
Synthesis of Demissidine Analogues from Tigogenin via Imine Intermediates †



Abstract
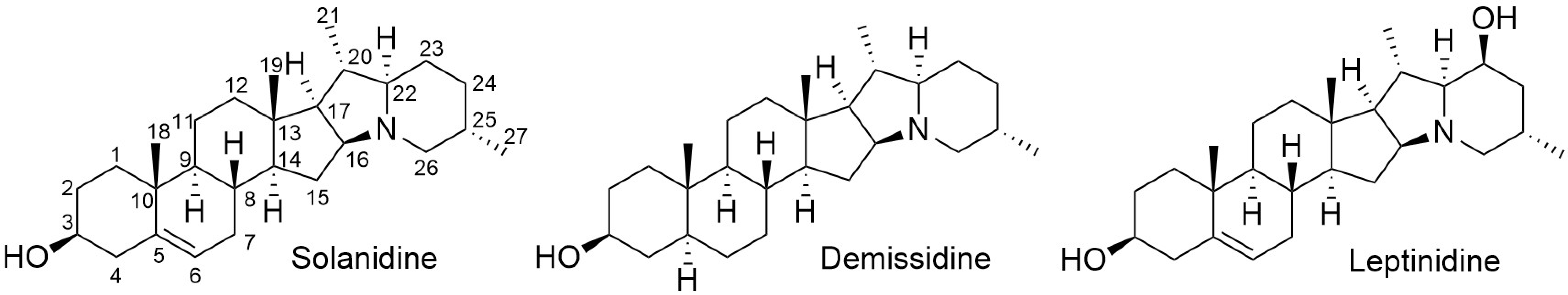
:1. Introduction
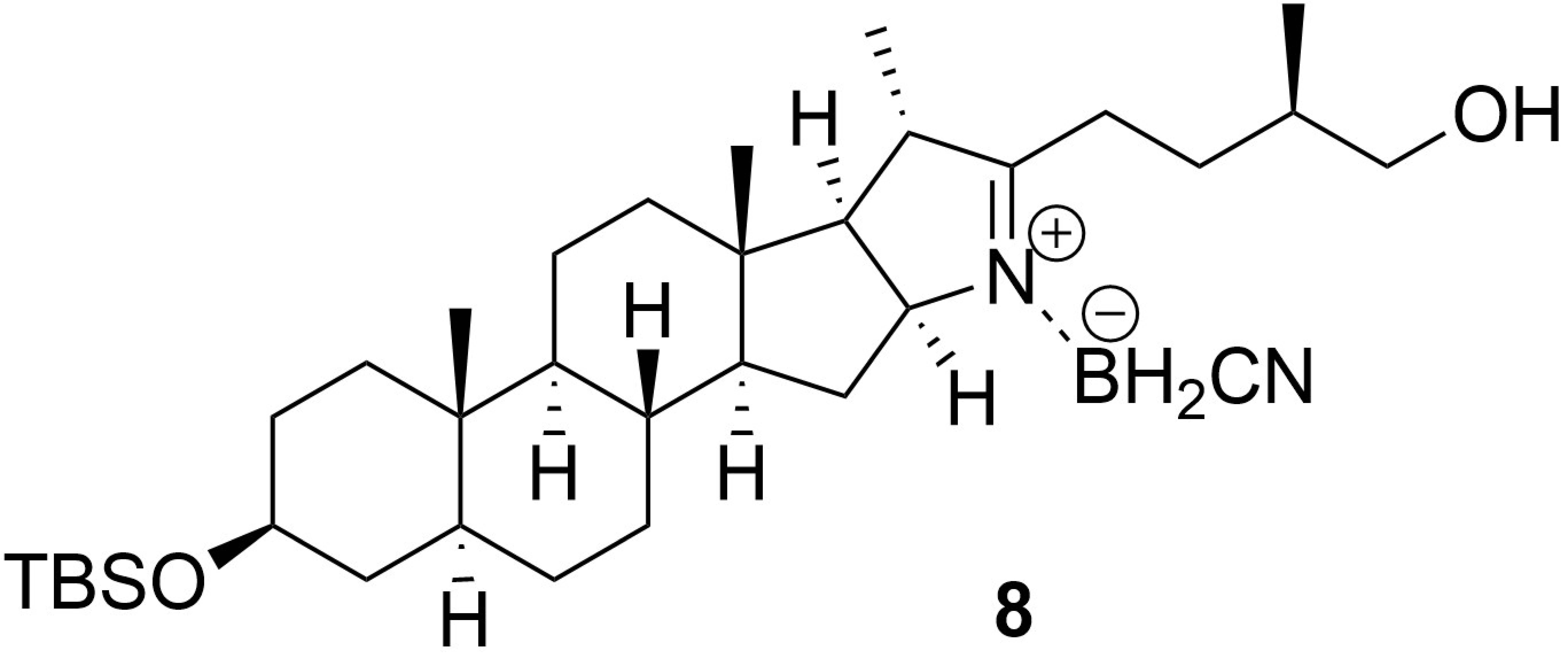
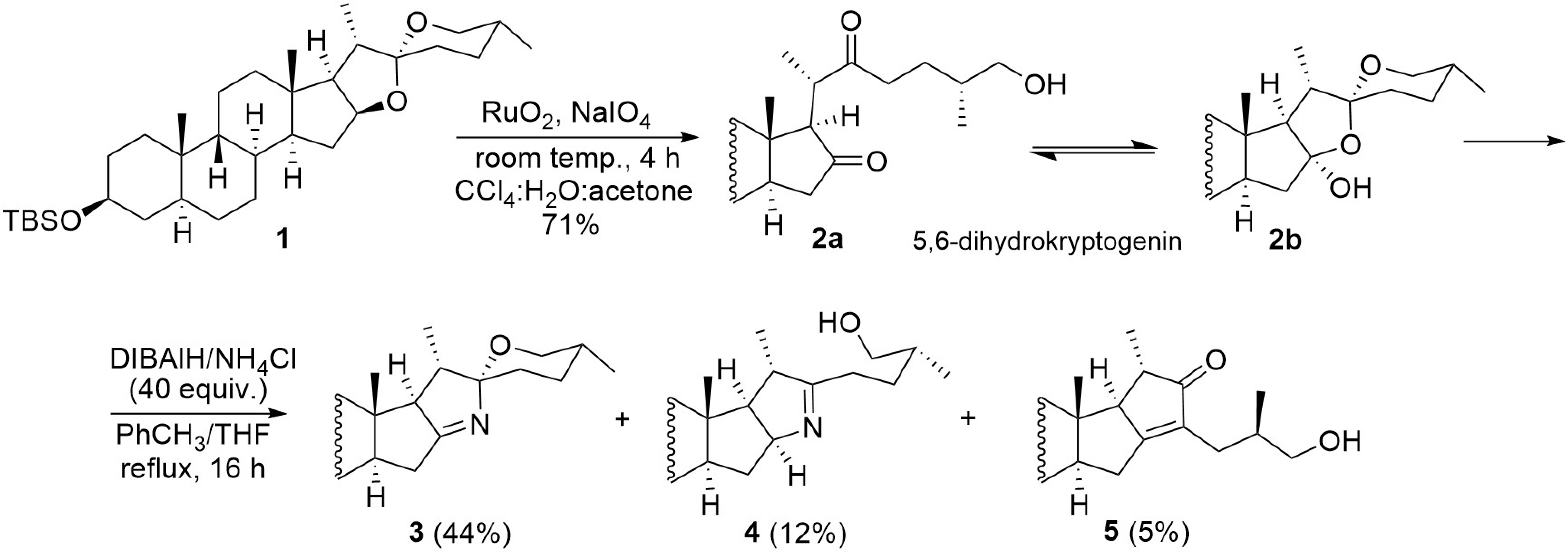
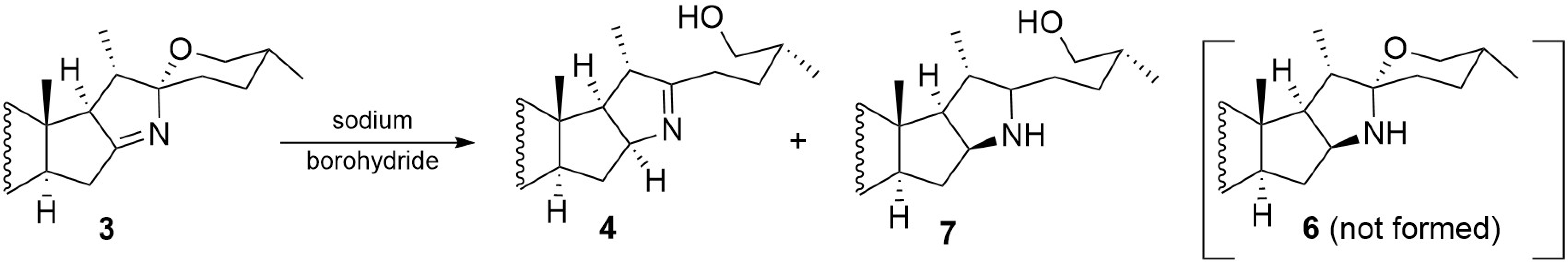
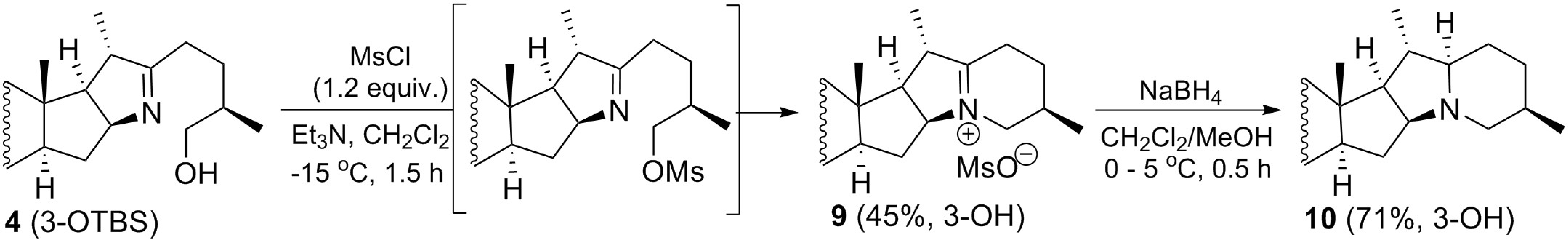
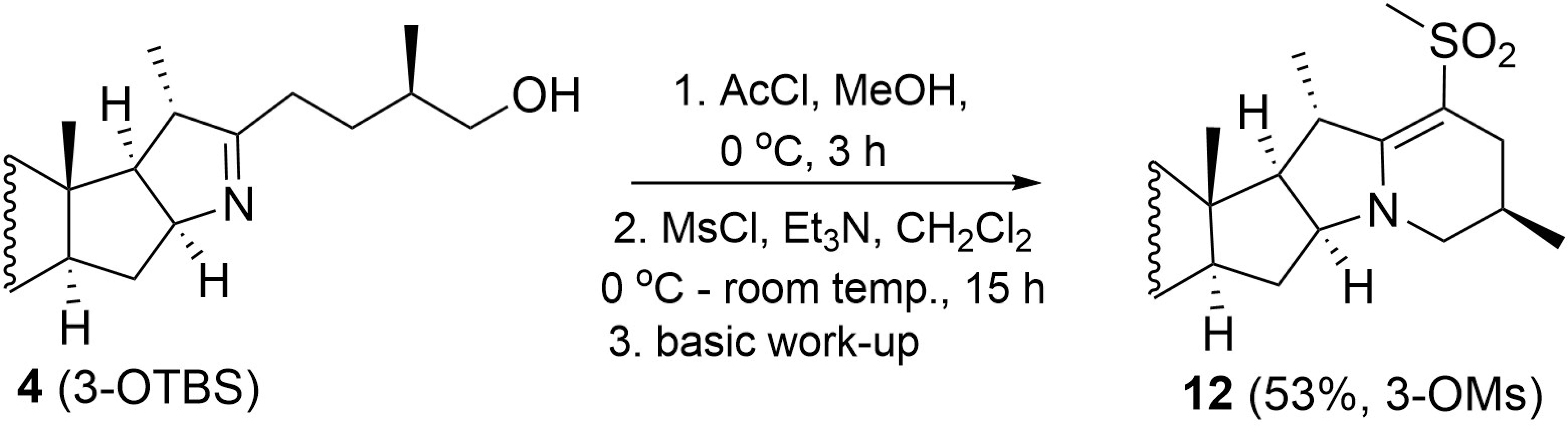
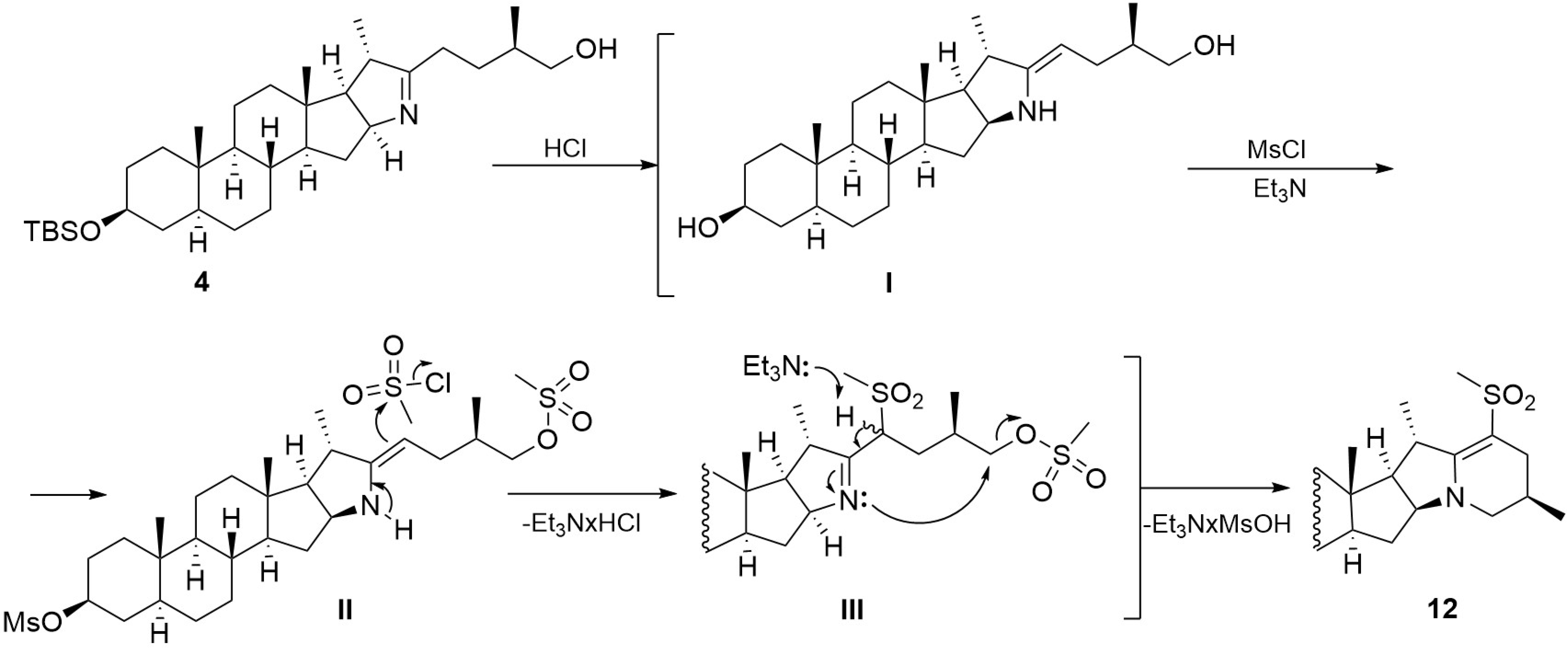
2. Results and Discussion
3. Materials and Methods
3.1. General
3.2. Chemical Synthesis
3.2.1. Oxidation of 3-TBS Tigogenin (1) with RuO4/NaIO4
3.2.2. Synthesis of (25R)-3β-t-butyldimethylsililoxy-16-aza-spirost-16(N)-ene (3)
3.2.3. General Procedure for Imine 3 Reduction with Complex Sodium Hydride
3.2.4. Synthesis of Compound 9
3.2.5. Synthesis of 25-epidemissidine (10)
3.2.6. Synthesis of Compound 12
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Attaur, R.; Choudhary, M.I. Diterpenoid and steroidal alkaloids. Nat. Prod. Rep. 1997, 14, 191–203. [Google Scholar] [CrossRef]
- Li, H.J.; Jiang, Y.; Li, P. Chemistry, bioactivity and geographical diversity of steroidal alkaloids from the Liliaceae family. Nat. Prod. Rep. 2006, 23, 735–752. [Google Scholar] [CrossRef]
- Jiang, Q.-W.; Chen, M.-W.; Cheng, K.-J.; Yu, P.-Z.; Wei, X.; Shi, Z. Therapeutic potential of steroidal alkaloids in cancer and other diseases. Med. Res. Rev. 2016, 36, 119–143. [Google Scholar] [CrossRef] [PubMed]
