
Convergent Canonical Pathways in Autism Spectrum Disorder from Proteomic, Transcriptomic and DNA Methylation Data


Abstract
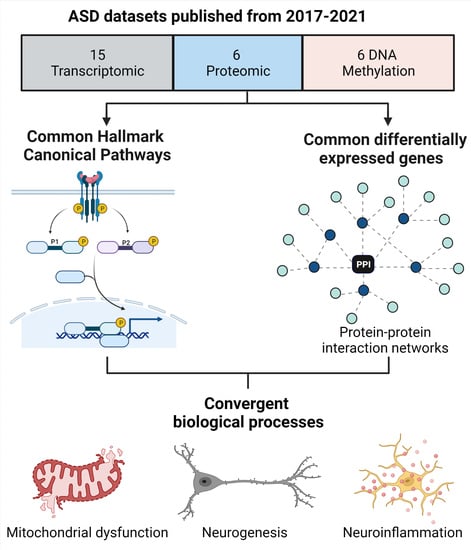
:
1. Introduction
2. Methods
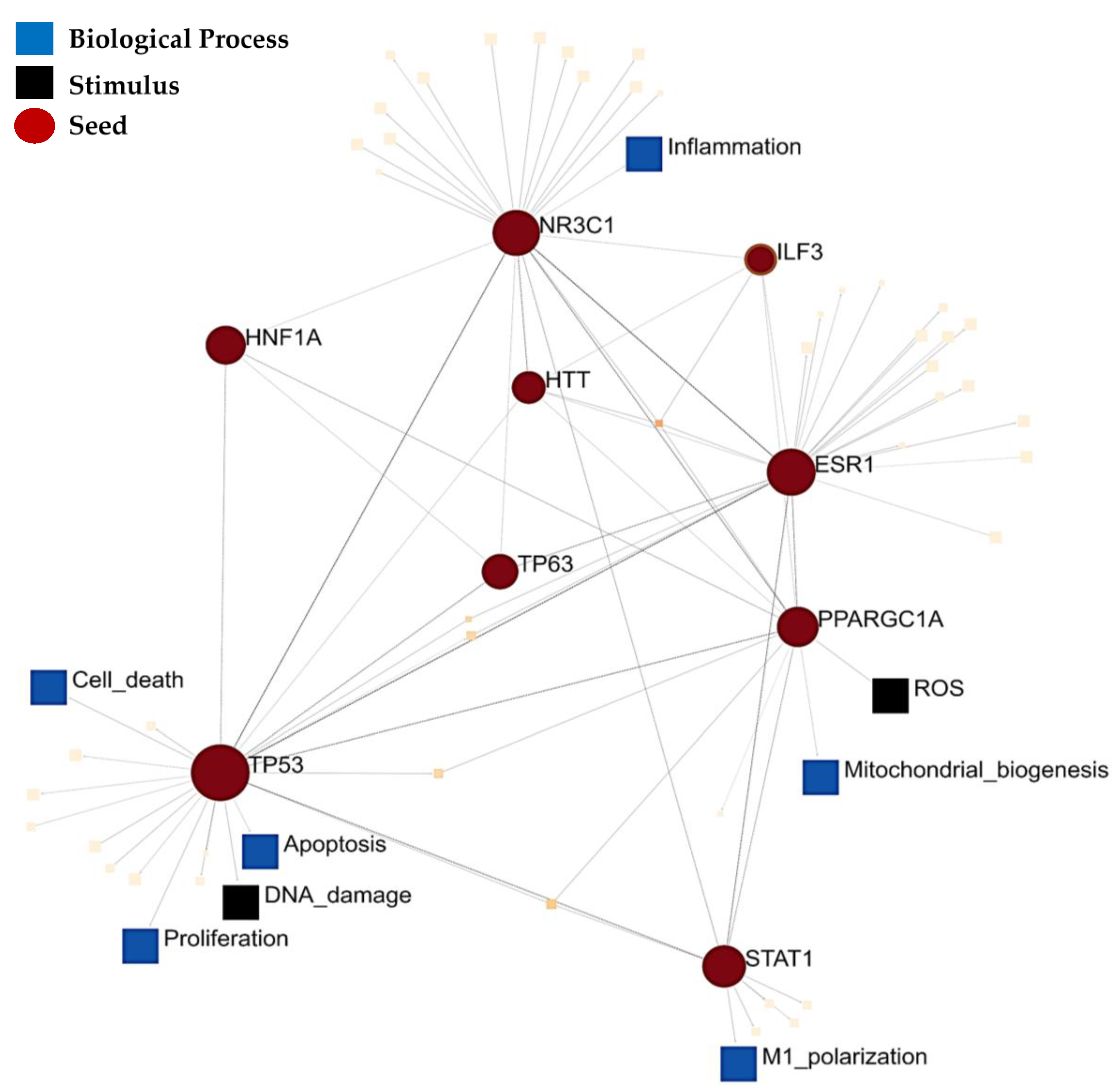
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wiśniowiecka-Kowalnik, B.; Nowakowska, B.A. Genetics and epigenetics of autism spectrum disorder—current evidence in the field. J. Appl. Genet. 2019, 60, 37–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, S.; Karlsson, H.; Dalman, C.; Widman, L.; Rai, D.; Gardner, R.M.; Magnusson, C.; Sandin, S.; Tabb, L.P.; Newschaffer, C.J.; et al. The Familial Risk of Autism Spectrum Disorder with and without Intellectual Disability. Autism Res. 2020, 13, 2242–2250. [Google Scholar] [CrossRef]
- Bai, D.; Yip, B.H.K.; Windham, G.C.; Sourander, A.; Francis, R.; Yoffe, R.; Glasson, E.; Mahjani, B.; Suominen, A.; Leonard, H.; et al. Association of Genetic and Environmental Factors with Autism in a 5-Country Cohort. JAMA Psychiatry 2019, 76, 1035–1043. [Google Scholar] [CrossRef]
- Pintacuda, G.; Martín, J.M.; Eggan, K.C. Mind the translational gap: Using iPS cell models to bridge from genetic discoveries to perturbed pathways and therapeutic targets. Mol. Autism 2021, 12, 10. [Google Scholar] [CrossRef]
- Torre-Ubieta, L.; de la Won, H.; Stein, J.L.; Geschwind, D.H. Advancing the understanding of autism disease mechanisms through genetics. Nat. Med. 2016, 224, 345–361. [Google Scholar] [CrossRef]
- Duffney, L.J.; Valdez, P.; Tremblay, M.W.; Cao, X.; Montgomery, S.; McConkie-Rosell, A.; Jiang, Y. Epigenetics and autism spectrum disorder: A report of an autism case with mutation in H1 linker histone HIST1H1E and literature review. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2018, 177, 426–433. [Google Scholar] [CrossRef]
- Choi, L.; An, J.-Y. Genetic architecture of autism spectrum disorder: Lessons from large-scale genomic studies. Neurosci. Biobehav. Rev. 2021, 128, 244–257. [Google Scholar] [CrossRef] [PubMed]
- Manoli, D.S.; State, M.W. Autism spectrum disorder genetics and the search for pathological mechanisms. Am. J. Psychiatry 2021, 178, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Comes, A.L.; Papiol, S.; Mueller, T.; Geyer, P.E.; Mann, M.; Schulze, T.G. Proteomics for blood biomarker exploration of severe mental illness: Pitfalls of the past and potential for the future. Transl. Psychiatry 2018, 81, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Shukla, H.; Wu, C.; Saxena, S. Challenges and Solutions in Proteomics. Curr. Genom. 2007, 8, 21–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, L.; Feng, C.; Zhang, K.; Chen, Y.; Gao, Y.; Ke, J.; Chen, X.; Lin, J.; Li, C.; Iqbal, J.; et al. Proteomics Study of Peripheral Blood Mononuclear Cells (PBMCs) in Autistic Children. Front. Cell. Neurosci. 2019, 13, 105. [Google Scholar] [CrossRef] [PubMed]
- Ruggeri, B.; Sarkans, U.; Schumann, G.; Persico, A.M. Biomarkers in autism spectrum disorder: The old and the new. Psychopharmacology 2013, 2316, 1201–1216. [Google Scholar] [CrossRef]
- Higdon, R.; Earl, R.K.; Stanberry, L.; Hudac, C.M.; Montague, E.; Stewart, E.; Janko, I.; Choiniere, J.; Broomall, W.; Kolker, N.; et al. The promise of multi-omics and clinical data integration to identify and target personalized healthcare approaches in autism spectrum disorders. OMICS 2015, 19, 197–208. [Google Scholar] [CrossRef] [Green Version]
- Hewitson, L.; Mathews, J.A.; Devlin, M.; Schutte, C.; Lee, J.; German, D.C. Blood biomarker discovery for autism spectrum disorder: A proteomic analysis. PLoS ONE 2021, 16, e0246581. [Google Scholar] [CrossRef]
- Shen, L.; Zhang, K.; Feng, C.; Chen, Y.; Li, S.; Iqbal, J.; Liao, L.; Zhao, Y.; Zhai, J. iTRAQ-Based Proteomic Analysis Reveals Protein Profile in Plasma from Children with Autism. Proteom.-Clin. Appl. 2018, 12, 1700085. [Google Scholar] [CrossRef]
- Yang, J.; Chen, Y.; Xiong, X.; Zhou, X.; Han, L.; Ni, L.; Wang, W.; Wang, X.; Zhao, L.; Shao, D.; et al. Peptidome Analysis Reveals Novel Serum Biomarkers for Children with Autism Spectrum Disorder in China. Proteom.-Clin. Appl. 2018, 12, 1700164. [Google Scholar] [CrossRef] [PubMed]
- Yao, F.; Zhang, K.; Feng, C.; Gao, Y.; Shen, L.; Liu, X. Protein Biomarkers of Autism Spectrum Disorder Identified by Computational and Experimental Methods. Front. Psychiatry 2021, 12, 1–13. [Google Scholar] [CrossRef]
- Abraham, J.R.; Szoko, N.; Natowicz, M.R. Proteomic Investigations of Autism Brain Identify Known and Novel Pathogenetic Processes. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Tylee, D.S.; Hess, J.L.; Quinn, T.P.; Barve, R.; Huang, H.; Zhang-James, Y.; Chang, J.; Stamova, B.S.; Sharp, F.R.; Hertz-Picciotto, I.; et al. Blood transcriptomic comparison of individuals with and without autism spectrum disorder: A combined-samples mega-analysis. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2017, 174, 181–201. [Google Scholar] [CrossRef] [Green Version]
