QTL Mapping and Candidate Gene Identification of Swollen Root Formation in Turnip

 ,
,  , and
, and
Abstract
:1. Introduction
2. Results
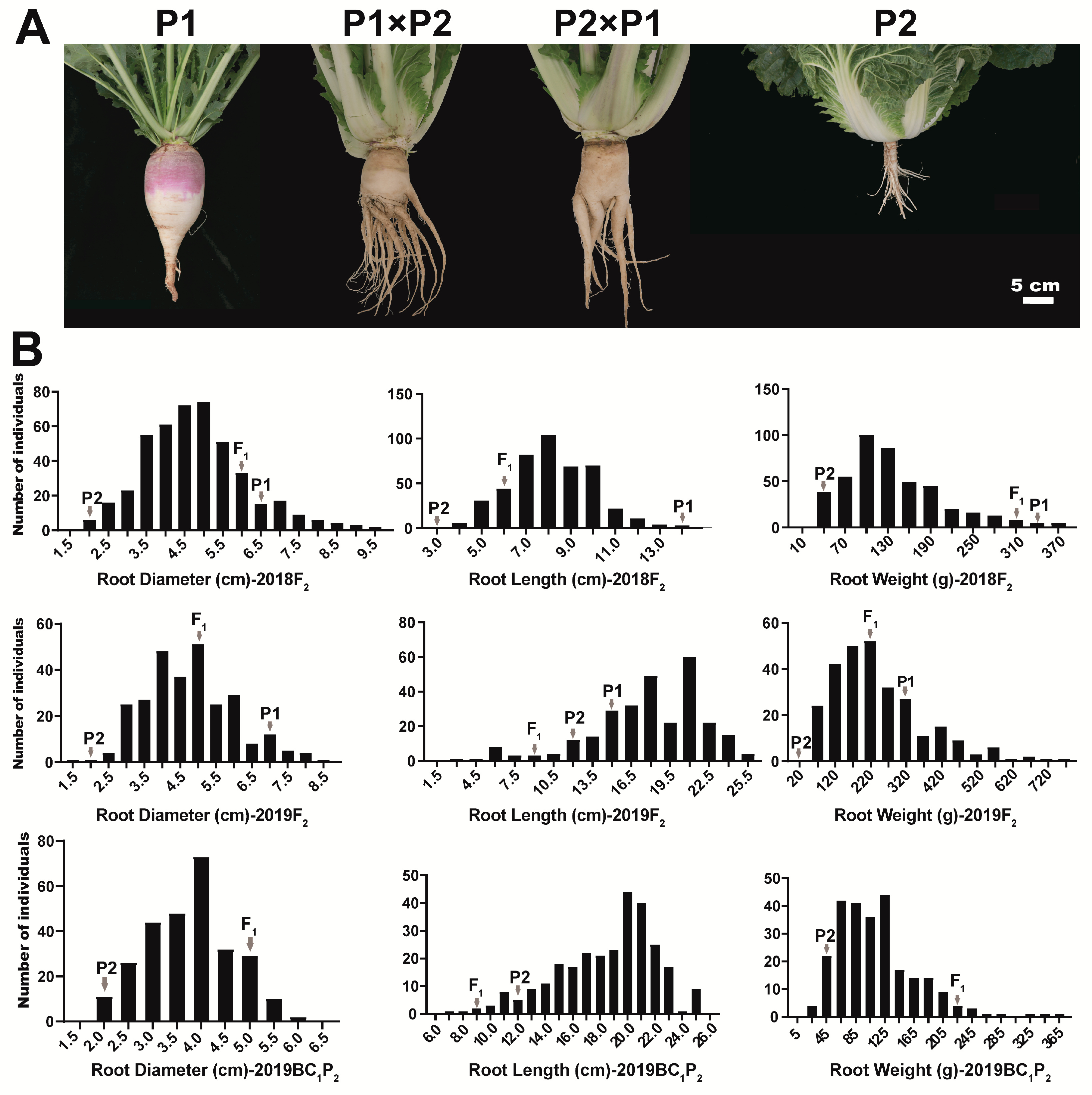
2.1. Inheritance of Swollen Root in B. rapa
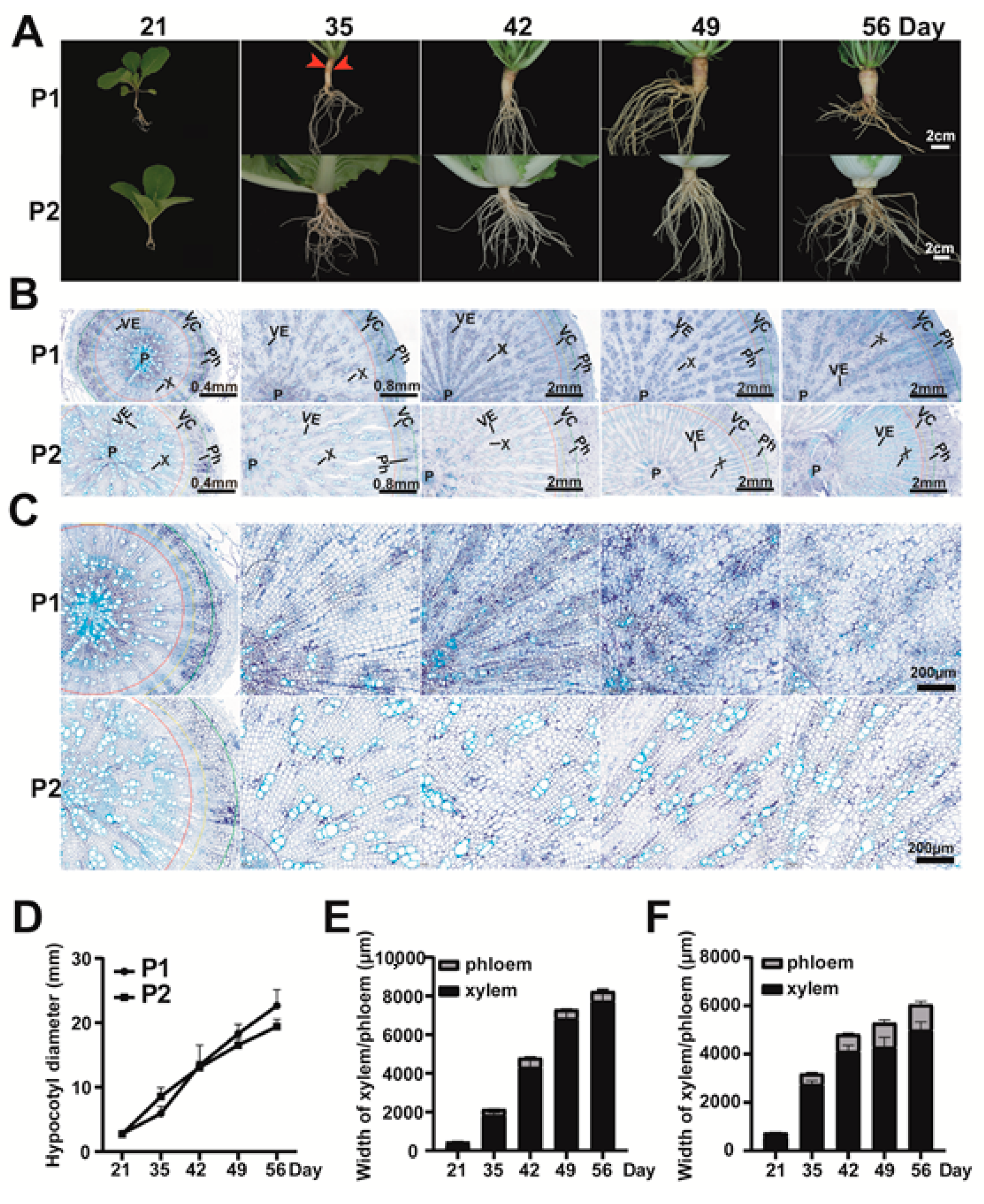
2.2. Phenotypic and Anatomical Observation of the Hypocotyl and Root from Parents
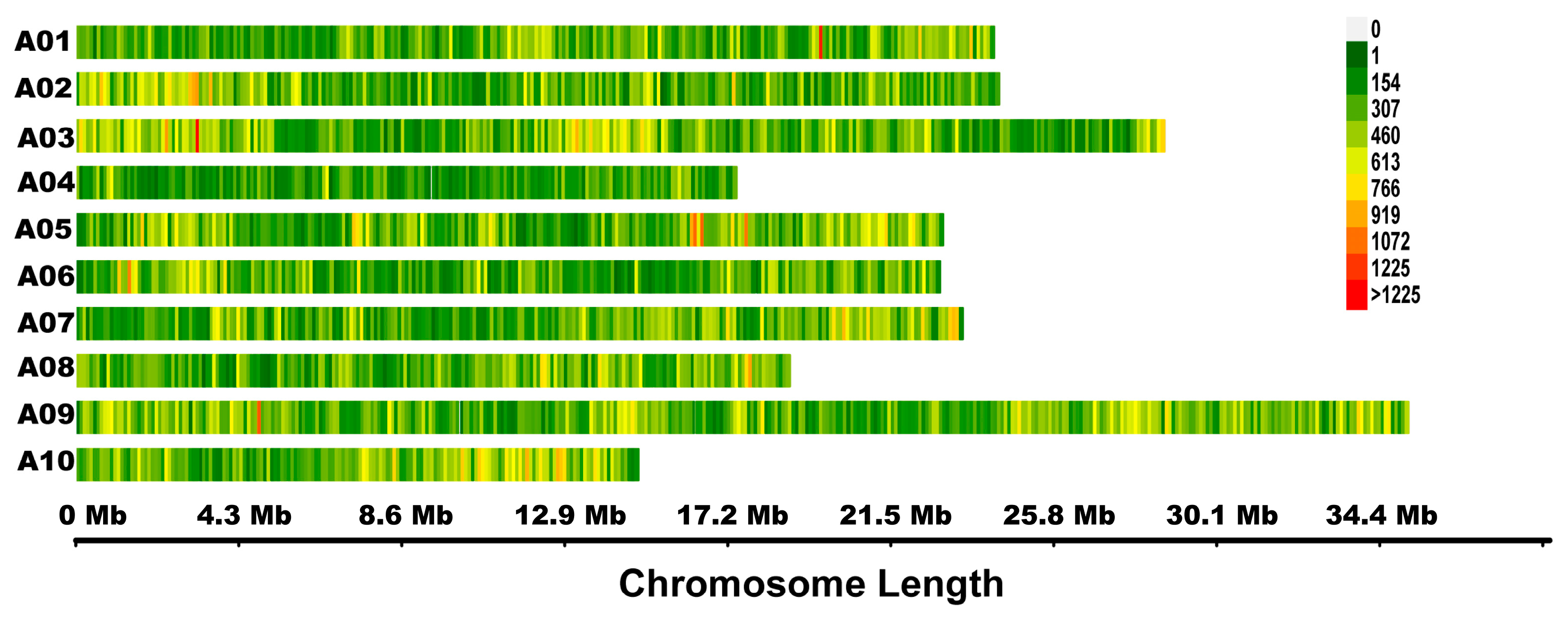
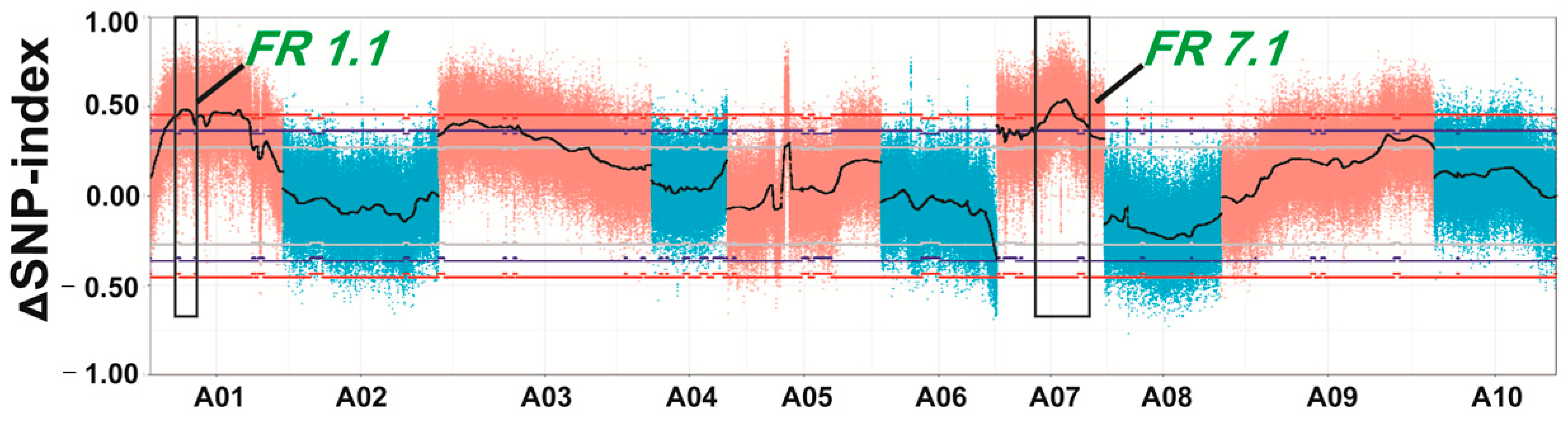
2.3. Quantitative Trait Loci (QTL)-seq Predicted Candidate Regions for Controlling the Swollen Root Traits
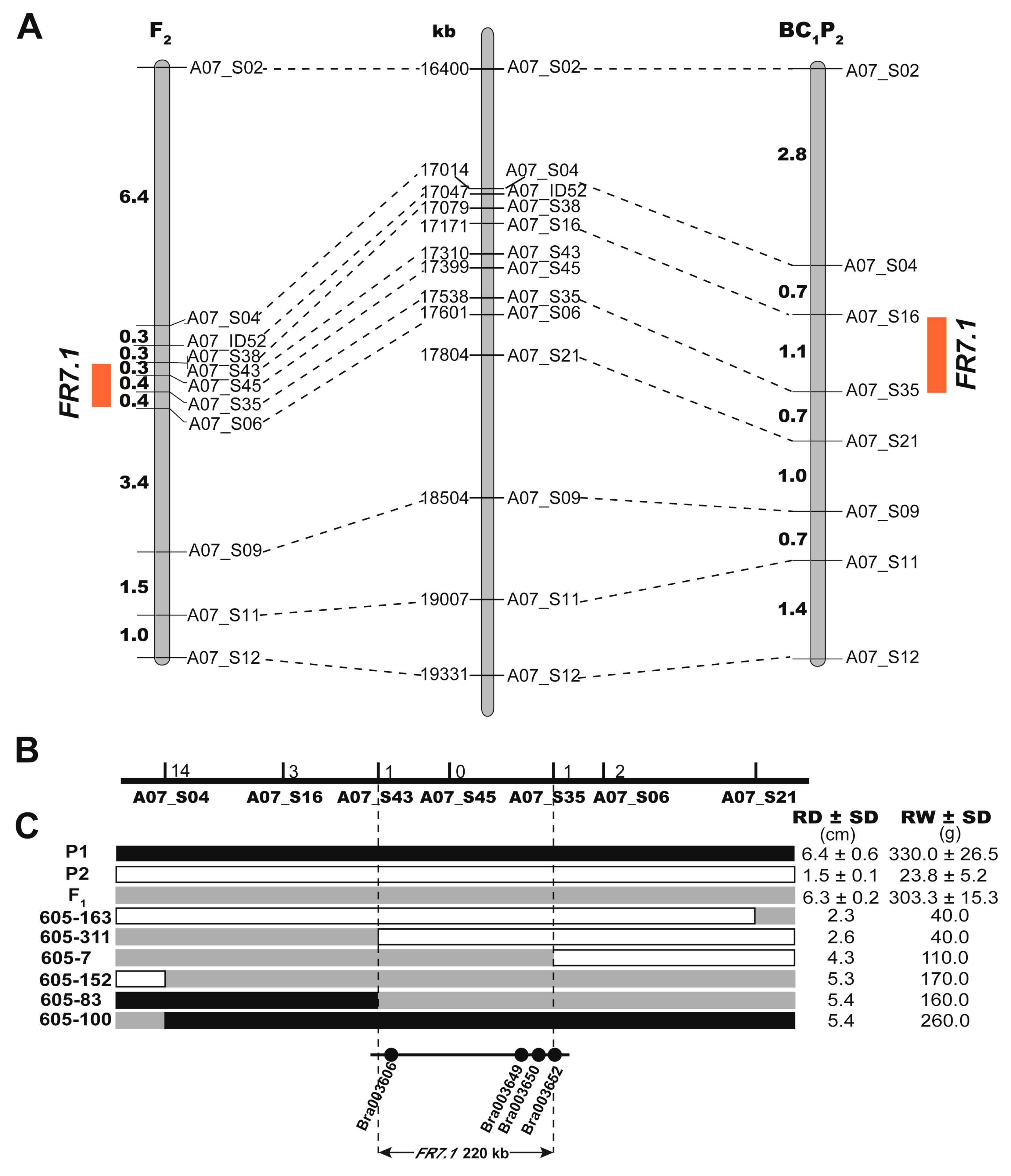
2.4. Confirmation and Fine-Mapping of the Two QTLs: FR1.1 and FR7.1
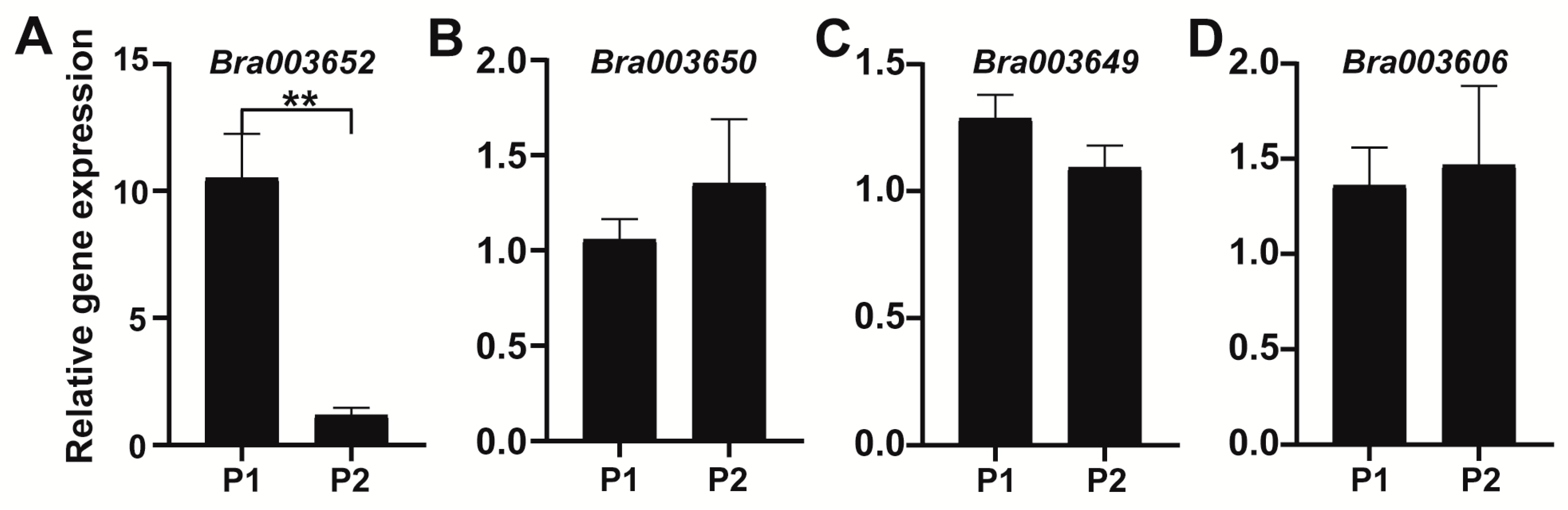
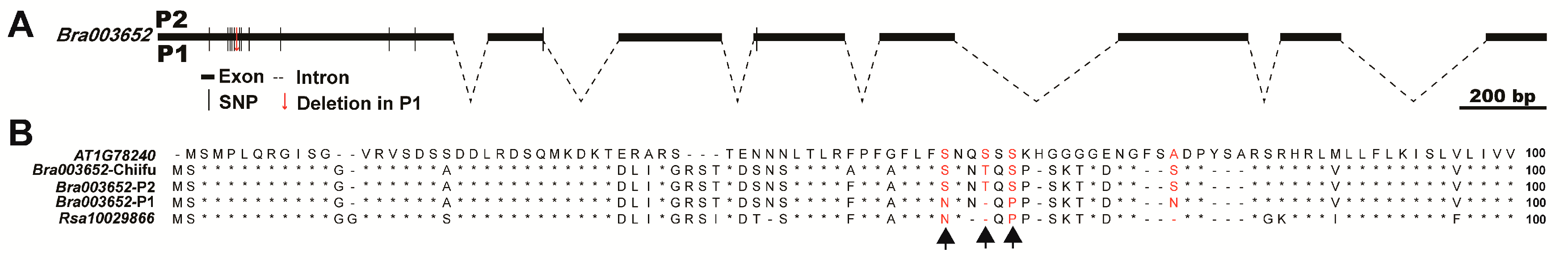
2.5. Identification of Candidate Genes for FR7.1 in Turnip
3. Discussion
3.1. QTL-seq Technology Accelerated the Research of Root-Related Traits
3.2. The Authenticity and Reliability of FR1.1 and FR7.1
3.3. Analysis and Determination of Candidate Genes
3.4. Root-Related Traits Positively Affect the Yield of Root Crops
4. Plant Materials and Methods
4.1. Parent Materials and Population Construction
4.2. Phenotype Assessment
4.3. QTL-seq Analysis
4.4. Molecular Marker Development, Linkage Analysis, and QTL Mapping
4.5. RNA Extraction and Quantitative Real-Time PCR
4.6. Histological Analysis of Hypocotyls
4.7. Sequence Analysis of Candidate Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| QTL | Quantitative trait loci |
| LOD | Likelihood of odd |
| qRT-PCR | Quantitative real-time polymerase chain reaction |
| SNP | Single nucleotide polymorphisms |
| InDel | Insertion and deletion |
| DNA | Deoxyribonucleic acid |
| RNA | Ribonucleic acid |
References