- Friedman, M.; Lee, K.R.; Kim, H.J.; Lee, I.S.; Kozukue, N. Anticarcinogenic effects of glycoalkaloids from potatoes against human cervical, liver, lymphoma, and stomach cancer cells. J. Agric. Food Chem. 2005, 53, 6162–6169. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.R.; Kozukue, N.; Han, J.S.; Park, J.H.; Chang, E.Y.; Baek, E.J.; Chang, J.S.; Friedman, M. Glycoalkaloids and metabolites inhibit the growth of human colon (HT29) and liver (HepG2) cancer cells. J. Agric. Food. Chem. 2004, 52, 2832–2839. [Google Scholar] [CrossRef]
- Tingey, W.M. Glycoalkaloids as pest resistance factors. Am. J. Potato. Res. 1984, 61, 157–167. [Google Scholar] [CrossRef]
- Schreiber, K. Steroid alkaloids: The Solanum group. In The Alkaloids: Chemistry and Physiology; Manske, R.H.F., Ed.; Academic Press: New York, NY, USA, 1968; Volume 10, pp. 1–192. [Google Scholar]
- Roddick, J.G. The acetylcholinesterase-inhibitory activity of steroidal glycoalkaloids and their aglycones. Phytochemistry 1989, 28, 2631–2634. [Google Scholar] [CrossRef]
- Roddick, J.G.; Weissenberg, M.; Leonard, A.L. Membrane disruption and enzyme inhibition by naturally-occurring and modified chacotriose-containing Solanum steroidal glycoalkaloids. Phytochemistry 2001, 56, 603–610. [Google Scholar] [CrossRef]
- Mótyán, G.; Kovács, F.; Wölfling, J.; Gyovai, A.; Zupkó, I.; Frank, É. Microwave-assisted stereoselective approach to novel steroidal ring D-fused 2-pyrazolines and an evaluation of their cell-growth inhibitory effects in vitro. Steroids 2016, 112, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Abdalla, M.M.; Al-Omar, M.A.; Bhat, M.A.; Amr, A.-G.E.; Al-Mohizeae, A.M. Steroidal pyrazolines evaluated as aromatase and quinone reductase-2 inhibitors for chemoprevention of cancer. Int. J. Biol. Macromol. 2012, 50, 1127–1132. [Google Scholar] [CrossRef]
