
Skin Barrier Dysregulation in Psoriasis



Abstract
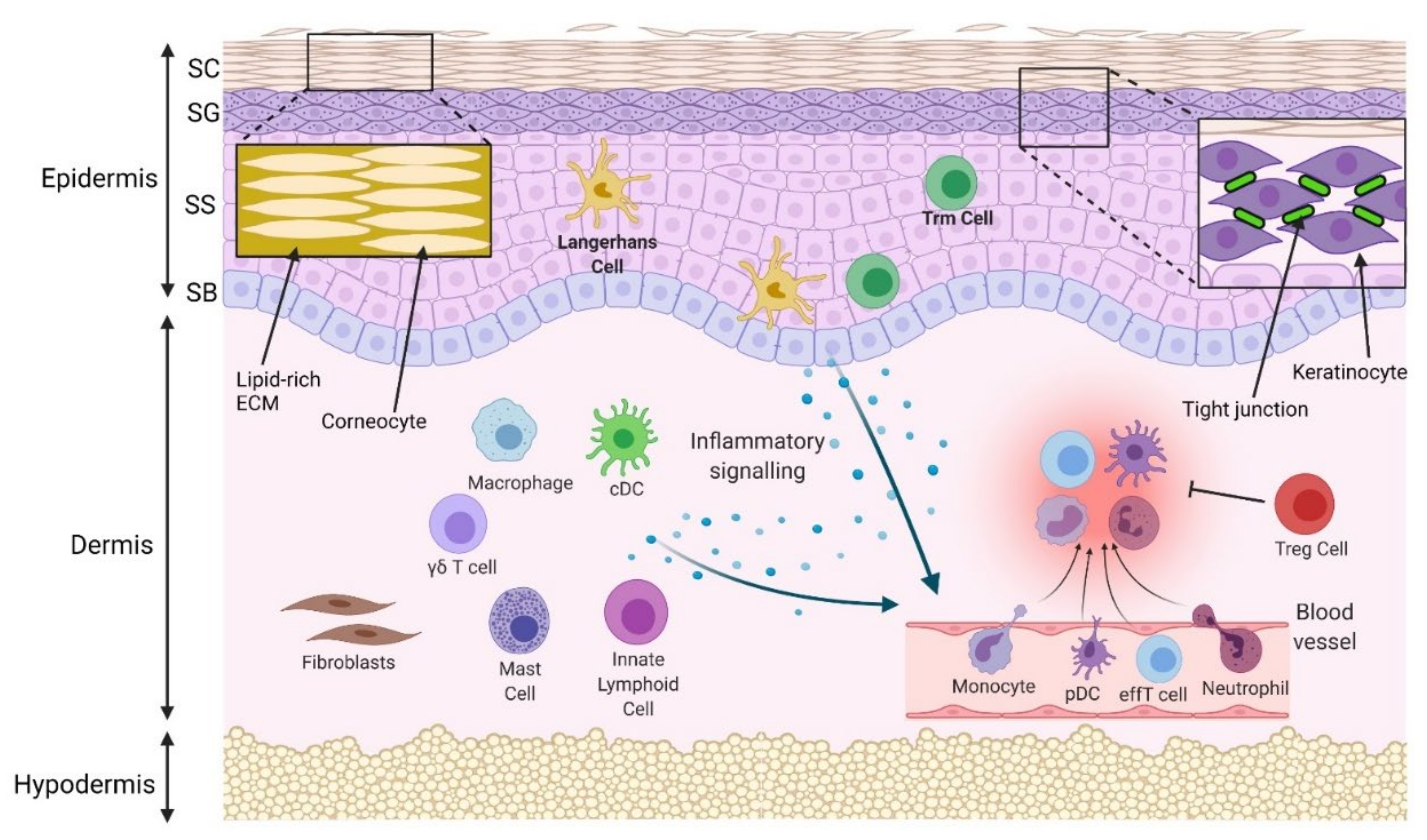
:1. Introduction
2. Psoriasis
3. Barrier Aberration in Psoriasis
3.1. Physical Barrier Disruption
3.1.1. Disruption of Keratinocyte Proliferation and Differentiation
3.1.2. Disruption of Intercellular Connections
3.1.3. Dysregulation of the Lipid-Rich ECM of the Stratum Corneum
3.1.4. Other Contributing Factors
3.2. Immune Barrier Dysregulation
3.2.1. Keratinocytes Promote Inflammation
3.2.2. Inflammatory Immune Cells
Altered Innate Immune Cell Function
Excessive Activation of Adaptive Immune Response
3.2.3. Immunosuppressing Cells
4. Contributors to Barrier Dysregulation in Psoriasis
4.1. Genetic Aberrations
4.1.1. Susceptible Genes

{kind=link}
{kind=link}
| Biological Pathway | Representative Susceptibility Genes | Possible Role/s in Psoriasis | Target Drugs |
|---|---|---|---|
| HLA mediated antigen presentation | HLA-C*06:02, HLA-A, HLA-B and HLA-DQ [176,177]. | Facilitates presentation of autoantigens [171,172]. | No targeted drug currently. HLA-C*06:02+ individuals respond better to the anti-IL-12/IL-23 biologic ustekinumab [178]. |
| NF-kB signalling | FASLG, IKBKE, NFKBIA, REL, SLC44A2, TNFAIP3, TNIP1, TRAF3IP2 [179] and CARD14 [180]. | Elevates innate immune responses, activates T helper cells and reduces keratinocyte death [181]. | Fumarate and apremilast inhibit NF-kB activation [182]. |
| Th17 cell activation | IL23R, IL23A, IL12B [183]. | Compels keratinocyte proliferation and promote psoriatic inflammation [132]. | Biologics targeting IL-23 (tildrakizumab, guselkumab, risankizumab, and ustekinumab), novel RORγ inhibitors and JAK inhibitors [184,185]. |
| Skin structure proteins | The LCE3 gene cluster, KLF4, COL6A5 and COL8A1 [173,175,186]. | LCE3 and KLF4 genes facilitate cornified envelope production, and their variants contribute to barrier dysfunction [187]. COL6A5 regulates cell adhesion and proliferation and COL8A1 mediates vascularisation [186]. | Topical calcitriol may operate by upregulating LCE genes [188]. |
| Keratinocyte proliferation and differentiation | Keratins 6, 10, 14, 16 and 17 [26,189]. PDCD5, PTEN and CHUK [179,190]. | Keratin 10, 14, 16 and 17 variants, reduced keratin 1 and 10 levels and elevated keratin 6, 16 and 17 levels associate with keratinocyte hyperproliferation and aberrant differentiation [23,26,189]. PDCD5 is hypermethylated, reducing its expression and capacity to facilitate apoptosis [190]. IKKa (the protein CHUK encodes) and PTEN typically regulate differentiation and proliferation, respectively [191,192]. | Topical calcineurin inhibitors and vitamin D receptor agonists, such as calcitriol and retinoids, prevent atypical keratinocyte proliferation and differentiation [193]. |
| Type 1 IFN signalling | DDX58, IFIH1 and RNF114 variants [180,194]. | Sensitises keratinocytes to IL-22, induces the maturation of cDCs and facilitates the differentiation of naïve CD4+ cells [6,90]. | UVB phototherapy downregulates IFN signalling pathways [195]. Novel IL-36 inhibitors may modulate IFN responses [196]. |
4.1.2. Epigenetic Modifications
4.2. Environmental Factors
4.2.1. Imbalances in the Gut/Skin Microbiome
4.2.2. Infections
4.2.3. Skin Damage
5. Psoriasis Current Treatment and Future Directions
5.1. Current Treatments
5.2. Future Directions of Treatment
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wong, R.; Geyer, S.; Weninger, W.; Guimberteau, J.-C.; Wong, J.K. The Dynamic Anatomy and Patterning of Skin. Exp. Dermatol. 2016, 25, 92–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madison, K.C. Barrier Function of the Skin: “La Raison d’Être” of the Epidermis. J. Investig. Dermatol. 2003, 121, 231–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bäsler, K.; Bergmann, S.; Heisig, M.; Naegel, A.; Zorn-Kruppa, M.; Brandner, J.M. The Role of Tight Junctions in Skin Barrier Function and Dermal Absorption. J. Control. Release 2016, 242, 105–118. [Google Scholar] [CrossRef]
- Bäsler, K.; Brandner, J.M. Tight Junctions in Skin Inflammation. Pflügers Archiv. 2017, 469, 3–14. [Google Scholar] [CrossRef]
- West, H.C.; Bennett, C.L. Redefining the Role of Langerhans Cells as Immune Regulators within the Skin. Front. Immunol. 2018, 8, 1941. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.; Bai, Y. Dendritic Cells: The driver of Psoriasis. J. Dermatol. 2020, 47, 104–113. [Google Scholar] [CrossRef]
- Chambers, E.S.; Vukmanovic-Stejic, M. Skin Barrier Immunity and Ageing. Immunology 2020, 160, 116–125. [Google Scholar] [CrossRef] [Green Version]
- Kabashima, K.; Honda, T.; Ginhoux, F.; Egawa, G. The Immunological Anatomy of the skin. Nat. Rev. Immunol. 2019, 19, 19–30. [Google Scholar] [CrossRef]
- Ali, N.; Rosenblum, M.D. Regulatory T Cells in Skin. Immunology 2017, 152, 372–381. [Google Scholar] [CrossRef]
- Greb, J.E.; Goldminz, A.M.; Elder, J.T.; Lebwohl, M.G.; Gladman, D.D.; Wu, J.J.; Mehta, N.N.; Finlay, A.Y.; Gottlieb, A.B. Psoriasis. Nat. Rev. Dis. Primers 2016, 2, 16082. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.W.; Read, C. Pathophysiology, Clinical Presentation, and Treatment of Psoriasis: A Review. JAMA 2020, 323, 1945–1960. [Google Scholar] [CrossRef]
- Obradors, M.; Blanch, C.; Comellas, M.; Figueras, M.; Lizan, L. Health-Related Quality of Life in Patients with Psoriasis: A Systematic Review of the European Literature. Qual. Life Res. 2016, 25, 2739–2754. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, J.; Grewal, S.; Langan, S.M.; Mehta, N.N.; Ogdie, A.; Van Voorhees, A.S.; Gelfand, J.M. Psoriasis and Comorbid Diseases: Implications for Management. J. Am. Acad. Dermatol. 2017, 76, 393–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- AlQassimi, S.; AlBrashdi, S.; Galadari, H.; Hashim, M.J. Global Burden of Psoriasis-Comparison of Regional and Global Epidemiology, 1990 to 2017. Int. J. Dermatol. 2020, 59, 566–571. [Google Scholar] [CrossRef] [PubMed]
- Parisi, R.; Iskandar, I.Y.K.; Kontopantelis, E.; Augustin, M.; Griffiths, C.E.M.; Ashcroft, D.M. National, Regional, and Worldwide Epidemiology of Psoriasis: Systematic Analysis and Modelling Study. BMJ 2020, 369, m1590. [Google Scholar] [CrossRef]
- Michalek, I.M.; Loring, B.; John, S.M. A Systematic Review of Worldwide Epidemiology of Psoriasis. J. Eur. Acad. Dermatol. Venereol. 2017, 31, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Raychaudhuri, S.K.; Maverakis, E.; Raychaudhuri, S.P. Diagnosis and Classification of Psoriasis. Autoimmun. Rev. 2014, 13, 490–495. [Google Scholar] [CrossRef]
- Ogawa, E.; Okuyama, R.; Seki, T.; Kobayashi, A.; Oiso, N.; Muto, M.; Nakagawa, H.; Kawada, A. Epidemiological Survey of Patients with Psoriasis in Matsumoto City, Nagano Prefecture, Japan. J. Dermatol. 2018, 45, 314–317. [Google Scholar] [CrossRef] [Green Version]
- Boehncke, W.-H.; Schön, M.P. Psoriasis. Lancet 2015, 386, 983–994. [Google Scholar] [CrossRef]
- Zhang, H.; Hou, W.; Henrot, L.; Schnebert, S.; Dumas, M.; Heusèle, C.; Yang, J. Modelling Epidermis homoeostasis and Psoriasis pathogenesis. J. R. Soc. Interface 2015, 12, 20141071. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.A.; Ryu, Y.W.; Kwon, J.I.; Choe, M.S.; Jung, J.W.; Cho, J.W. Differential Expression of Cyclin D1, Ki-67, pRb, and p53 in Psoriatic Skin Lesions and Normal Skin. Mol. Med. Report. 2018, 17, 735–742. [Google Scholar] [CrossRef] [Green Version]
- Hwang, Y.-J.; Na, J.-I.; Byun, S.-Y.; Kwon, S.-H.; Yang, S.-H.; Lee, H.-S.; Choi, H.-R.; Cho, S.; Youn, S.W.; Park, K.-C. Histone Deacetylase 1 and Sirtuin 1 Expression in Psoriatic Skin: A Comparison between Guttate and Plaque Psoriasis. Life 2020, 10, 157. [Google Scholar] [CrossRef]
- Zhang, X.; Yin, M.; Zhang, L.-j. Keratin 6, 16 and 17-Critical Barrier Alarmin Molecules in Skin Wounds and Psoriasis. Cells 2019, 8, 807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ota, T.; Takekoshi, S.; Takagi, T.; Kitatani, K.; Toriumi, K.; Kojima, T.; Kato, M.; Ikoma, N.; Mabuchi, T.; Ozawa, A. Notch Signaling May be Involved in the Abnormal Differentiation of Epidermal Keratinocytes in Psoriasis. Acta Histochem. Cytochem. 2014, 47, 175–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, M.; Wang, G. Keratin 17 as a Therapeutic Target for the Treatment of Psoriasis. J. Dermatol. Sci. 2012, 67, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Elango, T.; Sun, J.; Zhu, C.; Zhou, F.; Zhang, Y.; Sun, L.; Yang, S.; Zhang, X. Mutational Analysis of Epidermal and Hyperproliferative Type I Keratins in Mild and Moderate Psoriasis vulgaris Patients: A Possible Role in the Pathogenesis of Psoriasis along with Disease Severity. Hum. Genom. 2018, 12, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akhlaghi, M.; Karrabi, M.; Atabti, H.; Raoofi, A.; Mousavi Khaneghah, A. Investigation of the Role of IL18, IL-1β and NLRP3 Inflammasome in Reducing Expression of FLG-2 Protein in Psoriasis vulgaris Skin Lesions. Biotech. Histochem. 2021, 1–7. [Google Scholar] [CrossRef]