- Shattuck, V.I.; Kakuda, Y.; Shelp, B.J.; Kakuda, N. Chemical composition of turnip roots stored or intermittently grown at low temperature. J. Am. Soc. Hortic. Sci. 1991, 116, 818–822. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.K.; Singh, J.; Kaur, N. Sink development, sucrose metabolising enzymes and carbohydrate status in turnip (Brassica rapa L.). Acta Physiol. Plant. 2001, 23, 31–36. [Google Scholar] [CrossRef]
- Lu, G.; Cao, J.; Yu, X.; Xiang, X.; Chen, H. Mapping QTLs for root morphological traits in Brassica rapa L. based on AFLP and RAPD markers. J. Appl. Genet. 2008, 49, 23–31. [Google Scholar] [CrossRef]
- Vogl, C.R.; Reiner, H.; Vogl-Lukasser, B.J.E.R. The Turnip (Brassica rapa L. subsp. rapa) in Eastern Tyrol (Lienz district; Austria). Ethnobot. Res. Appl. 2007, 5, 305–317. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Zhao, J.; Lens, F.; de Visser, J.; Menamo, T.; Fang, W.; Xiao, D.; Bucher, J.; Basnet, R.K.; Lin, K.; et al. Morphology, carbohydrate composition and vernalization response in a genetically diverse collection of Asian and European turnips (Brassica rapa subsp. rapa). PLoS ONE 2014, 9, e114241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Bassetti, N.; Petrasch, S.; Zhang, N.; Bucher, J.; Shen, S.; Zhao, J.; Bonnema, G. What makes turnips: Anatomy, physiology and transcriptome during early stages of its hypocotyl-tuber development. Hortic. Res. 2019, 6, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubo, N.; Saito, M.; Tsukazaki, H.; Kondo, T.; Matsumoto, S.; Hirai, M. Detection of quantitative trait loci controlling morphological traits in Brassica rapa L. Breed. Sci. 2010, 60, 164–171. [Google Scholar] [CrossRef] [Green Version]
- Lou, P.; Zhao, J.; Kim, J.S.; Shen, S.; Del Carpio, D.P.; Song, X.; Jin, M.; Vreugdenhil, D.; Wang, X.; Koornneef, M.; et al. Quantitative trait loci for flowering time and morphological traits in multiple populations of Brassica rapa. J. Exp. Bot. 2007, 58, 4005–4016. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wang, H.; Wang, J.; Sun, R.; Wu, J.; Liu, S.; Bai, Y.; Mun, J.-H.; Bancroft, I.; Cheng, F.; et al. The genome of the mesopolyploid crop species Brassica rapa. Nat. Genet. 2011, 43, 1035–1039. [Google Scholar] [CrossRef] [Green Version]