- Schuster, D.; Wolber, G. Identification of bioactive natural products by pharmacophore-based virtual screening. Curr. Pharm. Des. 2010, 16, 1666–1681. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, R.; Low, I.; Trischmann, H. Uberfuhrung von tomatidin in demissidin. Angew. Chem. 1952, 64, 397–397. [Google Scholar] [CrossRef]
- Sato, Y.; Latham, H.G. Chemistry of dihydrotomatidines. J. Am. Chem. Soc. 1956, 78, 3146–3150. [Google Scholar] [CrossRef]
- Schreiber, K.; Ronsch, H. Solanum-alkaloide-LIV. Synthese von solanidin und 22-iso-solanidin aus tomatid-5-en-3β-ol. Tetrahedron 1965, 21, 645–650. [Google Scholar] [CrossRef]
- Adam, G.; Schreiber, K. Synthese des steroid alkaloids demissidin aus 3β-acetoxy-pregn-5-en-20-on; aufbau des solanidine-gerustes durch Hofmann-Loffler-Freytag-cyclisierung. Tetrahedron Lett. 1963, 4, 943–948. [Google Scholar] [CrossRef]
- Zhang, Z.; Giampa, G.M.; Draghici, C.; Huang, Q.; Brewer, M. Synthesis of demissidine by a ring fragmentation 1,3-dipolar cycloaddition approach. Org. Lett. 2013, 15, 2100–2103. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.-D.; Shi, Y.; Wu, J.-J.; Lin, J.-R.; Tian, W.-S. Synthesis of demissidine and solanidine. Org. Lett. 2016, 18, 3038–3040. [Google Scholar] [CrossRef]
- Hou, L.-L.; Shi, Y.; Zhang, Z.-D.; Wu, J.-J.; Yang, Q.-X.; Tian, W.-S. Divergent synthesis of solanidine and 22-epi-solanidine. J. Org. Chem. 2017, 82, 7463–7469. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, G.; Shi, Y.; Tian, W.-S.; Zhuang, C.; Chen, E.-F. Asymmetric synthesis of (−)-solanidine and (−)-tomatidenol. Org. Biomol. Chem. 2020, 18, 3169–3176. [Google Scholar] [CrossRef]
