, J.; Vaillancourt, R.; Zhu, P.; et al. Association mapping for wood quality and growth traits in Eucalyptus globulus ssp. globulus Labill identifies nine stable marker-trait associations for seven traits. Tree Genet. Genomes 2014, 10, 1661–1678. [Google Scholar] [CrossRef]
- Resende, R.T.; Resende, M.D.V.; Silva, F.F.; Azevedo, C.F.; Takahashi, E.K.; Silva-Junior, O.B.; Grattapaglia, D. Regional heritability mapping and genome-wide association identify loci for complex growth, wood and disease resistance traits in Eucalyptus. New Phytol. 2017, 213, 1287–1300. [Google Scholar] [CrossRef] [Green Version]
- Fahrenkrog, A.M.; Neves, L.G.; Resende, M.F.R.; Vazquez, A.I.; Campos, G.; Dervinis, C.; Sykes, R.; Davis, M.; Davenport, R.; Barbazuk, W.; et al. Genome-wide association study reveals putative regulators of bioenergy traits in Populus deltoides. New Phytol. 2016, 213, 799–811. [Google Scholar] [CrossRef]
- Baison, J.; Vidalis, A.; Zhou, L.; Chen, Z.Q.; Li, Z.; Sillanpää, M.J.; Bernhardsson, C.; Scoffield, D.; Forsberg, N.; Olsson, L.; et al. Association mapping identified novel candidate loci affecting wood formation in Norway spruce. bioRxiv 2018, 100, 83–100. [Google Scholar]
- Guerra, F.P.; Suren, H.; Holliday, J.; Richards, J.H.; Fiehn, O.; Famula, R.; Stanton, B.J.; Shuren, R.; Sykes, R.; Davis, M.F.; et al. Exome resequencing and GWAS for growth, ecophysiology, and chemical and metabolomic composition of wood of Populus trichocarpa. BMC Genom. 2019, 20, 875. [Google Scholar] [CrossRef] [Green Version]
- Valenzuela, C.E.; Ballesta, P.; Maldonado, C.; Baettig, R.; Arriagada, O.; Mafra, G.S.; Mora, F. Bayesian Mapping Reveals Large-Effect Pleiotropic QTLs for Wood Density and Slenderness Index in 17-Year-Old Trees of Eucalyptus cladocalyx. Forests 2019, 10, 241. [Google Scholar] [CrossRef] [Green Version]
- Daetwyler, H.D.; Calus, M.P.L.; Pong-Wong, R.; Campos, G.D.L.; Hickey, J.M. Genomic Prediction in Animals and Plants: Simulation of Data, Validation, Reporting, and Benchmarking. Genetics 2013, 193, 347–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Supple, M.A.; Bragg, J.G.; Broadhurst, L.M.; Nicotra, A.B.; Byrne, M.; Andrew, R.L.; Widdup, A.; Aitken, N.C.; Borevitz, J.O. Landscape genomic prediction for restoration of a Eucalyptus foundation species under climate change. eLife 2018, 24, e31835. [Google Scholar] [CrossRef] [PubMed]
- Arenas, S.; Cortés, A.J.; Mastretta-Yanes, A.; Jaramillo-Correa, J.P. Evaluating the accuracy of genomic prediction for the management and conservation of relictual natural tree populations. Tree Genet. Genomes 2021, 17, 1–19. [Google Scholar] [CrossRef]
- Tibshirani, R. Regression shrinkage and selection via the Lasso. J. R. Stat. Soc. Ser. B (Methodol.) 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Habier, D.; Fernando, R.L.; Kizilkaya, K.; Garrick, D.J. Extension of the Bayesian alphabet for genomic selection. BMC Bioinform. 2011, 12, 186. [Google Scholar] [CrossRef] [Green Version]
- VanRaden, P.M. Efficient Methods to Compute Genomic Predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef] [Green Version]
- Resende, M.F.R.; Muñoz, P.; Garrick, D.; Fernando, R.L.; Davis, J.M.; Jokela, E.J.; Martin, T.; Peter, G.F.; Kirst, M. Accuracy of Genomic Selection Methods in a Standard Data Set of Loblolly Pine (Pinus taeda L.). Genetics 2012, 190, 1503–1510. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Xu, Y.; Hu, Z.; Xu, C. Genomic selection methods for crop improvement: Current status and prospects. Crop J. 2018, 6, 330–340. [Google Scholar] [CrossRef]
- Heslot, N.; Yang, H.P.; Sorrells, M.E.; Jannink, J.L. Genomic selection in plant breeding: A comparison of models. Crop Sci. 2012, 52, 146–160. [Google Scholar] [CrossRef]
- Campos, G.D.L.; Hickey, J.M.; Pong-Wong, R.; Daetwyler, H.D.; Calus, M.P.L. Whole-Genome Regression and Prediction Methods Applied to Plant and Animal Breeding. Genetics 2013, 193, 327–345. [Google Scholar] [CrossRef] [Green Version]
- Gianola, D.; Fernando, R.L.; Stella, A. Genomic-Assisted Prediction of Genetic Value with Semiparametric Procedures. Genetics 2006, 173, 1761–1776. [Google Scholar] [CrossRef] [Green Version]
- Pérez, P.; Campos, G.D.L. Genome-Wide Regression and Prediction with the BGLR Statistical Package. Genetics 2014, 198, 483–495. [Google Scholar] [CrossRef]
- Du, C.; Wei, J.; Wang, S.; Jia, Z. Genomic selection using principal component regression. Heredity 2018, 121, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Su, G.; Janss, L.; Zhang, Y.; Lund, M. Model comparison on genomic predictions using high-density markers for different groups of bulls in the Nordic Holstein population. J. Dairy Sci. 2013, 96, 4678–4687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Lund, M.; Sun, D.; Zhang, Q.; Su, G. Impact of relationships between test and training animals and among training animals on reliability of genomic prediction. J. Anim. Breed. Genet. 2015, 132, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Pook, T.; Freudenthal, J.; Korte, A.; Simianer, H. Using Local Convolutional Neural Networks for Genomic Prediction. Front. Genet. 2020, 11, 1366. [Google Scholar] [CrossRef]
- Yin, B.; Balvert, M.A.; Van Der Spek, A.R.; Dutilh, E.B.; Bohté, S.; Veldink, J.; Schönhuth, A. Using the structure of genome data in the design of deep neural networks for predicting amyotrophic lateral sclerosis from genotype. Bioinformatics 2019, 35, i538–i547. [Google Scholar] [CrossRef] [Green Version]
- Glória, L.S.; Cruz, C.D.; Vieira, R.A.M.; de Resende, M.D.V.; Lopes, P.S.; de Siqueira, O.H.G.B.D.; Fonseca e Silva, F. Accessing marker effects and heritability estimates from genome prediction by Bayesian regularized neural networks. Livest. Sci. 2016, 191, 91–96. [Google Scholar] [CrossRef]
- Leung, M.K.K.; Delong, A.; Alipanahi, B.; Frey, B.J. Machine Learning in Genomic Medicine: A Review of Computational Problems and Data Sets. Proc. IEEE 2015, 104, 176–197. [Google Scholar] [CrossRef]
