
Role of Vascular Smooth Muscle Cell Phenotype Switching in Arteriogenesis



{kind=link}
{kind=link}
Abstract
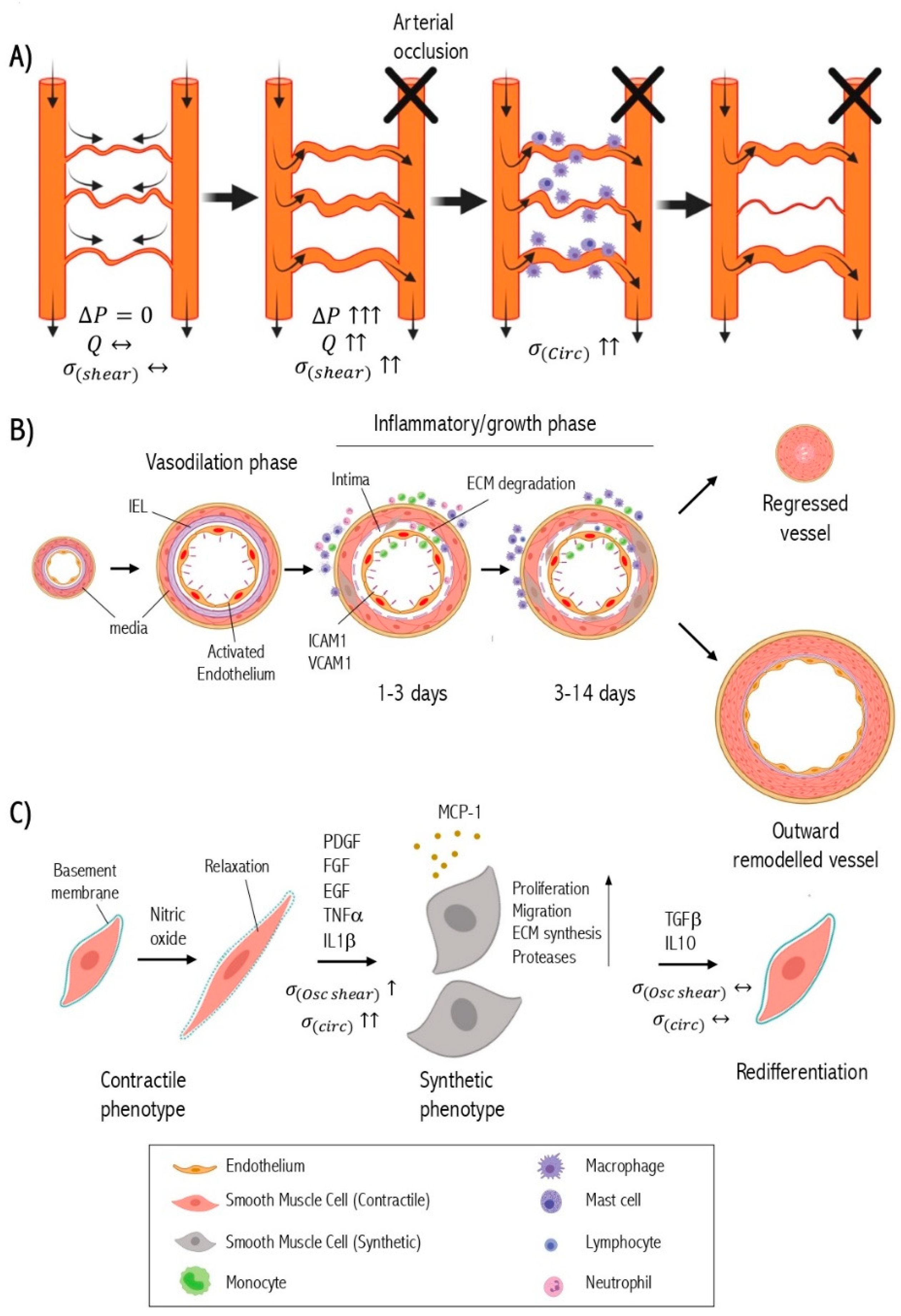
:1. The Pathophysiology of Arteriogenesis after Ischemia
2. Molecular Regulation of SMC Phenotype Switching
3. The Effect of Hemodynamics on Smooth Muscle Cell Phenotype during Arteriogenesis
4. The Role of Inflammation in SMC Phenotypic Change
5. Conclusions Remarks on Targeting SMC Phenotype Switching as Therapeutic Arteriogenesis
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bauersachs, R.; Zeymer, U.; Briere, J.-B.; Marre, C.; Bowrin, K.; Huelsebeck, M. Burden of Coronary Artery Disease and Peripheral Artery Disease: A Literature Review. Cardiovasc. Ther. 2019, 2019, 8295054. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, A.T.; Haskal, Z.J.; Hertzer, N.R.; Bakal, C.W.; Creager, M.A.; Halperin, J.L.; Hiratzka, L.F.; Murphy, W.R.; Olin, J.W.; Puschett, J.B.; et al. ACC/AHA 2005 practice guidelines for the management of patients with peripheral arterial disease (lower extremity, renal, mesenteric, and abdominal aortic): A collaborative report from the American Association for Vascular Surgery/Society for Vascular Surgery, Society for Cardiovascular Angiography and Interventions, Society for Vascular Medicine and Biology, Society of Interventional Radiology, and the ACC/AHA task force on practice guidelines (writing committee to develop guidelines for the management of patients with peripheral arterial disease): Endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation; National Heart, Lung, and Blood Institute; Society for Vascular Nursing; TransAtlantic Inter-Society Consensus; and Vascular Disease Foundation. Circulation 2006, 113, e463–e654. [Google Scholar] [PubMed] [Green Version]
- Johannesson, A.; Larsson, G.-U.; Ramstrand, N.; Turkiewicz, A.; Wiréhn, A.-B.; Atroshi, I. Incidence of Lower-Limb Amputation in the Diabetic and Nondiabetic General Population: A 10-year population-based cohort study of initial unilateral and contralateral amputations and reamputations. Diabetes Care 2009, 32, 275–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerhard-Herman, M.D.; Gornik, H.L.; Barrett, C.; Barshes, N.R.; Corriere, M.A.; Drachman, D.E.; Fleisher, L.A.; Fowkes, F.G.; Hamburg, N.M.; Kinlay, S.; et al. 2016 AHA/ACC guideline on the management of patients with lower extremity peripheral artery disease: A report of the american college of cardiology/american heart association task force on clinical practice guidelines. Circulation 2017, 135, e726–e779. [Google Scholar] [PubMed]
- Mohammed, M.; Gosch, K.; Safley, D.; Jelani, Q.-U.-A.; Aronow, H.D.; Mena, C.; Shishehbor, M.H.; Spertus, J.A.; Abbott, J.D.; Smolderen, K.G. Cilostazol and peripheral artery disease-specific health status in ambulatory patients with symptomatic PAD. Int. J. Cardiol. 2020, 316, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Lichtenberg, M. Peripheral artery disease: Endovascular therapy. Med. Mon. Pharm. 2017, 40, 102–106. [Google Scholar]
- Yao, H.Q.; Wang, F.J.; Kang, Z. Effects of endovascular interventions on vWF and Fb levels in type 2 diabetic patients with peripheral artery disease. Ann. Vasc. Surg. 2016, 33, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Bae, M.J.; Lee, J.G.; Chung, S.W.; Lee, C.W.; Kim, C.W. The factors affecting recurrence of symptoms after infrainguinal arterial endovascular angioplasty. Korean J. Thorac. Cardiovasc. Surg. 2014, 47, 517–522. [Google Scholar] [CrossRef]
- Meloni, M.; Izzo, V.; Giurato, L.; Del Giudice, C.; Da Ros, V.; Cervelli, V.; Gandini, R.; Uccioli, L. Recurrence of critical limb ischemia after endovascular intervention in patients with diabetic foot ulcers. Adv. Wound Care 2018, 7, 171–176. [Google Scholar] [CrossRef]
- Ingram, D.A.; Mead, L.E.; Moore, D.B.; Woodard, W.; Fenoglio, A.; Yoder, M.C. Vessel wall–derived endothelial cells rapidly proliferate because they contain a complete hierarchy of endothelial progenitor cells. Blood 2005, 105, 2783–2786. [Google Scholar] [CrossRef] [Green Version]
- Zengin, E.; Chalajour, F.; Gehling, U.M.; Ito, W.D.; Treede, H.; Lauke, H.; Weil, J.; Reichenspurner, H.; Kilic, N.; Ergün, S. Vascular wall resident progenitor cells: A source for postnatal vasculogenesis. Development 2006, 133, 1543–1551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higuchi, M.; Kato, T.; Yoshida, S.; Ueharu, H.; Nishimura, N.; Kato, Y. PRRX1- and PRRX2-positive mesenchymal stem/progenitor cells are involved in vasculogenesis during rat embryonic pituitary development. Cell Tissue Res. 2015, 361, 557–565. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P. Angiogenesis in health and disease. Nat. Med. 2003, 9, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Pugh, C.W.; Ratcliffe, P.J. Regulation of angiogenesis by hypoxia: Role of the HIF system. Nat. Med. 2003, 9, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Crivellato, E. “Sprouting angiogenesis”, a reappraisal. Dev. Biol. 2012, 372, 157–165. [Google Scholar] [CrossRef] [Green Version]
- Emanueli, C.; Madeddu, P. Angiogenesis gene therapy to rescue ischaemic tissues: Achievements and future directions. Br. J. Pharmacol. 2001, 133, 951–958. [Google Scholar] [CrossRef] [Green Version]
- Simons, M. Angiogenesis: Where do we stand now? Circulation 2005, 111, 1556–1566. [Google Scholar] [CrossRef] [Green Version]
