Targeting Abnormal Hematopoietic Stem Cells in Chronic Myeloid Leukemia and Philadelphia Chromosome-Negative Classical Myeloproliferative Neoplasms


Abstract
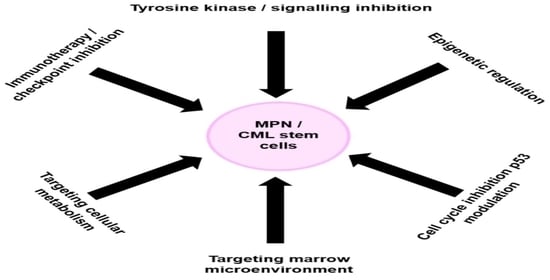
:
1. Introduction
2. Current Therapeutic Options in CML and Their Effects on CML Stem Cells
2.1. First and Second Generation TKIs
2.2. Ponatinib
2.3. Asciminib
2.4. Interferon-α
3. Current Therapeutic Options in MPN and Their Effects on MPN Stem Cells
3.1. IFNα
3.2. JAK Inhibitors
3.3. Allogeneic HSCT
4. Novel Therapy Targeting Signaling Pathways in CML Stem Cells
4.1. Novel Tyrosine Kinase Inhibitors
4.2. Targetting microRNAs
4.3. Targeting BCR-ABL1/Gab2/Grb2 Axis
4.4. Targeting MAPK/MNK1/2 Pathway
4.5. Targeting mTOR Pathway
4.6. Bcl-2 Targeting
4.7. JAK2 Inhibition
4.8. Targeting PPARγ/STAT5/HIF2α Axis
4.9. Prostaglandin E (PGE) 1 Analogue
4.10. Activation of Promyelocytic Leukaemia—Nuclear Bodies (PML-NB)
5. Targeting the CML Stem Cell Microenviroment, Survival and Self-Renewal
5.1. Dipeptidyl-Peptidase (DPP-4) Inhibition
5.2. E-Selectin Antagonist
5.3. Targeting SDF1/CXCR4/CXCR7 Axis
5.4. Hypoxia-Inducible Factor (HIF) Targeting
5.5. Targeting Hh Pathway
5.6. Targeting Wnt/β-Catenin Signalling
5.7. Targeting Protein Phosphatase 2A (PP2A)
6. Targeting CML Stem Cells via Epigenetic, Ribosomal and Transcriptional Regulation
6.1. Bromodomain and Extra-Terminal (BET) Inhibitor
6.2. EZH2 Inhibition
6.3. Histone Deacetylase (HDAC) Inhibitor
6.4. Protein Arginine Methyltransferase (PRMT5) Inhibitor
7. Targeting CML Stem Cells via P53 Modulation
7.1. Sirtuin 1 (SIRT1) Inhibition
7.2. Human Double Minute 2 Protein (HDM2) Inhibition
8. Targeting Autophagy in CML Stem Cells
8.1. Tigecycline
8.2. Chloroquine (CQ)
9. Immunotherapeutic Targeting of CML Stem Cell
Targeting PD-1/PD-L1 Axis
10. Novel Therapies Targeting MPN Stem Cells via Signaling, Apoptotic and Cell Cycle Pathways
10.1. Telomerase Inhibition
10.2. BET Inhibition
10.3. Mouse Double Minute 2 Homolog (MDM2) Inhibition
10.4. Heat Shock Protein (HSP) Inhibition
10.5. Poly-ADP-Ribose Polymerase (PARP) Inhibition
10.6. CD123 Targeting
10.7. Proviral Integration Site for Moloney Murine Leukemia Virus (PIM) Kinase Inhibition
10.8. PI3K/AKT/mTOR Inhibition
10.9. Bcl-xL Inhibition
11. Targeting MPN Stem Cells via Epigenetic Regulation
11.1. Lysine Specific Demethylase-1 (LSD-1) Inhibition
11.2. PRMT5 Inhibition
11.3. HDAC Inhibition
12. Targeting the MPN Stem Cell Niche and Marrow Microenvironment
12.1. β-3 Sympathomimetic Agonists
12.2. Targeting Transforming Growth Factor-β (TGF-β)
12.3. Aurora Kinase A (AURKA) Inhibition
12.4. Antifibrotic Therapy
13. Immunotherapeutic Targetting of MPN Stem Cells
13.1. Targeting PD-1/PD-L1 Pathway
13.2. Peptide Vaccination in CALR Exon 9
14. Other Potential Therapeutic Strategies Targeting MPN Stem Cells
15. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tefferi, A. Myeloproliferative neoplasms: A decade of discoveries and treatment advances. Am. J. Hematol. 2016, 91, 50–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shallis, R.M.; Wang, R.; Davidoff, A.; Ma, X.; Podoltsev, N.A.; Zeidan, A.M. Epidemiology of the classical myeloproliferative neoplasms: The four corners of an expansive and complex map. Blood Rev. 2020, 42, 100706. [Google Scholar] [CrossRef] [PubMed]