- Witzel, K.; Neugart, S.; Ruppel, S.; Schreiner, M.; Wiesner, M.; Baldermann, S. Recent progress in the use of ‘omics technologies in brassicaceous vegetables. Front. Plant Sci. 2015, 6, 244. [Google Scholar] [CrossRef] [Green Version]
- Cheng, F.; Sun, R.; Hou, X.; Zheng, H.; Zhang, F.; Zhang, Y.; Liu, B.; Liang, J.; Zhuang, M.; Liu, Y.; et al. Subgenome parallel selection is associated with morphotype diversification and convergent crop domestication in Brassica rapa and Brassica oleracea. Nat. Genet. 2016, 48, 1218–1224. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Fan, P.; Li, Y. High-throughput sequence analysis of small RNAs in skotomorphogenic seedlings of Brassica rapa ssp. rapa. Gene 2014, 548, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Hearn, D.J.; O’Brien, P.; Poulsen, T.M. Comparative transcriptomics reveals shared gene expression changes during independent evolutionary origins of stem and hypocotyl/root tubers in Brassica (Brassicaceae). PLoS ONE 2018, 13, e0197166. [Google Scholar] [CrossRef] [PubMed]
- Usuda, H.; Demura, T.; Shimogawara, K.; Fukuda, H. Development of sink capacity of the “Storage Root” in a radish cultivar with a high ratio of “Storage Root” to shoot. Plant Cell Physiol. 1999, 40, 369–377. [Google Scholar] [CrossRef] [Green Version]
- Luo, X.; Huang, Q. Relationships between leaf and stem soluble sugar content and tuberous root starch accumulation in cassava. J. Agric. Sci. 2011, 3. [Google Scholar] [CrossRef]
- Rouhier, H.; Usuda, H. Spatial and temporal distribution of sucrose synthase in the radish hypocotyl in relation to thickening growth. Plant Cell Physiol. 2001, 42, 583–593. [Google Scholar] [CrossRef] [Green Version]