- Gianola, D. Priors in whole-genome regression: The Bayesian alphabet returns. Genetics 2013, 194, 573–596. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Rodríguez, P.; Gianola, D.; González-Camacho, J.M.; Crossa, J.; Manès, Y.; Dreisigacker, S. Comparison Between Linear and Non-parametric Regression Models for Genome-Enabled Prediction in Wheat. G3 Genes Genomes Genet. 2012, 2, 1595–1605. [Google Scholar] [CrossRef] [Green Version]
- Zingaretti, L.M.; Gezan, S.A.; Ferrão, L.F.V.; Osorio, L.F.; Monfort, A.; Muñoz, P.R.; Whitaker, V.M.; Pérez-Enciso, M. Exploring Deep Learning for Complex Trait Genomic Prediction in Polyploid Outcrossing Species. Front. Plant Sci. 2020, 11, 25. [Google Scholar] [CrossRef] [Green Version]
- Alves, A.A.C.; Da Costa, R.M.; Bresolin, T.; Júnior, G.A.F.; Espigolan, R.; Ribeiro, A.M.F.; Carvalheiro, R.; De Albuquerque, L.G. Genome-wide prediction for complex traits under the presence of dominance effects in simulated populations using GBLUP and machine learning methods. J. Anim. Sci. 2020, 98, skaa179. [Google Scholar] [CrossRef]
- Resende, M.D.V.; Resende, M.F.R.; Sansaloni, C.P.; Petroli, C.D.; Missiaggia, A.A.; Aguiar, A.M.; Abad, J.M.; Takahashi, E.K.; Rosado, A.M.; Faria, D.A.; et al. Genomic selection for growth and wood quality in Eucalyptus: Capturing the missing heritability and accelerating breeding for complex traits in forest trees. New Phytol. 2012, 194, 116–128. [Google Scholar] [CrossRef]
- Weber, K.L.; Thallman, R.M.; Keele, J.W.; Snelling, W.M.; Bennett, G.L.; Smith, T.P.L.; McDaneld, T.G.; Allan, M.F.; Van Eenennaam, A.L.; Kuehn, L.A. Accuracy of genomic breeding values in multibreed beef cattle populations derived from deregressed breeding values and phenotypes1,2. J. Anim. Sci. 2012, 90, 4177–4190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pryce, J.; Johnston, J.; Hayes, B.; Sahana, G.; Weigel, K.; McParland, S.; Spurlock, D.; Krattenmacher, N.; Spelman, R.; Wall, E.; et al. Imputation of genotypes from low density (50,000 markers) to high density (700,000 markers) of cows from research herds in Europe, North America, and Australasia using 2 reference populations. J. Dairy Sci. 2014, 97, 1799–1811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isik, F.; Bartholomé, J.; Farjat, A.; Chancerel, E.; Raffin, A.; Sanchez, L.; Plomion, C.; Bouffier, L. Genomic selection in maritime pine. Plant Sci. 2016, 242, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Cappa, E.P.; de Lima, B.M.; da Silva-Junior, O.B.; Garcia, C.C.; Mansfield, S.D.; Grattapaglia, D. Improving genomic prediction of growth and wood traits in Eucalyptus using phenotypes from non-genotyped trees by single-step GBLUP. Plant Sci. 2019, 284, 9–15. [Google Scholar] [CrossRef]
- Ballesta, P.; Bush, D.; Silva, F.F.; Mora, F. Genomic Predictions Using Low-Density SNP Markers, Pedigree and GWAS Information: A Case Study with the Non-Model Species Eucalyptus cladocalyx. Plants 2020, 9, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, B.S.F.; Neves, L.G.; Filho, J.E.D.A.; Resende, M.F.R.; Muñoz, P.R.; Dos Santos, P.E.T.; Filho, E.P.; Kirst, M.; Grattapaglia, D. Genomic prediction in contrast to a genome-wide association study in explaining heritable variation of complex growth traits in breeding populations of Eucalyptus. BMC Genom. 2017, 18, 524. [Google Scholar] [CrossRef] [Green Version]
- Suontama, M.; Klápště, J.; Telfer, E.; Graham, N.; Stovold, T.; Low, C.; McKinley, R.; Dungey, H. Efficiency of genomic prediction across two Eucalyptus nitens seed orchards with different selection histories. Heredity 2019, 122, 370–379. [Google Scholar] [CrossRef] [Green Version]
- Thavamanikumar, S.; Arnold, R.J.; Luo, J.; Thumma, B.R. Genomic Studies Reveal Substantial Dominant Effects and Improved Genomic Predictions in an Open-Pollinated Breeding Population of Eucalyptus pellita. G3 Genes Genomes Genet. 2020, 10, 3751–3763. [Google Scholar] [CrossRef]
- Rambolarimanana, T.; Ramamonjisoa, L.; Verhaegen, D.; Tsy, J.-M.L.P.; Jacquin, L.; Cao-Hamadou, T.-V.; Makouanzi, G.; Bouvet, J.-M. Performance of multi-trait genomic selection for Eucalyptus robusta breeding program. Tree Genet. Genomes 2018, 14, 71. [Google Scholar] [CrossRef]
- Klápště, J.; Dungey, H.S.; Telfer, E.J.; Suontama, M.; Graham, N.J.; Li, Y.; McKinley, R. Marker Selection in Multivariate Genomic Prediction Improves Accuracy of Low Heritability Traits. Front. Genet. 2020, 11, 499094. [Google Scholar] [CrossRef]
- Tan, B.; Grattapaglia, D.; Martins, G.; Ferreira, K.Z.; Sundberg, B.; Ingvarsson, P.K. Evaluating the accuracy of genomic prediction of growth and wood traits in two Eucalyptus species and their F1 hybrids. BMC Plant Biol. 2017, 17, 110. [Google Scholar] [CrossRef] [Green Version]
- Cappa, E.P.; El-Kassaby, Y.A.; Muñoz, F.; Garcia, M.N.; Villalba, P.V.; Klápště, J.; Poltri, S.N.M. Genomic-based multiple-trait evaluation in Eucalyptus grandis using dominant DArT markers. Plant Sci. 2018, 271, 27–33. [Google Scholar] [CrossRef]
- Durán, R.; Isik, F.; Zapata-Valenzuela, J.; Balocchi, C.; Valenzuela, S. Genomic predictions of breeding values in a cloned Eucalyptus globulus population in Chile. Tree Genet. Genomes 2017, 13, 74. [Google Scholar] [CrossRef]
- Jurcic, E.J.; Villalba, P.V.; Pathauer, P.S.; Palazzini, D.A.; Oberschelp, G.P.J.; Harrand, L.; Garcia, M.N.; Aguirre, N.C.; Acuña, C.V.; Martínez, M.C.; et al. Single-step genomic prediction of Eucalyptus dunnii using different identity-by-descent and identity-by-state relationship matrices. Heredity 2021, 18, 1–4. [Google Scholar]