- Mordaunt, C.E.; Newschaffer, C.J.; Volk, H.E.; Ozonoff, S.; Hertz-Picciotto, I.; Lasalle, J.M.; Schmidt, R.J.; Fallin, M.D. A meta-analysis of two high-risk prospective cohort studies reveals autism-specific transcriptional changes to chromatin, autoimmune, and environmental response genes in umbilical cord blood. Mol. Autism 2019, 10, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, H.; Zhong, J.; Huang, Q.; Wu, X.; Mo, X.; Lu, L.; Liang, H. Integrated Systems Analysis Explores Dysfunctional Molecular Modules and Regulatory Factors in Children with Autism Spectrum Disorder. J. Mol. Neurosci. 2020, 71, 358–368. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Zhou, Y.; Ma, W.; Wang, J. An integrated transcriptomic analysis of autism spectrum disorder. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forés-Martos, J.; Catalá-López, F.; Sánchez-Valle, J.; Ibáñez, K.; Tejero, H.; Palma-Gudiel, H.; Climent, J.; Pancaldi, V.; Fañanás, L.; Arango, C.; et al. Transcriptomic metaanalyses of autistic brains reveals shared gene expression and biological pathway abnormalities with cancer. Mol. Autism 2019, 10, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, M.R.; Petralia, M.C.; Ciurleo, R.; Bramanti, A.; Fagone, P.; Shahjaman, M.; Wu, L.; Sun, Y.; Turanli, B.; Arga, K.Y.; et al. Comprehensive Analysis of RNA-Seq Gene Expression Profiling of Brain Transcriptomes Reveals Novel Genes, Regulators, and Pathways in Autism Spectrum Disorder. Brain Sci. 2020, 10, 747. [Google Scholar] [CrossRef]
- Wright, C.; Shin, J.H.; Rajpurohit, A.; Deep-Soboslay, A.; Collado-Torres, L.; Brandon, N.J.; Hyde, T.M.; Kleinman, J.E.; Jaffe, A.E.; Cross, A.J.; et al. Altered expression of histamine signaling genes in autism spectrum disorder. Transl. Psychiatry 2017, 7, e1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramaswami, G.; Won, H.; Gandal, M.J.; Haney, J.; Wang, J.C.; Wong, C.C.Y.; Sun, W.; Prabhakar, S.; Mill, J.; Geschwind, D.H. Integrative genomics identifies a convergent molecular subtype that links epigenomic with transcriptomic differences in autism. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Mordaunt, C.E.; Jianu, J.M.; Laufer, B.I.; Zhu, Y.; Hwang, H.; Dunaway, K.W.; Bakulski, K.M.; Feinberg, J.I.; Volk, H.E.; Lyall, K.; et al. Cord blood DNA methylome in newborns later diagnosed with autism spectrum disorder reflects early dysregulation of neurodevelopmental and X-linked genes. Genome Med. 2020, 12, 1–25. [Google Scholar] [CrossRef]
- Hu, V.W.; Hong, Y.; Xu, M.; Shu, H.T. Altered DNA methylation in a severe subtype of idiopathic autism: Evidence for sex differences in affected metabolic pathways. Autism 2020, 25, 887–910. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.C.Y.; Smith, R.G.; Hannon, E.; Ramaswami, G.; Parikshak, N.N.; Assary, E.; Troakes, C.; Poschmann, J.; Schalkwyk, L.C.; Sun, W.; et al. Genome-wide DNA methylation profiling identifies convergent molecular signatures associated with idiopathic and syndromic autism in post-mortem human brain tissue. Hum. Mol. Genet. 2019, 28, 2201–2211. [Google Scholar] [CrossRef] [Green Version]
- Stathopoulos, S.; Gaujoux, R.; Lindeque, Z.; Mahony, C.; Van Der Colff, R.; Van Der Westhuizen, F.; O’Ryan, C. DNA Methylation Associated with Mitochondrial Dysfunction in a South African Autism Spectrum Disorder Cohort. Autism Res. 2020, 13, 1079–1093. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 27, 1653–1659. [Google Scholar] [CrossRef] [Green Version]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef] [PubMed]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Subramanian, A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; Mesirov, J.P. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14. [Google Scholar] [CrossRef] [Green Version]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Licata, L.; Lo Surdo, P.; Iannuccelli, M.; Palma, A.; Micarelli, E.; Perfetto, L.; Peluso, D.; Calderone, A.; Castagnoli, L.; Cesareni, G. SIGNOR 2.0, the SIGnaling Network Open Resource 2.0: 2019 update. Nucleic Acids Res. 2020, 48, D504–D510. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Gill, E.E.; Hancock, R.E.W. NetworkAnalyst for statistical, visual and network-based meta-analysis of gene expression data. Nat. Protoc. 2015, 10, 823–844. [Google Scholar] [CrossRef]
- Lake, N.J.; Bird, M.J.; Isohanni, P.; Paetau, A. Leigh Syndrome Neuropathology and Pathogenesis. J. Neuropathol. Exp. Neurol. 2015, 74, 482–492. [Google Scholar] [CrossRef] [Green Version]
- Tremblay, M.W.; Jiang, Y. DNA Methylation and Susceptibility to Autism Spectrum Disorder. Annu. Rev. Med. 2019, 70, 151–166. [Google Scholar] [CrossRef]
- Siddiqui, M.F.; Elwell, C.; Johnson, M.H. Mitochondrial Dysfunction in Autism Spectrum Disorders. Autism. Open Access 2016, 6, 1000190. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, K.K.; Levy, R.J. Evidence of Mitochondrial Dysfunction in Autism: Biochemical Links, Genetic-Based Associations, and Non-Energy-Related Mechanisms. Oxid. Med. Cell. Longev. 2017, 2017, 4314025. [Google Scholar] [CrossRef]
- Rose, S.; Niyazov, D.M.; Rossignol, D.A.; Goldenthal, M.; Kahler, S.G.; Frye, R.E. Clinical and Molecular Characteristics of Mitochondrial Dysfunction in Autism Spectrum Disorder. Molecular Diagn. Ther. 2018, 22, 571–593. [Google Scholar] [CrossRef] [Green Version]
- Castora, F.J. Mitochondrial function and abnormalities implicated in the pathogenesis of ASD. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2019, 92, 83–108. [Google Scholar] [CrossRef] [PubMed]
- Citrigno, L.; Muglia, M.; Qualtieri, A.; Spadafora, P.; Cavalcanti, F.; Pioggia, G.; Cerasa, A. The Mitochondrial Dysfunction Hypothesis in Autism Spectrum Disorders: Current Status and Future Perspectives. Int. J. Mol. Sci. 2020, 21, 5785. [Google Scholar] [CrossRef] [PubMed]
- Thorsen, M. Oxidative stress, metabolic and mitochondrial abnormalities associated with autism spectrum disorder. Prog. Mol. Biol. Transl. Sci. 2020, 173, 331–354. [Google Scholar] [PubMed]