- Mitsui, Y.; Shimomura, M.; Komatsu, K.; Namiki, N.; Shibata-Hatta, M.; Imai, M.; Katayose, Y.; Mukai, Y.; Kanamori, H.; Kurita, K.; et al. The radish genome and comprehensive gene expression profile of tuberous root formation and development. Sci. Rep. 2015, 5, 10835. [Google Scholar] [CrossRef] [Green Version]
- Fan, M.; Liu, Z.; Zhou, L.; Lin, T.; Liu, Y.; Luo, L. Effects of plant growth regulators and saccharide on in vitro plant and tuberous root regeneration of cassava (Manihot esculenta Crantz). J. Plant Growth Regul. 2010, 30, 11–19. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, L.; Guan, Y.; Wang, Z. Endogenous hormone concentration in developing tuberous roots of different sweet potato genotypes. Agric. Sci. China 2006, 5, 919–927. [Google Scholar] [CrossRef]
- Roumeliotis, E.; Kloosterman, B.; Oortwijn, M.; Lange, T.; Visser, R.G.; Bachem, C.W. Down regulation of StGA3ox genes in potato results in altered GA content and affect plant and tuber growth characteristics. J. Plant Physiol. 2013, 170, 1228–1234. [Google Scholar] [CrossRef]
- Gao, J.; Cao, X.; Shi, S.; Ma, Y.; Wang, K.; Liu, S.; Chen, D.; Chen, Q.; Ma, H. Genome-wide survey of Aux/IAA gene family members in potato (Solanum tuberosum): Identification, expression analysis, and evaluation of their roles in tuber development. Biochem. Biophys. Res. Commun. 2016, 471, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Koda, Y.; Ohkawa-Takahashi, K.; Kikuta, Y. Stimulation of root thickening and inhibition of bolting by jasmonic acid in beet plants. Plant Prod. Sci. 2015, 4, 131–135. [Google Scholar] [CrossRef]