- Buschmann, I.; Schaper, W. The pathophysiology of the collateral circulation (arteriogenesis). J. Pathol. 2000, 190, 338–342. [Google Scholar] [CrossRef]
- Faber, J.E.; Chilian, W.M.; Deindl, E.; van Royen, N.; Simons, M. A Brief Etymology of the Collateral Circulation. Arter. Thromb. Vasc. Biol. 2014, 34, 1854–1859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaper, W. Collateral circulation: Past and present. Basic Res. Cardiol. 2009, 104, 5–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherman, J.A.; Hall, A.; Malenka, D.J.; De Muinck, E.D.; Simons, M. Humoral and cellular factors responsible for coronary collateral formation. Am. J. Cardiol. 2006, 98, 1194–1197. [Google Scholar] [CrossRef]
- Clayton, J.A.; Chalothorn, D.; Faber, J.E. Vascular endothelial growth factor-a specifies formation of native collaterals and regulates collateral growth in Ischemia. Circ. Res. 2008, 103, 1027–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Prabhakar, P.; Sealock, R.; E Faber, J. Wide genetic variation in the native pial collateral circulation is a major determinant of variation in severity of stroke. J. Cereb. Blood Flow Metab. 2010, 30, 923–934. [Google Scholar] [CrossRef] [PubMed]
- Helisch, A.; Schaper, W. Arteriogenesis: The development and growth of collateral arteries. Microcirculation 2003, 10, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Scholz, D.; Ziegelhoeffer, T.; Helisch, A.; Wagner, S.; Friedrich, C.; Podzuweit, T.; Schaper, W. Contribution of arteriogenesis and angiogenesis to postocclusive hindlimb perfusion in mice. J. Mol. Cell. Cardiol. 2002, 34, 775–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziegler, M.A.; DiStasi, M.R.; Bills, R.G.; Miller, S.J.; Alloosh, M.; Murphy, M.P.; Akingba, A.G.; Sturek, M.; Dalsing, M.C.; Unthank, J.L. Marvels, mysteries, and misconceptions of vascular compensation to peripheral artery occlusion. Microcirculation 2010, 17, 3–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herzog, S.; Sager, H.; Khmelevski, E.; Deylig, A.; Ito, W.D. Collateral arteries grow from preexisting anastomoses in the rat hindlimb. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H2012–H2020. [Google Scholar] [CrossRef] [Green Version]
- Resnick, N.; Einav, S.; Chen-Konak, L.; Zilberman, M.; Yahav, H.; Shay-Salit, A. Hemodynamic forces as a stimulus for arteriogenesis. Endothelium 2003, 10, 197–206. [Google Scholar] [CrossRef]
- Park, B.; Hoffman, A.; Yang, Y.; Yan, J.; Tie, G.; Bagshahi, H.; Nowicki, P.T.; Messina, L.M. Endothelial nitric oxide synthase affects both early and late collateral arterial adaptation and blood flow recovery after induction of hind limb ischemia in mice. J. Vasc. Surg. 2010, 51, 165–173. [Google Scholar] [CrossRef] [Green Version]
- van Royen, N.; Piek, J.J.; Buschmann, I.; Hoefer, I.; Voskuil, M.; Schaper, W. Stimulation of arteriogenesis; a new concept for the treatment of arterial occlusive disease. Cardiovasc. Res. 2001, 49, 543–553. [Google Scholar] [CrossRef]
- Bruce, A.C.; Kelly-Goss, M.R.; Heuslein, J.L.; Meisner, J.K.; Price, R.J.; Peirce, S.M. Monocytes are recruited from venules during arteriogenesis in the murine spinotrapezius ligation model. Arter. Thromb. Vasc. Biol. 2014, 34, 2012–2022. [Google Scholar] [CrossRef] [Green Version]
- van Royen, N.; Hoefer, I.; Buschmann, I.; Heil, M.; Kostin, S.; Deindl, E.; Vogel, S.; Korff, T.; Augustin, H.; Bode, C.; et al. Exogenous application of transforming growth factor beta 1 stimulates arteriogenesis in the peripheral circulation. FASEB J. 2002, 16, 432–434. [Google Scholar] [CrossRef]
- Hoefer, I.E.; Van Royen, N.; Rectenwald, J.E.; Bray, E.J.; Abouhamze, Z.; Moldawer, L.L.; Voskuil, M.; Piek, J.J.; Buschmann, I.R.; Ozaki, C.K. Direct evidence for tumor necrosis factor-α signaling in arteriogenesis. Circulation 2002, 105, 1639–1641. [Google Scholar] [CrossRef]
- Belmadani, S.; Matrougui, K.; Kolz, C.; Pung, Y.F.; Palen, D.; Prockop, D.J.; Chilian, W.M. Amplification of coronary arteriogenic capacity of multipotent stromal cells by epidermal growth factor. Arter. Thromb. Vasc. Biol. 2009, 29, 802–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deindl, E.; Hoefer, I.E.; Fernandez, B.; Barancik, M.; Heil, M.; Strniskova, M.; Schaper, W. Involvement of the fibroblast growth factor system in adaptive and chemokine-induced arteriogenesis. Circ. Res. 2003, 92, 561–568. [Google Scholar] [CrossRef] [Green Version]
- Vågesjö, E.; Parv, K.; Ahl, D.; Seignez, C.; Hidalgo, C.H.; Giraud, A.; Amoêdo-Leite, C.; Korsgren, O.; Wallén, H.; Juusola, G.; et al. Perivascular macrophages regulate blood flow following tissue damage. Circ. Res. 2021, 128, 1694–1707. [Google Scholar] [CrossRef] [PubMed]
- Buschmann, I.; Heil, M.; Jost, M.; Schaper, W. Influence of inflammatory cytokines on arteriogenesis. Microcirculation 2003, 10, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Scholz, D.; Ito, W.; Fleming, I.; Deindl, E.; Sauer, A.; Wiesnet, M.; Busse, R.; Schaper, J. Ultrastructure and molecular histology of rabbit hind-limb collateral artery growth (arteriogenesis). Virchows Archiv. 2000, 436, 257–270. [Google Scholar] [CrossRef]
- Ungerleider, J.L.; Johnson, T.D.; Hernandez, M.J.; Elhag, D.I.; Braden, R.L.; Dzieciatkowska, M.; Osborn, K.G.; Hansen, K.C.; Mahmud, E.; Christman, K.L. Extracellular matrix hydrogel promotes tissue remodeling, arteriogenesis, and perfusion in a rat hindlimb ischemia model. JACC Basic Transl. Sci. 2016, 1, 32–44. [Google Scholar] [CrossRef] [Green Version]