- Hultcrantz, M.; Ravn Landtblom, A.; Andréasson, B.; Samuelsson, J.; Dickman, P.W.; Kristinsson, S.Y.; Björkholm, M.; Andersson, T.M. Incidence of myeloproliferative neoplasms—Trends by subgroup and age in a population-based study in Sweden. J. Intern. Med. 2020, 287, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Pellicano, F.; Park, L.; Hopcroft, L.E.M.; Shah, M.M.; Jackson, L.; Scott, M.T.; Clarke, C.J.; Sinclair, A.; Abraham, S.A.; Hair, A.; et al. hsa-mir183/EGR1-mediated regulation of E2F1 is required for CML stem/progenitor cell survival. Blood 2018, 131, 1532–1544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kvasnicka, H.M. The differential diagnosis of classical myeloproliferative neoplasms (MPN): The updated WHO criteria. Rinsho Ketsueki 2019, 60, 1166–1175. [Google Scholar]
- Passamonti, F.; Maffioli, M. Update from the latest WHO classification of MPNs: A user’s manual. Hematology 2016, 2016, 534–542. [Google Scholar] [CrossRef] [Green Version]
- Pophali, P.A.; Patnaik, M.M. The Role of New Tyrosine Kinase Inhibitors in Chronic Myeloid Leukemia. Cancer J. 2016, 22, 40–50. [Google Scholar] [CrossRef] [Green Version]
- Vetrie, D.; Helgason, G.V.; Copland, M. The leukaemia stem cell: Similarities, differences and clinical prospects in CML and AML. Nat. Rev. Cancer 2020, 20, 158–173. [Google Scholar] [CrossRef]
- Mead, A.J.; Mullally, A. Myeloproliferative neoplasm stem cells. Blood 2017, 129, 1607–1616. [Google Scholar] [CrossRef] [Green Version]
- Nangalia, J.; Green, A.R. Myeloproliferative neoplasms: From origins to outcomes. Blood 2017, 130, 2475–2483. [Google Scholar] [CrossRef]
- Grabek, J.; Straube, J.; Bywater, M.; Lane, S.W. MPN: The Molecular Drivers of Disease Initiation, Progression and Transformation and their Effect on Treatment. Cells 2020, 9, 1901. [Google Scholar] [CrossRef] [PubMed]
- Vainchenker, W.; Kralovics, R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood 2017, 129, 667–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, M.-A.; Choi, C.W. Recent insights regarding the molecular basis of myeloproliferative neoplasms. Korean J. Intern. Med. 2020, 35, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allegra, A.; Pioggia, G.; Tonacci, A.; Casciaro, M.; Musolino, C.; Gangemi, S. Synergic Crosstalk between Inflammation, Oxidative Stress, and Genomic Alterations in BCR–ABL-Negative Myeloproliferative Neoplasm. Antioxidants 2020, 9, 1037. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Xu, R. Leukemia stem cells: The root of chronic myeloid leukemia. Protein Cell 2015, 6, 403–412. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, H.; Sadovnik, I.; Cerny-Reiterer, S.; Rülicke, T.; Stefanzl, G.; Willmann, M.; Hoermann, G.; Bilban, M.; Blatt, K.; Herndlhofer, S.; et al. Dipeptidylpeptidase IV (CD26) defines leukemic stem cells (LSC) in chronic myeloid leukemia. Blood 2014, 123, 3951–3962. [Google Scholar] [CrossRef] [Green Version]