- Jackson, S.D. Multiple signaling pathways control tuber induction in potato. Plant Physiol. 1999, 119, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannapel, D.J.; Sharma, P.; Lin, T.; Banerjee, A.K. The multiple signals that control tuber formation. Plant Physiol. 2017, 174, 845–856. [Google Scholar] [CrossRef] [Green Version]
- Kumar, D.; Wareing, P.F. Studies on tuberization in Solanum andigena: I. Evidence for the existence and movement of a specific tuberization stimulus. New Phytol. 1973, 72, 283–287. [Google Scholar] [CrossRef]
- Xie, Y.; Xu, L.; Wang, Y.; Fan, L.; Chen, Y.; Tang, M.; Luo, X.; Liu, L. Comparative proteomic analysis provides insight into a complex regulatory network of taproot formation in radish (Raphanus sativus L.). Hortic. Res. 2018, 5, 51. [Google Scholar] [CrossRef] [Green Version]
- Yu, R.; Wang, Y.; Xu, L.; Zhu, X.; Zhang, W.; Wang, R.; Gong, Y.; Limera, C.; Liu, L. Transcriptome profiling of root microRNAs reveals novel insights into taproot thickening in radish (Raphanus sativus L.). BMC Plant. Biol. 2015, 15, 30. [Google Scholar] [CrossRef] [Green Version]
- Aksenova, N.P.; Konstantinova, T.N.; Golyanovskaya, S.A.; Sergeeva, L.I.; Romanov, G.A. Hormonal regulation of tuber formation in potato plants. Russ. J. Plant Physiol. 2012, 59, 451–466. [Google Scholar] [CrossRef]
- Zhang, L.; Li, Z.; Garraway, J.; Cai, Q.; Zhou, Y.; Li, X.; Hu, Z.; Zhang, M.; Yang, J. The casein kinase 2 beta subunit CK2B1 is required for swollen stem formation via cell cycle control in vegetable Brassica juncea. Plant J. 2020, 104, 706–717. [Google Scholar] [CrossRef]
- Krupkova, E.; Immerzeel, P.; Pauly, M.; Schmulling, T. The TUMOROUS SHOOT DEVELOPMENT2 gene of Arabidopsis encoding a putative methyltransferase is required for cell adhesion and co-ordinated plant development. Plant J. 2007, 50, 735–750. [Google Scholar] [CrossRef]
- Qin, Y.; Cheng, P.; Cheng, Y.; Feng, Y.; Huang, D.; Huang, T.; Song, X.; Ying, J. QTL-Seq identified a major QTL for grain length and weight in rice using near isogenic F2 population. Rice Sci. 2018, 25, 121–131. [Google Scholar] [CrossRef]