- Lenz, P.R.N.; Nadeau, S.; Azaiez, A.; Gérardi, S.; DesLauriers, M.; Perron, M.; Isabel, N.; Beaulieu, J.; Bousquet, J. Genomic prediction for hastening and improving efficiency of forward selection in conifer polycross mating designs: An example from white spruce. Heredity 2020, 124, 562–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaulieu, J.; Nadeau, S.; Ding, C.; Celedon, J.M.; Azaiez, A.; Ritland, C.; Laverdière, J.P.; Deslauriers, M.; Adams, G.; Fullarton, M.; et al. Genomic selection for resistance to spruce budworm in white spruce and relationships with growth and wood quality traits. Evolut. Appl. 2020, 13, 2704–2722. [Google Scholar] [CrossRef] [PubMed]
- Lenz, P.R.; Beaulieu, J.; Mansfield, S.D.; Clément, S.; Desponts, M.; Bousquet, J. Factors affecting the accuracy of genomic selection for growth and wood quality traits in an advanced-breeding population of black spruce (Picea mariana). BMC Genom. 2017, 18, 335. [Google Scholar] [CrossRef]
- Ratcliffe, B.; El-Dien, O.G.; Cappa, E.P.; Porth, I.; Klápště, J.; Chen, C.; El-Kassaby, Y.A. Single-Step BLUP with Varying Genotyping Effort in Open-Pollinated Picea glauca. G3 Genes Genomes Genet. 2017, 7, 935–942. [Google Scholar] [CrossRef] [Green Version]
- Lenz, P.R.N.; Nadeau, S.; Mottet, M.J.; Perron, M.; Isabel, N.; Beaulieu, J.; Bousquet, J. Multi-trait genomic selection for weevil resistance, growth, and wood quality in Norway spruce. Evolut. Appl. 2020, 13, 76–94. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.-Q.; Baison, J.; Pan, J.; Karlsson, B.; Andersson, B.; Westin, J.; García-Gil, M.R.; Wu, H.X. Accuracy of genomic selection for growth and wood quality traits in two control-pollinated progeny trials using exome capture as the genotyping platform in Norway spruce. BMC Genom. 2018, 19, 946. [Google Scholar] [CrossRef] [Green Version]
- Ukrainetz, N.K.; Mansfield, S.D. Assessing the sensitivities of genomic selection for growth and wood quality traits in lodgepole pine using Bayesian models. Tree Genet. Genomes 2019, 16, 14. [Google Scholar] [CrossRef]
- Li, Y.; Klápště, J.; Telfer, E.; Wilcox, P.; Graham, N.; Macdonald, L.; Dungey, H.S. Genomic selection for non-key traits in radiata pine when the documented pedigree is corrected using DNA marker information. BMC Genom. 2019, 20, 1026. [Google Scholar] [CrossRef] [PubMed]
- Calleja-Rodriguez, A.; Pan, J.; Funda, T.; Chen, Z.; Baison, J.; Isik, F.; Abrahamsson, S.; Wu, H.X. Evaluation of the efficiency of genomic versus pedigree predictions for growth and wood quality traits in Scots pine. BMC Genom. 2020, 21, 796. [Google Scholar] [CrossRef] [PubMed]
- Cros, D.; Mbo-Nkoulou, L.; Bell, J.M.; Oum, J.; Masson, A.; Soumahoro, M.; Tran, D.M.; Achour, Z.; Le Guen, V.; Clement-Demange, A. Within-family genomic selection in rubber tree (Hevea brasiliensis) increases genetic gain for rubber production. Ind. Crop. Prod. 2019, 138, 111464. [Google Scholar] [CrossRef]
- Souza, L.M.; Francisco, F.R.; Gonçalves, P.S.; Junior, E.J.S.; Le Guen, V.; Fritsche-Neto, R.; Souza, A.P. Genomic Selection in Rubber Tree Breeding: A Comparison of Models and Methods for Managing G×E Interactions. Front. Plant Sci. 2019, 10, 1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rincent, R.; Charpentier, J.-P.; Faivre-Rampant, P.; Paux, E.; Le Gouis, J.; Bastien, C.; Segura, V. Phenomic Selection Is a Low-Cost and High-Throughput Method Based on Indirect Predictions: Proof of Concept on Wheat and Poplar. G3 Genes Genomes Genet. 2018, 8, 3961–3972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thistlethwaite, F.R.; Ratcliffe, B.; Klápště, J.; Porth, I.; Chen, C.; Stoehr, M.U.; El-Kassaby, Y.A. Genomic prediction accuracies in space and time for height and wood density of Douglas-fir using exome capture as the genotyping platform. BMC Genom. 2017, 18, 930. [Google Scholar] [CrossRef] [Green Version]
- Brown, M.D.; Glazner, C.G.; Zheng, C.; Thompson, A.E. Inferring Coancestry in Population Samples in the Presence of Linkage Disequilibrium. Genetics 2012, 190, 1447–1460. [Google Scholar] [CrossRef] [Green Version]
- Slavov, G.T.; DiFazio, S.P.; Martin, J.; Schackwitz, W.; Muchero, W.; Rodgers-Melnick, E.; Lipphardt, M.F.; Pennacchio, C.P.; Hellsten, U.; Pennacchio, L.A.; et al. Genome resequencing reveals multiscale geographic structure and extensive linkage disequilibrium in the forest tree Populus trichocarpa. New Phytol. 2012, 196, 713–725. [Google Scholar] [CrossRef]
- Lu, M.; Krutovsky, K.V.; Nelson, C.D.; Koralewski, T.E.; Byram, T.D.; Loopstra, C.A. Exome genotyping, linkage disequilibrium and population structure in loblolly pine (Pinus taeda L.). BMC Genom. 2016, 17, 730. [Google Scholar] [CrossRef] [Green Version]
- Gupta, P.K.; Rustgi, S.; Kulwal, P.L. Linkage disequilibrium and association studies in higher plants: Present status and future prospects. Plant Mol. Biol. 2005, 57, 461–485. [Google Scholar] [CrossRef]
- Neale, D.B.; Kremer, A. Forest tree genomics: Growing resources and applications. Nat. Rev. Genet. 2011, 12, 111–122. [Google Scholar] [CrossRef]
- Thavamanikumar, S.; Southerton, S.G.; Bossinger, G.; Thumma, B.R. Dissection of complex traits in forest trees—opportunities for marker-assisted selection. Tree Genet. Genomes 2013, 9, 627–639. [Google Scholar] [CrossRef]
- Olson, M.S.; Robertson, A.L.; Takebayashi, N.; Silim, S.; Schroeder, W.R.; Tiffin, P. Nucleotide diversity and linkage disequilibrium in balsam poplar (Populus balsamifera). New Phytol. 2010, 186, 526–536. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, C.T.; Wilkin, J.; Zhuang, J.; Cortés, A.J.; Perez-Quintero, A.L.; Gallagher, T.F.; Bohlmann, J.; Douglas, C.J.; Ellis, B.E.; Ritland, K. SNP discovery, gene diversity, and linkage disequilibrium in wild populations of Populus tremuloides. Tree Genet. Genomes 2012, 8, 821–829. [Google Scholar] [CrossRef]
- Guerra, F.P.; Wegrzyn, J.L.; Sykes, R.; Davis, M.F.; Stanton, B.J.; Neale, D.B. Association genetics of chemical wood properties in black poplar (Populus nigra). New Phytol. 2012, 197, 162–176. [Google Scholar] [CrossRef] [PubMed]