- Houshmand, M.; Simonetti, G.; Circosta, P.; Gaidano, V.; Cignetti, A.; Martinelli, G.; Saglio, G.; Gale, R.P. Chronic myeloid leukemia stem cells. Leukemia 2019, 33, 1543–1556. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, R. Novel approaches to therapy in CML. Hematol. Am. Soc. Hematol. Educ. Program 2017, 2017, 115–120. [Google Scholar] [CrossRef] [Green Version]
- Hehlmann, R. Chronic Myeloid Leukemia in 2020. Hemasphere 2020, 4, e468. [Google Scholar] [CrossRef]
- Ye, H.; Adane, B.; Khan, N.; Sullivan, T.; Minhajuddin, M.; Gasparetto, M.; Stevens, B.; Pei, S.; Balys, M.; Ashton, J.M.; et al. Leukemic Stem Cells Evade Chemotherapy by Metabolic Adaptation to an Adipose Tissue Niche. Cell Stem Cell 2016, 19, 23–37. [Google Scholar] [CrossRef] [Green Version]
- Valent, P.; Sadovnik, I.; Eisenwort, G.; Bauer, K.; Herrmann, H.; Gleixner, K.V.; Schulenburg, A.; Rabitsch, W.; Sperr, W.R.; Wolf, D. Immunotherapy-Based Targeting and Elimination of Leukemic Stem Cells in AML and CML. Int. J. Mol. Sci. 2019, 20, 4233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, Y.; Fukushima, T.; Mikami, K.; Adachi, K.; Fukuyama, T.; Goyama, S.; Kitamura, T. Efficacy of tyrosine kinase inhibitors on a mouse chronic myeloid leukemia model and chronic myeloid leukemia stem cells. Exp. Hematol. 2020, 90, 46–51.e2. [Google Scholar] [CrossRef] [PubMed]
- Houshmand, M.; Blanco, T.M.; Circosta, P.; Yazdi, N.; Kazemi, A.; Saglio, G.; Zarif, M.N. Bone marrow microenvironment: The guardian of leukemia stem cells. World J. Stem Cells 2019, 11, 476–490. [Google Scholar] [CrossRef] [PubMed]
- Helgason, G.V.; Mukhopadhyay, A.; Karvela, M.; Salomoni, P.; Calabretta, B.; Holyoake, T.L. Autophagy in chronic myeloid leukaemia: Stem cell survival and implication in therapy. Curr. Cancer Drug Targets 2013, 13, 724–734. [Google Scholar] [CrossRef] [PubMed]
- Arrigoni, E.; Del Re, M.; Galimberti, S.; Restante, G.; Rofi, E.; Crucitta, S.; Baratè, C.; Petrini, M.; Danesi, R.; Di Paolo, A. Concise Review: Chronic Myeloid Leukemia: Stem Cell Niche and Response to Pharmacologic Treatment. Stem Cells Transl. Med. 2018, 7, 305–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baquero, P.; Dawson, A.; Mukhopadhyay, A.; Kuntz, E.M.; Mitchell, R.; Olivares, O.; Ianniciello, A.; Scott, M.T.; Dunn, K.; Nicastri, M.C.; et al. Targeting quiescent leukemic stem cells using second generation autophagy inhibitors. Leukemia 2019, 33, 981–994. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Mak, P.Y.; Mu, H.; Mak, D.H.; Zeng, Z.; Cortes, J.; Liu, Q.; Andreeff, M.; Carter, B.Z. Combined inhibition of β-catenin and Bcr-Abl synergistically targets tyrosine kinase inhibitor-resistant blast crisis chronic myeloid leukemia blasts and progenitors in vitro and in vivo. Leukemia 2017, 31, 2065–2074. [Google Scholar] [CrossRef]
- Zhang, H.; Li, H.; Xi, H.S.; Li, S. HIF1α is required for survival maintenance of chronic myeloid leukemia stem cells. Blood 2012, 119, 2595–2607. [Google Scholar] [CrossRef] [Green Version]