- Shu, J.; Liu, Y.; Zhang, L.; Li, Z.; Fang, Z.; Yang, L.; Zhuang, M.; Zhang, Y.; Lv, H. QTL-seq for rapid identification of candidate genes for flowering time in broccoli × cabbage. Theor. Appl. Genet. 2018, 131, 917–928. [Google Scholar] [CrossRef]
- Takagi, H.; Abe, A.; Yoshida, K.; Kosugi, S.; Natsume, S.; Mitsuoka, C.; Uemura, A.; Utsushi, H.; Tamiru, M.; Takuno, S.; et al. QTL-seq: Rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J. 2013, 74, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Chen, H.; Liu, J.; Luo, W.; Xie, D.; Luo, S.; Wu, T.; Akram, W.; Zhong, Y. A high-density genetic map developed by specific-locus amplified fragment (SLAF) sequencing and identification of a locus controlling anthocyanin pigmentation in stalk of Zicaitai (Brassica rapa L. ssp. chinensis var. purpurea). BMC Genom. 2019, 20, 343. [Google Scholar] [CrossRef] [PubMed]
- Semagn, K.; Babu, R.; Hearne, S.; Olsen, M.J.M.B. Single nucleotide polymorphism genotyping using Kompetitive Allele Specific PCR (KASP): Overview of the technology and its application in crop improvement. Mol. Breed. 2014, 33, 1–14. [Google Scholar] [CrossRef]
- Noh, S.A.; Lee, H.S.; Huh, E.J.; Huh, G.H.; Paek, K.H.; Shin, J.S.; Bae, J.M. SRD1 is involved in the auxin-mediated initial thickening growth of storage root by enhancing proliferation of metaxylem and cambium cells in sweetpotato (Ipomoea batatas). J. Exp. Bot. 2010, 61, 1337–1349. [Google Scholar] [CrossRef] [PubMed]
- Qu, L.; Wu, C.; Zhang, F.; Wu, Y.; Fang, C.; Jin, C.; Liu, X.; Luo, J. Rice putative methyltransferase gene OsTSD2 is required for root development involving pectin modification. J. Exp. Bot. 2016, 67, 5349–5362. [Google Scholar] [CrossRef] [Green Version]