- Kim, B.E.; Howell, M.D.; Guttman, E.; Gilleaudeau, P.M.; Cardinale, I.R.; Boguniewicz, M.; Krueger, J.G.; Leung, D.Y.M. TNF-a; Downregulates Filaggrin and Loricrin through c-Jun N-terminal Kinase: Role for TNF-a; Antagonists to Improve Skin Barrier. J. Investig. Dermatol. 2011, 131, 1272–1279. [Google Scholar] [CrossRef] [Green Version]
- Lowes, M.A.; Bowcock, A.M.; Krueger, J.G. Pathogenesis and Therapy of Psoriasis. Nature 2007, 445, 866–873. [Google Scholar] [CrossRef] [PubMed]
- Blauvelt, A.; Chiricozzi, A. The Immunologic Role of IL-17 in Psoriasis and Psoriatic Arthritis Pathogenesis. Clin. Rev. Allergy Immunol. 2018, 55, 379–390. [Google Scholar] [CrossRef] [Green Version]
- Furue, M.; Furue, K.; Tsuji, G.; Nakahara, T. Interleukin-17A and Keratinocytes in Psoriasis. Int. J. Mol. Sci. 2020, 21, 1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Cai, G.; Liu, C.; Zhao, J.; Gu, C.; Wu, L.; Hamilton, T.A.; Zhang, C.-J.; Ko, J.; Zhu, L.; et al. IL-17R-EGFR Axis Links Wound Healing to Tumorigenesis in Lrig1(+) Stem Cells. J. Exp. Med. 2019, 216, 195–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Liu, X.; Liu, N.; Chen, H. Prediction of Crucial Epigenetically-Associated, Differentially Expressed Genes by Integrated Bioinformatics Analysis and the Identification of S100A9 as a Novel Biomarker in Psoriasis. Int. J. Mol. Med. 2020, 45, 93–102. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, L.; Ma, W.; Yan, J.; Zhong, H. Evaluation of the Effects of IL-22 on the Proliferation and Differentiation of Keratinocytes In Vitro. Mol. Med. Rep. 2020, 22, 2715–2722. [Google Scholar] [CrossRef]
- Ekman, A.-K.; Bivik Eding, C.; Rundquist, I.; Enerbäck, C. IL-17 and IL-22 Promote Keratinocyte Stemness in the Germinative Compartment in Psoriasis. J. Investig. Dermatol. 2019, 139, 1564–1573. [Google Scholar] [CrossRef] [Green Version]
- Piro, M.C.; Ventura, A.; Smirnov, A.; Saggini, A.; Lena, A.M.; Mauriello, A.; Bianchi, L.; Melino, G.; Candi, E. Transglutaminase 3 Reduces the Severity of Psoriasis in Imiquimod-Treated Mouse Skin. Int. J. Mol. Sci. 2020, 21, 1566. [Google Scholar] [CrossRef] [Green Version]
- Matsuki, M.; Yamashita, F.; Ishida-Yamamoto, A.; Yamada, K.; Kinoshita, C.; Fushiki, S.; Ueda, E.; Morishima, Y.; Tabata, K.; Yasuno, H.; et al. Defective Stratum corneum and Early Neonatal Death in Mice Lacking the Gene for Transglutaminase 1 (Keratinocyte Transglutaminase). Proc. Natl. Acad. Sci. USA 1998, 95, 1044–1049. [Google Scholar] [CrossRef] [Green Version]
- Peltonen, S.; Riehokainen, J.; Pummi, K.; Peltonen, J. Tight Junction Components Occludin, ZO-1, and Claudin-1, -4 and -5 in Active and Healing Psoriasis. Br. J. Dermatol. 2007, 156, 466–472. [Google Scholar] [CrossRef]
- Kirschner, N.; Poetzl, C.; von den Driesch, P.; Wladykowski, E.; Moll, I.; Behne, M.J.; Brandner, J.M. Alteration of Tight Junction Proteins is an Early Event in Psoriasis: Putative Involvement of Proinflammatory Cytokines. Am. J. Pathol. 2009, 175, 1095–1106. [Google Scholar] [CrossRef] [Green Version]
- Visconti, B.; Paolino, G.; Carotti, S.; Pendolino, A.L.; Morini, S.; Richetta, A.G.; Calvieri, S. Immunohistochemical Expression of VDR is Associated with Reduced Integrity of Tight Junction Complex in Psoriatic Skin. J. Eur. Acad. Dermatol. Venereol. 2015, 29, 2038–2042. [Google Scholar] [CrossRef] [PubMed]
- Montero-Vilchez, T.; Segura-Fernández-Nogueras, M.-V.; Pérez-Rodríguez, I.; Soler-Gongora, M.; Martinez-Lopez, A.; Fernández-González, A.; Molina-Leyva, A.; Arias-Santiago, S. Skin Barrier Function in Psoriasis and Atopic Dermatitis: Transepidermal Water Loss and Temperature as Useful Tools to Assess Disease Severity. J. Clin. Med. 2021, 10, 359. [Google Scholar] [CrossRef] [PubMed]
- Watson, R.E.B.; Poddar, R.; Walker, J.M.; McGuill, I.; Hoare, L.M.; Griffiths, C.E.M.; O’Neill, C.A. Altered Claudin Expression is a Feature of Chronic Plaque Psoriasis. J. Pathol. 2007, 212, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Gutowska-Owsiak, D.; Schaupp, A.L.; Salimi, M.; Selvakumar, T.A.; McPherson, T.; Taylor, S.; Ogg, G.S. IL-17 Downregulates Filaggrin and Affects Keratinocyte Expression of Genes Associated with Cellular Adhesion. Exp. Dermatol. 2012, 21, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Chung, E.; Cook, P.W.; Parkos, C.A.; Park, Y.-K.; Pittelkow, M.R.; Coffey, R.J. Amphiregulin Causes Functional Downregulation of Adherens Junctions in Psoriasis. J. Investig. Dermatol. 2005, 124, 1134–1140. [Google Scholar] [CrossRef] [Green Version]
- Allen, M.; Ishida-Yamamoto, A.; McGrath, J.; Davison, S.; Iizuka, H.; Simon, M.; Guerrin, M.; Hayday, A.; Vaughan, R.; Serre, G.; et al. Corneodesmosin Expression in Psoriasis Vulgaris Differs from Normal Skin and Other Inflammatory Skin Disorders. Lab. Investig. 2001, 81, 969–976. [Google Scholar] [CrossRef] [Green Version]
- Simon, M.; Tazi-Ahnini, R.; Jonca, N.; Caubet, C.; Cork, M.J.; Serre, G. Alterations in the Desquamation-Related Proteolytic Cleavage of Corneodesmosin and other Corneodesmosomal Proteins in Psoriatic Lesional Epidermis. Br. J. Dermatol. 2008, 159, 77–85. [Google Scholar] [CrossRef]
- Labarthe, M.-P.; Saurat, J.-H.; Salomon, D.; Bosco, D.; Meda, P. Upregulation of Connexin 26 between Keratinocytes of Psoriatic Lesions. J. Investig. Dermatol. 1998, 111, 72–76. [Google Scholar] [CrossRef] [Green Version]
- Stylianaki, E.-A.; Karpouzis, A.; Tripsianis, G.; Veletza, S. Assessment of Gap Junction Protein Beta-2 rs3751385 Gene Polymorphism in Psoriasis Vulgaris. J. Clin. Med. Res. 2019, 11, 642–650. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.-D.; Cheng, H.; Wang, Z.-X.; Zhang, A.-P.; Wang, P.-G.; Xu, J.-H.; Zhu, Q.-X.; Zhou, H.-S.; Ellinghaus, E.; Zhang, F.-R.; et al. Association Analyses Identify Six New Psoriasis Susceptibility Loci in the Chinese Population. Nat. Genet. 2010, 42, 1005–1009. [Google Scholar] [CrossRef] [Green Version]
- O’Shaughnessy, E.M.; Duffy, W.; Garcia-Vega, L.; Hussey, K.; Burden, A.D.; Zamiri, M.; Martin, P.E. Dysregulation of Connexin Expression Plays a Pivotal Role in Psoriasis. Int. J. Mol. Sci. 2021, 22, 6060. [Google Scholar] [CrossRef]
- Yokose, U.; Ishikawa, J.; Morokuma, Y.; Naoe, A.; Inoue, Y.; Yasuda, Y.; Tsujimura, H.; Fujimura, T.; Murase, T.; Hatamochi, A. The Ceramide [NP]/[NS] Ratio in the Stratum corneum is a Potential Marker for Skin Properties and Epidermal Differentiation. BMC Dermatol. 2020, 20, 6. [Google Scholar] [CrossRef] [PubMed]
- Łuczaj, W.; Wroński, A.; Domingues, P.; Domingues, M.R.; Skrzydlewska, E. Lipidomic Analysis Reveals Specific Differences between Fibroblast and Keratinocyte Ceramide Profile of Patients with Psoriasis Vulgaris. Molecules 2020, 25, 630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakajima, K.; Terao, M.; Takaishi, M.; Kataoka, S.; Goto-Inoue, N.; Setou, M.; Horie, K.; Sakamoto, F.; Ito, M.; Azukizawa, H.; et al. Barrier Abnormality Due to Ceramide Deficiency Leads to Psoriasiform Inflammation in a Mouse Model. J. Investig. Dermatol. 2013, 133, 2555–2565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietrzak, A.; Michalak-Stoma, A.; Chodorowska, G.; Szepietowski, J.C. Lipid Disturbances in Psoriasis: An Update. Mediators Inflamm. 2010, 2010, 535612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varshney, P.; Narasimhan, A.; Mittal, S.; Malik, G.; Sardana, K.; Saini, N. Transcriptome Profiling Unveils the Role of Cholesterol in IL-17A Signaling in Psoriasis. Sci. Rep. 2016, 6, 19295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knox, S.; O’Boyle, N.M. Skin Lipids in Health and Disease: A Review. Chem. Phys. Lipids 2021, 236, 105055. [Google Scholar] [CrossRef]
- Farwanah, H.; Raith, K.; Neubert, R.H.H.; Wohlrab, J. Ceramide Profiles of the Uninvolved Skin in Atopic Dermatitis and Psoriasis are Comparable to Those of Healthy Skin. Arch. Dermatol. Res. 2005, 296, 514–521. [Google Scholar] [CrossRef]
- Patel, R.; Kevin Heard, L.; Chen, X.; Bollag, W.B. Aquaporins in the Skin. In Aquaporins; Yang, B., Ed.; Springer: Dordrecht, The Netherlands, 2017; pp. 173–191. [Google Scholar]
- Voss, K.E.; Bollag, R.J.; Fussell, N.; By, C.; Sheehan, D.J.; Bollag, W.B. Abnormal Aquaporin-3 Protein Expression in Hyperproliferative Skin Disorders. Arch. Dermatol. Res. 2011, 303, 591–600. [Google Scholar] [CrossRef] [Green Version]
- Ramadan, W.; Gheida, S.; El-Ashmawy, A.; Shareef, M. Aquaporin-3 Expression in Common Hyperproliferative Skin Disorders: An Immunohistochemical Study. J. Egypt. Women’s Dermatol. Soc. 2017, 14, 128–136. [Google Scholar] [CrossRef]
- Lee, Y.; Je, Y.-J.; Lee, S.-S.; Li, Z.J.; Choi, D.-K.; Kwon, Y.-B.; Sohn, K.-C.; Im, M.; Seo, Y.J.; Lee, J.H. Changes in Transepidermal Water Loss and Skin Hydration According to Expression of Aquaporin-3 in Psoriasis. Ann. Dermatol. 2012, 24, 168–174. [Google Scholar] [CrossRef]
- Krueger, G.G.; Jorgensen, C.M. Experimental Models for Psoriasis. J. Investig. Dermatol. 1990, 95, 56–58. [Google Scholar] [CrossRef] [Green Version]
- Barygina, V.; Becatti, M.; Prignano, F.; Lotti, T.; Taddei, N.; Fiorillo, C. Fibroblasts to Keratinocytes Redox Signaling: The Possible Role of ROS in Psoriatic Plaque Formation. Antioxidants 2019, 8, 566. [Google Scholar] [CrossRef] [Green Version]