- Rivas-Loaiza, J.A.; Baj, A.; López, Y.; Witkowski, S.; Wojtkielewicz, A.; Morzycki, J.W. Synthesis of solanum alkaloid demissidine stereoisomers and analogues. J. Org. Chem. 2021, 86, 1575–1582. [Google Scholar] [CrossRef]
- Cheng, M.S.; Wang, Q.L.; Tian, Q.; Song, Y.H.; Liu, Y.X.; Li, Q.; Xu, X.; Miao, H.D.; Yao, X.S.; Yang, Z. Total synthesis of methyl protodioscin: A potent agent with antitumor activity. J. Org. Chem. 2003, 68, 3658–3662. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Cao, H.; Fuchs, P.L. Ruthenium-catalyzed mild C–H oxyfunctionalization of cyclic steroidal ethers. J. Org. Chem. 2007, 72, 5820–5823. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Liao, J.; Zhang, J.; Hui, Y. The first synthetic route to furostan saponins. Tetrahedron Lett. 2001, 42, 77–79. [Google Scholar] [CrossRef]
- Bovicelli, P.; Lupettelli, P.; Fracas, D. Sapogenins and dimethyldioxirane: A new entry to cholestanes functionalized at the side chain. Tetrahedron Lett. 1994, 35, 935–938. [Google Scholar] [CrossRef]
- Overman, L.E.; Sugai, S. A Convenient Method for obtaining trans-2-aminocyclohexanol and trans-2-aminocyclopentanol in enantiomerically pure form. J. Org. Chem. 1985, 50, 4154–4155. [Google Scholar] [CrossRef]
- Overman, L.E.; Flippin, L.A. Facile aminolysis of epoxides with diethylaluminum amides. Tetrahedron Lett. 1981, 22, 195–198. [Google Scholar] [CrossRef]