- Scholz, D.; Cai, W.; Schaper, W. Arteriogenesis, a new concept of vascular adaptation in occlusive disease. Angiogenesis 2001, 4, 247–257. [Google Scholar] [CrossRef]
- Ma, T.; Bai, Y.P. The hydromechanics in arteriogenesis. Aging Med. 2020, 3, 169–177. [Google Scholar] [CrossRef] [Green Version]
- Rzucidlo, E.M.; Martin, K.A.; Powell, R.J. Regulation of vascular smooth muscle cell differentiation. J. Vasc. Surg. 2007, 45, A25–A32. [Google Scholar] [CrossRef] [Green Version]
- Heil, M.; Schaper, W. Influence of mechanical, cellular, and molecular factors on collateral artery growth (arteriogenesis). Circ. Res. 2004, 95, 449–458. [Google Scholar] [CrossRef] [Green Version]
- Hoefer, I.E.; Van Royen, N.; Buschmann, I.R.; Piek, J.J.; Schaper, W. Time course of arteriogenesis following femoral artery occlusion in the rabbit. Cardiovasc. Res. 2001, 49, 609–617. [Google Scholar] [CrossRef]
- Shi, N.; Mei, X.; Chen, S.Y. Smooth muscle cells in vascular remodeling. Arterioscler. Thromb. Vasc. Biol. 2019, 39, e247–e252. [Google Scholar] [CrossRef] [PubMed]
- Leopold, J.A. Vascular calcification: Mechanisms of vascular smooth muscle cell calcification. Trends Cardiovasc. Med. 2015, 25, 267–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, P.; Xia, C.; Duan, C.; Li, S.; Mei, Z. Biological characteristics of foam cell formation in smooth muscle cells derived from bone marrow stem cells. Int. J. Biol. Sci. 2011, 7, 937–946. [Google Scholar] [CrossRef]
- Hegner, B.; Schaub, T.; Catar, R.; Kusch, A.; Wagner, P.; Essin, K.; Lange, C.; Riemekasten, G.; Dragun, D. Intrinsic deregulation of vascular smooth muscle and myofibroblast differentiation in mesenchymal stromal cells from patients with systemic sclerosis. PLoS ONE 2016, 11, e0153101. [Google Scholar] [CrossRef]
- Owens, G.K.; Kumar, M.S.; Wamhoff, B.R. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 2004, 84, 767–801. [Google Scholar] [CrossRef]
- Bennett, M.R.; Sinha, S.; Owens, G.K. Vascular smooth muscle cells in atherosclerosis. Circ. Res. 2016, 118, 692–702. [Google Scholar] [CrossRef]
- Chen, P.Y.; Qin, L.; Li, G.; Malagon-Lopez, J.; Wang, Z.; Bergaya, S.; Gujja, S.; Caulk, A.W.; Murtada, S.-I.; Zhang, X.; et al. Smooth muscle cell reprogramming in aortic aneurysms. Cell Stem Cell 2020, 26, 542–557. [Google Scholar] [CrossRef]
- Régent, A.; Ly, K.H.; Lofek, S.; Clary, G.; Tamby, M.; Tamas, N.; Federici, C.; Broussard, C.; Chafey, P.; Liaudet-Coopman, E.; et al. Proteomic analysis of vascular smooth muscle cells in physiological condition and in pulmonary arterial hypertension: Toward contractile versus synthetic phenotypes. Proteomics 2016, 16, 2637–2649. [Google Scholar] [CrossRef]
- Acampora, K.B.; Nagatomi, J.; Langan E.M., III; LaBerge, M. Increased synthetic phenotype behavior of smooth muscle cells in response to in vitro balloon angioplasty injury model. Ann. Vasc. Surg. 2010, 24, 116–126. [Google Scholar] [CrossRef]
- Wang, G.; Jacquet, L.; Karamariti, E.; Xu, Q. Origin and differentiation of vascular smooth muscle cells. J. Physiol. 2015, 593, 3013–3030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabbiani, G.; Schmid, E.; Winter, S.; Chaponnier, C.; de Ckhastonay, C.; Vandekerckhove, J.; Weber, K.; Franke, W.W. Vascular smooth muscle cells differ from other smooth muscle cells: Predominance of vimentin filaments and a specific alpha-type actin. Proc. Natl. Acad. Sci. USA 1981, 78, 298–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miano, J.; Cserjesi, P.; Ligon, K.L.; Periasamy, M.; Olson, E.N. Smooth muscle myosin heavy chain exclusively marks the smooth muscle lineage during mouse embryogenesis. Circ. Res. 1994, 75, 803–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duband, J.-L.; Gimona, M.; Scatena, M.; Sartore, S.; Small, J.V. Calponin and SM22 as differentiation markers of smooth muscle: Spatiotemporal distribution during avian embryonic development. Differentiation 1993, 55, 1–11. [Google Scholar] [CrossRef]
- van der Loop, F.T.; Schaart, G.; Timmer, E.D.; Ramaekers, F.C.; van Eys, G.J. Smoothelin, a novel cytoskeletal protein specific for smooth muscle cells. J. Cell Biol. 1996, 134, 401–411. [Google Scholar] [CrossRef] [Green Version]
- Chamley-Campbell, J.; Campbell, G.R.; Ross, R. The smooth muscle cell in culture. Physiol. Rev. 1979, 59, 1–61. [Google Scholar] [CrossRef]
- García-Miguel, M.; Riquelme, J.A.; Norambuena-Soto, I.; Morales, P.E.; Sanhueza-Olivares, F.; Núñez-Soto, C.; Mondaca-Ruff, D.; Cancino-Arenas, N.; Martín, A.S.; Chiong, M. Autophagy mediates tumor necrosis factor-α-induced phenotype switching in vascular smooth muscle A7r5 cell line. PLoS ONE 2018, 13, e0197210. [Google Scholar] [CrossRef] [Green Version]
- Yu, B.; Wong, M.M.; Potter, C.M.F.; Simpson, R.M.L.; Karamariti, E.; Zhang, Z.; Zeng, L.; Warren, D.; Hu, Y.; Wang, W.; et al. Vascular stem/progenitor cell migration induced by smooth muscle cell-derived chemokine (C-C Motif) ligand 2 and chemokine (C-X-C motif) ligand 1 contributes to neointima formation. Stem Cells 2016, 34, 2368–2380. [Google Scholar] [CrossRef] [Green Version]