- Iwata, H.; Haga, N.; Ujiie, H. Possible Role of Epiregulin from Dermal Fibroblasts in the Keratinocyte Hyperproliferation of Psoriasis. J. Dermatol. 2021, 48, 1433–1438. [Google Scholar] [CrossRef]
- Gęgotek, A.; Domingues, P.; Wroński, A.; Skrzydlewska, E. Changes in Proteome of Fibroblasts Isolated from Psoriatic Skin Lesions. Int. J. Mol. Sci. 2020, 21, 5363. [Google Scholar] [CrossRef]
- Ten Bergen, L.L.; Petrovic, A.; Aarebrot, A.K.; Appel, S. Current Knowledge on Autoantigens and Autoantibodies in Psoriasis. Scand. J. Immunol. 2020, 92, e12945. [Google Scholar] [CrossRef]
- Lou, F.; Sun, Y.; Xu, Z.; Niu, L.; Wang, Z.; Deng, S.; Liu, Z.; Zhou, H.; Bai, J.; Yin, Q.; et al. Excessive Polyamine Generation in Keratinocytes Promotes Self-RNA Sensing by Dendritic Cells in Psoriasis. Immunity 2020, 53, 204–216.e210. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Samperio, P. The Human Cathelicidin hCAP18/LL-37: A Multifunctional Peptide Involved in Mycobacterial Infections. Peptides 2010, 31, 1791–1798. [Google Scholar] [CrossRef] [PubMed]
- Lande, R.; Gregorio, J.; Facchinetti, V.; Chatterjee, B.; Wang, Y.-H.; Homey, B.; Cao, W.; Wang, Y.-H.; Su, B.; Nestle, F.O.; et al. Plasmacytoid Dendritic Cells Sense Self-DNA Coupled with Antimicrobial Peptide. Nature 2007, 449, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, D.; Chamilos, G.; Lande, R.; Gregorio, J.; Meller, S.; Facchinetti, V.; Homey, B.; Barrat, F.J.; Zal, T.; Gilliet, M. Self-RNA-Antimicrobial Peptide Complexes Activate Human Dendritic Cells through TLR7 and TLR8. J. Exp. Med. 2009, 206, 1983–1994. [Google Scholar] [CrossRef] [PubMed]
- Morizane, S.; Yamasaki, K.; Mühleisen, B.; Kotol, P.F.; Murakami, M.; Aoyama, Y.; Iwatsuki, K.; Hata, T.; Gallo, R.L. Cathelicidin Antimicrobial Peptide LL-37 in Psoriasis Enables Keratinocyte Reactivity against TLR9 Ligands. J. Investig. Dermatol. 2012, 132, 135–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lande, R.; Botti, E.; Jandus, C.; Dojcinovic, D.; Fanelli, G.; Conrad, C.; Chamilos, G.; Feldmeyer, L.; Marinari, B.; Chon, S.; et al. The Antimicrobial Peptide LL37 is a T-Cell Autoantigen in Psoriasis. Nat. Commun. 2014, 5, 5621. [Google Scholar] [CrossRef]
- Bader, H.L.; Wang, L.W.; Ho, J.C.; Tran, T.; Holden, P.; Fitzgerald, J.; Atit, R.P.; Reinhardt, D.P.; Apte, S.S. A Disintegrin-Like and Metalloprotease Domain Containing Thrombospondin Type 1 Motif-Like 5 (ADAMTSL5) is a Novel Fibrillin-1-, Fibrillin-2-, and Heparin-Binding Member of the ADAMTS Superfamily Containing a Netrin-Like Module. Matrix Biol. 2012, 31, 398–411. [Google Scholar] [CrossRef] [Green Version]
- Arakawa, A.; Siewert, K.; Stöhr, J.; Besgen, P.; Kim, S.-M.; Rühl, G.; Nickel, J.; Vollmer, S.; Thomas, P.; Krebs, S.; et al. Melanocyte Antigen Triggers Autoimmunity in Human Psoriasis. J. Exp. Med. 2015, 212, 2203–2212. [Google Scholar] [CrossRef]
- Chiba, H.; Michibata, H.; Wakimoto, K.; Seishima, M.; Kawasaki, S.; Okubo, K.; Mitsui, H.; Torii, H.; Imai, Y. Cloning of a Gene for a Novel Epithelium-Specific Cytosolic Phospholipase A2, cPLA2δ, Induced in Psoriatic Skin. J. Biol. Chem. 2004, 279, 12890–12897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, K.L.; Jarrett, R.; Subramaniam, S.; Salimi, M.; Gutowska-Owsiak, D.; Chen, Y.-L.; Hardman, C.; Xue, L.; Cerundolo, V.; Ogg, G. Psoriatic T Cells Recognize Neolipid Antigens Generated by Mast Cell Phospholipase Delivered by Exosomes and Presented by CD1a. J. Exp. Med. 2016, 213, 2399–2412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bragulla, H.H.; Homberger, D.G. Structure and Functions of Keratin Proteins in Simple, Stratified, Keratinized and Cornified Epithelia. J. Anat. 2009, 214, 516–559. [Google Scholar] [CrossRef] [PubMed]
- Johnston, A.; Gudjonsson, J.E.; Sigmundsdottir, H.; Love, T.J.; Valdimarsson, H. Peripheral Blood T Cell Responses to Keratin Peptides that Share Sequences with Streptococcal M Proteins are Largely Restricted to Skin-Homing CD8(+) T Cells. Clin. Exp. Immunol. 2004, 138, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Besgen, P.; Trommler, P.; Vollmer, S.; Prinz, J.C. Ezrin, Maspin, Peroxiredoxin 2, and Heat Shock Protein 27: Potential Targets of a Streptococcal-Induced Autoimmune Response in Psoriasis. J. Immunol. 2010, 184, 5392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jean-Philippe, J.; Paz, S.; Caputi, M. hnRNP A1: The Swiss Army Knife of Gene Expression. Int. J. Mol. Sci. 2013, 14, 18999–19024. [Google Scholar] [CrossRef] [Green Version]
- Guarneri, C.; Aguennouz, M.; Guarneri, F.; Polito, F.; Benvenga, S.; Cannavò, S.P. Autoimmunity to Heterogeneous Nuclear Ribonucleoprotein A1 in Psoriatic Patients and Correlation with Disease Severity. J. Dtsch. Dermatol. Ges. 2018, 16, 1103–1107. [Google Scholar] [CrossRef]
- Niyonsaba, F.; Ogawa, H. Protective Roles of the Skin against Infection: Implication of Naturally Occurring Human Antimicrobial Agents β-Defensins, Cathelicidin LL-37 and Lysozyme. J. Dermatol. Sci. 2005, 40, 157–168. [Google Scholar] [CrossRef]
- Lande, R.; Chamilos, G.; Ganguly, D.; Demaria, O.; Frasca, L.; Durr, S.; Conrad, C.; Schröder, J.; Gilliet, M. Cationic antimicrobial Peptides in Psoriatic Skin Cooperate to Break Innate Tolerance to Self-DNA. Eur. J. Immunol. 2015, 45, 203–213. [Google Scholar] [CrossRef]
- Takahashi, T.; Yamasaki, K. Psoriasis and Antimicrobial Peptides. Int. J. Mol. Sci. 2020, 21, 6791. [Google Scholar] [CrossRef]
- Albanesi, C.; Madonna, S.; Gisondi, P.; Girolomoni, G. The Interplay between Keratinocytes and Immune Cells in the Pathogenesis of Psoriasis. Front. Immunol. 2018, 9, 1549. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.; Xue, F.; Quan, C.; Qu, M.; Liu, N.; Zhang, Y.; Fleming, C.; Hu, X.; Zhang, H.-G.; Weichselbaum, R.; et al. A Critical Role of the IL-1β-IL-1R Signaling Pathway in Skin Inflammation and Psoriasis Pathogenesis. J. Investig. Dermatol. 2019, 139, 146–156. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Fang, H.; Shao, S.; Dang, E.; Zhang, J.; Qiao, P.; Yang, A.; Wang, G. Keratinocyte Exosomes Activate Neutrophils and Enhance Skin Inflammation in Psoriasis. FASEB J. 2019, 33, 13241–13253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orlik, C.; Deibel, D.; Küblbeck, J.; Balta, E.; Ganskih, S.; Habicht, J.; Niesler, B.; Schröder-Braunstein, J.; Schäkel, K.; Wabnitz, G.; et al. Keratinocytes Costimulate Naive Human T Cells via CD2: A Potential Target to Prevent the Development of Proinflammatory Th1 Cells in the Skin. Cell. Mol. Immunol. 2020, 17, 380–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albanesi, C.; Scarponi, C.; Pallotta, S.; Daniele, R.; Bosisio, D.; Madonna, S.; Fortugno, P.; Gonzalvo-Feo, S.; Franssen, J.-D.; Parmentier, M.; et al. Chemerin Expression Marks Early Psoriatic Skin Lesions and Correlates with Plasmacytoid Dendritic Cell Recruitment. J. Exp. Med. 2009, 206, 249–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tohyama, M.; Yang, L.; Hanakawa, Y.; Dai, X.; Shirakata, Y.; Sayama, K. IFN-α Enhances IL-22 Receptor Expression in Keratinocytes: A Possible Role in the Development of Psoriasis. J. Investig. Dermatol. 2012, 132, 1933–1935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Keeffe, M.; Mok, W.H.; Radford, K.J. Human Dendritic Cell Subsets and Function in Health and Disease. Cell. Mol. Life Sci. 2015, 72, 4309–4325. [Google Scholar] [CrossRef]
- Kim, T.-G.; Jee, H.; Fuentes-Duculan, J.; Wu, W.H.; Byamba, D.; Kim, D.-S.; Kim, D.-Y.; Lew, D.-H.; Yang, W.-I.; Krueger, J.G.; et al. Dermal Clusters of Mature Dendritic Cells and T Cells Are Associated with the CCL20/CCR6 Chemokine System in Chronic Psoriasis. J. Investig. Dermatol. 2014, 134, 1462–1465. [Google Scholar] [CrossRef] [Green Version]
- Chiricozzi, A.; Romanelli, P.; Volpe, E.; Borsellino, G.; Romanelli, M. Scanning the Immunopathogenesis of Psoriasis. Int. J. Mol. Sci. 2018, 19, 179. [Google Scholar] [CrossRef] [Green Version]
- Terhorst, D.; Chelbi, R.; Wohn, C.; Malosse, C.; Tamoutounour, S.; Jorquera, A.; Bajenoff, M.; Dalod, M.; Malissen, B.; Henri, S. Dynamics and Transcriptomics of Skin Dendritic Cells and Macrophages in an Imiquimod-Induced, Biphasic Mouse Model of Psoriasis. J. Immunol. 2015, 195, 4953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glitzner, E.; Korosec, A.; Brunner, P.M.; Drobits, B.; Amberg, N.; Schonthaler, H.B.; Kopp, T.; Wagner, E.F.; Stingl, G.; Holcmann, M.; et al. Specific Roles for Dendritic Cell Subsets during Initiation and Progression of Psoriasis. EMBO Mol. Med. 2014, 6, 1312–1327. [Google Scholar] [CrossRef]
- Lee, M.; Kim, S.H.; Kim, T.-G.; Park, J.; Lee, J.W.; Lee, M.-G. Resident and Monocyte-Derived Langerhans Cells are Required for Imiquimod-Induced Psoriasis-Like Dermatitis Model. J. Dermatol. Sci. 2018, 91, 52–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, T.; Zhao, W.; Li, H.; Xiao, S.; Hu, R.; Han, M.; Liu, H.; Liu, Y.; Otsu, K.; Liu, X.; et al. p38α Signaling in Langerhans cells Promotes the Development of IL-17–Producing T Cells and Psoriasiform Skin Inflammation. Sci. Signal. 2018, 11, eaao1685. [Google Scholar] [CrossRef] [Green Version]