- Ashworth, I.W.; Bowden, M.C.; Dembofsky, B.; Levin, D.; Moss, W.; Robinson, E.; Szczur, N.; Virica, J. A new route for manufacture of 3-cyano-1-naphthalenecarboxylic acid. Org. Process. Res. Dev. 2003, 7, 74–81. [Google Scholar] [CrossRef]
- Veerasamy, N.; Carlson, E.C.; Collett, N.D.; Saha, M.; Carter, R.G. Enantioselective approach to quinolizidines: Total synthesis of cermizine D and formal syntheses of senepodine G and cermizine C. J. Org. Chem. 2013, 78, 4779–4800. [Google Scholar] [CrossRef] [Green Version]
- Bakunova, S.M.; Bakunov, S.A.; Wenzler, T.; Barszcz, T.; Werbovetz, K.A.; Brun, R.; Hall, J.E.; Tidwell, R.R. Synthesis and in vitro antiprotozoal activity of bisbenzofuran cations. J. Med. Chem. 2007, 50, 5807–5823. [Google Scholar] [CrossRef]
- Huang, S.-Q.; Zheng, X.; Deng, X.-M. DIBAL-H-H2NR and DIBAL-H-HNR1R2·HCl complexes for efficient conversion of lactones and esters to amides. Tetrahedron Lett. 2001, 42, 9039–9041. [Google Scholar] [CrossRef]
- Wojtkielewicz, A.; Łotowski, Z.; Morzycki, J.W. One-step synthesis of nitriles from acids, esters and amides using DIBAL-H and ammonium chloride. Synlett 2015, 26, 2288–2292. [Google Scholar] [CrossRef]
- Bechara, W.S.; Charette, A.B.; Na, R.; Wang, W.; Zheng, C. Diethyl 1,4-dihydro-2,6-dimethyl-3,5-pyridinedicarboxylate. In Encyclopedia of Reagents for Organic Synthesis 2020; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2020. [Google Scholar] [CrossRef]
- Czajkowska-Szczykowska, D.; Jastrzebska, I.; Rode, J.E.; Morzycki, J.W. Revision of the structure of N,O-diacetylsolasodine. Unusual epimerization at the spiro carbon atom during acetylation of solasodine. J. Nat. Prod. 2019, 82, 59–65. [Google Scholar] [CrossRef]