- Larsson, H.; Källman, T.; Gyllenstrand, N.; Lascoux, M. Distribution of Long-Range Linkage Disequilibrium and Tajima’s D Values in Scandinavian Populations of Norway Spruce (Picea abies). G3 Genes Genomes Genet. 2013, 3, 795–806. [Google Scholar] [CrossRef] [Green Version]
- Müller, D.; Schopp, P.; Melchinger, A.E. Selection on Expected Maximum Haploid Breeding Values Can Increase Genetic Gain in Recurrent Genomic Selection. G3 Genes Genomes Genet. 2018, 8, 1173–1181. [Google Scholar] [CrossRef] [Green Version]
- Thavamanikumar, S.; McManus, L.J.; Tibbits, J.F.; Bossinger, G. The significance of single nucleotide polymorphisms (SNPs) in Eucalyptus globulus breeding programs. Aust. For. 2011, 74, 23–29. [Google Scholar] [CrossRef]
- Stejskal, J.; Lstibůrek, M.; Klápště, J.; Čepl, J.; El-Kassaby, Y.A. Effect of genomic prediction on response to selection in forest tree breeding. Tree Genet. Genomes 2018, 14, 74. [Google Scholar] [CrossRef]
- Grattapaglia, D.; Resende, M.D.V. Genomic selection in forest tree breeding. Tree Genet. Genomes 2010, 7, 241–255. [Google Scholar] [CrossRef]
- Lorenz, A.J.; Smith, K.P. Adding Genetically Distant Individuals to Training Populations Reduces Genomic Prediction Accuracy in Barley. Crop Sci. 2015, 55, 2657–2667. [Google Scholar] [CrossRef] [Green Version]
- Lorenz, A.J.; Smith, K.; Jannink, J. Potential and Optimization of Genomic Selection for Fusarium Head Blight Resistance in Six-Row Barley. Crop Sci. 2012, 52, 1609–1621. [Google Scholar] [CrossRef]
- Thorwarth, P.; Ahlemeyer, J.; Bochard, A.-M.; Krumnacker, K.; Blümel, H.; Laubach, E.; Knöchel, N.; Cselényi, L.; Ordon, F.; Schmid, K.J. Genomic prediction ability for yield-related traits in German winter barley elite material. Theor. Appl. Genet. 2017, 130, 1669–1683. [Google Scholar] [CrossRef] [PubMed]
- Sapkota, S.; Boyles, R.; Cooper, E.; Brenton, Z.; Myers, M.; Kresovich, S. Impact of sorghum racial structure and diversity on genomic prediction of grain yield components. Crop Sci. 2020, 60, 132–148. [Google Scholar] [CrossRef] [Green Version]
- Gienapp, P.; Fior, S.; Guillaume, F.; Lasky, J.R.; Sork, V.L.; Csilléry, K. Genomic Quantitative Genetics to Study Evolution in the Wild. Trends Ecol. Evol. 2017, 32, 897–908. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Pressoir, G.; Briggs, W.H.; Bi, I.V.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S.; Nielsen, D.M.; Holland, J.; et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 2005, 38, 203–208. [Google Scholar] [CrossRef]
- Yamamoto, T.; Nagasaki, H.; Yonemaru, J.-I.; Ebana, K.; Nakajima, M.; Shibaya, T.; Yano, M. Fine definition of the pedigree haplotypes of closely related rice cultivars by means of genome-wide discovery of single-nucleotide polymorphisms. BMC Genom. 2010, 11, 267. [Google Scholar] [CrossRef] [Green Version]
- Voorrips, R.E.; Bink, M.C.A.M.; Kruisselbrink, J.W.; Putten, H.J.J.K.-V.; Van De Weg, W.E. PediHaplotyper: Software for consistent assignment of marker haplotypes in pedigrees. Mol. Breed. 2016, 36, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Pikunova, A.; Madduri, M.; Sedov, E.; Noordijk, Y.; Peil, A.; Troggio, M.; Bus, V.; Visser, R.G.; van de Weg, E. ‘Schmidt’s Antonovka’ is identical to ‘Common Antonovka’, an apple cultivar widely used in Russia in breeding for biotic and abiotic stresses. Tree Genet. Genomes 2013, 10, 261–271. [Google Scholar] [CrossRef]
- Di Guardo, M.; Micheletti, D.; Bianco, L.; Putten, H.J.J.K.-V.; Longhi, S.; Costa, F.; Aranzana, M.J.; Velasco, R.; Arús, P.; Troggio, M.; et al. ASSIsT: An automatic SNP scoring tool for in- and outbreeding species. Bioinformatics 2015, 31, 3873–3874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordborg, M.; Tavaré, S. Linkage disequilibrium: What history has to tell us. Trends Genet. 2002, 18, 83–90. [Google Scholar] [CrossRef]
- Machiela, M.J.; Chanock, S.J. LDlink: A web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics 2015, 31, 3555–3557. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.R.; Lübberstedt, T. Functional markers in plants. Trends Plant Sci. 2003, 8, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Hodgkinson, K.; Pullman, D. Duty to warn and genetic disease. Canadian journal of cardiovascular nursing. Can. J. Cardiovasc. Nurs. 2010, 20, 12–15. [Google Scholar] [PubMed]
- Sun, Y.; Lu, Y.; Xie, L.; Deng, Y.; Li, S.; Qin, X. Interferon gamma polymorphisms and hepatitis B virus-related liver cirrhosis risk in a Chinese population. Cancer Cell Int. 2015, 15, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Roos, A.; Schrooten, C.; Druet, T. Genomic breeding value estimation using genetic markers, inferred ancestral haplotypes, and the genomic relationship matrix. J. Dairy Sci. 2011, 94, 4708–4714. [Google Scholar] [CrossRef] [Green Version]
- Boichard, D.; Guillaume, F.; Baur, A.; Croiseau, P.; Rossignol, M.N.; Boscher, M.Y.; Druet, T.; Genestout, L.; Colleau, J.J.; Journaux, L.; et al. Genomic selection in French dairy cattle. Anim. Prod. Sci. 2012, 52, 115–120. [Google Scholar] [CrossRef] [Green Version]
- Edriss, V.; Fernando, R.L.; Su, G.; Lund, M.S.; Guldbrandtsen, B. The effect of using genealogy-based haplotypes for genomic prediction. Genet. Sel. Evol. 2013, 45, 5. [Google Scholar] [CrossRef] [Green Version]
- Cuyabano, B.C.; Su, G.; Lund, M.S. Genomic prediction of genetic merit using LD-based haplotypes in the Nordic Holstein population. BMC Genom. 2014, 15, 1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jónás, D.; Ducrocq, V.; Croiseau, P. Short communication: The combined use of linkage disequilibrium–based haploblocks and allele frequency–based haplotype selection methods enhances genomic evaluation accuracy in dairy cattle. J. Dairy Sci. 2017, 100, 2905–2908. [Google Scholar] [CrossRef] [Green Version]