- Singh, T.P.; Zhang, H.H.; Borek, I.; Wolf, P.; Hedrick, M.N.; Singh, S.P.; Kelsall, B.L.; Clausen, B.E.; Farber, J.M. Monocyte-derived Inflammatory Langerhans Cells and Dermal Dendritic Cells Mediate Psoriasis-Like Inflammation. Nat. Commun. 2016, 7, 13581. [Google Scholar] [CrossRef] [Green Version]
- Fanoni, D.; Venegoni, L.; Vergani, B.; Tavecchio, S.; Cattaneo, A.; Leone, B.E.; Berti, E.; Marzano, A.V. Evidence for a Role of Autoinflammation in Early-Phase Psoriasis. Clin. Exp. Immunol. 2019, 198, 283–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, C.-C.; Cheng, W.-J.; Korinek, M.; Lin, C.-Y.; Hwang, T.-L. Neutrophils in Psoriasis. Front. Immunol. 2019, 10, 2376. [Google Scholar] [CrossRef] [PubMed]
- Mutua, V.; Gershwin, L.J. A Review of Neutrophil Extracellular Traps (NETs) in Disease: Potential Anti-NETs Therapeutics. Clin. Rev. Allergy Immunol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Lambert, S.; Hambro, C.A.; Johnston, A.; Stuart, P.E.; Tsoi, L.C.; Nair, R.P.; Elder, J.T. Neutrophil Extracellular Traps Induce Human Th17 Cells: Effect of Psoriasis-Associated TRAF3IP2 Genotype. J. Investig. Dermatol. 2019, 139, 1245–1253. [Google Scholar] [CrossRef] [Green Version]
- Herster, F.; Bittner, Z.; Archer, N.K.; Dickhöfer, S.; Eisel, D.; Eigenbrod, T.; Knorpp, T.; Schneiderhan-Marra, N.; Löffler, M.W.; Kalbacher, H.; et al. Neutrophil Extracellular Trap-Associated RNA and LL37 Enable Self-Amplifying Inflammation in Psoriasis. Nat. Commun. 2020, 11, 105. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Edelmayer, R.; Wetter, J.; Salte, K.; Gauvin, D.; Leys, L.; Paulsboe, S.; Su, Z.; Weinberg, I.; Namovic, M.; et al. Monocytes/Macrophages Play a Pathogenic Role in IL-23 Mediated Psoriasis-Like Skin Inflammation. Sci. Rep. 2019, 9, 5310. [Google Scholar] [CrossRef] [Green Version]
- Yawalkar, N.; Karlen, S.; Hunger, R.; Brand, C.U.; Braathen, L.R. Expression of Interleukin-12 is Increased in Psoriatic Skin. J. Investig. Dermatol. 1998, 111, 1053–1057. [Google Scholar] [CrossRef]
- Fuentes-Duculan, J.; Suárez-Fariñas, M.; Zaba, L.C.; Nograles, K.E.; Pierson, K.C.; Mitsui, H.; Pensabene, C.A.; Kzhyshkowska, J.; Krueger, J.G.; Lowes, M.A. A Subpopulation of CD163-Positive Macrophages is Classically Activated in Psoriasis. J. Investig. Dermatol. 2010, 130, 2412–2422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Y.; Shen, X.; Ding, C.; Qi, C.; Li, K.; Li, X.; Jala, V.R.; Zhang, H.-g.; Wang, T.; Zheng, J.; et al. Pivotal Role of Dermal IL-17-Producing γδ T Cells in Skin Inflammation. Immunity 2011, 35, 596–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Y.; Zhu, L.; Tian, H.; Sun, H.-X.; Wang, R.; Zhang, L.; Zhao, Y. IL-23-Induced Macrophage Polarization and its Pathological Roles in Mice with Imiquimod-Induced Psoriasis. Protein Cell 2018, 9, 1027–1038. [Google Scholar] [CrossRef]
- Lin, A.M.; Rubin, C.J.; Khandpur, R.; Wang, J.Y.; Riblett, M.; Yalavarthi, S.; Villanueva, E.C.; Shah, P.; Kaplan, M.J.; Bruce, A.T. Mast Cells and Neutrophils Release IL-17 through Extracellular Trap Formation in Psoriasis. J. Immunol. 2011, 187, 490–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mashiko, S.; Bouguermouh, S.; Rubio, M.; Baba, N.; Bissonnette, R.; Sarfati, M. Human Mast Cells are Major IL-22 Producers in Patients with Psoriasis and Atopic Dermatitis. J. Allergy Clin. Immunol. 2015, 136, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Peres, L.P.; Oliveira, F.B.; Cartell, A.; Mazzotti, N.G.; Cestari, T.F. Density of Mast Cells and Intensity of Pruritus in Psoriasis Vulgaris: A Cross Sectional Study. An. Bras. Dermatol. 2018, 93, 368–372. [Google Scholar] [CrossRef] [PubMed]
- Keren, A.; Shemer, A.; Ginzburg, A.; Ullmann, Y.; Schrum, A.G.; Paus, R.; Gilhar, A. Innate Lymphoid Cells 3 Induce Psoriasis in Xenotransplanted Healthy Human Skin. J. Allergy Clin. Immunol. 2018, 142, 305–308.e306. [Google Scholar] [CrossRef] [Green Version]
- Bielecki, P.; Riesenfeld, S.J.; Hütter, J.-C.; Torlai Triglia, E.; Kowalczyk, M.S.; Ricardo-Gonzalez, R.R.; Lian, M.; Amezcua Vesely, M.C.; Kroehling, L.; Xu, H.; et al. Skin-Resident Innate Lymphoid Cells Converge on a Pathogenic Effector State. Nature 2021, 592, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Teunissen, M.B.M.; Munneke, J.M.; Bernink, J.H.; Spuls, P.I.; Res, P.C.M.; te Velde, A.; Cheuk, S.; Brouwer, M.W.D.; Menting, S.P.; Eidsmo, L.; et al. Composition of Innate Lymphoid Cell Subsets in the Human Skin: Enrichment of NCR + ILC3 in Lesional Skin and Blood of Psoriasis Patients. J. Investig. Dermatol. 2014, 134, 2351–2360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, Y.; Ogawa, E.; Okuyama, R. Role of Innate Immune Cells in Psoriasis. Int. J. Mol. Sci. 2020, 21, 6604. [Google Scholar] [CrossRef] [PubMed]
- Polese, B.; Zhang, H.; Thurairajah, B.; King, I.L. Innate Lymphocytes in Psoriasis. Front. Immunol. 2020, 11, 242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laggner, U.; Di Meglio, P.; Perera, G.K.; Hundhausen, C.; Lacy, K.E.; Ali, N.; Smith, C.H.; Hayday, A.C.; Nickoloff, B.J.; Nestle, F.O. Identification of a Novel Proinflammatory Human Skin-Homing Vγ9Vδ2 T Cell Subset with a Potential Role in Psoriasis. J. Immunol. 2011, 187, 2783–2793. [Google Scholar] [CrossRef]
- Plužarić, V.; Štefanić, M.; Mihalj, M.; Tolušić Levak, M.; Muršić, I.; Glavaš-Obrovac, L.; Petrek, M.; Balogh, P.; Tokić, S. Differential Skewing of Circulating MR1-Restricted and γδ T Cells in Human Psoriasis Vulgaris. Front. Immunol. 2020, 11, 572924. [Google Scholar] [CrossRef]
- Ramírez-Valle, F.; Gray, E.E.; Cyster, J.G. Inflammation Induces Dermal Vγ4+ γδT17 Memory-Like Cells that Travel to Distant Skin and Accelerate Secondary IL-17-Driven Responses. Proc. Natl. Acad. Sci. USA 2015, 112, 8046–8051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartwig, T.; Pantelyushin, S.; Croxford, A.L.; Kulig, P.; Becher, B. Dermal IL-17-Producing γδ T Cells Establish Long-Lived Memory in the Skin. Eur. J. Immunol. 2015, 45, 3022–3033. [Google Scholar] [CrossRef]
- Sumaria, N.; Roediger, B.; Ng, L.G.; Qin, J.; Pinto, R.; Cavanagh, L.L.; Shklovskaya, E.; Fazekas de St Groth, B.; Triccas, J.A.; Weninger, W. Cutaneous Immunosurveillance by Self-Renewing Dermal Gammadelta T Cells. J. Exp. Med. 2011, 208, 505–518. [Google Scholar] [CrossRef] [Green Version]
- Bos, J.D.; Teunissen, M.B.M.; Cairo, I.; Krieg, S.R.; Kapsenberg, M.L.; Das, P.K.; Borst, J. T-Cell Receptor γδ Bearing Cells in Normal Human Skin. J. Investig. Dermatol. 1990, 94, 37–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebert, L.M.; Meuter, S.; Moser, B. Homing and Function of Human Skin γδ T Cells and NK Cells: Relevance for Tumor Surveillance. J. Immunol. 2006, 176, 4331. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Zhu, J. CD4 T Helper Cell Subsets and Related Human Immunological Disorders. Int. J. Mol. Sci. 2020, 21, 8011. [Google Scholar] [CrossRef] [PubMed]
- Raphael, I.; Nalawade, S.; Eagar, T.N.; Forsthuber, T.G. T Cell Subsets and Their Signature Cytokines in Autoimmune and Inflammatory Diseases. Cytokine 2015, 74, 5–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Austin, L.M.; Ozawa, M.; Kikuchi, T.; Walters, I.B.; Krueger, J.G. The Majority of Epidermal T Cells in Psoriasis Vulgaris Lesions can Produce Type 1 Cytokines, Interferon-γ, Interleukin-2, and Tumor Necrosis Factor-α, Defining TC1 (Cytotoxic T Lymphocyte) and TH1 Effector Populations: A Type 1 Differentiation Bias is also Measured in Circulating Blood T Cells in Psoriatic Patients. J. Investig. Dermatol. 1999, 113, 752–759. [Google Scholar] [CrossRef] [Green Version]
- Furiati, S.C.; Catarino, J.S.; Silva, M.V.; Silva, R.F.; Estevam, R.B.; Teodoro, R.B.; Pereira, S.L.; Ataide, M.; Rodrigues, V., Jr.; Rodrigues, D.B.R. Th1, Th17, and Treg Responses are Differently Modulated by TNF-α Inhibitors and Methotrexate in Psoriasis Patients. Sci. Rep. 2019, 9, 7526. [Google Scholar] [CrossRef] [Green Version]
- Reich, K.; Reich, J.L.K.; Falk, T.M.; Blödorn-Schlicht, N.; Mrowietz, U.; von Kiedrowski, R.; Pfeiffer, C.; Niesmann, J.; Frambach, Y.; Warren, R.B. Clinical Response of Psoriasis to Subcutaneous Methotrexate Correlates with Inhibition of Cutaneous T Helper 1 and 17 Inflammatory Pathways. Br. J. Dermatol. 2019, 181, 859–862. [Google Scholar] [CrossRef]
- Priyadarssini, M.; Chandrashekar, L.; Rajappa, M. Effect of Methotrexate Monotherapy on T-Cell Subsets in the Peripheral Circulation in Psoriasis. Clin. Exp. Dermatol. 2019, 44, 491–497. [Google Scholar] [CrossRef]
- Chiricozzi, A.; Guttman-Yassky, E.; Suárez-Fariñas, M.; Nograles, K.E.; Tian, S.; Cardinale, I.; Chimenti, S.; Krueger, J.G. Integrative Responses to IL-17 and TNF-α in Human Keratinocytes Account for Key Inflammatory Pathogenic Circuits in Psoriasis. J. Investig. Dermatol. 2011, 131, 677–687. [Google Scholar] [CrossRef]