- Gładysz, D.; Krzywdzińska, A.; Hozyasz, K.K. Immune Abnormalities in Autism Spectrum Disorder—Could They Hold Promise for Causative Treatment? Mol. Neurobiol. 2018, 55, 6387–6435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, X.; Liu, Y.; Fu, X.; Li, Y. Postmortem Studies of Neuroinflammation in Autism Spectrum Disorder: A Systematic Review. Mol. Neurobiol. 2020, 57, 3424–3438. [Google Scholar] [CrossRef]
- Pangrazzi, L.; Balasco, L.; Bozzi, Y. Oxidative Stress and Immune System Dysfunction in Autism Spectrum Disorders. Int. J. Mol. Sci. 2020, 21, 3293. [Google Scholar] [CrossRef]
- Götz, M.; Huttner, W.B. The cell biology of neurogenesis. Nat. Rev. Mol. Cell Biol. 2005, 610, 777–788. [Google Scholar] [CrossRef]
- Vieira, M.S.; Santos, A.K.; Vasconcellos, R.; Goulart, V.A.M.; Parreira, R.C.; Kihara, A.H.; Ulrich, H.; Resende, R.R. Neural stem cell differentiation into mature neurons: Mechanisms of regulation and biotechnological applications. Biotechnol. Adv. 2018, 36, 1946–1970. [Google Scholar] [CrossRef]
- Castro-Torres, R.D.; Busquets, O.; Parcerisas, A.; Verdaguer, E.; Olloquequi, J.; Ettcheto, M.; Beas-Zarate, C.; Folch, J.; Camins, A.; Auladell, C. Involvement of JNK1 in Neuronal Polarization During Brain Development. Cells 2020, 9, 1897. [Google Scholar] [CrossRef]
- Sierra, A.; Encinas, J.M.; Deudero, J.J.P.; Chancey, J.H.; Enikolopov, G.; Overstreet-Wadiche, L.S.; Tsirka, S.E.; Maletic-Savatic, M. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell 2010, 7, 483–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganesan, H.; Balasubramanian, V.; Iyer, M.; Venugopal, A.; Subramaniam, M.D.; Cho, S.-G.; Vellingiri, B. mTOR signalling pathway—A root cause for idiopathic autism? BMB Rep. 2019, 52, 424. [Google Scholar] [CrossRef] [Green Version]
- Boksha, I.S.; Prokhorova, T.A.; Tereshkina, E.B.; Savushkina, O.K.; Burbaeva, G.S. Protein Phosphorylation Signaling Cascades in Autism: The Role of mTOR Pathway. Biochem. 2021, 865, 577–596. [Google Scholar]
- Geschwind, D. Genetics of autism spectrum disorders. Trends Cogn. Sci. 2011, 15, 409–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Parada, L.F. PTEN signaling in autism spectrum disorders. Curr. Opin. Neurobiol. 2012, 22, 873–879. [Google Scholar] [CrossRef] [PubMed]
- Masini, E.; Loi, E.; Vega-Benedetti, A.F.; Carta, M.; Doneddu, G.; Fadda, R.; Zavattari, P. An Overview of the Main Genetic, Epigenetic and Environmental Factors Involved in Autism Spectrum Disorder Focusing on Synaptic Activity. Int. J. Mol. Sci. 2020, 21, 8290. [Google Scholar] [CrossRef]
- Kwon, C.H.; Luikart, B.W.; Powell, C.M.; Zhou, J.; Matheny, S.A.; Zhang, W.; Li, Y.; Baker, S.J.; Parada, L.F. Pten Regulates Neuronal Arborization and Social Interaction in Mice. Neuron 2006, 50, 377–388. [Google Scholar] [CrossRef] [Green Version]
- Poduri, A.; Evrony, G.D.; Cai, X.; Elhosary, P.C.; Beroukhim, R.; Lehtinen, M.K.; Hills, B.L.; Heinzen, E.L.; Hill, A.; Hill, S.R.; et al. Somatic Activation of AKT3 Causes Hemispheric Developmental Brain Malformations. Neuron 2012, 74, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Tsai, P.T.; Hull, C.; Chu, Y.; Greene-Colozzi, E.; Sadowski, A.R.; Leech, J.M.; Steinberg, J.; Crawley, J.N.; Regehr, W.G.; Sahin, M. Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature 2012, 488, 647–651. [Google Scholar] [CrossRef] [Green Version]
- Tang, G.; Gudsnuk, K.; Kuo, S.H.; Cotrina, M.L.; Rosoklija, G.; Sosunov, A.; Sonders, M.S.; Kanter, E.; Castagna, C.; Yamamoto, A.; et al. Loss of mTOR-Dependent Macroautophagy Causes Autistic-like Synaptic Pruning Deficits. Neuron 2014, 83, 1131–1143. [Google Scholar] [CrossRef] [Green Version]
- Lieberman, O.J.; Cartocci, V.; Pigulevskiy, I.; Molinari, M.; Carbonell, J.; Broseta, M.B.; Post, M.R.; Sulzer, D.; Borgkvist, A.; Santini, E. mTOR Suppresses Macroautophagy During Striatal Postnatal Development and Is Hyperactive in Mouse Models of Autism Spectrum Disorders. Front. Cell. Neurosci. 2020, 14, 70. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Ma, L.; Yang, M.; Shao, Q.; Xu, J.; Lu, Z.; Zhao, Z.; Chen, R.; Chai, Y.; Chen, J.-F. Cerebral organoid and mouse models reveal a RAB39b–PI3K–mTOR pathway-dependent dysregulation of cortical development leading to macrocephaly/autism phenotypes. Genes Dev. 2020, 34, 580–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basilico, B.; Morandell, J.; Novarino, G. Molecular mechanisms for targeted ASD treatments. Curr. Opin. Genet. Dev. 2020, 65, 126–137. [Google Scholar] [CrossRef]
- Khacho, M.; Harris, R.; Slack, R.S. Mitochondria as central regulators of neural stem cell fate and cognitive function. Nat. Rev. Neurosci. 2019, 20, 34–48. [Google Scholar] [CrossRef] [PubMed]
- Beckervordersandforth, R. Mitochondrial Metabolism-Mediated Regulation of Adult Neurogenesis. Brain Plast. 2017, 3, 73–87. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Boyer, L.; Jin, M.; Mertens, J.; Kim, Y.; Ma, L.; Ma, L.; Hamm, M.; Gage, F.H.; Hunter, T. Metabolic reprogramming during neuronal differentiation from aerobic glycolysis to neuronal oxidative phosphorylation. Elife 2016, 5, e13374. [Google Scholar] [CrossRef]
- Maffezzini, C.; Calvo-Garrido, J.; Wredenberg, A.; Freyer, C. Metabolic regulation of neurodifferentiation in the adult brain. Cell. Mol. Life Sci. 2020, 77, 2483–2496. [Google Scholar] [CrossRef] [Green Version]
- Licausi, F.; Hartman, N.W. Role of mTOR complexes in neurogenesis. Int. J. Mol. Sci. 2018, 19, 1544. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, J.T.; Rodgers, J.T.; Arlow, D.H.; Vazquez, F.; Mootha, V.K.; Puigserver, P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1α transcriptional complex. Nature 2007, 450, 736–740. [Google Scholar] [CrossRef] [PubMed]