- Okada, Y.; Katsuda, S.; Matsui, Y.; Watanabe, H.; Nakanishi, I. Collagen Synthesis by Cultured Arterial Smooth Muscle Cells during Spontaneous Phenotypic Modulation. Pathol. Int. 1990, 40, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, R.N.; Kashiwagi, K.; Kashiwagi, F.; Tsukahara, S.; Lindsey, J.D. Prostaglandins increase matrix metalloproteinase release from human ciliary smooth muscle cells. Investig. Ophthalmol. Vis. Sci. 1997, 38, 2772–2780. [Google Scholar]
- Yoshida, T.; Sinha, S.; Dandre, F.; Wamhoff, B.R.; Hoofnagle, M.H.; Kremer, B.E.; Wang, D.Z.; Olson, E.N.; Owens, G.K. Myocardin is a key regulator of CArG-dependent transcription of multiple smooth muscle marker genes. Circ. Res. 2003, 92, 856–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mack, C.P.; Hinson, J.S. Regulation of smooth muscle differentiation by the myocardin family of serum response factor co-factors. J. Thromb. Haemost. 2005, 3, 1976–1984. [Google Scholar] [CrossRef]
- Mack, C.P.; Owens, G.K. Regulation of smooth muscle alpha-actin expression in vivo is dependent on CArG elements within the 5’ and first intron promoter regions. Circ. Res. 1999, 84, 852–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miano, J.M. Serum response factor: Toggling between disparate programs of gene expression. J. Mol. Cell. Cardiol. 2003, 35, 577–593. [Google Scholar] [CrossRef]
- Wang, D.Z.; Chang, P.S.; Wang, Z.; Sutherland, L.; Richardson, J.A.; Small, E.; Krieg, P.A.; Olson, E.N. Activation of cardiac gene expression by myocardin, a transcriptional cofactor for serum response factor. Cell 2001, 105, 851–862. [Google Scholar] [CrossRef] [Green Version]
- Cenik, B.K.; Liu, N.; Chen, B.; Bezprozvannaya, S.; Olson, E.N.; Bassel-Duby, R. Myocardin-related transcription factors are required for skeletal muscle development. Development 2016, 143, 2853–2861. [Google Scholar] [CrossRef] [Green Version]
- Esnault, C.; Gualdrini, F.; Horswell, S.; Kelly, G.; Stewart, A.; East, P.; Matthews, N.; Treisman, R. ERK-induced activation of TCF family of SRF cofactors initiates a chromatin modification cascade associated with transcription. Mol. Cell 2017, 65, 1081–1095. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Shi, W. Rho/ROCK-MYOCD in regulating airway smooth muscle growth and remodeling. Am. J. Physiol. Lung Cell Mol. Physiol. 2021, 321, L1–L5. [Google Scholar] [CrossRef]
- Esnault, C.; Stewart, A.; Gualdrini, F.; East, P.; Horswell, S.; Matthews, N.; Treisman, R. Rho-actin signaling to the MRTF coactivators dominates the immediate transcriptional response to serum in fibroblasts. Genes Dev. 2014, 28, 943–958. [Google Scholar] [CrossRef] [Green Version]
- A Hipskind, R.; Buscher, D.; Nordheim, A.; Baccarini, M. Ras/MAP kinase-dependent and -independent signaling pathways target distinct ternary complex factors. Genes Dev. 1994, 8, 1803–1816. [Google Scholar] [CrossRef] [Green Version]
- Dennler, S.; Itoh, S.; Vivien, D.; ten Dijke, P.; Huet, S.; Gauthier, J.M. Direct binding of Smad3 and Smad4 to critical TGF beta-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. 1998, 17, 3091–3100. [Google Scholar] [CrossRef] [Green Version]
- Jia, G.; Mitra, A.K.; Gangahar, D.M.; Agrawal, D.K. Insulin-like growth factor-1 induces phosphorylation of PI3K-Akt/PKB to potentiate proliferation of smooth muscle cells in human saphenous vein. Exp. Mol. Pathol. 2010, 89, 20–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.-P.; Wang, Z.; Yanagisawa, H.; Olson, E.N. Phenotypic modulation of smooth muscle cells through interaction of Foxo4 and myocardin. Dev. Cell 2005, 9, 261–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dandre, F.; Owens, G.K. Platelet-derived growth factor-BB and Ets-1 transcription factor negatively regulate transcription of multiple smooth muscle cell differentiation marker genes. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H2042–H2051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Wang, D.-Z.; Hockemeyer, D.; McAnally, J.; Nordheim, A.; Olson, E.N. Myocardin and ternary complex factors compete for SRF to control smooth muscle gene expression. Nat. Cell Biol. 2004, 428, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Salmon, M.; Gomez, D.; Greene, E.; Shankman, L.; Owens, G.K. Cooperative binding of KLF4, pELK-1, and HDAC2 to a G/C repressor element in the SM22α promoter mediates transcriptional silencing during SMC phenotypic switching in vivo. Circ. Res. 2012, 111, 685–696. [Google Scholar] [CrossRef]
- Kawai-Kowase, K.; Owens, G.K. Multiple repressor pathways contribute to phenotypic switching of vascular smooth muscle cells. Am. J. Physiol. Cell Physiol. 2007, 292, C59–C69. [Google Scholar] [CrossRef]
- Alexander, M.R.; Owens, G.K. Epigenetic control of smooth muscle cell differentiation and phenotypic switching in vascular development and disease. Annu. Rev. Physiol. 2012, 74, 13–40. [Google Scholar] [CrossRef]
- Qiu, P.; Ritchie, R.P.; Gong, X.Q.; Hamamori, Y.; Li, L. Dynamic changes in chromatin acetylation and the expression of histone acetyltransferases and histone deacetylases regulate the SM22alpha transcription in response to Smad3-mediated TGFbeta1 signaling. Biochem. Biophys. Res. Commun. 2006, 348, 351–358. [Google Scholar] [CrossRef]
- Hiltunen, M.O.; Turunen, M.P.; Häkkinen, T.P.; Rutanen, J.; Hedman, M.; Mäkinen, K.; Turunen, A.M.; Aalto-Setalä, K.; Ylä-Herttuala, S. DNA hypomethylation and methyltransferase expression in atherosclerotic lesions. Vasc. Med. 2002, 7, 5–11. [Google Scholar] [CrossRef] [Green Version]