- Nograles, K.E.; Zaba, L.C.; Guttman-Yassky, E.; Fuentes-Duculan, J.; Suárez-Fariñas, M.; Cardinale, I.; Khatcherian, A.; Gonzalez, J.; Pierson, K.C.; White, T.R.; et al. Th17 Cytokines Interleukin (IL)-17 and IL-22 Modulate Distinct Inflammatory and Keratinocyte-Response Pathways. Br. J. Dermatol. 2008, 159, 1092–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, N.J.; Boniface, K.; Chan, J.R.; McKenzie, B.S.; Blumenschein, W.M.; Mattson, J.D.; Basham, B.; Smith, K.; Chen, T.; Morel, F.; et al. Development, Cytokine Profile and Function of Human Interleukin 17–Producing Helper T Cells. Nat. Immunol. 2007, 8, 950–957. [Google Scholar] [CrossRef]
- Harper, E.G.; Guo, C.; Rizzo, H.; Lillis, J.V.; Kurtz, S.E.; Skorcheva, I.; Purdy, D.; Fitch, E.; Iordanov, M.; Blauvelt, A. Th17 Cytokines Stimulate CCL20 Expression in Keratinocytes In Vitro and In Vivo: Implications for Psoriasis pathogenesis. J. Investig. Dermatol. 2009, 129, 2175–2183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matos, T.R.; O’Malley, J.T.; Lowry, E.L.; Hamm, D.; Kirsch, I.R.; Robins, H.S.; Kupper, T.S.; Krueger, J.G.; Clark, R.A. Clinically Resolved Psoriatic Lesions Contain Psoriasis-Specific IL-17-Producing αβ T Cell Clones. J. Clin. Investig. 2017, 127, 4031–4041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkes, J.E.; Yan, B.Y.; Chan, T.C.; Krueger, J.G. Discovery of the IL-23/IL-17 Signaling Pathway and the Treatment of Psoriasis. J. Immunol. 2018, 201, 1605–1613. [Google Scholar] [CrossRef] [PubMed]
- Stockinger, B.; Omenetti, S. The Dichotomous Nature of T Helper 17 Cells. Nat. Rev. Immunol. 2017, 17, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Flores, C.; Castro-Escamilla, O.; Ortega-Rocha, E.M.; Maldonado-García, C.; Jurado-Santa Cruz, F.; Pérez-Montesinos, G.; Lemini-López, A.; Bonifaz, L.C. Association of Pathogenic Th17 Cells with the Disease Severity and Its Potential Implication for Biological Treatment Selection in Psoriasis Patients. Mediat. Inflamm. 2020, 2020, 8065147. [Google Scholar] [CrossRef]
- Wu, L.; Hollinshead, K.E.R.; Hao, Y.; Au, C.; Kroehling, L.; Ng, C.; Lin, W.-Y.; Li, D.; Silva, H.M.; Shin, J.; et al. Niche-Selective Inhibition of Pathogenic Th17 Cells by Targeting Metabolic Redundancy. Cell 2020, 182, 641–654. [Google Scholar] [CrossRef]
- Ortega, C.; Fernández, A.S.; Carrillo, J.M.; Romero, P.; Molina, I.J.; Moreno, J.C.; Santamaría, M. IL-17-Producing CD8+ T Lymphocytes from Psoriasis Skin Plaques are Cytotoxic Effector Cells that Secrete Th17-Related Cytokines. J. Leukoc. Biol. 2009, 86, 435–443. [Google Scholar] [CrossRef]
- Hijnen, D.; Knol, E.F.; Gent, Y.Y.; Giovannone, B.; Beijn, S.J.P.; Kupper, T.S.; Bruijnzeel-Koomen, C.A.F.M.; Clark, R.A. CD8(+) T Cells in the Lesional Skin of Atopic Dermatitis and Psoriasis Patients Are an Important Source of IFN-γ, IL-13, IL-17, and IL-22. J. Investig. Dermatol. 2013, 133, 973–979. [Google Scholar] [CrossRef] [Green Version]
- Vičić, M.; Kaštelan, M.; Sotošek Tokmadžić, V.; Prpić Massari, L. Systemic and Local Increase of Granulysin Expression in Cytotoxic Lymphocytes in Severe Psoriasis. Acta Derm. Venereol. 2019, 99, 1136–1142. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Chang, H.-W.; Huang, Z.-M.; Nakamura, M.; Sekhon, S.; Ahn, R.; Munoz-Sandoval, P.; Bhattarai, S.; Beck, K.M.; Sanchez, I.M.; et al. Single-Cell RNA Sequencing of Psoriatic Skin Identifies Pathogenic Tc17 Cell Subsets and Reveals Distinctions between CD8+ T Cells in Autoimmunity and Cancer. J. Allergy Clin. Immunol. 2021, 147, 2370–2380. [Google Scholar] [CrossRef]
- Watanabe, R. Protective and Pathogenic Roles of Resident Memory T Cells in Human Skin Disorders. J. Dermatol. Sci. 2019, 95, 2–7. [Google Scholar] [CrossRef] [Green Version]
- Kurihara, K.; Fujiyama, T.; Phadungsaksawasdi, P.; Ito, T.; Tokura, Y. Significance of IL-17A-Producing CD8+CD103+ Skin Resident Memory T Cells in Psoriasis Lesion and Their Possible Relationship to Clinical Course. J. Dermatol. Sci. 2019, 95, 21–27. [Google Scholar] [CrossRef]
- Choi, C.W.; Kim, B.R.; Yang, S.; Kim, Y.; Kang, J.S.; Youn, S.W. Regulatory T Cells Suppress Skin Inflammation in the Imiquimod-Induced Psoriasis-Like Mouse Model. J. Dermatol. Sci. 2020, 98, 199–202. [Google Scholar] [CrossRef]
- Stockenhuber, K.; Hegazy, A.N.; West, N.R.; Ilott, N.E.; Stockenhuber, A.; Bullers, S.J.; Thornton, E.E.; Arnold, I.C.; Tucci, A.; Waldmann, H.; et al. Foxp3(+) T Reg Cells Control Psoriasiform Inflammation by Restraining an IFN-I-Driven CD8(+) T Cell Response. J. Exp. Med. 2018, 215, 1987–1998. [Google Scholar] [CrossRef]
- Hau, C.S.; Shimizu, T.; Tada, Y.; Kamata, M.; Takeoka, S.; Shibata, S.; Mitsui, A.; Asano, Y.; Sugaya, M.; Kadono, T.; et al. The Vitamin D3 Analog, Maxacalcitol, Reduces Psoriasiform Skin Inflammation by Inducing Regulatory T Cells and Downregulating IL-23 and IL-17 Production. J. Dermatol. Sci. 2018, 92, 117–126. [Google Scholar] [CrossRef] [Green Version]
- Sulaimani, J.; Cluxton, D.; Clowry, J.; Petrasca, A.; Molloy, O.E.; Moran, B.; Sweeney, C.M.; Malara, A.; McNicholas, N.; McGuigan, C.; et al. Dimethyl Fumarate Modulates the Treg–Th17 Cell Axis in Patients with Psoriasis. Br. J. Dermatol. 2021, 184, 495–503. [Google Scholar] [CrossRef]
- Nussbaum, L.; Chen, Y.L.; Ogg, G.S. Role of Regulatory T Cells in Psoriasis Pathogenesis and Treatment. Br. J. Dermatol. 2021, 184, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, H.; Gyulai, R.; Toichi, E.; Garaczi, E.; Shimada, S.; Stevens, S.R.; McCormick, T.S.; Cooper, K.D. Dysfunctional Blood and Target Tissue CD4 + CD25 High Regulatory T Cells in Psoriasis: Mechanism Underlying Unrestrained Pathogenic Effector T Cell Proliferation. J. Immunol. 2005, 174, 164–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Lei, J.; Yang, L.; Gao, C.; Dang, E.; Cao, T.; Xue, K.; Zhuang, Y.; Shao, S.; Zhi, D.; et al. Dysregulation of Akt-FOXO1 Pathway Leads to Dysfunction of Regulatory T Cells in Patients with Psoriasis. J. Investig. Dermatol. 2019, 139, 2098–2107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodman, W.A.; Levine, A.D.; Massari, J.V.; Sugiyama, H.; McCormick, T.S.; Cooper, K.D. IL-6 Signaling in Psoriasis Prevents Immune Suppression by Regulatory T Cells. J. Immunol. 2009, 183, 3170–3176. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Chen, Z.; Zhao, Z.; Yu, Y.; Fan, H.; Xu, X.; Bu, X.; Gu, J. IL-21 Induces an Imbalance of Th17/Treg Cells in Moderate-to-Severe Plaque Psoriasis Patients. Front. Immunol. 2019, 10, 1865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Li, B.; Dang, E.; Jin, L.; Fan, X.; Wang, G. Impaired Function of Regulatory T Cells in Patients with Psoriasis is Mediated by Phosphorylation of STAT3. J. Dermatol. Sci. 2016, 81, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Grän, F.; Kerstan, A.; Serfling, E.; Goebeler, M.; Muhammad, K. Current Developments in the Immunology of Psoriasis. Yale J. Biol. Med. 2020, 93, 97–110. [Google Scholar] [PubMed]
- Hayashi, M.; Yanaba, K.; Umezawa, Y.; Yoshihara, Y.; Kikuchi, S.; Ishiuji, Y.; Saeki, H.; Nakagawa, H. IL-10-Producing Regulatory B Cells Are Decreased in Patients with Psoriasis. J. Dermatol. Sci. 2016, 81, 93–100. [Google Scholar] [CrossRef]
- Mizumaki, K.; Horii, M.; Kano, M.; Komuro, A.; Matsushita, T. Suppression of IL-23-Mediated Psoriasis-Like Inflammation by Regulatory B Cells. Sci. Rep. 2021, 11, 2106. [Google Scholar] [CrossRef]
- Yanaba, K.; Kamata, M.; Ishiura, N.; Shibata, S.; Asano, Y.; Tada, Y.; Sugaya, M.; Kadono, T.; Tedder, T.F.; Sato, S. Regulatory B Cells Suppress Imiquimod-Induced, Psoriasis-Like Skin Inflammation. J. Leukoc. Biol. 2013, 94, 563–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Tan, L.; Zhu, W.; Lei, L.; Kuang, Y.; Liu, P.; Li, J.; Chen, X.; Peng, C. Targeting Myeloid-Derived Suppressor Cells Is a Novel Strategy for Anti-Psoriasis Therapy. Mediat. Inflamm. 2020, 2020, 8567320. [Google Scholar] [CrossRef] [PubMed]
- Ilkovitch, D.; Ferris, L.K. Myeloid-Derived Suppressor Cells Are Elevated in Patients with Psoriasis and Produce Various Molecules. Mol. Med. Report. 2016, 14, 3935–3940. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.Y.; Chung, J.-S.; Teshima, T.; Feigenbaum, L.; Cruz, P.D., Jr.; Jacobe, H.T.; Chong, B.F.; Ariizumi, K. Myeloid-Derived Suppressor Cells in Psoriasis Are an Expanded Population Exhibiting Diverse T-Cell-Suppressor Mechanisms. J. Investig. Dermatol. 2016, 136, 1801–1810. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.-H.; Yoo, J.K.; Jeon, S.H.; Lim, C.-Y.; Lee, J.-H.; Koo, D.-B.; Park, M.-Y. Anti-Psoriatic Effect of Myeloid-Derived Suppressor Cells on Imiquimod-Induced Skin Inflammation in Mice. Scand. J. Immunol. 2019, 89, e12742. [Google Scholar] [CrossRef] [PubMed]
- Afonina, I.S.; Van Nuffel, E.; Beyaert, R. Immune Responses and Therapeutic Options in Psoriasis. Cell. Mol. Life Sci. 2021, 78, 2709–2727. [Google Scholar] [CrossRef]
- Wong, V.W.; Akaishi, S.; Longaker, M.T.; Gurtner, G.C. Pushing Back: Wound Mechanotransduction in Repair and Regeneration. J. Investig. Dermatol. 2011, 131, 2186–2196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lønnberg, A.S.; Skov, L.; Skytthe, A.; Kyvik, K.O.; Pedersen, O.B.; Thomsen, S.F. Heritability of Psoriasis in a Large Twin Sample. Br. J. Dermatol. 2013, 169, 412–416. [Google Scholar] [CrossRef] [PubMed]