- Chagnon, F.; Guay, I.; Bonin, M.-A.; Mitchell, G.; Bouarab, K.; Malouin, F.; Marsault, É. Unraveling the structure-reactivity relationship of tomatidine, a steroid alkaloid with unique antibiotic properties against persistent forms of Staphylococcus aureus. Eur. J. Med. Chem. 2014, 80, 605–620. [Google Scholar] [CrossRef]
- Zha, X.M.; Zhang, F.R.; Shan, J.Q.; Zhang, Y.H.; Liu, J.O.; Sun, H.B. Synthesis and evaluation of in vitro anticancer activity of novel solasodine derivatives. Chin. Chem. Lett. 2010, 21, 1087–1090. [Google Scholar] [CrossRef]
- Uhle, F.C.; Sallmann, F. The synthesis and transformations of a steroid pyrroline Derivative. J. Am. Chem. Soc. 1960, 82, 1190–1199. [Google Scholar] [CrossRef]
- Reddy, C.R.; Ramesh, P.; Latha, B. Formal syntheses of 5,8-disubstituted indolizidine alkaloids (–)-205A, (–)-207A, and (–)-235B. Synlett 2016, 27, 481–484. [Google Scholar] [CrossRef]
- Donohoe, T.J.; Sintim, H.O.; Hollinshead, J. A Noncarbohydrate Based Approach to Polyhydroxylated. J. Org. Chem. 2005, 70, 7297–7304. [Google Scholar] [CrossRef]
- Mátyus, P.; Szilágyi, G.; Kasztreiner, E.; Sohár, P. Studies on pyridazine derivatives. IV. Mesylation reaction of pyridazinylpyrazoles. J. Heterocycl. Chem. 1980, 17, 781–783. [Google Scholar] [CrossRef]
- Back, T.G.; Hu, N.-X. Synthesis of Antibiotic A25822 B. Tetrahedron 1993, 49, 337–348. [Google Scholar] [CrossRef]
- Cheng, S.; Du, Y.; Mac, B.; Tanc, D. Total synthesis of a furostan saponin, timosaponin BII. Org. Biomol. Chem. 2009, 7, 3112–3118. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