- Connelly, J.J.; Cherepanova, O.A.; Doss, J.F.; Karaoli, T.; Lillard, T.S.; Markunas, C.; Nelson, S.; Wang, T.; Ellis, P.D.; Langford, C.F.; et al. Epigenetic regulation of COL15A1 in smooth muscle cell replicative aging and atherosclerosis. Hum. Mol. Genet. 2013, 22, 5107–5120. [Google Scholar] [CrossRef] [Green Version]
- Pipp, F.; Boehm, S.; Cai, W.-J.; Adili, F.; Ziegler, B.; Karanovic, G.; Ritter, R.; Balzer, J.; Scheler, C.; Schaper, W.; et al. Elevated fluid shear stress enhances postocclusive collateral artery growth and gene expression in the pig hind limb. Arter. Thromb. Vasc. Biol. 2004, 24, 1664–1668. [Google Scholar] [CrossRef] [Green Version]
- Gruionu, G.; Hoying, J.B.; Pries, A.R.; Secomb, T. Structural remodeling of the mouse gracilis artery: Coordinated changes in diameter and medial area maintain circumferential stress. Microcirculation 2012, 19, 610–618. [Google Scholar] [CrossRef]
- Sakamoto, N.; Ohashi, T.; Sato, M. Effect of fluid shear stress on migration of vascular smooth muscle cells in cocultured model. Ann. Biomed. Eng. 2006, 34, 408–415. [Google Scholar] [CrossRef]
- Zhang, H.; Chalothorn, D.; Faber, J.E. Collateral vessels have unique endothelial and smooth muscle cell phenotypes. Int. J. Mol. Sci. 2019, 20, 3608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagher, P.; Beleznai, T.; Kansui, Y.; Mitchell, R.; Garland, C.J.; Dora, K.A. Low intravascular pressure activates endothelial cell TRPV4 channels, local Ca2+ events, and IKCa channels, reducing arteriolar tone. Proc. Natl. Acad. Sci. USA 2012, 109, 18174–18179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Rivers, R.J. Measurement of membrane potential and intracellular Ca(2+) of arteriolar endothelium and smooth muscle in vivo. Microvasc. Res. 2001, 62, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.A.; Sung, J.Y.; Woo, C.-H.; Choi, H.C. Laminar shear stress suppresses vascular smooth muscle cell proliferation through nitric oxide-AMPK pathway. Biochem. Biophys. Res. Commun. 2017, 490, 1369–1374. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.-C.; Chen, L.; Zhou, J.; Tang, Z.; Hsu, T.-F.; Wang, Y.; Shih, Y.-T.; Peng, H.-H.; Wang, N.; Guan, Y.; et al. Shear stress induces synthetic-to-contractile phenotypic modulation in smooth muscle cells via peroxisome proliferator-activated receptor alpha/delta activations by prostacyclin released by sheared endothelial cells. Circ. Res. 2009, 105, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Talotta-Altenburg, L.M.; Silimperi, K.A.; Ciabattoni, G.O.; Lowe-Krentz, L.J. Endothelial nitric oxide synthase activation is required for heparin receptor effects on vascular smooth muscle cells. Am. J. Physiol. Physiol. 2020, 318, C463–C475. [Google Scholar] [CrossRef] [PubMed]
- Lowry, J.L.; Brovkovych, V.; Zhang, Y.; Skidgel, R.A. Endothelial nitric-oxide synthase activation generates an inducible nitric-oxide synthase-like output of nitric oxide in inflamed endothelium. J. Biol. Chem. 2013, 288, 4174–4193. [Google Scholar] [CrossRef] [Green Version]
- Tabatabaei, S.N.; Girouard, H. Nitric oxide and cerebrovascular regulation. Vitam. Horm. 2014, 96, 347–385. [Google Scholar] [PubMed]
- Zuckerbraun, B.S.; Stoyanovsky, D.A.; Sengupta, R.; Shapiro, R.A.; Ozanich, B.A.; Rao, J.; Barbato, J.E.; Tzeng, E. Nitric oxide-induced inhibition of smooth muscle cell proliferation involves S-nitrosation and inactivation of RhoA. Am. J. Physiol. Physiol. 2007, 292, C824–C831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, S.; Katoh, Y.; Konishi, H.; Takaya, N.; Kimura, T.; Periasamy, M.; Yamaguchi, H. Nitric oxide regulates smooth-muscle-specific myosin heavy chain gene expression at the transcriptional level—Possible role of SRF and YY1 through CArG element. J. Mol. Cell. Cardiol. 2001, 33, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Boerth, N.J.; Dey, N.B.; Cornwell, T.L.; Lincoln, T.M. Cyclic GMP-dependent protein kinase regulates vascular smooth muscle cell phenotype. J. Vasc. Res. 1997, 34, 245–259. [Google Scholar] [CrossRef]
- Lincoln, T.M.; Sellak, H.; Dey, N.; Browner, N.; Choi, C.S.; Dostmann, W.W. Regulation of vascular smooth muscle cell gene expression and phenotype by cyclic GMP and cyclic GMP-dependent protein kinase. BMC News Views 2003, 3, 356–367. [Google Scholar] [CrossRef]
- Dey, N.B.; Foley, K.F.; Lincoln, T.M.; Dostmann, W.R. Inhibition of cGMP-dependent protein kinase reverses phenotypic modulation of vascular smooth muscle cells. J. Cardiovasc. Pharmacol. 2005, 45, 404–413. [Google Scholar] [CrossRef]
- Zhou, W.; Dasgupta, C.; Negash, S.; Raj, J.U. Modulation of pulmonary vascular smooth muscle cell phenotype in hypoxia: Role of cGMP-dependent protein kinase. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 292, L1459–L1466. [Google Scholar] [CrossRef] [PubMed]
- Mees, B.; Wagner, S.; Ninci, E.; Tribulova, S.; Martin, S.; Van Haperen, R.; Kostin, S.; Heil, M.; De Crom, R.; Schaper, W. Endothelial nitric oxide synthase activity is essential for vasodilation during blood flow recovery but not for arteriogenesis. Arter. Thromb. Vasc. Biol. 2007, 27, 1926–1933. [Google Scholar] [CrossRef] [Green Version]