- Kaur, I.; Singh, R.; Singh, D. Correlation and path coefficient analysis for yield components and quality traits in radish (Raphanus sativus L.). Agric. Res. J. 2017, 54. [Google Scholar] [CrossRef]
- Gurmu, F.; Shimelis, H.A.; Laing, M.D. Correlation and path-coefficient analyses of root yield and related traits among selected sweetpotato genotypes. S. Afr. J. Plant Soil. 2017, 35, 179–186. [Google Scholar] [CrossRef]
- Murray, M.G.; Thompson, W.F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 1980, 8, 4321–4325. [Google Scholar] [CrossRef] [Green Version]
- Dai, M.; Thompson, R.C.; Maher, C.; Contreras-Galindo, R.; Kaplan, M.H.; Markovitz, D.M.; Omenn, G.; Meng, F. NGSQC: Cross-platform quality analysis pipeline for deep sequencing data. BMC Genomics 2010, 11 (Suppl. 4), S7. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, A.; Kosugi, S.; Yoshida, K.; Natsume, S.; Takagi, H.; Kanzaki, H.; Matsumura, H.; Yoshida, K.; Mitsuoka, C.; Tamiru, M.; et al. Genome sequencing reveals agronomically important loci in rice using MutMap. Nat. Biotechnol. 2012, 30, 174–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Ooijen, J.W.; van’t Verlaat, J.W.; van Tol, J.; Dalen, J.; Buren, J.; van der Meer, J.; van Krieken, J.H.; van Kessel, J.; Voorips, R.; van den Heuvel, L. JoinMap 4.0: Software for the Calculation of Genetic Linkage Maps in Experimental Population; Kyazma BV, Netherlands: Wageningen, The Netherlands, 2006; Available online: https://www.scienceopen.com/document?vid=baa76c8c-fb55-4c13-a6ca-24c71002ab5a (accessed on 10 January 2021).
- Van Ooijen, J.W.; Boer, M.P.; Jansen, R.C.; Maliepaard, C. MapQTL 4.0: Software for the calculation of QTL positions on genetic maps (user manual). Plant Res. Int. 2000. Available online: https://research.wur.nl/en/publications/mapqtl-40-software-for-the-calculation-of-qtl-positions-on-geneti (accessed on 10 January 2021).