- Grjibovski, A.M.; Olsen, A.O.; Magnus, P.; Harris, J.R. Psoriasis in Norwegian Twins: Contribution of Genetic and Environmental Effects. J. Eur. Acad. Dermatol. Venereol. 2007, 21, 1337–1343. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, K.; Okada, Y. The Current Landscape of Psoriasis Genetics in 2020. J. Dermatol. Sci. 2020, 99, 2–8. [Google Scholar] [CrossRef]
- Gudjonsson, J.E.; Karason, A.; Hjaltey Runarsdottir, E.; Antonsdottir, A.A.; Hauksson, V.B.; Jónsson, H.H.; Gulcher, J.; Stefansson, K.; Valdimarsson, H. Distinct Clinical Differences between HLA-Cw*0602 Positive and Negative Psoriasis Patients-An Analysis of 1019 HLA-C-And HLA-B-Typed Patients. J. Investig. Dermatol. 2006, 126, 740–745. [Google Scholar] [CrossRef] [Green Version]
- Gudjonsson, J.E.; Thorarinsson, A.M.; Sigurgeirsson, B.; Kristinsson, K.G.; Valdimarsson, H. Streptococcal Throat Infections and Exacerbation of Chronic Plaque Psoriasis: A Prospective Study. Br. J. Dermatol. 2003, 149, 530–534. [Google Scholar] [CrossRef]
- Prinz, J.C. Melanocytes: Target Cells of an HLA-C*06:02–Restricted Autoimmune Response in Psoriasis. J. Investig. Dermatol. 2017, 137, 2053–2058. [Google Scholar] [CrossRef] [Green Version]
- Mabuchi, T.; Hirayama, N. Binding Affinity and Interaction of LL-37 with HLA-C*06:02 in Psoriasis. J. Investig. Dermatol. 2016, 136, 1901–1903. [Google Scholar] [CrossRef] [Green Version]
- Wei, P.; Yang, Y.; Liu, Z.; Luo, Z.; Tu, W.; Han, J.; Deng, Y.; Yin, L. Characterization of Autoantigen Presentation by HLA-C*06:02 in Psoriasis. J. Investig. Dermatol. 2017, 137, 2238–2241. [Google Scholar] [CrossRef] [Green Version]
- Ray-Jones, H.; Duffus, K.; McGovern, A.; Martin, P.; Shi, C.; Hankinson, J.; Gough, O.; Yarwood, A.; Morris, A.P.; Adamson, A.; et al. Mapping DNA Interaction Landscapes in Psoriasis Susceptibility Loci Highlights KLF4 as a Target Gene in 9q31. BMC Biol. 2020, 18, 47. [Google Scholar] [CrossRef]
- Segre, J.A.; Bauer, C.; Fuchs, E. Klf4 is a Transcription Factor Required for Establishing the Barrier Function of the Skin. Nat. Genet. 1999, 22, 356–360. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-J.; Huang, W.; Yang, S.; Sun, L.-D.; Zhang, F.-Y.; Zhu, Q.-X.; Zhang, F.-R.; Zhang, C.; Du, W.-H.; Pu, X.-M.; et al. Psoriasis Genome-Wide Association Study Identifies Susceptibility Variants within LCE Gene Cluster at 1q21. Nat. Genet. 2009, 41, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Low, H.Q.; Wang, L.; Li, Y.; Ellinghaus, E.; Han, J.; Estivill, X.; Sun, L.; Zuo, X.; Shen, C.; et al. Genome-Wide Meta-Analysis Identifies Multiple Novel Associations and Ethnic Heterogeneity of Psoriasis Susceptibility. Nat. Commun. 2015, 6, 6916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirata, J.; Hirota, T.; Ozeki, T.; Kanai, M.; Sudo, T.; Tanaka, T.; Hizawa, N.; Nakagawa, H.; Sato, S.; Mushiroda, T.; et al. Variants at HLA-A, HLA-C, and HLA-DQB1 Confer Risk of Psoriasis Vulgaris in Japanese. J. Investig. Dermatol. 2018, 138, 542–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Vugt, L.J.; van den Reek, J.M.P.A.; Hannink, G.; Coenen, M.J.H.; de Jong, E.M.G.J. Association of HLA-C*06:02 Status with Differential Response to Ustekinumab in Patients with Psoriasis: A Systematic Review and Meta-Analysis. JAMA Dermatol. 2019, 155, 708–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsoi, L.C.; Stuart, P.E.; Tian, C.; Gudjonsson, J.E.; Das, S.; Zawistowski, M.; Ellinghaus, E.; Barker, J.N.; Chandran, V.; Dand, N.; et al. Large Scale Meta-Analysis Characterizes Genetic Architecture for Common Psoriasis Associated Variants. Nat. Commun. 2017, 8, 15382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsoi, L.C.; Spain, S.L.; Knight, J.; Ellinghaus, E.; Stuart, P.E.; Capon, F.; Ding, J.; Li, Y.; Tejasvi, T.; Gudjonsson, J.E.; et al. Identification of 15 New Psoriasis Susceptibility Loci Highlights the Role of Innate Immunity. Nat. Genet. 2012, 44, 1341–1348. [Google Scholar] [CrossRef] [Green Version]
- Goldminz, A.M.; Au, S.C.; Kim, N.; Gottlieb, A.B.; Lizzul, P.F. NF-κB: An Essential Transcription Factor in Psoriasis. J. Dermatol. Sci. 2013, 69, 89–94. [Google Scholar] [CrossRef]
- Tseng, J.-C.; Chang, Y.-C.; Huang, C.-M.; Hsu, L.-C.; Chuang, T.-H. Therapeutic Development Based on the Immunopathogenic Mechanisms of Psoriasis. Pharmaceutics 2021, 13, 1064. [Google Scholar] [CrossRef] [PubMed]
- Nair, R.P.; Duffin, K.C.; Helms, C.; Ding, J.; Stuart, P.E.; Goldgar, D.; Gudjonsson, J.E.; Li, Y.; Tejasvi, T.; Feng, B.-J.; et al. Genome-Wide Scan Reveals Association of Psoriasis with IL-23 and NF-kappaB Pathways. Nat. Genet. 2009, 41, 199–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokuyama, M.; Mabuchi, T. New Treatment Addressing the Pathogenesis of Psoriasis. Int. J. Mol. Sci. 2020, 21, 7488. [Google Scholar] [CrossRef] [PubMed]
- Kvist-Hansen, A.; Hansen, P.R.; Skov, L. Systemic Treatment of Psoriasis with JAK Inhibitors: A Review. Dermatol. Ther. 2020, 10, 29–42. [Google Scholar] [CrossRef] [Green Version]
- Caputo, V.; Strafella, C.; Termine, A.; Campione, E.; Bianchi, L.; Novelli, G.; Giardina, E.; Cascella, R. RNAseq-Based Prioritization Revealed COL6A5, COL8A1, COL10A1 and MIR146A as Common and Differential Susceptibility Biomarkers for Psoriasis and Psoriatic Arthritis: Confirmation from Genotyping Analysis of 1417 Italian Subjects. Int. J. Mol. Sci. 2020, 21, 2740. [Google Scholar] [CrossRef]
- Bergboer, J.G.M.; Tjabringa, G.S.; Kamsteeg, M.; van Vlijmen-Willems, I.M.J.J.; Rodijk-Olthuis, D.; Jansen, P.A.M.; Thuret, J.-Y.; Narita, M.; Ishida-Yamamoto, A.; Zeeuwen, P.L.J.M.; et al. Psoriasis Risk Genes of the Late Cornified Envelope-3 Group Are Distinctly Expressed Compared with Genes of Other LCE Groups. Am. J. Pathol. 2011, 178, 1470–1477. [Google Scholar] [CrossRef]
- Hoss, E.; Austin, H.R.; Batie, S.F.; Jurutka, P.W.; Haussler, M.R.; Whitfield, G.K. Control of Late Cornified Envelope Genes Relevant to Psoriasis Risk: Upregulation by 1,25-Dihydroxyvitamin D3 and Plant-Derived Delphinidin. Arch. Dermatol. Res. 2013, 305, 867–878. [Google Scholar] [CrossRef] [Green Version]
- Caputo, V.; Strafella, C.; Termine, A.; Dattola, A.; Mazzilli, S.; Lanna, C.; Cosio, T.; Campione, E.; Novelli, G.; Giardina, E.; et al. Overview of the Molecular Determinants Contributing to the Expression of Psoriasis and Psoriatic Arthritis phenotypes. J. Cell. Mol. Med. 2020, 24, 13554–13563. [Google Scholar] [CrossRef]
- Zhang, P.; Zhao, M.; Liang, G.; Yin, G.; Huang, D.; Su, F.; Zhai, H.; Wang, L.; Su, Y.; Lu, Q. Whole-Genome DNA Methylation in Skin Lesions from Patients with Psoriasis Vulgaris. J. Autoimmun. 2013, 41, 17–24. [Google Scholar] [CrossRef]
- Hu, Y.; Baud, V.; Oga, T.; Kim, K.I.; Yoshida, K.; Karin, M. IKKα Controls Formation of the Epidermis Independently of NF-κB. Nature 2001, 410, 710–714. [Google Scholar] [CrossRef]
- Yu, N.; Yang, Y.; Li, X.; Zhang, M.; Huang, J.; Wang, X.; Long, X. MiR-26a Inhibits Proliferation and Migration of HaCaT Keratinocytes through Regulating PTEN Expression. Gene 2016, 594, 117–124. [Google Scholar] [CrossRef]
- Verallo-Rowell, V.M.; Katalbas, S.S.; Evangelista, M.T.P.; Dayrit, J.F. Review Update on Topical Therapy for Psoriasis. Curr. Dermatol. Rep. 2018, 7, 24–36. [Google Scholar] [CrossRef]
- Bijlmakers, M.-J.; Kanneganti, S.K.; Barker, J.N.; Trembath, R.C.; Capon, F. Functional Analysis of the RNF114 Psoriasis Susceptibility Gene Implicates Innate Immune Responses to Double-Stranded RNA in Disease Pathogenesis. Hum. Mol. Genet. 2011, 20, 3129–3137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rácz, E.; Prens, E.P.; Kurek, D.; Kant, M.; de Ridder, D.; Mourits, S.; Baerveldt, E.M.; Ozgur, Z.; van Ijcken, W.F.J.; Laman, J.D.; et al. Effective Treatment of Psoriasis with Narrow-Band UVB Phototherapy Is Linked to Suppression of the IFN and Th17 Pathways. J. Investig. Dermatol. 2011, 131, 1547–1558. [Google Scholar] [CrossRef] [Green Version]
- Catapano, M.; Vergnano, M.; Romano, M.; Mahil, S.K.; Choon, S.-E.; Burden, A.D.; Young, H.S.; Carr, I.M.; Lachmann, H.J.; Lombardi, G.; et al. IL-36 Promotes Systemic IFN-I Responses in Severe Forms of Psoriasis. J. Investig. Dermatol. 2020, 140, 816–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandra, A.; Senapati, S.; Roy, S.; Chatterjee, G.; Chatterjee, R. Epigenome-Wide DNA Methylation Regulates Cardinal Pathological Features of Psoriasis. Clin. Epigenetics 2018, 10, 108. [Google Scholar] [CrossRef]
- Chen, M.; Wang, Y.; Yao, X.; Li, C.; Jiang, M.; Cui, P.; Wang, B. Hypermethylation of HLA-C May Be an Epigenetic Marker in Psoriasis. J. Dermatol. Sci. 2016, 83, 10–16. [Google Scholar] [CrossRef]
- Zhou, F.; Shen, C.; Hsu, Y.-H.; Gao, J.; Dou, J.; Ko, R.; Zheng, X.; Sun, L.; Cui, Y.; Zhang, X. DNA Methylation-Based Subclassification of Psoriasis in the Chinese Han Population. Front. Med. 2018, 12, 717–725. [Google Scholar] [CrossRef]
- Li, F.; Yuan, C.W.; Xu, S.; Zu, T.; Woappi, Y.; Lee, C.A.A.; Abarzua, P.; Wells, M.; Ramsey, M.R.; Frank, N.Y.; et al. Loss of the Epigenetic Mark 5-hmC in Psoriasis: Implications for Epidermal Stem Cell Dysregulation. J. Investig. Dermatol. 2020, 140, 1266–1275.e1263. [Google Scholar] [CrossRef]