- Curtis, D.; North, B.V.; Sham, P.C. Use of an artificial neural network to detect association between a disease and multiple marker genotypes. Ann. Hum. Genet. 2001, 65, 95–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Schmidt, R.H.; Reif, J.C. Haplotype-Based Genome-Wide Prediction Models Exploit Local Epistatic Interactions among Markers. G3 Genes Genomes Genet. 2018, 8, 1687–1699. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Schmidt, R.H.; Reif, J.C.; Jiang, Y. Selecting Closely-Linked SNPs Based on Local Epistatic Effects for Haplotype Construction Improves Power of Association Mapping. G3 Genes Genomes Genet. 2019, 9, 4115–4126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Q.; Wei, Z.; Zhao, X.; Yang, X.; Ci, N.; Zhang, D. Dissection of additive, dominant, epistatic roles of allelic variation within heat shock factor genes in Chinese indigenous poplar (Populus simonii). Tree Genet. Genomes 2016, 12, 91. [Google Scholar] [CrossRef]
- Hickey, J.M.; Dreisigacker, S.; Crossa, J.; Hearne, S.; Babu, R.; Prasanna, B.M.; Grondona, M.; Zambelli, A.D.; Windhausen, V.S.; Mathews, K.; et al. Evaluation of Genomic Selection Training Population Designs and Genotyping Strategies in Plant Breeding Programs Using Simulation. Crop Sci. 2014, 54, 1476–1488. [Google Scholar] [CrossRef] [Green Version]
- Calus, M.P.; Meuwissen, T.H.; Windig, J.J.; Knol, E.F.; Schrooten, C.; Vereijken, A.L.; Veerkamp, R.F. Effects of the number of markers per haplotype and clustering of haplotypes on the accuracy of QTL mapping and prediction of genomic breeding values. Genet. Sel. Evol. 2009, 178, 553–561. [Google Scholar] [CrossRef] [Green Version]
- Ødegård, J.; Moen, T.; Santi, N.; Korsvoll, S.A.; Kjøglum, S.; Meuwissen, T.H.E. Genomic prediction in an admixed population of Atlantic salmon (Salmo salar). Front. Genet. 2014, 5, 402. [Google Scholar] [CrossRef] [Green Version]
- Villumsen, T.; Janss, L.; Lund, M. The importance of haplotype length and heritability using genomic selection in dairy cattle. J. Anim. Breed. Genet. 2009, 126, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Rutkoski, J.; Poland, J.; Mondal, S.; Autrique, E.; Pérez, L.G.; Crossa, J.; Reynolds, M.P.; Singh, R. Canopy Temperature and Vegetation Indices from High-Throughput Phenotyping Improve Accuracy of Pedigree and Genomic Selection for Grain Yield in Wheat. G3 Genes Genomes Genet. 2016, 6, 2799–2808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Z.; Cogan, N.O.I.; Pembleton, L.W.; Spangenberg, G.C.; Forster, J.W.; Hayes, B.; Daetwyler, H.D. Genetic Gain and Inbreeding from Genomic Selection in a Simulated Commercial Breeding Program for Perennial Ryegrass. Plant Genome 2016, 130, 969–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eynard, E.S.; Croiseau, P.; Laloë, D.; Fritz, S.; Calus, M.P.L.; Restoux, G. Which Individuals to Choose To Update the Reference Population? Minimizing the Loss of Genetic Diversity in Animal Genomic Selection Programs. G3 Genes Genomes Genet. 2018, 8, 113–121. [Google Scholar] [CrossRef] [Green Version]
- Doublet, A.-C.; Croiseau, P.; Fritz, S.; Michenet, A.; Hozé, C.; Danchin-Burge, C.; Laloë, D.; Restoux, G. The impact of genomic selection on genetic diversity and genetic gain in three French dairy cattle breeds. Genet. Sel. Evol. 2019, 51, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Z.; Shi, F.; Hayes, B.; Daetwyler, H.D. Mitigation of inbreeding while preserving genetic gain in genomic breeding programs for outbred plants. Theor. Appl. Genet. 2017, 130, 969–980. [Google Scholar] [CrossRef] [PubMed]
- Sonesson, A.K.; Woolliams, A.J.; Meuwissen, T.H. Genomic selection requires genomic control of inbreeding. Genet. Sel. Evol. 2012, 44, 27. [Google Scholar] [CrossRef] [Green Version]
- Daetwyler, H.D.; Hayden, M.; Spangenberg, G.C.; Hayes, B. Selection on Optimal Haploid Value Increases Genetic Gain and Preserves More Genetic Diversity Relative to Genomic Selection. Genetics 2015, 200, 1341–1348. [Google Scholar] [CrossRef] [Green Version]
- Leng, P.F.; Lübberstedt, T.; Xu, M.L. Genomics-assisted breeding—A revolutionary strategy for crop improvement. J. Integr. Agric. 2017, 16, 2674–2685. [Google Scholar] [CrossRef]
- McKown, A.D.; Klápště, J.; Guy, R.D.; El-Kassaby, Y.A.; Mansfield, S.D. Ecological genomics of variation in bud-break phenology and mechanisms of response to climate warming inPopulus trichocarpa. New Phytol. 2018, 220, 300–316. [Google Scholar] [CrossRef] [Green Version]
- Apuli, R.-P.; Richards, T.; Rendón-Anaya, M.; Karacic, A.; Rönnberg-Wästljung, A.-C.; Ingvarsson, P.K. The genetic basis of adaptation in phenology in an introduced population of Black Cottonwood (Populus trichocarpa, Torr. & Gray). BMC Plant Biol. 2021, 21, 317. [Google Scholar] [CrossRef]
- Lamara, M.; Raherison, E.; Lenz, P.; Beaulieu, J.; Bousquet, J.; MacKay, J. Genetic architecture of wood properties based on association analysis and co-expression networks in white spruce. New Phytol. 2015, 210, 240–255. [Google Scholar] [CrossRef] [Green Version]
- Hiraoka, Y.; Fukatsu, E.; Mishima, K.; Hirao, T.; Teshima, K.; Tamura, M.; Tsubomura, M.; Iki, T.; Kurita, M.; Takahashi, M.; et al. Potential of Genome-Wide Studies in Unrelated Plus Trees of a Coniferous Species, Cryptomeria japonica (Japanese Cedar). Front. Plant Sci. 2018, 9, 1322. [Google Scholar] [CrossRef]
- Gong, C.; Du, Q.; Xie, J.; Quan, M.; Chen, B.; Zhang, D. Dissection of Insertion–Deletion Variants within Differentially Expressed Genes Involved in Wood Formation in Populus. Front. Plant Sci. 2018, 8, 2199. [Google Scholar] [CrossRef] [Green Version]
- Chhetri, H.; Furches, A.; Macaya-Sanz, D.; Walker, A.R.; Kainer, D.; Jones, P.; Harman-Ware, A.E.; Tschaplinski, T.J.; Jacobson, D.; Tuskan, G.A.; et al. Genome-Wide Association Study of Wood Anatomical and Morphological Traits in Populus trichocarpa. Front. Plant Sci. 2020, 11, 1391. [Google Scholar] [CrossRef]