- Xiao, D.; Zhang, N.; Zhao, J.; Bonnema, G.; Hou, X. Validation of reference genes for real-time quantitative PCR normalisation in non-heading Chinese cabbage. Funct. Plant. Biol. 2012, 39, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 2000, 408, 796–815. [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Yue, Z.; Mei, S.; Qiu, Y.; Yang, X.; Chen, X.; Cheng, F.; Wu, Z.; Sun, Y.; Jing, Y.; et al. A de novo genome of a Chinese radish cultivar. Hortic. Plant J. 2015, 1, 155–164. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | Year | P1 | P2 | F1 | F2 | BC1P2 | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mean ± SE | Mean ± SE | Mean ± SE | Mean ± SE | Range | CV% | Mean ± SE | Range | CV% | ||
| RD (cm) | 2018 | 6.4 ± 0.2 A | 1.5 ± 0.1 B | 6.3 ± 0.1 A | 4.6 ± 0.1 | 1.8–11.2 | 30.2 | − | − | − |
| 2019 | 7.0 ± 0.4 A | 1.9 ± 0.1 C | 4.8 ± 0.04 B | 4.6 ± 0.1 | 1.5–8.5 | 25.5 | 3.6 ± 0.1 | 1.6–6.0 | 24.7 | |
| RW (g) | 2018 | 330.0 ± 8.4 A | 23.8 ± 1.8 B | 303.3 ± 8.8 A | 137.5 ± 4.3 | 10.0–680.0 | 66.8 | − | − | − |
| 2019 | 316.0 ± 16.6 A | 30.8 ± 2.5 C | 198.0 ± 22.1 B | 220.7 ± 7.8 | 30.0–690.0 | 58.6 | 102.2 ± 3.5 | 10.0–340.0 | 53.3 | |
| RL (cm) | 2018 | 13.5 ± 0.5 A | 2.5 ± 0.2 C | 5.5 ± 0.8 B | 7.7 ± 0.1 | 3.1–17.2 | 24.5 | − | − | − |
| 2019 | 14.1 ± 1.5 A | 12.0 ± 0.6 A | 8.3 ± 0.5 B | 17.2 ± 0.3 | 3.0–25.0 | 24.5 | 18.6 ± 0.2 | 8.0–25.0 | 19.0 | |
| Primer Name | Physical Position (bp) a | F2-2018 | BC1P2-2019 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| RD | RW | RD | RW | ||||||
| LOD | %Exp b | LOD | %Exp | LOD | %Exp | LOD | %Exp | ||
| A07_S04 | 17014037-17014237 | 0.08 | 0.20 | 0.08 | 0.20 | 0.10 | 0.30 | 0.95 | 2.80 |
| A07_ID52 | 17046755-17047055 | 0.08 | 0.20 | 0.04 | 0.10 | − | − | − | − |
| A07_S38 | 17079762-17079962 | 0.12 | 0.30 | 0.07 | 0.10 | − | − | − | − |
| A07_S16 | 17171621-17171821 | − | − | − | − | 4.59 | 13.10 | 5.00 | 16.00 |
| A07_S43 | 17310743-17310943 | 0.12 | 0.30 | 0.07 | 0.10 | − | − | − | − |
| A07_S45 | 17399470-17399670 | 9.38 | 23.00 | 13.27 | 31.00 | − | − | − | − |
| A07_S35 | 17538617-17538817 | 9.38 | 23.00 | 13.27 | 31.00 | 4.40 | 12.60 | 4.38 | 14.20 |
| A07_S06 | 17601827-17602027 | 0.76 | 1.60 | 0.56 | 1.10 | − | − | − | − |
| A07_S21 | 17804469-17804669 | − | − | − | − | 1.31 | 3.40 | 0.20 | 0.60 |
| Primer Name | Physical Position (bp) | RD | |
|---|---|---|---|
| LOD Value | %Exp | ||
| A01_ID03 | A01_4623486-4623786 | 0.86 | 1.90 |
| A01_ID27 | A01_4798250-4798550 | 6.22 | 15.40 |
| A01_ID04 | A01_4831809-4832109 | 7.01 | 17.20 |
| A01_ID31 | A01_5291260-5291560 | 0.28 | 0.60 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Y.; Zhang, S.; Zhang, H.; Li, F.; Li, G.; Fan, C.; Sun, R.; Zhang, S. QTL Mapping and Candidate Gene Identification of Swollen Root Formation in Turnip. Int. J. Mol. Sci. 2021, 22, 653. https://doi.org/10.3390/ijms22020653
Wu Y, Zhang S, Zhang H, Li F, Li G, Fan C, Sun R, Zhang S. QTL Mapping and Candidate Gene Identification of Swollen Root Formation in Turnip. International Journal of Molecular Sciences. 2021; 22(2):653. https://doi.org/10.3390/ijms22020653
Chicago/Turabian StyleWu, Yudi, Shifan Zhang, Hui Zhang, Fei Li, Guoliang Li, Chuchuan Fan, Rifei Sun, and Shujiang Zhang. 2021. "QTL Mapping and Candidate Gene Identification of Swollen Root Formation in Turnip" International Journal of Molecular Sciences 22, no. 2: 653. https://doi.org/10.3390/ijms22020653
APA StyleWu, Y., Zhang, S., Zhang, H., Li, F., Li, G., Fan, C., Sun, R., & Zhang, S. (2021). QTL Mapping and Candidate Gene Identification of Swollen Root Formation in Turnip. International Journal of Molecular Sciences, 22(2), 653. https://doi.org/10.3390/ijms22020653