- Zeng, C.; Tsoi, L.C.; Gudjonsson, J.E. Dysregulated Epigenetic Modifications in Psoriasis. Exp. Dermatol. 2021, 30, 1156–1166. [Google Scholar] [CrossRef]
- Shao, S.; Gudjonsson, J.E. Epigenetics of Psoriasis. In Epigenetics in Allergy and Autoimmunity; Chang, C., Lu, Q., Eds.; Springer: Singapore, 2020; pp. 209–221. [Google Scholar]
- Dei-Cas, I.; Giliberto, F.; Luce, L.; Dopazo, H.; Penas-Steinhardt, A. Metagenomic Analysis of Gut Microbiota in Non-Treated Plaque Psoriasis Patients Stratified by Disease Severity: Development of a New Psoriasis-Microbiome Index. Sci. Rep. 2020, 10, 12754. [Google Scholar] [CrossRef]
- Kim, C.H. Control of Lymphocyte Functions by Gut Microbiota-Derived Short-Chain Fatty Acids. Cell. Mol. Immunol. 2021, 18, 1161–1171. [Google Scholar] [CrossRef]
- Zhang, X.; Shi, L.; Sun, T.; Guo, K.; Geng, S. Dysbiosis of Gut Microbiota and Its Correlation with Dysregulation of Cytokines in Psoriasis Patients. BMC Microbiol. 2021, 21, 78. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, J.; Cohen, N.A.; Shalev, V.; Uzan, A.; Koren, O.; Maharshak, N. Psoriatic Patients Have a Distinct Structural and Functional Fecal Microbiota Compared with Controls. J. Dermatol. 2019, 46, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, A.; Philippsen, R.; Schwarz, T. Induction of Regulatory T Cells and Correction of Cytokine Disbalance by Short-Chain Fatty Acids: Implications for Psoriasis Therapy. J. Investig. Dermatol. 2021, 141, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.-W.; Yan, D.; Singh, R.; Liu, J.; Lu, X.; Ucmak, D.; Lee, K.; Afifi, L.; Fadrosh, D.; Leech, J.; et al. Alteration of the Cutaneous Microbiome in Psoriasis and Potential Role in Th17 Polarization. Microbiome 2018, 6, 154. [Google Scholar] [CrossRef]
- Fyhrquist, N.; Muirhead, G.; Prast-Nielsen, S.; Jeanmougin, M.; Olah, P.; Skoog, T.; Jules-Clement, G.; Feld, M.; Barrientos-Somarribas, M.; Sinkko, H.; et al. Microbe-Host Interplay in Atopic Dermatitis and Psoriasis. Nat. Commun. 2019, 10, 4703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quan, C.; Chen, X.-Y.; Li, X.; Xue, F.; Chen, L.-H.; Liu, N.; Wang, B.; Wang, L.-Q.; Wang, X.-P.; Yang, H.; et al. Psoriatic Lesions are Characterized by Higher Bacterial Load and Imbalance between Cutibacterium and Corynebacterium. J. Am. Acad. Dermatol. 2020, 82, 955–961. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Li, J.; Zhu, W.; Kuang, Y.; Liu, T.; Zhang, W.; Chen, X.; Peng, C. Skin and Gut Microbiome in Psoriasis: Gaining Insight Into the Pathophysiology of It and Finding Novel Therapeutic Strategies. Front. Microbiol. 2020, 11, 3201. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.Y.; Huang, Y.H.; Chu, C.F.; Wu, T.C.; Liu, S.H. Risks for Staphylococcus aureus Colonization in Patients with Psoriasis: A Systematic Review and Meta-Analysis. Br. J. Dermatol. 2017, 177, 967–977. [Google Scholar] [CrossRef]
- Abbas, I.; Al-Mohana, A.; Abbas, E.; Al-Dhalimi, M. Cytokine Serum Level Association with Superantigen Production by Staphylococcus aureus in Psoriasis vulgaris. Res. J. Pharm. Biol. Chem. Sci. 2016, 7, 172–180. [Google Scholar]
- Polak, K.; Bergler-Czop, B.; Szczepanek, M.; Wojciechowska, K.; Frątczak, A.; Kiss, N. Psoriasis and Gut Microbiome-Current State of Art. Int. J. Mol. Sci. 2021, 22, 4529. [Google Scholar] [CrossRef]
- Myers, B.; Brownstone, N.; Reddy, V.; Chan, S.; Thibodeaux, Q.; Truong, A.; Bhutani, T.; Chang, H.-W.; Liao, W. The Gut Microbiome in Psoriasis and Psoriatic Arthritis. Best Pract. Res. Clin. Rheumatol. 2019, 33, 101494. [Google Scholar] [CrossRef]
- Chen, Y.-J.; Ho, H.J.; Tseng, C.-H.; Lai, Z.-L.; Shieh, J.-J.; Wu, C.-Y. Intestinal Microbiota Profiling and Predicted Metabolic Dysregulation in Psoriasis Patients. Exp. Dermatol. 2018, 27, 1336–1343. [Google Scholar] [CrossRef]
- Telfer, N.R.; Chalmers, R.J.G.; Whale, K.; Colman, G. The Role of Streptococcal Infection in the Initiation of Guttate Psoriasis. Arch. Dermatol. 1992, 128, 39–42. [Google Scholar] [CrossRef]
- Rachakonda, T.D.; Dhillon, J.S.; Florek, A.G.; Armstrong, A.W. Effect of Tonsillectomy on Psoriasis: A Systematic Review. J. Am. Acad. Dermatol. 2015, 72, 261–275. [Google Scholar] [CrossRef]
- Haapasalo, K.; Koskinen, L.L.E.; Suvilehto, J.; Jousilahti, P.; Wolin, A.; Suomela, S.; Trembath, R.; Barker, J.; Vuopio, J.; Kere, J.; et al. The Psoriasis Risk Allele HLA-C*06:02 Shows Evidence of Association with Chronic or Recurrent Streptococcal Tonsillitis. Infect. Immun. 2018, 86, e00304–e00318. [Google Scholar] [CrossRef] [Green Version]
- Yong, W.C.; Upala, S.; Sanguankeo, A. Association between Psoriasis and Helicobacter Pylori Infection: A Systematic Review and Meta-Analysis. Indian J. Dermatol. 2018, 63, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Pietrzak, A.; Grywalska, E.; Socha, M.; Roliński, J.; Franciszkiewicz-Pietrzak, K.; Rudnicka, L.; Rudzki, M.; Krasowska, D. Prevalence and Possible Role of Candida Species in Patients with Psoriasis: A Systematic Review and Meta-Analysis. Mediat. Inflamm. 2018, 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, A.S.; Al-Najjar, A.H.; Alshalahi, H.; Altowayan, W.M.; Elgharabawy, R.M. Clinical Significance of Helicobacter Pylori Infection on Psoriasis Severity. J. Interferon Cytokine Res. 2021, 41, 44–51. [Google Scholar] [CrossRef] [PubMed]
- De Jesús-Gil, C.; Sans-de San Nicolàs, L.; Ruiz-Romeu, E.; Ferran, M.; Soria-Martínez, L.; García-Jiménez, I.; Chiriac, A.; Casanova-Seuma, J.M.; Fernández-Armenteros, J.M.; Owens, S.; et al. Interplay between Humoral and CLA(+) T Cell Response against Candida albicans in Psoriasis. Int. J. Mol. Sci. 2021, 22, 1519. [Google Scholar] [CrossRef] [PubMed]
- Camargo, C.M.d.S.; Brotas, A.M.; Ramos-e-Silva, M.; Carneiro, S. Isomorphic Phenomenon of Koebner: Facts and Controversies. Clin. Dermatol. 2013, 31, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.-J. Type1 Interferons Potential Initiating Factors Linking Skin Wounds with Psoriasis Pathogenesis. Front. Immunol. 2019, 10, 1440. [Google Scholar] [CrossRef]
- Ni, X.; Lai, Y. Keratinocyte: A trigger or an Executor of Psoriasis? J. Leukoc. Biol. 2020, 108, 485–491. [Google Scholar] [CrossRef]
- Qiao, P.; Guo, W.; Ke, Y.; Fang, H.; Zhuang, Y.; Jiang, M.; Zhang, J.; Shen, S.; Qiao, H.; Dang, E.; et al. Mechanical Stretch Exacerbates Psoriasis by Stimulating Keratinocyte Proliferation and Cytokine Production. J. Investig. Dermatol. 2019, 139, 1470–1479. [Google Scholar] [CrossRef] [Green Version]
- Elewski, B.; Alexis, A.F.; Lebwohl, M.; Stein Gold, L.; Pariser, D.; Del Rosso, J.; Yosipovitch, G. Itch: An Under-Recognized Problem in Psoriasis. J. Eur. Acad. Dermatol. Venereol. 2019, 33, 1465–1476. [Google Scholar] [CrossRef]
- Kamiya, K.; Kishimoto, M.; Sugai, J.; Komine, M.; Ohtsuki, M. Risk Factors for the Development of Psoriasis. Int. J. Mol. Sci. 2019, 20, 4347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nosbaum, A.; Dahel, K.; Goujon, C.; Nicolas, J.-F.; Mengeaud, V.; Vocanson, M. Psoriasis is a Disease of the Entire Skin: Non-Lesional Skin Displays a Prepsoriasis Phenotype. Eur. J. Dermatol. 2021, 31, 143–154. [Google Scholar] [CrossRef]
- Mack, M.R.; Kim, B.S. The Itch–Scratch Cycle: A Neuroimmune Perspective. Trends Immunol. 2018, 39, 980–991. [Google Scholar] [CrossRef]
- Dogra, S.; Kamat, D. Drug-Induced Psoriasis. Indian J. Rheumatol. 2019, 14, 37–43. [Google Scholar] [CrossRef]
- Shi, C.R.; Nambudiri, V.E. Widespread Psoriasis Flare Following Influenza Vaccination. Vaccine 2017, 35, 4785–4786. [Google Scholar] [CrossRef]
- Choudhry, A.; Mathena, J.; Albano, J.D.; Yacovone, M.; Collins, L. Safety Evaluation of Adenovirus Type 4 and Type 7 Vaccine Live, Oral in Military Recruits. Vaccine 2016, 34, 4558–4564. [Google Scholar] [CrossRef] [Green Version]
- Barrea, L.; Savanelli, M.C.; Di Somma, C.; Napolitano, M.; Megna, M.; Colao, A.; Savastano, S. Vitamin D and Its Role in Psoriasis: An Overview of the Dermatologist and Nutritionist. Rev. Endocr. Metab. Disord. 2017, 18, 195–205. [Google Scholar] [CrossRef] [Green Version]
- Brownstone, N.; Mosca, M.; Hong, J.; Hadeler, E.; Bhutani, T. Phototherapy for Psoriasis: New Research and Insights. Curr. Dermatol. Rep. 2021, 10, 16–20. [Google Scholar] [CrossRef]
- Sbidian, E.; Chaimani, A.; Garcia-Doval, I.; Doney, L.; Dressler, C.; Hua, C.; Hughes, C.; Naldi, L.; Afach, S.; Le Cleach, L. Systemic Pharmacological Treatments for Chronic Plaque Psoriasis: A Network Meta-Analysis. Cochrane Database Syst. Rev. 2021. [Google Scholar] [CrossRef]
- Nogueira, M.; Puig, L.; Torres, T. JAK Inhibitors for Treatment of Psoriasis: Focus on Selective TYK2 Inhibitors. Drugs 2020, 80, 341–352. [Google Scholar] [CrossRef]
- Florek, A.G.; Wang, C.J.; Armstrong, A.W. Treatment Preferences and Treatment Satisfaction among Psoriasis Patients: A Systematic Review. Arch. Dermatol. Res. 2018, 310, 271–319. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, C.E.M.; Armstrong, A.W.; Gudjonsson, J.E.; Barker, J.N.W.N. Psoriasis. Lancet 2021, 397, 1301–1315. [Google Scholar] [CrossRef]