- Vadlakonda, L.; Pasupuleti, M.; Pallu, R. Role of PI3K-AKT-mTOR and Wnt signaling pathways in transition of G1-S phase of cell cycle in cancer cells. Front. Oncol. 2013, 3, 85. [Google Scholar] [CrossRef] [Green Version]
- Vallée, A.; Lecarpentier, Y.; Guillevin, R.; Vallée, J. Opposite Interplay Between the Canonical WNT/β-Catenin Pathway and PPAR Gamma: A Potential Therapeutic Target in Gliomas. Neurosci. Bull. 2018, 34, 573–588. [Google Scholar] [CrossRef]
- Faigle, R.; Song, H. Signaling mechanisms regulating adult neural stem cells and neurogenesis. Biochim. Biophys. Acta-Gen. Subj. 2013, 1830, 2435–2448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, T.P.; Kühl, M. An Updated Overview on Wnt Signaling Pathways. Circ. Res. 2010, 106, 1798–1806. [Google Scholar] [CrossRef] [PubMed]
- López-Tobón, A.; Villa, C.E.; Cheroni, C.; Trattaro, S.; Caporale, N.; Conforti, P.; Iennaco, R.; Lachgar, M.; Rigoli, M.T.; Marcó de la Cruz, B.; et al. Human Cortical Organoids Expose a Differential Function of GSK3 on Cortical Neurogenesis. Stem Cell Rep. 2019, 13, 847–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussaini, S.M.Q.; Choi, C., II; Cho, C.H.; Kim, H.J.; Jun, H.; Jang, M.H. Wnt signaling in neuropsychiatric disorders: Ties with adult hippocampal neurogenesis and behavior. Neurosci. Biobehav. Rev. 2014, 47, 369–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caglayan, A.O. Genetic causes of syndromic and non-syndromic autism. Dev. Med. Child Neurol. 2010, 52, 130–138. [Google Scholar] [CrossRef]
- Mulligan, K.A.; Cheyette, B.N.R. Neurodevelopmental Perspectives on Wnt Signaling in Psychiatry. Mol. Neuropsychiatry 2016, 2, 219–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iakoucheva, L.M.; Muotri, A.R.; Sebat, J. Getting to the Cores of Autism. Cell 2019, 178, 1287–1298. [Google Scholar] [CrossRef] [PubMed]
- Caracci, M.O.; Avila, M.E.; De Ferrari, G.V. Synaptic Wnt/GSK3 β Signaling Hub in Autism. Neural Plast. 2016, 2016, 9603751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwan, V.; Unda, B.K.; Singh, K.K. Wnt signaling networks in autism spectrum disorder and intellectual disability. J. Neurodev. Disord. 2016, 8, 45. [Google Scholar] [CrossRef] [Green Version]
- Perera, R.J.; Marcusson, E.G.; Koo, S.; Kang, X.; Kim, Y.; White, N.; Dean, N.M. Identification of novel PPARγ target genes in primary human adipocytes. Gene 2006, 369, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Panov, A.; Orynbayeva, Z.; Vavilin, V.; Lyakhovich, V. Fatty acids in energy metabolism of the central nervous system. Biomed. Res. Int. 2014, 2014, 472459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, K.; Turcotte, R.; Cui, J.; Zimmerman, S.E.; Pinho, S.; Mizoguchi, T.; Arai, F.; Runnels, J.M.; Alt, C.; Teruya-Feldstein, J.; et al. Self-renewal of a purified Tie2+ hematopoietic stem cell population relies on mitochondrial clearance. Science 2016, 354, 1156–1160. [Google Scholar] [CrossRef] [Green Version]
- Longo, N.; Frigeni, M.; Pasquali, M. Carnitine transport and fatty acid oxidation. Biochim. Biophys. Acta-Mol. Cell Res. 2016, 1863, 2422–2435. [Google Scholar] [CrossRef]
- Knobloch, M.; Pilz, G.A.; Ghesquière, B.; Kovacs, W.J.; Wegleiter, T.; Moore, D.L.; Hruzova, M.; Zamboni, N.; Carmeliet, P.; Jessberger, S. A Fatty Acid Oxidation-Dependent Metabolic Shift Regulates Adult Neural Stem Cell Activity. Cell Rep. 2017, 20, 2144–2155. [Google Scholar] [CrossRef] [Green Version]
- Xie, Z.; Jones, A.; Deeney, J.T.; Hur, S.K.; Bankaitis, V.A. Inborn Errors of Long-Chain Fatty Acid β-Oxidation Link Neural Stem Cell Self-Renewal to Autism. Cell Rep. 2016, 14, 991–999. [Google Scholar] [CrossRef] [Green Version]
- Ristori, M.V.; Mortera, S.L.; Marzano, V.; Guerrera, S.; Vernocchi, P.; Ianiro, G.; Gardini, S.; Torre, G.; Valeri, G.; Vicari, S. Proteomics and Metabolomics Approaches towards a Functional Insight onto AUTISM Spectrum Disorders: Phenotype Stratification and Biomarker Discovery. Int. J. Mol. Sci. 2020, 21, 6274. [Google Scholar] [CrossRef]
- Shen, L.X.K.; Zhang, H.; Lin, J.; Feng, C.; Iqbal, J. Biomarkers in autism spectrum disorders: Current progress. Clin. Chim. Acta 2020, 502, 41–54. [Google Scholar] [CrossRef]
- Barone, R.; Rizzo, R.; Tabbí, G.; Malaguarnera, M.; Frye, R.E.; Bastin, J. Nuclear Peroxisome Proliferator-Activated Receptors (PPARs) as therapeutic targets of resveratrol for autism spectrum disorder. Int. J. Mol. Sci. 2019, 20, 1878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallée, A.; Vallée, J.N.; Lecarpentier, Y. PPARγ agonists: Potential treatment for autism spectrum disorder by inhibiting the canonical WNT/β-catenin pathway. Mol. Psychiatry 2019, 24, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Beckervordersandforth, R.; Ebert, B.; Schäffner, I.; Moss, J.; Fiebig, C.; Shin, J.; Moore, D.L.; Ghosh, L.; Trinchero, M.F.; Stockburger, C.; et al. Role of Mitochondrial Metabolism in the Control of Early Lineage Progression and Aging Phenotypes in Adult Hippocampal Neurogenesis. Neuron 2017, 93, 560–573.e6. [Google Scholar] [CrossRef] [Green Version]