- Quan, M.; Liu, X.; Du, Q.; Xiao, L.; Lu, W.; Fang, Y.; Li, P.; Ji, L.; Zhang, D. Erratum to: Genome-wide association studies reveal the coordinated regulatory networks underlying photosynthesis and wood formation in Populus. J. Exp. Bot. 2021, 72, 5777–5780. [Google Scholar] [CrossRef] [PubMed]
- Müller, B.S.F.; Filho, J.E.D.A.; Lima, B.M.; Garcia, C.C.; Missiaggia, A.; Aguiar, A.M.; Takahashi, E.; Kirst, M.; Gezan, S.A.; Silva-Junior, O.B.; et al. Independent and Joint- GWAS for growth traits in Eucalyptus by assembling genome-wide data for 3373 individuals across four breeding populations. New Phytol. 2018, 221, 818–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, Q.; Cai, Y.; He, B.; Liu, W.; Pan, Q.; Zhang, Q. Core set construction and association analysis of Pinus massoniana from Guangdong province in southern China using SLAF-seq. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wu, H.; Yang, W.; Zhao, W.; Tong, C. Multivariate linear mixed model enhanced the power of identifying genome-wide association to poplar tree heights in a randomized complete block design. G3 Genes Genomes Genet. 2021, 11, jkaa053. [Google Scholar] [CrossRef]
- Liu, J.; Sniezko, R.A.; Zamany, A.; Williams, H.; Wang, N.; Kegley, A.; Savin, D.P.; Chen, H.; Sturrock, R.N. Saturated genic SNP mapping identified functional candidates and selection tools for the Pinus monticola Cr2 locus controlling resistance to white pine blister rust. Plant Biotechnol. J. 2017, 15, 1149–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muchero, W.; Sondreli, K.L.; Chen, J.G.; Urbanowicz, B.R.; Zhang, J.; Singan, V.; Yang, Y.; Brueggeman, R.S.; Franco-Coronado, J.; Abraham, N.; et al. Association mapping, transcriptomics, and transient expression identify candidate genes mediating plant-pathogen interactions in a tree. Proc. Natl. Acad. Sci. USA 2018, 115, 11573–11578. [Google Scholar] [CrossRef] [Green Version]
- De Lange, W.J.; Veldtman, R.; Allsopp, M.H. Valuation of pollinator forage services provided by Eucalyptus cladocalyx. J. Environ. Manag. 2013, 125, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Elfstrand, M.; Baison, J.; Lundén, K.; Zhou, L.; Vos, I.; Capador, H.D.; Åslund, M.S.; Chen, Z.; Chaudhary, R.; Olson, Å.; et al. Association genetics identifies a specifically regulated Norway spruce laccase gene, PaLAC5, linked to Heterobasidion parviporum resistance. Plant Cell Environ. 2020, 43, 1779–1791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, D.; Hallingbäck, H.R.; Wu, H.X. Estimation of number and size of QTL effects in forest tree traits. Tree Genet. Genomes 2016, 12, 110. [Google Scholar] [CrossRef] [Green Version]
- Thumma, B.R.; Southerton, S.G.; Bell, J.C.; Owen, J.V.; Henery, M.L.; Moran, G.F. Quantitative trait locus (QTL) analysis of wood quality traits in Eucalyptus nitens. Tree Genet. Genomes 2010, 6, 305–317. [Google Scholar] [CrossRef]
- Du, Q.; Lu, W.; Quan, M.; Xiao, L.; Song, F.; Li, P.; Zhou, D.; Xie, J.; Wang, L.; Zhang, D. Genome-Wide Association Studies to Improve Wood Properties: Challenges and Prospects. Front. Plant Sci. 2018, 9, 1912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dort, E.N.; Tanguay, P.; Hamelin, R.C. CRISPR/Cas9 Gene Editing: An Unexplored Frontier for Forest Pathology. Front. Plant Sci. 2020, 11, 1126. [Google Scholar] [CrossRef]
- Elorriaga, E.; Klocko, A.L.; Ma, C.; Strauss, S.H. Variation in Mutation Spectra among CRISPR/Cas9 Mutagenized Poplars. Front. Plant Sci. 2018, 9, 594. [Google Scholar] [CrossRef] [Green Version]
- Strauss, S.H.; Costanza, A.; Séguin, A. Genetically engineered trees: Paralysis from good intentions. Science 2015, 349, 794–795. [Google Scholar] [CrossRef]
- Strauss, S.H.; Jones, K.N.; Lu, H.; Petit, J.D.; Klocko, A.L.; Betts, M.G.; Brosi, B.J.; Fletcher Jr, R.J.; Needham, M.D. Reproductive modification in forest plantations: Impacts on biodiversity and society. New Phytol. 2017, 213, 1000–1021. [Google Scholar] [CrossRef] [Green Version]
- Mussolino, C.; Cathomen, T. RNA guides genome engineering. Nat. Biotechnol. 2013, 31, 208–209. [Google Scholar] [CrossRef]
- Koonin, E.V.; Makarova, K.S.; Zhang, F. Diversity, classification and evolution of CRISPR-Cas systems. Curr. Opin. Microbiol. 2017, 37, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Demirci, Y.; Zhang, B.; Unver, T. CRISPR/Cas9: An RNA-guided highly precise synthetic tool for plant genome editing. J. Cell. Physiol. 2017, 233, 1844–1859. [Google Scholar] [CrossRef] [PubMed]
- Fernandez i Marti, A.F.; Dodd, R.S. Using CRISPR as a gene editing tool for validating adaptive gene function in tree landscape genomics. Front. Ecol. Evol. 2018, 6, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Fan, D.; Liu, T.; Li, C.; Jiao, B.; Li, S.; Hou, Y.; Luo, K. Efficient CRISPR/Cas9-mediated Targeted Mutagenesis in Populus in the First Generation. Sci. Rep. 2015, 5, 12217. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Murovec, J.; Pirc, Ž.; Yang, B. New variants of CRISPR RNA-guided genome editing enzymes. Plant Biotechnol. J. 2017, 15, 917–926. [Google Scholar] [CrossRef] [Green Version]
- Mao, Y.; Zhang, H.; Xu, N.; Zhang, B.; Gou, F.; Zhu, J.-K. Application of the CRISPR–Cas System for Efficient Genome Engineering in Plants. Mol. Plant 2013, 6, 2008–2011. [Google Scholar] [CrossRef] [Green Version]
- Charrier, A.; Vergne, E.; Dousset, N.; Richer, A.; Petiteau, A.; Chevreau, E. Efficient Targeted Mutagenesis in Apple and First Time Edition of Pear Using the CRISPR-Cas9 System. Front. Plant Sci. 2019, 10, 40. [Google Scholar] [CrossRef] [Green Version]
- Mao, Y.; Botella, J.R.; Liu, Y.; Zhu, J.-K. Gene editing in plants: Progress and challenges. Natl. Sci. Rev. 2019, 6, 421–437. [Google Scholar] [CrossRef] [Green Version]
- Samanta, M.K.; Dey, A.; Gayen, S. CRISPR/Cas9: An advanced tool for editing plant genomes. Transgenic Res. 2016, 25, 561–573. [Google Scholar] [CrossRef]
- Ren, C.; Liu, X.; Zhang, Z.; Wang, Y.; Duan, W.; Li, S.; Liang, Z. CRISPR/Cas9-mediated efficient targeted mutagenesis in Chardonnay (Vitis vinifera L.). Sci. Rep. 2016, 6, 32289. [Google Scholar] [CrossRef]