- Fluhr, J.W.; Cavallotti, C.; Berardesca, E. Emollients, Moisturizers, and Keratolytic Agents in Psoriasis. Clin. Dermatol. 2008, 26, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Jacobi, A.; Mayer, A.; Augustin, M. Keratolytics and Emollients and Their Role in the Therapy of Psoriasis: A Systematic Review. Dermatol. Ther. 2015, 5, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Maroto-Morales, D.; Montero-Vilchez, T.; Arias-Santiago, S. Study of Skin Barrier Function in Psoriasis: The Impact of Emollients. Life 2021, 11, 651. [Google Scholar] [CrossRef]
- Pfaff, C.M.; Marquardt, Y.; Fietkau, K.; Baron, J.M.; Lüscher, B. The Psoriasis-Associated IL-17A Induces and Cooperates with IL-36 Cytokines to Control Keratinocyte Differentiation and Function. Sci. Rep. 2017, 7, 15631. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Yu, X.; Wu, C.; Jin, H. IL-36γ Inhibits Differentiation and Induces Inflammation of Keratinocyte via Wnt Signaling Pathway in Psoriasis. Int. J. Med. Sci. 2017, 14, 1002–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miura, S.; Garcet, S.; Salud-Gnilo, C.; Gonzalez, J.; Li, X.; Murai-Yamamura, M.; Yamamura, K.; Rambhia, D.; Kunjravia, N.; Guttman-Yassky, E.; et al. IL-36 and IL-17A Cooperatively Induce a Psoriasis-Like Gene Expression Response in Human Keratinocytes. J. Investig. Dermatol. 2021, 141, 2086–2090. [Google Scholar] [CrossRef]
- Bachelez, H.; Choon, S.-E.; Marrakchi, S.; Burden, A.D.; Tsai, T.-F.; Morita, A.; Turki, H.; Hall, D.B.; Shear, M.; Baum, P.; et al. Inhibition of the Interleukin-36 Pathway for the Treatment of Generalized Pustular Psoriasis. N. Engl. J. Med. 2019, 380, 981–983. [Google Scholar] [CrossRef] [PubMed]
- Choon, S.E.; Lebwohl, M.G.; Marrakchi, S.; Burden, A.D.; Tsai, T.-F.; Morita, A.; Navarini, A.A.; Zheng, M.; Xu, J.; Turki, H.; et al. Study Protocol of the Global Effisayil 1 Phase II, Multicentre, Randomised, Double-Blind, Placebo-Controlled Trial of Spesolimab in Patients with Generalized Pustular Psoriasis Presenting with an Acute Flare. BMJ Open 2021, 11, e043666. [Google Scholar] [CrossRef] [PubMed]
- Mrowietz, U.; Burden, A.D.; Pinter, A.; Reich, K.; Schäkel, K.; Baum, P.; Datsenko, Y.; Deng, H.; Padula, S.J.; Thoma, C.; et al. Spesolimab, an Anti-Interleukin-36 Receptor Antibody, in Patients with Palmoplantar Pustulosis: Results of a Phase IIa, Multicenter, Double-Blind, Randomized, Placebo-Controlled Pilot Study. Dermatol. Ther. 2021, 11, 571–585. [Google Scholar] [CrossRef]
- Tsai, Y.-C.; Tsai, T.-F. Anti-Interleukin and Interleukin Therapies for Psoriasis: Current Evidence and Clinical Usefulness. Ther. Adv. Musculoskelet. Dis. 2017, 9, 277–294. [Google Scholar] [CrossRef]
- Harden, J.L.; Johnson-Huang, L.M.; Chamian, M.F.; Lee, E.; Pearce, T.; Leonardi, C.L.; Haider, A.; Lowes, M.A.; Krueger, J.G. Humanized Anti-IFN-γ (HuZAF) in the Treatment of Psoriasis. J. Allergy Clin. Immunol. 2015, 135, 553–556. [Google Scholar] [CrossRef]
- Mease, P.J.; Gottlieb, A.B.; Berman, A.; Drescher, E.; Xing, J.; Wong, R.; Banerjee, S. The Efficacy and Safety of Clazakizumab, an Anti–Interleukin-6 Monoclonal Antibody, in a Phase IIb Study of Adults with Active Psoriatic Arthritis. Arthritis Rheumatol. 2016, 68, 2163–2173. [Google Scholar] [CrossRef]
- Furue, K.; Ito, T.; Tsuji, G.; Nakahara, T.; Furue, M. The CCL20 and CCR6 Axis in Psoriasis. Scand. J. Immunol. 2020, 91, e12846. [Google Scholar] [CrossRef] [Green Version]
- Bouma, G.; Zamuner, S.; Hicks, K.; Want, A.; Oliveira, J.; Choudhury, A.; Brett, S.; Robertson, D.; Felton, L.; Norris, V.; et al. CCL20 Neutralization by a Monoclonal Antibody in Healthy Subjects Selectively Inhibits Recruitment of CCR6(+) Cells in an Experimental Suction Blister. Br. J. Clin. Pharmacol. 2017, 83, 1976–1990. [Google Scholar] [CrossRef] [Green Version]
- Laffan, S.B.; Thomson, A.S.; Mai, S.; Fishman, C.; Kambara, T.; Nistala, K.; Raymond, J.T.; Chen, S.; Ramani, T.; Pageon, L.; et al. Immune Complex Disease in a Chronic Monkey Study with a Humanised, Therapeutic Antibody against CCL20 is Associated with Complement-Containing Drug Aggregates. PLoS ONE 2020, 15, e0231655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heidenreich, R.; Röcken, M.; Ghoreschi, K. Angiogenesis Drives Psoriasis Pathogenesis. Int. J. Exp. Pathol. 2009, 90, 232–248. [Google Scholar] [CrossRef] [PubMed]
- Luengas-Martinez, A.; Hardman-Smart, J.; Paus, R.; Young, H.S. Vascular Endothelial Growth Factor-A as a Promising Therapeutic Target for the Management of Psoriasis. Exp. Dermatol. 2020, 29, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Hsu, M.-C.; Chen, C.-C. Psoriasis Flare-Ups Following Sorafenib Therapy: A Rare Case. Dermatol. Sin. 2016, 34, 148–150. [Google Scholar] [CrossRef] [Green Version]
- Adachi, T.; Hiraoka, A.; Okazaki, H.; Nagamatsu, K.; Izumoto, H.; Yoshino, T.; Tsuruta, M.; Aibiki, T.; Okudaira, T.; Yamago, H.; et al. Exacerbation of Psoriasis vulgaris by Sorafenib Treatment for Hepatocellular Carcinoma. Clin. J. Gastroenterol. 2020, 13, 891–895. [Google Scholar] [CrossRef]
- Ohashi, T.; Yamamoto, T. Exacerbation of Psoriasis with Pustulation by Sorafenib in a Patient with Metastatic Hepatocellular Carcinoma. Indian J. Dermatol. 2019, 64, 75–77. [Google Scholar] [CrossRef]
- Dumet, C.; Pottier, J.; Gouilleux, V.; Watier, H. New Structural Formats of Therapeutic Antibodies for Rheumatology. Jt. Bone Spine 2018, 85, 47–52. [Google Scholar] [CrossRef]
- Papp, K.A.; Weinberg, M.A.; Morris, A.; Reich, K. IL17A/F Nanobody Sonelokimab in Patients with Plaque Psoriasis: A Multicentre, Randomised, Placebo-Controlled, Phase 2b Study. Lancet 2021, 397, 1564–1575. [Google Scholar] [CrossRef]
- Membrive Jiménez, C.; Pérez Ramírez, C.; Sánchez Martín, A.; Vieira Maroun, S.; Arias Santiago, S.A.; Ramírez Tortosa, M.D.C.; Jiménez Morales, A. Influence of Genetic Polymorphisms on Response to Biologics in Moderate-to-Severe Psoriasis. J. Pers. Med. 2021, 11, 293. [Google Scholar] [CrossRef]
- Dand, N.; Duckworth, M.; Baudry, D.; Russell, A.; Curtis, C.J.; Lee, S.H.; Evans, I.; Mason, K.J.; Alsharqi, A.; Becher, G.; et al. HLA-C*06:02 Genotype is a Predictive Biomarker of Biologic Treatment Response in Psoriasis. J. Allergy Clin. Immunol. 2019, 143, 2120–2130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konrad, R.J.; Higgs, R.E.; Rodgers, G.H.; Ming, W.; Qian, Y.-W.; Bivi, N.; Mack, J.K.; Siegel, R.W.; Nickoloff, B.J. Assessment and Clinical Relevance of Serum IL-19 Levels in Psoriasis and Atopic Dermatitis Using a Sensitive and Specific Novel Immunoassay. Sci. Rep. 2019, 9, 5211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, S.; Reddy, V.; Myers, B.; Thibodeaux, Q.; Brownstone, N.; Liao, W. Machine Learning in Dermatology: Current Applications, Opportunities, and Limitations. Dermatol. Ther. 2020, 10, 365–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]


| Autoantigen | Traditional Function | Autoantigenic Function in Psoriasis |
|---|---|---|
| Cathelicidin (LL-37) | Antimicrobial peptide that induces innate immune cell response [68]. | Activates DCs to release IFN-α [69,70]. Stimulates keratinocytes to produce IFN-α and IFN-β [71]. Activates CD4+ and CD8+ T cells [72]. |
| ADAMTSL5 | Regulates extracellular matrix microfibrils [73]. | Activates CD8+ T cells, compelling IFN-γ and IL-17 production [74]. |
| Lipids produced by PLA2G4D | PLA2G4D metabolizes lipids, producing linoleic acid [75]. | Stimulate T cells to produce IL-17 and IL-22 [76]. |
| Keratin-17 | Plays roles in wound healing and tissue development [77]. | Induces CD8+ T cells from psoriasis patients to release IFN-γ [78]. |
| Ezrin, Maspin, HSP27, PRDX2 (keratinocyte produced proteins) | Modulate cytoskeleton regulation, inhibit proteases, promote chaperoning and enhance antioxidation, respectively [79]. | Autoantibodies against these proteins have been identified. Maspin and PRDX2 induce psoriatic T cell IFN-γ release. Autoantigenic function may stem from sequence homology with streptococcal peptides [79]. |
| hnRNP-A1 | Regulates mRNA transcription and processing [80]. | Provokes an autoantibody response [81]. Facilitates immunogenic RNA–amine complex entry into DCs [67]. |
| Lysozyme, β-Defensins 2/3 | Antimicrobial agents [82]. | Bind self-DNA and activate pDCs to produce IFN-α [83]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orsmond, A.; Bereza-Malcolm, L.; Lynch, T.; March, L.; Xue, M. Skin Barrier Dysregulation in Psoriasis. Int. J. Mol. Sci. 2021, 22, 10841. https://doi.org/10.3390/ijms221910841
Orsmond A, Bereza-Malcolm L, Lynch T, March L, Xue M. Skin Barrier Dysregulation in Psoriasis. International Journal of Molecular Sciences. 2021; 22(19):10841. https://doi.org/10.3390/ijms221910841
Chicago/Turabian StyleOrsmond, Andreas, Lara Bereza-Malcolm, Tom Lynch, Lyn March, and Meilang Xue. 2021. "Skin Barrier Dysregulation in Psoriasis" International Journal of Molecular Sciences 22, no. 19: 10841. https://doi.org/10.3390/ijms221910841
APA StyleOrsmond, A., Bereza-Malcolm, L., Lynch, T., March, L., & Xue, M. (2021). Skin Barrier Dysregulation in Psoriasis. International Journal of Molecular Sciences, 22(19), 10841. https://doi.org/10.3390/ijms221910841