- Abruzzo, P.M.; Matté, A.; Bolotta, A.; Federti, E.; Ghezzo, A.; Guarnieri, T.; Marini, M.; Posar, A.; Siciliano, A.; De Franceschi, L.; et al. Plasma peroxiredoxin changes and inflammatory cytokines support the involvement of neuro-inflammation and oxidative stress in Autism Spectrum Disorder. J. Transl. Med. 2019, 171, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bjørklund, G.; Meguid, N.A.; El-Bana, M.A.; Tinkov, A.A.; Saad, K.; Dadar, M.; Hemimi, M.; Skalny, A.V.; Hosnedlová, B.; Kizek, R.; et al. Oxidative Stress in Autism Spectrum Disorder. Mol. Neurobiol. 2020, 57, 2314–2332. [Google Scholar] [CrossRef]
- Hu, T.; Dong, Y.; He, C.; Zhao, M.; He, Q. The Gut Microbiota and Oxidative Stress in Autism Spectrum Disorders (ASD). Oxid. Med. Cell. Longev. 2020, 2020, 8396708. [Google Scholar] [CrossRef]
- Manivasagam, T.; Arunadevi, S.; Essa, M.M.; SaravanaBabu, C.; Borah, A.; Thenmozhi, A.J.; Qoronfleh, M.W. Role of Oxidative Stress and Antioxidants in Autism. Adv. Neurobiol. 2020, 24, 193–206. [Google Scholar]
- Morimoto, M.; Hashimoto, T.; Tsuda, Y.; Nakatsu, T.; Kitaoka, T.; Kyotani, S. Assessment of oxidative stress in autism spectrum disorder using reactive oxygen metabolites and biological antioxidant potential. PLoS ONE 2020, 15, e0233550. [Google Scholar] [CrossRef]
- Chen, L.; Shi, X.-J.; Liu, H.; Mao, X.; Gui, L.-N.; Wang, H.; Cheng, Y. Oxidative stress marker aberrations in children with autism spectrum disorder: A systematic review and meta-analysis of 87 studies (N = 9109). Transl. Psychiatry 2021, 11, 1–10. [Google Scholar] [CrossRef]
- Le Belle, J.E.; Orozco, N.M.; Paucar, A.A.; Saxe, J.P.; Mottahedeh, J.; Pyle, A.D.; Wu, H.; Kornblum, H.I. Proliferative Neural Stem Cells Have High Endogenous ROS Levels that Regulate Self-Renewal and Neurogenesis in a PI3K/Akt-Dependant Manner. Cell Stem Cell 2011, 8, 59–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prozorovski, T.; Schneider, R.; Berndt, C.; Hartung, H.P.; Aktas, O. Redox-regulated fate of neural stem progenitor cells. Biochim. Et Biophys. Acta-Gen. Subj. 2015, 1850, 1543–1554. [Google Scholar] [CrossRef] [PubMed]
- Prozorovski, T.; Schulze-Topphoff, U.; Glumm, R.; Baumgart, J.; Schröter, F.; Ninnemann, O.; Siegert, E.; Bendix, I.; Brüstle, O.; Nitsch, R.; et al. Sirt1 contributes critically to the redox-dependent fate of neural progenitors. Nat. Cell Biol. 2008, 10, 385–394. [Google Scholar] [CrossRef]
- Hou, Y.; Ouyang, X.; Wan, R.; Cheng, H.; Mattson, M.P.; Cheng, A. Mitochondrial superoxide production negatively regulates neural progenitor proliferation and cerebral cortical development. Stem Cells 2012, 30, 2535–2547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, T.T.; Zou, Y.; Corniola, R. Oxidative stress and adult neurogenesis-Effects of radiation and superoxide dismutase deficiency. Semin. Cell Dev. Biol. 2012, 23, 738–744. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Cao, L.; Chen, J.; Song, S.; Lee, I.H.; Quijano, C.; Liu, H.; Keyvanfar, K.; Chen, H.; Cao, L.-Y.; et al. Bmi1 regulates mitochondrial function and the DNA damage response pathway. Nature 2009, 459, 387–392. [Google Scholar] [CrossRef]
- Fasano, C.A.; Phoenix, T.N.; Kokovay, E.; Lowry, N.; Elkabetz, Y.; Dimos, J.T.; Lemischka, I.R.; Studer, L.; Temple, S. Bmi-1 cooperates with Foxgl to maintain neural stem cell self-renewal in the forebrain. Genes Dev. 2009, 23, 561–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Liang, C.C.; Bian, Z.C.; Zhu, Y.; Guan, J.L. FIP200 is required for maintenance and differentiation of postnatal neural stem cells. Nat. Neurosci. 2013, 16, 532–542. [Google Scholar] [CrossRef] [Green Version]
- Chatoo, W.; Abdouh, M.; Duparc, R.H.; Bernier, G. Bmi1 distinguishes immature retinal progenitor/stem cells from the main progenitor cell population and is required for normal retinal development. Stem Cells 2010, 28, 1412–1423. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.K.; Squier, M.V. Neuropathology and pathogenesis of mitochondrial diseases. J. Inherit. Metab. Dis. 1996, 19, 553–572. [Google Scholar] [CrossRef] [PubMed]
- Chow, C.W.; Thorburn, D.R. Morphological correlates of mitochondrial dysfunction in children. Hum. Reprod. 2000, 15, 68–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saneto, R.P.; Friedman, S.D.; Shaw, D.W.W. Neuroimaging of mitochondrial disease. Mitochondrion 2008, 8, 396–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiménez-Caballero, P.E.; Mollejo-Villanueva, M.; Álvarez-Tejerina, A. Mitochondrial encephalopathy due to complex i deficiency. brain tissue biopsy findings and clinical course following pharmacological. Rev. Neurol. 2008, 47, 27–30. [Google Scholar] [PubMed]
- Johnson, S.C.; Yanos, M.E.; Kayser, E.-B.; Quintana, A.; Sangesland, M.; Castanza, A.; Uhde, L.; Hui, J.; Wall, V.Z.; Gagnidze, A.; et al. mTOR Inhibition Alleviates Mitochondrial Disease in a Mouse Model of Leigh Syndrome. Science 2013, 342, 1524–1528. [Google Scholar] [CrossRef] [Green Version]
- Wen, Y.; Li, W.; Choudhury, G.R.; He, R.; Yang, T.; Liu, R.; Jin, K.; Yang, S.H. Astroglial PTEN loss disrupts neuronal lamination by dysregulating radial glia-guided neuronal migration. Aging Dis. 2013, 4, 113–126. [Google Scholar]
- Sarn, N.; Jaini, R.; Thacker, S.; Lee, H.; Dutta, R.; Eng, C. Cytoplasmic-predominant Pten increases microglial activation and synaptic pruning in a murine model with autism-like phenotype. Mol. Psychiatry 2020, 26, 1458–1471. [Google Scholar] [CrossRef] [Green Version]
- Vallée, A.; Lecarpentier, Y. Crosstalk Between Peroxisome Proliferator-Activated Receptor Gamma and the Canonical WNT/β-Catenin Pathway in Chronic Inflammation and Oxidative Stress During Carcinogenesis. Front. Immunol. 2018, 9, 1. [Google Scholar] [CrossRef] [Green Version]