- Wilstein, Z.; Alligood, D.M.; McLure, V.L.; Miller, A.C. Mathematical model of hypertension-induced arterial remodeling: A chemo-mechanical approach. Math. Biosci. 2018, 303, 10–25. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Clark, J.W.; Bryan, R.M.; Robertson, C.S. Mathematical modeling of the nitric oxide/cGMP pathway in the vascular smooth muscle cell. Am. J. Physiol. Circ. Physiol. 2005, 289, H886–H897. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, S.L.; Kan, J.S.; Mitchell, S.E.; Flaherty, J.T.; White, R.I. Embolization of systemic to pulmonary artery collaterals in the management of hemoptysis in pulmonary atresia. Am. J. Cardiol. 1986, 58, 1130–1132. [Google Scholar] [CrossRef]
- Davies, P.F. Hemodynamic shear stress and the endothelium in cardiovascular pathophysiology. Nat. Clin. Pr. Neurol. 2008, 6, 16–26. [Google Scholar] [CrossRef] [Green Version]
- Shi, Z.D.; Tarbell, J.M. Fluid flow mechanotransduction in vascular smooth muscle cells and fibroblasts. Ann. Biomed. Eng. 2011, 39, 1608–1619. [Google Scholar] [CrossRef] [Green Version]
- Ziegelhoeffer, T.; Scholz, D.; Friedrich, C.; Helisch, A.; Wagner, S.; Fernandez, B.; Schaper, W. Inhibition of collateral artery growth by mibefradil: Possible role of volume-regulated chloride channels. Endothelium 2003, 10, 237–246. [Google Scholar] [CrossRef]
- Swain, S.M.; Liddle, R.A. Piezo1 acts upstream of TRPV4 to induce pathological changes in endothelial cells due to shear stress. J. Biol. Chem. 2021, 296, 100171. [Google Scholar] [CrossRef]
- Sieve, I.; Münster-Kühnel, A.K.; Hilfiker-Kleiner, D. Regulation and function of endothelial glycocalyx layer in vascular diseases. Vasc. Pharmacol. 2018, 100, 26–33. [Google Scholar] [CrossRef]
- Pahakis, M.Y.; Kosky, J.R.; Dull, R.; Tarbell, J.M. The role of endothelial glycocalyx components in mechanotransduction of fluid shear stress. Biochem. Biophys. Res. Commun. 2007, 355, 228–233. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Rubin, J.; Tzima, E. Role of PECAM-1 in arteriogenesis and specification of preexisting collaterals. Circ. Res. 2010, 107, 1355–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Z.D.; Ji, X.Y.; Berardi, D.E.; Qazi, H.; Tarbell, J.M. Interstitial flow induces MMP-1 expression and vascular SMC migration in collagen I gels via an ERK1/2-dependent and c-Jun-mediated mechanism. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H127–H135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.; Liu, J.; Sun, A.; Liu, X.; Fan, Y.; Deng, X. Vascular smooth muscle cell glycocalyx mediates shear stress-induced contractile responses via a Rho kinase (ROCK)-myosin light chain phosphatase (MLCP) pathway. Sci. Rep. 2017, 7, 42092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, M.A.; Schaller, M.D.; Ginsberg, M.H. Integrins: Emerging paradigms of signal transduction. Annu Rev. Cell Dev. Biol. 1995, 11, 549–599. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhou, Y.; Liu, S.; Li, C. Biomechanical signal communication in vascular smooth muscle cells. J. Cell Commun. Signal. 2020, 14, 357–376. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Böck, G.; Wick, G.; Xu, Q. Activation of PDGF receptor α in vascular smooth muscle cells by mechanical stress. FASEB J. 1998, 12, 1135–1142. [Google Scholar] [CrossRef]
- Li, C.; Xu, Q. Mechanical stress-initiated signal transductions in vascular smooth muscle cells. Cell Signal. 2000, 12, 435–445. [Google Scholar] [CrossRef]
- Arnold, C.; Feldner, A.; Pfisterer, L.; Hödebeck, M.; Troidl, K.; Genové, G.; Wieland, T.; Hecker, M.; Korff, T. RGS 5 promotes arterial growth during arteriogenesis. EMBO Mol. Med. 2014, 6, 1075–1089. [Google Scholar] [CrossRef]
- Shi, Z.D.; Abraham, G.; Tarbell, J.M. Shear stress modulation of smooth muscle cell marker genes in 2-D and 3-D depends on mechanotransduction by heparan sulfate proteoglycans and ERK1/2. PLoS ONE 2010, 5, e12196. [Google Scholar] [CrossRef]
- Dardik, A.; Yamashita, A.; Aziz, F.; Asada, H.; Sumpio, B.E. Shear stress-stimulated endothelial cells induce smooth muscle cell chemotaxis via platelet-derived growth factor-BB and interleukin-1α. J. Vasc. Surg. 2005, 41, 321–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, M.; Matsumori, A.; Ono, K.; Furukawa, Y.; Shioi, T.; Iwasaki, A.; Matsushima, K.; Sasayama, S. Cyclic stretch upregulates production of interleukin-8 and monocyte chemotactic and activating factor/monocyte chemoattractant protein-1 in human endothelial cells. Arter. Thromb. Vasc. Biol. 1998, 18, 894–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demicheva, E.; Hecker, M.; Korff, T. Stretch-induced activation of the transcription factor activator protein-1 controls monocyte chemoattractant protein-1 expression during arteriogenesis. Circ. Res. 2008, 103, 477–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korff, T.; Braun, J.; Pfaff, D.; Augustin, H.G.; Hecker, M. Role of ephrinB2 expression in endothelial cells during arteriogenesis: Impact on smooth muscle cell migration and monocyte recruitment. Blood 2008, 112, 73–81. [Google Scholar] [CrossRef] [Green Version]
- O’Callaghan, C.J.; Williams, B. Mechanical strain-induced extracellular matrix production by human vascular smooth muscle cells: Role of TGF-beta(1). Hypertension 2000, 36, 319–324. [Google Scholar] [CrossRef] [Green Version]
- Parker, S.B.; Dobrian, A.D.; Wade, S.S.; Prewitt, R.L. AT1 receptor inhibition does not reduce arterial wall hypertrophy or PDGF-A expression in renal hypertension. Am. J. Physiol. Circ. Physiol. 2000, 278, H613–H622. [Google Scholar] [CrossRef] [Green Version]