| Entry | Reagent (equiv.) | Conditions | Product 4 Yield (%) | Product 7 Yield (%) | Substrate Recovery (%) |
| 1 | NaBH4 (2) | MeOH/CH2Cl2, −18 °C, 1 h | 64 | 30 | <1 |
| 2 | NaBH4 (1) | MeOH/CH2Cl2, −18 °C, 1 h | 58 | <5 | 21 |
| 3 | NaBH4 (1) | MeOH/CH2Cl2, 0 °C, 1 h | 67 | 13 | <5 |
| 4 | NaBH4 (1) | NaOAc (1 equiv.), MeOH/CH2Cl2, 0 °C–room temp., 2 h | 65 | <5 | 30 |
| 5 | NaBH4 (4) | I2 (4 equiv.), MeOH/ CH2Cl2, 0 °C–reflux, 16 h | 78 | 9 | <1 |
| 6 | NaBH3CN (2) | AcOH (2 equiv.), THF, room temp., 1 h | complex 8 (48) | nd * | <5 |
| 7 | NaBH3CN (2) | THF/MeOH, room temp., 2 h | 28 | nd * | 30 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wojtkielewicz, A.; Kiełczewska, U.; Baj, A.; Morzycki, J.W. Synthesis of Demissidine Analogues from Tigogenin via Imine Intermediates. Int. J. Mol. Sci. 2021, 22, 10879. https://doi.org/10.3390/ijms221910879
Wojtkielewicz A, Kiełczewska U, Baj A, Morzycki JW. Synthesis of Demissidine Analogues from Tigogenin via Imine Intermediates. International Journal of Molecular Sciences. 2021; 22(19):10879. https://doi.org/10.3390/ijms221910879
Chicago/Turabian StyleWojtkielewicz, Agnieszka, Urszula Kiełczewska, Aneta Baj, and Jacek W. Morzycki. 2021. "Synthesis of Demissidine Analogues from Tigogenin via Imine Intermediates" International Journal of Molecular Sciences 22, no. 19: 10879. https://doi.org/10.3390/ijms221910879
APA StyleWojtkielewicz, A., Kiełczewska, U., Baj, A., & Morzycki, J. W. (2021). Synthesis of Demissidine Analogues from Tigogenin via Imine Intermediates. International Journal of Molecular Sciences, 22(19), 10879. https://doi.org/10.3390/ijms221910879