- Scuderi, C.; Jaini, R.; Thacker, S.; Lee, H.; Dutta, R.; Eng, C. Palmitoylethanolamide controls reactive gliosis and exerts neuroprotective functions in a rat model of Alzheimer’s disease. Cell Death Dis. 2014, 59, e1419. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Xu, B.; Wang, Z.X.; Tan, G.W.; Shen, S.H. Inhibition of Wnt/β-catenin signal is alleviated reactive gliosis in rats with hydrocephalus. Child Nerv. Syst. 2015, 31, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Lee, S.; Chang, S.-C.; Lee, J. Significant roles of neuroinflammation in Parkinson’s disease: Therapeutic targets for PD prevention. Arch. Pharmacal Res. 2019, 425, 416–425. [Google Scholar] [CrossRef]
- Mendrysa, S.M.; Ghassemifar, S.; Malek, R. p53 in the CNS: Perspectives on Development, Stem Cells, and Cancer. Genes Cancer 2011, 2, 431. [Google Scholar] [CrossRef] [Green Version]
- Silva-Adaya, D.; Garza-Lombó, C.; Gonsebatt, M.E. Xenobiotic transport and metabolism in the human brain. Neurotoxicology 2021, 86, 125–138. [Google Scholar] [CrossRef]
- Sánchez-Alegría, K.; Flores-León, M.; Avila-Muñoz, E.; Rodríguez-Corona, N.; Arias, C. PI3K signaling in neurons: A central node for the control of multiple functions. Int. J. Mol. Sci. 2018, 19, 3725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, J.-J.; Wang, W.; Li, X.; Deokar, H.; Buolamwini, J.K.; Zhang, R. Inhibiting β-Catenin by β-Carboline-Type MDM2 Inhibitor for Pancreatic Cancer Therapy. Front. Pharmacol. 2018, 9, 5. [Google Scholar] [CrossRef]
- Forsberg, K.; Wuttke, A.; Quadrato, G.; Chumakov, P.M.; Wizenmann, A.; Di Giovanni, S. The tumor suppressor p53 fine-tunes reactive oxygen species levels and neurogenesis via PI3 kinase signaling. J. Neurosci. 2013, 33, 14318–14330. [Google Scholar] [CrossRef]
- Sofroniew, M.V. Astrocyte barriers to neurotoxic inflammation. Nat. Rev. Neurosci. 2015, 16, 249–263. [Google Scholar] [CrossRef] [Green Version]
- Petrelli, F.; Pucci, L.; Bezzi, P. Astrocytes and microglia and their potential link with autism spectrum disorders. Front. Cell. Neurosci. 2016, 10, 21. [Google Scholar] [CrossRef] [Green Version]
- Mekori-Domachevsky, E.; Segal-Gavish, H.; Gross, R. Neuroinflammation and neuroprotection in schizophrenia and autism spectrum disorder. In Neuroprotection in Autism, Schizophrenia and Alzheimer’s Disease; Elsevier: Amsterdam, The Netherlands, 2019; pp. 101–122. [Google Scholar] [CrossRef]
- Liao, X.; Yang, J.; Wang, H.; Li, Y. Microglia mediated neuroinflammation in autism spectrum disorder. J. Psychiatr. Res. 2020, 130, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Li, Y. Nuclear Factor Kappa B in Autism Spectrum Disorder: A Systematic Review. Pharmacol. Res. 2020, 159, 104918. [Google Scholar] [CrossRef]
- Voineagu, I. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 2011, 474, 380–384. [Google Scholar] [CrossRef]
- Laurence, J.A.; Fatemi, S.H. Glial fibrillary acidic protein is elevated in superior frontal, parietal and cerebellar cortices of autistic subjects. Cerebellum 2005, 4, 206–210. [Google Scholar] [CrossRef]
- Edmonson, C.; Ziats, M.N.; Rennert, O.M. Altered glial marker expression in autistic post-mortem prefrontal cortex and cerebellum. Mol. Autism 2014, 5, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fatemi, S.H.; Folsom, T.D.; Reutiman, T.J.; Lee, S. Expression of astrocytic markers aquaporin 4 and connexin 43 is altered in brains of subjects with autism. Synapse 2008, 62, 501. [Google Scholar] [CrossRef] [Green Version]
- Wegiel, J.; Kuchna, I.; Nowicki, K.; Imaki, H.; Wegiel, J.; Marchi, E.; Ma, S.Y.; Chauhan, A.; Chauhan, V.; Bobrowicz, T.; et al. The neuropathology of autism: Defects of neurogenesis and neuronal migration, and dysplastic changes. Acta Neuropathol. 2010, 119, 755–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, L.E.; Barres, B.A. Emerging roles of astrocytes in neural circuit development. Nat. Rev. Neurosci. 2013, 14, 311–321. [Google Scholar] [CrossRef] [Green Version]
- Sahlender, D.A.; Savtchouk, I.; Volterra, A. What do we know about gliotransmitter release from astrocytes? Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130592. [Google Scholar] [CrossRef] [Green Version]
- Lauro, C.; Limatola, C. Metabolic Reprograming of Microglia in the Regulation of the Innate Inflammatory Response. Front. Immunol. 2020, 11, 493. [Google Scholar] [CrossRef] [Green Version]
- Lively, S.; Schlichter, L.C. Microglia responses to pro-inflammatory stimuli (LPS, IFNγ+TNFα) and reprogramming by resolving cytokines (IL-4, IL-10). Front. Cell. Neurosci. 2018, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loving, B.A.; Bruce, K.D. Lipid and Lipoprotein Metabolism in Microglia. Front. Physiol. 2020, 11, 393. [Google Scholar] [CrossRef]
- Chausse, B.; Kakimoto, P.A.; Kann, O. Microglia and lipids: How metabolism controls brain innate immunity. Semin. Cell Dev. Biol. 2021, 112, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Baik, S.H.; Kang, S.; Lee, W.; Choi, H.; Chung, S.; Kim, J., II; Mook-Jung, I. A Breakdown in Metabolic Reprogramming Causes Microglia Dysfunction in Alzheimer’s Disease. Cell Metab. 2019, 30, 493–507.e6. [Google Scholar] [CrossRef] [PubMed]
- Bernier, L.P.; York, E.M.; MacVicar, B.A. Immunometabolism in the Brain: How Metabolism Shapes Microglial Function. Trends Neurosci. 2020, 43, 854–869. [Google Scholar] [CrossRef]