- Etz, C.D.; Kari, F.A.; Mueller, C.S.; Brenner, R.M.; Lin, H.-M.; Griepp, R.B. The collateral network concept: Remodeling of the arterial collateral network after experimental segmental artery sacrifice. J. Thorac. Cardiovasc. Surg. 2011, 141, 1029–1036. [Google Scholar] [CrossRef] [Green Version]
- Amaya, R.; Pierides, A.; Tarbell, J.M. The interaction between fluid wall shear stress and solid circumferential strain affects endothelial gene expression. PLoS ONE 2015, 10, e0129952. [Google Scholar] [CrossRef] [Green Version]
- Orr, A.; Hastings, N.E.; Blackman, B.R.; Wamhoff, B.R. Complex regulation and function of the inflammatory smooth muscle cell phenotype in atherosclerosis. J. Vasc. Res. 2010, 47, 168–180. [Google Scholar] [CrossRef] [Green Version]
- Doran, A.C.; Meller, N.; McNamara, C.A. Role of smooth muscle cells in the initiation and early progression of atherosclerosis. Arter. Thromb. Vasc. Biol. 2008, 28, 812–819. [Google Scholar] [CrossRef] [Green Version]
- Sorokin, V.; Vickneson, K.; Kofidis, T.; Woo, C.C.; Lin, X.Y.; Foo, R.; Shanahan, C.M. Role of vascular smooth muscle cell plasticity and interactions in vessel wall inflammation. Front. Immunol. 2020, 11, 3053. [Google Scholar] [CrossRef]
- Nossent, A.Y.; Bastiaansen, A.J.N.M.; Peters, E.A.B.; de Vries, M.R.; Aref, Z.; Welten, S.M.J.; de Jager, S.C.A.; van der Pouw Kraan, T.C.T.M.; Quax, P.H.A. CCR7-CCL19/CCL21 axis is essential for effective arteriogenesis in a murine model of hindlimb ischemia. J. Am. Heart Assoc. 2017, 6, e005281. [Google Scholar] [CrossRef] [Green Version]
- Kadl, A.; Leitinger, N. The role of endothelial cells in the resolution of acute inflammation. Antioxid. Redox Signal. 2005, 7, 1744–1754. [Google Scholar] [CrossRef]
- Moraes, F.; Paye, J.; Mac Gabhann, F.; Zhuang, Z.W.; Zhang, J.; Lanahan, A.A.; Simons, M. Endothelial cell–dependent regulation of arteriogenesis. Circ. Res. 2013, 113, 1076–1086. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.C.; Pan, M.; Zhu, L.P.; Sun, Q.; Zhou, Z.S.; Li, C.C.; Zhang, G.G. NFAT5 promotes arteriogenesis via MCP-1-dependent monocyte recruitment. J. Cell Mol. Med. 2020, 24, 2052–2063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denger, S.; Jahn, L.; Wende, P.; Watson, L.; Gerber, S.H.; Kübler, W.; Kreuzer, J. Expression of monocyte chemoattractant protein-1 cDNA in vascular smooth muscle cells: Induction of the synthetic phenotype: A possible clue to SMC differentiation in the process of atherogenesis. Atherosclerosis 1999, 144, 15–23. [Google Scholar] [CrossRef]
- Li, C.; Xu, Q. Mechanical stress-initiated signal transduction in vascular smooth muscle cells in vitro and in vivo. Cell Signal. 2007, 19, 881–891. [Google Scholar] [CrossRef] [PubMed]
- Heil, M.; Ziegelhoeffer, T.; Wagner, S.; Fernández, B.; Helisch, A.; Martin, S.; Tribulova, S.; Kuziel, W.A.; Bachmann, G.; Schaper, W. Collateral artery growth (arteriogenesis) after experimental arterial occlusion is impaired in mice lacking CC-chemokine receptor-2. Circ. Res. 2004, 94, 671–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fung, E.; Helisch, A. Macrophages in collateral arteriogenesis. Front. Physiol. 2012, 3, 353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kratofil, R.M.; Kubes, P.; Deniset, J.F. Monocyte conversion during inflammation and injury. Arter. Thromb. Vasc. Biol. 2017, 37, 35–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cochain, C.; Rodero, M.; Vilar, J.; Recalde, A.; Richart, A.L.; Loinard, C.; Zouggari, Y.; Guérin, C.; Duriez, M.; Combadière, B.; et al. Regulation of monocyte subset systemic levels by distinct chemokine receptors controls post-ischaemic neovascularization. Cardiovasc. Res. 2010, 88, 186–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Rourke, S.A.; Dunne, A.; Monaghan, M. The role of macrophages in the infarcted myocardium: Orchestrators of ECM remodeling. Front. Cardiovasc. Med. 2019, 6, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nahrendorf, M.; Swirski, F.K.; Aikawa, E.; Stangenberg, L.; Wurdinger, T.; Figueiredo, J.-L.; Libby, P.; Weissleder, R.; Pittet, M.J. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med. 2007, 204, 3037–3047. [Google Scholar] [CrossRef] [Green Version]
- Rappolee, D.A.; Werb, Z. Macrophage-derived growth factors. Curr. Top. Microbiol. Immunol. 1992, 181, 87–140. [Google Scholar] [PubMed]
- Macarie, R.D.; Vadana, M.; Ciortan, L.; Tucureanu, M.M.; Ciobanu, A.; Vinereanu, D.; Manduteanu, I.; Simionescu, M.; Butoi, E. The expression of MMP-1 and MMP-9 is up-regulated by smooth muscle cells after their cross-talk with macrophages in high glucose conditions. J. Cell Mol. Med. 2018, 22, 4366–4376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butoi, E.; Gan, A.; Tucureanu, M.; Stan, D.; Macarie, R.; Constantinescu, C.; Calin, M.; Simionescu, M.; Manduteanu, I. Cross-talk between macrophages and smooth muscle cells impairs collagen and metalloprotease synthesis and promotes angiogenesis. Biochim. Biophys. Acta (BBA) Bioenerg. 2016, 1863, 1568–1578. [Google Scholar] [CrossRef]
- Ntokou, A.; Dave, J.M.; Kauffman, A.C.; Sauler, M.; Ryu, C.; Hwa, J.; Herzog, E.L.; Singh, I.; Saltzman, W.M.; Greif, D.M. Macrophage-derived PDGF-B induces muscularization in murine and human pulmonary hypertension. JCI Insight 2021, 6, e139067. [Google Scholar] [CrossRef]