- Van Zeijl, A.; Wardhani, T.; Kalhor, M.S.; Rutten, L.; Bu, F.; Hartog, M.; Linders, S.; Fedorova, E.; Bisseling, T.; Kohlen, W.; et al. CRISPR/Cas9-Mediated Mutagenesis of Four Putative Symbiosis Genes of the Tropical Tree Parasponia andersonii Reveals Novel Phenotypes. Front. Plant Sci. 2018, 9, 284. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Jacobs, T.B.; Xue, L.; Harding, S.A.; Tsai, C. Exploiting SNP s for biallelic CRISPR mutations in the outcrossing woody perennial Populus reveals 4-coumarate: CoA ligase specificity and redundancy. New Phytol. 2015, 208, 298–301. [Google Scholar] [CrossRef]
- Bewg, W.P.; Ci, D.; Tsai, C.-J. Genome Editing in Trees: From Multiple Repair Pathways to Long-Term Stability. Front. Plant Sci. 2018, 9, 1732. [Google Scholar] [CrossRef]
- Tsai, C.-J.; Xue, L.-J. CRISPRing into the woods. GM Crop. Food 2015, 6, 206–215. [Google Scholar] [CrossRef]
- Boerjan, W.; Ralph, J.; Baucher, M. Lignin biosynthesis. Ann. Rev. Plant Boil. 2003, 54, 519–546. [Google Scholar] [CrossRef]
- Xu, C.; Fu, X.; Liu, R.; Guo, L.; Ran, L.; Li, C.; Tian, Q.; Jiao, B.; Wang, B.; Luo, K. PtoMYB170 positively regulates lignin deposition during wood formation in poplar and confers drought tolerance in transgenic Arabidopsis. Tree Physiol. 2017, 37, 1713–1726. [Google Scholar] [CrossRef] [Green Version]
- Wan, S.; Li, C.; Ma, X.; Luo, K. PtrMYB57 contributes to the negative regulation of anthocyanin and proanthocyanidin biosynthesis in poplar. Plant Cell Rep. 2017, 36, 1263–1276. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Li, Y.; Xu, D.; Yang, C.; Li, C.; Luo, K. Molecular cloning and characterization of a brassinosteriod biosynthesis-related gene PtoDWF4 from Populus tomentosa. Tree Physiol. 2018, 38, 1424–1436. [Google Scholar] [CrossRef] [PubMed]
- Elorriaga, E.; Klocko, A.L.; Ma, C.; du Plessis, M.; An, X.; Myburg, A.A.; Strauss, S.H. Genetic containment in vegetatively propagated forest trees: CRISPR disruption of LEAFY function in Eucalyptus gives sterile indeterminate inflorescences and normal juvenile development. Plant Biotechnol. J. 2021, 19, 1743–1755. [Google Scholar] [CrossRef]
- Sow, M.D.; Allona, I.; Ambroise, C.; Conde, D.; Fichot, R.; Gribkova, S.; Jorge, V.; Le-Provost, G.; Pâques, L.; Plomion, C.; et al. Epigenetics in forest trees: State of the art and potential implications for breeding and management in a context of climate change. Adv. Bot. Res. 2018, 88, 387–453. [Google Scholar] [CrossRef]
- Ding, L.; Chen, Y.; Ma, Y.; Wang, H.; Wei, J. Effective reduction in chimeric mutants of poplar trees produced by CRISPR/Cas9 through a second round of shoot regeneration. Plant Biotechnol. Rep. 2020, 14, 549–558. [Google Scholar] [CrossRef]
- Bruegmann, T.; Deecke, K.; Fladung, M. Evaluating the Efficiency of gRNAs in CRISPR/Cas9 Mediated Genome Editing in Poplars. Int. J. Mol. Sci. 2019, 20, 3623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grattapaglia, D.; Silva-Junior, O.B.; Resende, R.T.; Cappa, E.P.; Müller, B.; Tan, B.; Isik, F.; Ratcliffe, B.; El-Kassaby, Y.A. Quantitative Genetics and Genomics Converge to Accelerate Forest Tree Breeding. Front. Plant Sci. 2018, 9, 1693. [Google Scholar] [CrossRef]
- Rennie, E.A.; Hansen, S.F.; Baidoo, E.E.; Hadi, M.Z.; Keasling, J.; Scheller, H.V. Three Members of the Arabidopsis Glycosyltransferase Family 8 Are Xylan Glucuronosyltransferases. Plant Physiol. 2012, 159, 1408–1417. [Google Scholar] [CrossRef] [Green Version]
- Comeau, A.M.; Dufour, J.; Bouvet, G.F.; Jacobi, V.; Nigg, M.; Henrissat, B.; Laroche, J.; Levesque, R.C.; Bernier, L. Functional Annotation of the Ophiostoma novo-ulmi Genome: Insights into the Phytopathogenicity of the Fungal Agent of Dutch Elm Disease. Genome Biol. Evol. 2014, 7, 410–430. [Google Scholar] [CrossRef] [Green Version]
- Forgetta, V.; Leveque, G.; Dias, J.; Grove, D.; Lyons Jr, R.; Genik, S.; Wright, C.; Singh, S.; Peterson, N.; Zianni, M. Sequencing of the Dutch elm disease fungus genome using the Roche/454 GS-FLX Titanium System in a comparison of multiple genomics core facilities. J. Biomol. Tech. JBT 2013, 24, 39. [Google Scholar] [CrossRef] [PubMed]
- An, Y.; Geng, Y.; Yao, J.; Fu, C.; Lu, M.; Wang, C.; Du, J. Efficient Genome Editing in Populus Using CRISPR/Cas12a. Front. Plant Sci. 2020, 11, 593938. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Doudna, J.A. CRISPR–Cas9 Structures and Mechanisms. Annu. Rev. Biophys. 2017, 46, 505–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osakabe, Y.; Sugano, S.S.; Osakabe, K. Genome engineering of woody plants: Past, present and future. J. Wood Sci. 2016, 62, 217–225. [Google Scholar] [CrossRef] [Green Version]
- Luo, G.; Palmgren, M. GRF-GIF Chimeras Boost Plant Regeneration. Trends Plant Sci. 2020, 26, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Debernardi, J.M.; Tricoli, D.M.; Ercoli, M.F.; Hayta, S.; Ronald, P.; Palatnik, J.F.; Dubcovsky, J. A GRF–GIF chimeric protein improves the regeneration efficiency of transgenic plants. Nat. Biotechnol. 2020, 38, 1274–1279. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.; Martin-Ortigosa, S.; Finer, J.; Orchard, N.; Gunadi, A.; Batts, L.A.; Thakare, D.; Rush, B.; Schmitz, O.; Stuiver, M.; et al. Overexpression of the Transcription Factor Growth-Regulating Factor5 Improves Transformation of Dicot and Monocot Species. Front. Plant Sci. 2020, 11, 572319. [Google Scholar] [CrossRef] [PubMed]
- Chiurugwi, T.; Kemp, S.; Powell, W.; Hickey, L.T.; Powell, W. Speed breeding orphan crops. Theor. Appl. Genet. 2018, 132, 607–616. [Google Scholar] [CrossRef]
- Ghosh, S.; Watson, A.; Gonzalez-Navarro, O.E.; Ramirez-Gonzalez, R.H.; Yanes, L.; Mendoza-Suárez, M.; Simmonds, J.; Wells, R.; Rayner, T.; Green, P.; et al. Speed breeding in growth chambers and glasshouses for crop breeding and model plant research. Nat. Protoc. 2018, 13, 2944–2963. [Google Scholar] [CrossRef] [Green Version]