- Revuelta, M.; Scheuer, T.; Chew, L.J.; Schmitz, T. Glial Factors Regulating White Matter Development and Pathologies of the Cerebellum. Neurochem. Res. 2020, 45, 643–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markiewski, M.M.; Nilsson, B.; Ekdahl, K.N.; Mollnes, T.E.; Lambris, J.D. Complement and coagulation: Strangers or partners in crime? Trends Immunol. 2007, 28, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Amara, U.; Rittirsch, D.; Flierl, M.; Bruckner, U.; Klos, A.; Gebhard, F.; Lambris, J.D.; Huber-Lang, M. Interaction Between the Coagulation and Complement System. Adv. Exp. Med. Biol. 2008, 632, 71. [Google Scholar] [PubMed] [Green Version]
- Amara, U.; Flierl, M.A.; Rittirsch, D.; Klos, A.; Chen, H.; Acker, B.; Brückner, U.B.; Nilsson, B.; Gebhard, F.; Lambris, J.D.; et al. Molecular Intercommunication between the Complement and Coagulation Systems. J. Immunol. 2010, 185, 5628–5636. [Google Scholar] [CrossRef] [Green Version]
- Orsini, F.; De Blasio, D.; Zangari, R.; Zanier, E.R.; De Simoni, M.-G. Versatility of the complement system in neuroinflammation, neurodegeneration and brain homeostasis. Front. Cell. Neurosci. 2014, 8, 380. [Google Scholar] [CrossRef] [Green Version]
- Hammad, A.; Westacott, L.; Zaben, M. The role of the complement system in traumatic brain injury: A review. J. Neuroinflammation 2018, 15, 24. [Google Scholar] [CrossRef] [Green Version]
- Fletcher-Sandersjöö, A.; Maegele, M.; Bellander, B.-M. Does Complement-Mediated Hemostatic Disturbance Occur in Traumatic Brain Injury? A Literature Review and Observational Study Protocol. Int. J. Mol. Sci. 2020, 21, 1596. [Google Scholar] [CrossRef] [Green Version]
- De Luca, C.; Virtuoso, A.; Maggio, N.; Papa, M. Neuro-Coagulopathy: Blood Coagulation Factors in Central Nervous System Diseases. Int. J. Mol. Sci. 2017, 18, 2128. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, M.Z.; Adamiak, M.; Kucia, M.; Tse, W.; Ratajczak, J.; Wiktor-Jedrzejczak, W. The emerging link between the complement cascade and purinergic signaling in stress hematopoiesis. Front. Immunol. 2018, 9, 1295. [Google Scholar] [CrossRef] [Green Version]
- Gorelik, A.; Sapir, T.; Haffner-Krausz, R.; Olender, T.; Woodruff, T.M.; Reiner, O. Developmental activities of the complement pathway in migrating neurons. Nat. Commun. 2017, 81, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Magdalon, J.; Mansur, F.; Teles e Silva, A.L.; de Goes, V.A.; Reiner, O.; Sertié, A.L. Complement System in Brain Architecture and Neurodevelopmental Disorders. Front. Neurosci. 2020, 14, 23. [Google Scholar] [CrossRef] [PubMed]
- Ziabska, K.; Ziemka-Nalecz, M.; Pawelec, P.; Sypecka, J.; Zalewska, T. Aberrant complement system activation in neurological disorders. Int. J. Mol. Sci. 2021, 22, 4675. [Google Scholar] [CrossRef] [PubMed]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia Sculpt Postnatal Neural Circuits in an Activity and Complement-Dependent Manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Reynolds, K.; Ji, Y.; Gu, R.; Rai, S.; Zhou, C.J. Impaired neurodevelopmental pathways in autism spectrum disorder: A review of signaling mechanisms and crosstalk. J. Neurodev. Disord. 2019, 11, 1–14. [Google Scholar] [CrossRef]
- Arredondo, S.B.; Valenzuela-Bezanilla, D.; Mardones, M.D.; Varela-Nallar, L. Role of Wnt Signaling in Adult Hippocampal Neurogenesis in Health and Disease. Front. Cell Dev. Biol. 2020, 8, 860. [Google Scholar] [CrossRef]




{kind=link}
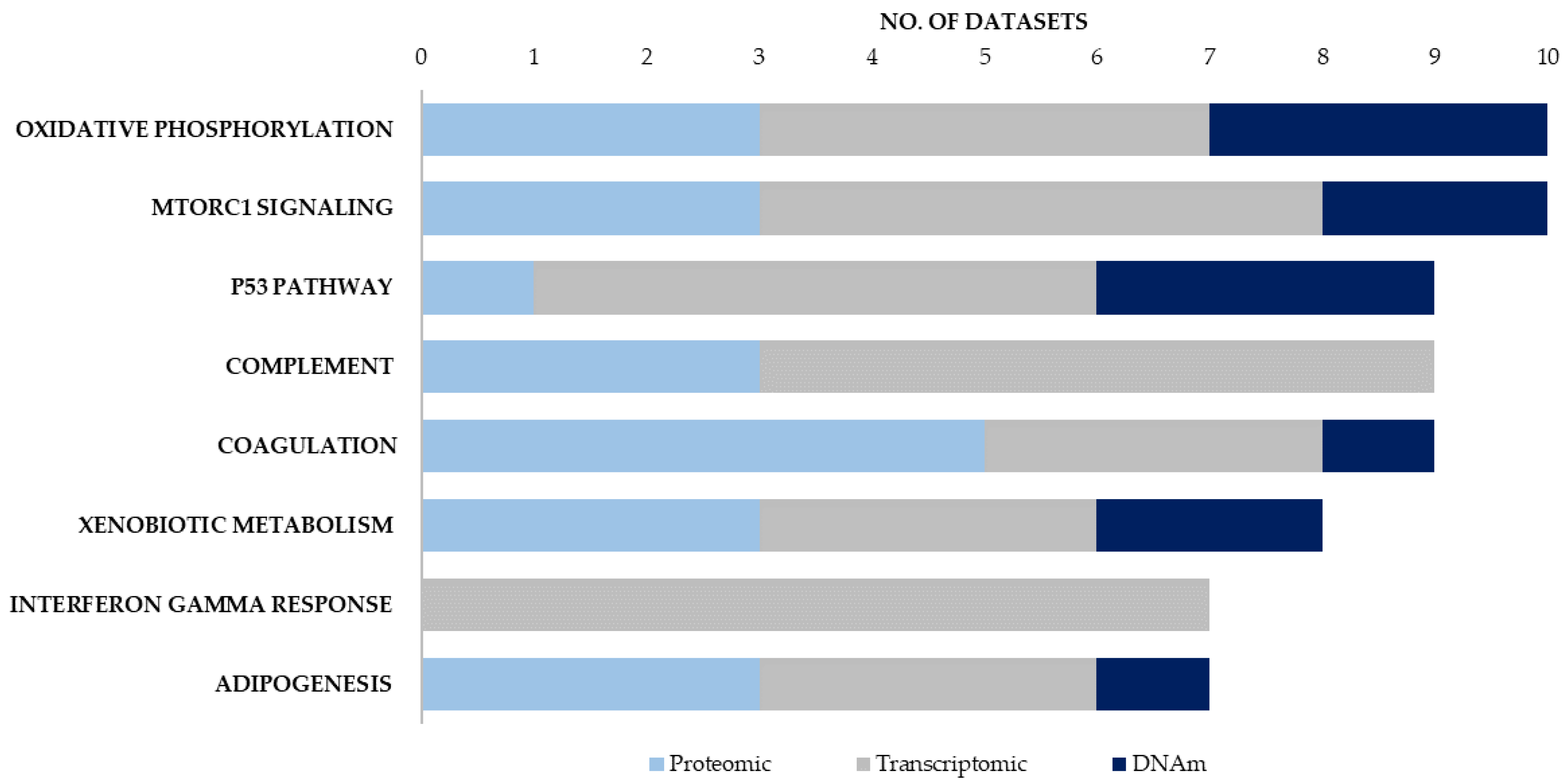{kind=link}
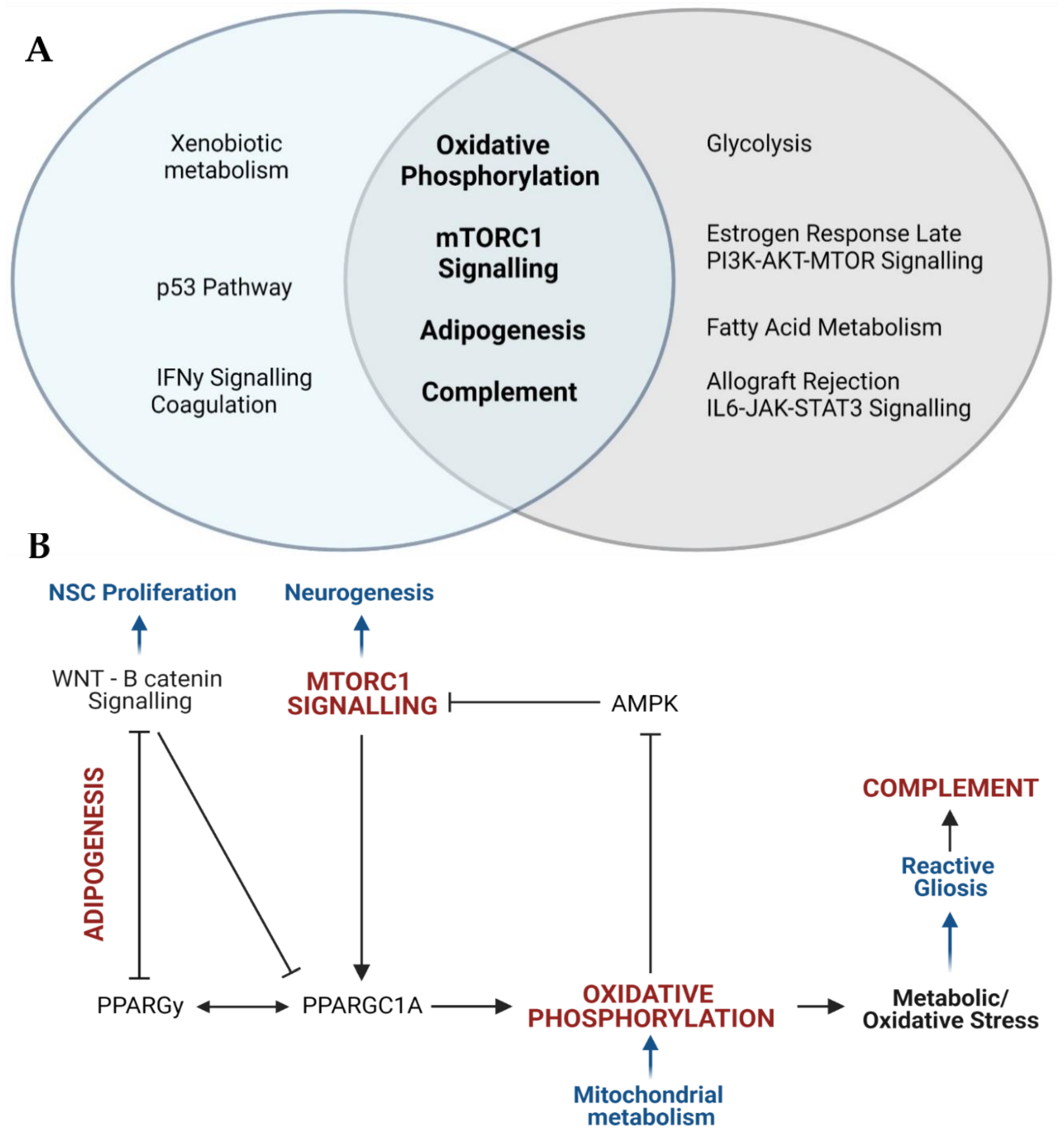{kind=link}
{kind=link}
| Proteomics Analysis | ||||
|---|---|---|---|---|
| Shen et al., 2019 [11] | 41 DE proteins in ASD | 24 male and 6 female ASD (2–6 yrs) and age/gender-matched controls | Blood | Blood PBMCs |
| Hewitson et al., 2021 [14] | 86 downregulated, 52 upregulated proteins in ASD (FDR < 0.05) | 76 ASD (boys) and 78 controls (boys), 18 months to 8 yrs | Blood serum | |
| Shen et al., 2018 [15] | 24 DE proteins in ASD | 24 male and 6 female autistic patients (2–6 yrs) and age/gender-matched controls | Blood plasma | |
| Yang et al., 2018 [16] | Eight biomarker peaks with higher expression in ASD | Han Chinese children: 68 ASD (average age = 12.4 yrs) and 80 age-matched controls (average age 14.3 yrs) | Blood serum | |
| Yao et al., 2021 [17] | 59 genes predicted to encode ASD-related blood-secretory proteins; six proteins were validated using an ELISA | 79 brain tissue samples from 19 ASD and 17 controls; ELISA analysis of 20 ASD, and 20 age/gender-matched controls. The average age of the patients and controls were 24 yrs (ranged from 2 to 56) and 34.6 yrs (ranged from 16 to 56) respectively | Brain tissue | Blood samples |
| Abraham et al., 2019 [18] | 146 DE proteins from BA19 between ASD and controls (p < 0.05) | 9 ASD cases (2–60 yrs) and 9 age- and gender-matched controls (1–60 yrs) | Cerebellum (CB) and Brodmann area 19 (BA19) | |
| Transcriptomic Meta-Analysis | ||||
| Reference | Dataset Used in Analysis | Cohort | Tissue | |
| Tylee et al., 2017 [19] | 90 DE genes in ASD (p < 0.05) | 626 ASD and 447 controls across seven independent studies; mean age and SD of 5 ± 3.8 yrs | Blood | Ex vivo peripheral blood samples or isolated leukocyte samples derived from peripheral blood |
| Mordaunt et al., 2019 [20] | 172 DE genes in ASD (p < 0.01) | 59 ASD, 92 non-typically developing, 120 typically developing controls | Umbilical cord blood samples from both the Markers of Autism Risk in Babies-Learning Early Signs (MARBLES) and the Early Autism Risk Longitudinal Investigation (EARLI) high-risk pregnancy cohorts | |
| Gao et al., 2020 [21] | 3624 DE genes in ASD | 96 ASD and 42 controls age range: 2–18 yrs | Peripheral blood samples (GSE18123 and GSE6575) | |