- Xiong, W.; Frasch, S.C.; Thomas, S.M.; Bratton, D.L.; Henson, P.M. Induction of TGF-beta1 synthesis by macrophages in response to apoptotic cells requires activation of the scavenger receptor CD36. PLoS ONE 2013, 8, e72772. [Google Scholar] [CrossRef]
- Ji, Y.; Lisabeth, E.M.; Neubig, R.R. Transforming growth factor beta1 increases expression of contractile genes in human pulmonary arterial smooth muscle cells by potentiating sphingosine-1-phosphate signaling. Mol. Pharmacol. 2021, 100, 53–60. [Google Scholar] [CrossRef]
- Elkington, P.T.; Green, J.A.; Friedland, J.S. Analysis of Matrix Metalloproteinase Secretion by Macrophages. Adv. Struct. Saf. Stud. 2009, 531, 253–265. [Google Scholar]
- Hobeika, M.J.; Edlin, R.S.; Muhs, B.E.; Sadek, M.; Gagne, P.J. Matrix metalloproteinases in critical limb ischemia. J. Surg. Res. 2008, 149, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.-J.; Koltai, S.; Kocsis, E.; Scholz, D.; Schaper, W.; Schaper, J. Connexin37, not Cx40 and Cx43, is induced in vascular smooth muscle cells during coronary arteriogenesis. J. Mol. Cell. Cardiol. 2001, 33, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.L. Matrix metalloproteinases: Influence on smooth muscle cells and atherosclerotic plaque stability. Expert Rev. Cardiovasc. Ther. 2007, 5, 265–282. [Google Scholar] [CrossRef] [PubMed]
- Bagi, Z. Impaired coronary collateral growth: miR-shaken neutrophils caught in the act. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H1321–H1322. [Google Scholar] [CrossRef] [Green Version]
- Bot, I.; Velden, D.V.; Bouwman, M.; Kroner, M.J.; Kuiper, J.; Quax, P.H.A.; de Vries, M.R. Local mast cell activation promotes neovascularization. Cells 2020, 9, 701. [Google Scholar] [CrossRef] [Green Version]
- Stabile, E.; Kinnaird, T.; la Sala, A.; Hanson, S.K.; Watkins, C.; Campia, U.; Shou, M.; Zbinden, S.; Fuchs, S.; Kornfeld, H.; et al. CD8 + T lymphocytes regulate the arteriogenic response to ischemia by infiltrating the site of collateral vessel development and recruiting CD4 + mononuclear cells through the expression of interleukin-16. Circulation 2006, 113, 118–124. [Google Scholar] [CrossRef] [Green Version]
- Chillo, O.; Kleinert, E.C.; Lautz, T.; Lasch, M.; Pagel, J.-I.; Heun, Y.; Troidl, K.; Fischer, S.; Caballero-Martinez, A.; Mauer, A.; et al. Perivascular mast cells govern shear stress-induced arteriogenesis by orchestrating leukocyte function. Cell Rep. 2016, 16, 2197–2207. [Google Scholar] [CrossRef] [Green Version]
- Stabile, E.; Burnett, M.S.; Watkins, C.; Kinnaird, T.; Bachis, A.; la Sala, A.; Miller, J.M.; Shou, M.; Epstein, S.E.; Fuchs, S. Impaired arteriogenic response to acute hindlimb ischemia in CD4-knockout mice. Circulation 2003, 108, 205–210. [Google Scholar] [CrossRef] [Green Version]
- van Weel, V.; Toes, R.E.; Seghers, L.; Deckers, M.M.; de Vries, M.R.; Eilers, P.H.; Sipkens, J.; Schepers, A.; Eefting, D.; van Hinsbergh, V.W.; et al. Natural killer cells and CD4+ T-cells modulate collateral artery development. Arterioscler Thromb. Vasc Biol. 2007, 27, 2310–2318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiter, M.S.; Van Golde, J.M.; Schaper, N.; Stehouwer, C.D.; Huijberts, M.S. Diabetes impairs arteriogenesis in the peripheral circulation: Review of molecular mechanisms. Clin. Sci. 2010, 119, 225–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eitenmüller, I.; Volger, O.; Kluge, A.; Troidl, K.; Barancik, M.; Cai, W.-J.; Heil, M.; Pipp, F.; Fischer, S.; Horrevoets, A.J.G.; et al. The range of adaptation by collateral vessels after femoral artery occlusion. Circ. Res. 2006, 99, 656–662. [Google Scholar] [CrossRef] [PubMed]
- Unger, E.F.; Banai, S.; Shou, M.; Lazarous, D.F.; Jaklitsch, M.T.; Scheinowitz, M.; Correa, R.; Klingbeil, C.; Epstein, S.E. Basic fibroblast growth factor enhances myocardial collateral flow in a canine model. Am. J. Physiol. Circ. Physiol. 1994, 266, H1588–H1595. [Google Scholar] [CrossRef] [PubMed]
- Yamada, N.; Li, W.; Ihaya, A.; Kimura, T.; Morioka, K.; Uesaka, T.; Takamori, A.; Handa, M.; Tanabe, S.; Tanaka, K. Platelet-derived endothelial cell growth factor gene therapy for limb ischemia. J. Vasc. Surg. 2006, 44, 1322–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schierling, W.; Troidl, K.; Troidl, C.; Schmitz-Rixen, T.; Schaper, W.; Eitenmüller, I.K. The role of angiogenic growth factors in arteriogenesis. J. Vasc. Res. 2009, 46, 365–374. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ashraf, J.V.; Al Haj Zen, A. Role of Vascular Smooth Muscle Cell Phenotype Switching in Arteriogenesis. Int. J. Mol. Sci. 2021, 22, 10585. https://doi.org/10.3390/ijms221910585
Ashraf JV, Al Haj Zen A. Role of Vascular Smooth Muscle Cell Phenotype Switching in Arteriogenesis. International Journal of Molecular Sciences. 2021; 22(19):10585. https://doi.org/10.3390/ijms221910585
Chicago/Turabian StyleAshraf, Jasni Viralippurath, and Ayman Al Haj Zen. 2021. "Role of Vascular Smooth Muscle Cell Phenotype Switching in Arteriogenesis" International Journal of Molecular Sciences 22, no. 19: 10585. https://doi.org/10.3390/ijms221910585
APA StyleAshraf, J. V., & Al Haj Zen, A. (2021). Role of Vascular Smooth Muscle Cell Phenotype Switching in Arteriogenesis. International Journal of Molecular Sciences, 22(19), 10585. https://doi.org/10.3390/ijms221910585