- Watson, A.; Ghosh, S.; Williams, M.J.; Cuddy, W.S.; Simmonds, J.; Rey, M.-D.; Hatta, M.A.M.; Hinchliffe, A.; Steed, A.; Reynolds, D.; et al. Speed breeding is a powerful tool to accelerate crop research and breeding. Nat. Plants 2018, 4, 23–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hickey, L.T.; Hafeez, N.A.; Robinson, H.; Jackson, S.A.; Leal-Bertioli, S.C.M.; Tester, M.; Gao, C.; Godwin, I.D.; Hayes, B.J.; Wulff, B.B.H. Breeding crops to feed 10 billion. Nat. Biotechnol. 2019, 37, 744–754. [Google Scholar] [CrossRef] [Green Version]
- Poovaiah, C.; Phillips, L.; Geddes, B.; Reeves, C.; Sorieul, M.; Thorlby, G. Genome Editing With CRISPR/Cas9 in Pinus radiata (D. Don). BMC Plant Boil. 2021, 11, 1734. [Google Scholar]


| Species | Genome Size a | N-SNP | Density (SNP/Mb) b | Reference |
|---|---|---|---|---|
| Eucalyptus spp. | 640 Mb | 60 K | 93.75 | [81] |
| Populus spp. | 420 Mb | 34 K | 91.5 | [87] |
| Populus nigra | 400–500 Mb | 12 K | 24–30 | [95] |
| Quercus spp. | 950–930 Mb | 7.9 K | 8 | [89] |
| Picea spp. | 20 Gb | 7.3 K and 9.6 K | 0.4–0.5 | [88] |
| Picea spp. | 20 Gb | 50 K | 2.5 | [93] |
| Araucaria angustifolia (Bertol.) Kuntze | - | 3 K | - | [90] |
| Pinus spp. | 20–30 Gb | 50 K | 1.6–2.5 | [91] |
| Pinus radiata D. Don | 20–30 Gb | 80–49 K | 2–4 | [96] |
| Pseudotsuga menziesii (Mirb.) Franco | 16 Gb | 28 K | 1.75 | [92] |
| Species | Traits | Population | N-Markers | Model | Reference |
|---|---|---|---|---|---|
| Eucalyptus pellita F. Muell | DBH, HT, VOL | OP | 19 K | GBLUP, BA, BB, BC, BL, BRR | [151] |
| E. pellita F. Muell | DBH, HT, VOL | OP | 2 K | GBLUP, ssGBLUP | [153] |
| E. robusta Sm. | VOL, LIG, HCEL | Provenance trial | 2.9 K | RKHS, GBLUP, EN | [154] |
| E. benthamii Maiden & Cambage | DBH, HT, VOL | OP | 13 K | GBLUP, BA, BB, BC, BL, BRR | [151] |
| E. nitens (H.Deane & Maiden) Maiden | WD, DBH, TS, GST | OP | 4.3 K | GBLUP | [26] |
| E. nitens (H. Deane & Maiden) Maiden | DBH, WD, WS, GST, TAS | OP | 9.7 K | GBLUP | [155] |
| E. nitens (H. Deane & Maiden) Maiden | DBH, HT, ST and 9 wood related traits | OP | 12 K | GBLUP | [152] |
| E. urophylla x E. grandis | HT, VOL, WD, PY, CBH | Go and G1 | 10 K | GBLUP, RRBLUP, BL, RKHS | [156] |
| E. grandis × E. urophylla | VOL, KL, HCEL, Wi, δ13C | Clones | 3.3 K | GBLUP | [7] |
| E. grandis × E. urophylla | DBH, VOL, HT, MAI, CELL, S:G, LIG, WD | Full-sibs | 33.4 k | ssGBLUP, GBLUP | [149] |
| E. grandis | DBH, HT, ST | OP | 2.8 K | GBLUP multitrait | [157] |
| E. grandis W. Hill | FL, FW, CELL, S:G, WD, DBH, HT | Full-sibs | 15 K | GBLUP | [65] |
| E. globulus Labill | BQ, DBH, ST, VOL, HT | Full-sibs and OP | 14 K | RRBLUP, RRBLUPB, BA, BB, BL, PCR, SPCR | [13] |
| E. globulus Labill | HT, DBH, ST, BQ, PP | Full-sibs and OP | 14 K | BA, BB, BC, BL, BRR | [36] |
| E. globulus Labill | VOL, WD | Clones | 12 K | GBLUP, BL, BB, BC | [158] |
| E. globulus Labill | PP, ST, HT, DBH, BQ | Full-sibs | 14 K | BRR, BL, BA, BB, BC, RKHS, GBLUP, DL, BRNN | [1] |
| E. dunni Maiden | DBH, ST | OP | 11 K | ssGBLUP | [159] |
| E. cladocalyx F. Muell | HT, DBH, ST, SLD, PP, FI, BHT | OP | 3.8 K | GSq, BA, BB, BC, BRR | [150] |
| Picea glauca (Moench) Voss | hat, DBH, VOL, AV, WD | Polycross, Full-sibs | 4 K | GBLUP | [160] |
| P. glauca (Moench) Voss | HT, DBH, VOL, AV, PIC, PUN, PINC | Full-sibs | 4.1 K | GBLUP | [161] |
| P. mariana (Mill.) Britton, Sterns & Poggenb | WD, DBH, HT, MFA | Full-sibs | 5 K | GBLUP | [162] |
| P. abies (L.) H. Karst | HT, WD | OP | 6.3 K | HBLUP | [163] |
| P. abies (L.) H. Karst | AV, WD, MFA, DBH, HT, SLD, WA | Polycross | 4 K | GBLUP, BRR, BC | [164] |
| P. abies (L.) H. Karst | WD, MFA, MOE, AV | OP | 130 K | GBLUP, BB, RKHS, RRBLUP | [64] |
| P. abies (L.) H. Karst | PP, AV, MOE, HT | Full-sibs | 116 K | BLASSO, BRR, GBLUP, RKHS, BRR | [165] |
| Pinus contorta Douglas ex Loudon | WD, MFA, HT | Full-sibs and OP | 19 K | GBLUP, BC | [166] |
| Pinus radiata D. Don | BCF, ST, ICH, ERB | Full-sibs and clones | 67 K | GBLUP | [167] |
| Pinus radiata D. Don | ST, DBH, WD, MOE | Full-sibs | 58.6 K | GBLUP | [155] |
| Pinus sylvestris Thunb. | HT, DBH, MFA, MOE, WD | Full-sibs | 8.7 K | GBLUP, BRR, BL | [168] |
| Hevea brasiliensis Muell. Arg | RB | Full-sibs | 0.3 K | RKHS, RRBLUP, BL | [169] |
| H. brasiliensis Muell. Arg | CBH (Two watering contrasting conditions) | Full-sibs | 30 K | GBLUP | [170] |
| Populus nigra L. | HT, CBH, BF, BS, RST | Clones | 8 K | GBLUP, BL | [171] |
| Pseudotsuga menziesii (Mirb.) Franco | JHT | Full-sibs | 70 K | RRBLUP, GRR, BB | [32] |
| Pseudotsuga menziesii (Mirb.) Franco | HT, WD, DBH | Full-sibs | 70 K | RRBLUP, GRR | [172] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmar, S.; Ballesta, P.; Ali, M.; Mora-Poblete, F. Achievements and Challenges of Genomics-Assisted Breeding in Forest Trees: From Marker-Assisted Selection to Genome Editing. Int. J. Mol. Sci. 2021, 22, 10583. https://doi.org/10.3390/ijms221910583
Ahmar S, Ballesta P, Ali M, Mora-Poblete F. Achievements and Challenges of Genomics-Assisted Breeding in Forest Trees: From Marker-Assisted Selection to Genome Editing. International Journal of Molecular Sciences. 2021; 22(19):10583. https://doi.org/10.3390/ijms221910583
Chicago/Turabian StyleAhmar, Sunny, Paulina Ballesta, Mohsin Ali, and Freddy Mora-Poblete. 2021. "Achievements and Challenges of Genomics-Assisted Breeding in Forest Trees: From Marker-Assisted Selection to Genome Editing" International Journal of Molecular Sciences 22, no. 19: 10583. https://doi.org/10.3390/ijms221910583
APA StyleAhmar, S., Ballesta, P., Ali, M., & Mora-Poblete, F. (2021). Achievements and Challenges of Genomics-Assisted Breeding in Forest Trees: From Marker-Assisted Selection to Genome Editing. International Journal of Molecular Sciences, 22(19), 10583. https://doi.org/10.3390/ijms221910583