| He et al., 2019 [22] | DE genes (p < 0.05) in ASD | 485 ASD and 398 controls | Five data sets from blood lymphoblastoid cell lines (LCLs) (GSE18123, GSE25507, GSE29691, GSE37772, GSE42133) | |
| He et al., 2019 [22] | DE genes (p < 0.05) in ASD | 109 ASD and 129 controls | Brain tissue | Three data sets from postmortem brain tissue (GSE28475, GSE28521, GSE38322) |
| Forés-Martos et al., 2019 [23] | 1055 DE genes in ASD (FDR p < 0.05) | 34 ASD cases and 130 controls across three studies. Mean age 20.3 yrs | Frontal cortex tissue | |
| Rahman et al., 2020 [24] | 1567 DE genes in ASD | 15 ASD and 15 controls across two studies | Post-mortem brain tissue (GSE30573 and GSE64018) | |
| Wright et al., 2017 [25] | 1463 DE genes in ASD across all moderately expressed Ensembl genes (13,011) at marginal statistical (p < 0.05) significance | 13 ASD (3F, 10M), average age 22 yrs (4 to 67) and 39 controls (9F, 30M), average age 22 yrs (2 to 69) | Postmortem brain tissue: dorsolateral prefrontal cortex | |
| Yao et al., 2021 [17] | 364 DE genes in ASD | 79 brain tissue samples from 19 ASD and 17 controls. Average age of the patients and controls were 24 yrs (2 to 56) and 34.6 yrs (16 to 56) respectively | Brain tissue: cerebellum, frontal cortex, and temporal cortex (GSE28521) | |
| Ramaswami et al., 2020 [26] | 5200 DE genes (FDR < 0.05) | 82 ASD samples and 74 control samples from 47 ASD and 44 control brains from (Parikshak et al.); mean age and SD were 28 (+/−17) yrs | Frontal and temporal cortex tissue from the Harvard Autism Tissue Program and NIH Neuro Brain Bank | |
| DNA Methylation Analysis | ||||
| Mordaunt et al., 2020 [27] | 537 DM genes in both discovery and replication sets in males | Discovery set = 74 males (35 ASD and 39 controls) and 32 females (15 ASD and 17 controls) in the MARBLES and EARLI studies. Replication set = 38 males (21 ASD and 17 controls) and 8 females (5 ASD and 3 controls) | Blood | Umbilical cord blood samples |
| Hu et al., 2020 [28] | 181 DM genes that overlap between the discovery and validation groups MALES | 21 ASD and 21 controls, average age 8.4 yrs | LCLs | |
| Wong et al., 2019 [29] | i)Top ranked iASD-associated DM probes identified in the cross-cortex model incorporating both prefrontal cortex and temporal cortex data | 43 ASD and 38 controls, average age at death 29.0 (+/−18.9) and 48.7 (+/−8.8) yrs respectively | Brain tissue | Post-mortem brain tissue from prefrontal cortex, temporal cortex and cerebellum |
| Ramaswami et al., 2020 [26] | DM genes (promoter or gene body; FDR < 0.05) | 56 ASD samples and 41 control samples from 33 ASD and 26 control brains. Mean age and SD = 34 (+/−15) yrs | Frontal and temporal cortex tissue from the Harvard Autism Tissue Program and NIH Neuro Brain Bank | |
| Stathopoulous et al., 2020 [30] | 898 DM genes in ASD | 48 boys (32 ASD and 16 controls, 6–12 yrs) | Buccal cells | Buccal DNA |
| No. of Enrichment Signatures | |||||
|---|---|---|---|---|---|
| Proteomic | Transcriptomic | DNAm | |||
| Hallmark Canonical Pathways | Blood/Brain | Hallmark Canonical Pathways | Blood/Brain | Hallmark Canonical Pathways | Blood/Brain/Buccal |
| COAGULATION | 3/2 | INTERFERON GAMMA RESPONSE | 3/6 | OXIDATIVE PHOSPHORYLATION | 0/2/1 |
| COMPLEMENT | 3/0 | COMPLEMENT | 4/4 | P53 PATHWAY | 1/1/1 |
| OXIDATIVE PHOSPHORYLATION | 1/2 | MTORC1 SIGNALING | 3/3 | MITOTIC SPINDLE | 1/1/1 |
| MTORC1 SIGNALING | 1/2 | P53 PATHWAY | 2/5 | MTORC1 SIGNALING | 0/1/1 |
| XENOBIOTIC METABOLISM | 1/2 | ALLOGRAFT REJECTION | 3/3 | XENOBIOTIC METABOLISM | 1/0/1 |
| ADIPOGENESIS | 1/2 | INTERFERON ALPHA RESPONSE | 4/4 | UV RESPONSE UP | 1/1/0 |
| UNFOLDED PROTEIN RESPONSE | 1/1 | TNFA SIGNALING VIA NFKB | 4/3 | UNFOLDED PROTEIN RESPONSE | 0/2/0 |
| MYOGENESIS | 1/1 | HYPOXIA | 2/3 | ESTROGEN RESPONSE EARLY | 1/1/0 |
| FATTY ACID METABOLISM | 1/1 | INFLAMMATORY RESPONSE | 3/4 | E2F TARGETS | 0/1/1 |
| MYC TARGETS V1 | 1/1 | APOPTOSIS | 3/4 | DNA REPAIR | 0/1/1 |
| ANGIOGENESIS | 2/0 | OXIDATIVE PHOSPHORYLATION | 1/3 | PEROXISOME | 0/2/0 |
| EPITHELIAL MESENCHYMAL TRANSITION | 1/4 | ||||
| KRAS SIGNALING UP | 0/4 | ||||
| ClinVar Disease Pathway | Disease Phenotype | p Value |
|---|---|---|
| Leigh Syndrome | Neurological disease | 0.0027 |
| Familial Partial Lipodystrophy | Metabolic disease | 0.0299 |
| Pyruvate Dehydrogenase Complex Deficiency | 0.0299 | |
| Mitochondrial DNA Deletion Syndrome | 0.0474 | |
| Autoimmune Lymphoproliferative Syndrome | Autoimmune disease | 0.0358 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahony, C.; O’Ryan, C. Convergent Canonical Pathways in Autism Spectrum Disorder from Proteomic, Transcriptomic and DNA Methylation Data. Int. J. Mol. Sci. 2021, 22, 10757. https://doi.org/10.3390/ijms221910757
Mahony C, O’Ryan C. Convergent Canonical Pathways in Autism Spectrum Disorder from Proteomic, Transcriptomic and DNA Methylation Data. International Journal of Molecular Sciences. 2021; 22(19):10757. https://doi.org/10.3390/ijms221910757
Chicago/Turabian StyleMahony, Caitlyn, and Colleen O’Ryan. 2021. "Convergent Canonical Pathways in Autism Spectrum Disorder from Proteomic, Transcriptomic and DNA Methylation Data" International Journal of Molecular Sciences 22, no. 19: 10757. https://doi.org/10.3390/ijms221910757
APA StyleMahony, C., & O’Ryan, C. (2021). Convergent Canonical Pathways in Autism Spectrum Disorder from Proteomic, Transcriptomic and DNA Methylation Data. International Journal of Molecular Sciences, 22(19), 10757. https://doi.org/10.3390/ijms221910757