- Carrà, G.; Cartellà, A.; Maffeo, B.; Morotti, A. Strategies For Targeting Chronic Myeloid Leukaemia Stem Cells. Blood Lymphat. Cancer 2019, 9, 45–52. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Li, L.; Ho, Y.; Li, M.; Marcucci, G.; Tong, W.; Bhatia, R. Heterogeneity of leukemia-initiating capacity of chronic myelogenous leukemia stem cells. J. Clin. Invest. 2016, 126, 975–991. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, H.; Cerny-Reiterer, S.; Gleixner, K.V.; Blatt, K.; Herndlhofer, S.; Rabitsch, W.; Jäger, E.; Mitterbauer-Hohendanner, G.; Streubel, B.; Selzer, E.; et al. CD34(+)/CD38(-) stem cells in chronic myeloid leukemia express Siglec-3 (CD33) and are responsive to the CD33-targeting drug gemtuzumab/ozogamicin. Haematologica 2012, 97, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Chaar, M.; Kamta, J.; Ait-Oudhia, S. Mechanisms, monitoring, and management of tyrosine kinase inhibitors-associated cardiovascular toxicities. Onco. Targets Ther. 2018, 11, 6227–6237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, Q.; Bi, L.; Ren, Y.; Song, S.; Wang, Q.; Wang, Y.-s. Advances in studies of tyrosine kinase inhibitors and their acquired resistance. Mol. Cancer 2018, 17, 36. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Wang, H.; Zhu, X.; Liu, W.; Zhou, S.; Geng, Z.; Xiao, Y.; Zou, P.; You, Y.; Li, Q.; et al. The Impact of Tyrosine Kinase Inhibitors on Chronic Myeloid Leukemia Stem Cells and the Implication in Discontinuation. Stem Cells Dev. 2019, 28, 1480–1485. [Google Scholar] [CrossRef] [PubMed]
- Hamad, A.; Sahli, Z.; El Sabban, M.; Mouteirik, M.; Nasr, R. Emerging Therapeutic Strategies for Targeting Chronic Myeloid Leukemia Stem Cells. Stem Cells Int. 2013, 2013, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Willmann, M.; Sadovnik, I.; Eisenwort, G.; Entner, M.; Bernthaler, T.; Stefanzl, G.; Hadzijusufovic, E.; Berger, D.; Herrmann, H.; Hoermann, G.; et al. Evaluation of cooperative antileukemic effects of nilotinib and vildagliptin in Ph + chronic myeloid leukemia. Exp. Hematol. 2018, 57, 50–59.e6. [Google Scholar] [CrossRef]
- Pungolino, E.; Rossi, G.; De Canal, G.; Trojani, A.; D’Adda, M.; Perego, A.; Orlandi, E.M.; Lunghi, F.; Turrini, M.; Borin, L.; et al. Nilotinib induced bone marrow CD34+/lin-Ph+ cells early clearance in newly diagnosed CP-chronic myeloid leukemia. Am. J. Hematol. 2018, 93, E162–E164. [Google Scholar] [CrossRef]
- Konig, H.; Holyoake, T.L.; Bhatia, R. Effective and selective inhibition of chronic myeloid leukemia primitive hematopoietic progenitors by the dual Src/Abl kinase inhibitor SKI-606. Blood 2008, 111, 2329–2338. [Google Scholar] [CrossRef]
- Ciarcia, R.; Vitiello, M.T.; Galdiero, M.; Pacilio, C.; Iovane, V.; d’Angelo, D.; Pagnini, D.; Caparrotti, G.; Conti, D.; Tomei, V.; et al. Imatinib treatment inhibit IL-6, IL-8, NF-KB and AP-1 production and modulate intracellular calcium in CML patients. J. Cell Physiol. 2012, 227, 2798–2803. [Google Scholar] [CrossRef]
- Corrado, C.; Saieva, L.; Raimondo, S.; Santoro, A.; De Leo, G.; Alessandro, R. Chronic myelogenous leukaemia exosomes modulate bone marrow microenvironment through activation of epidermal growth factor receptor. J. Cell Mol. Med. 2016, 20, 1829–1839. [Google Scholar] [CrossRef] [Green Version]
- Masamoto, Y.; Kurokawa, M. Targeting chronic myeloid leukemia stem cells: Can transcriptional program be a druggable target for cancers? Stem Cell Investig. 2018, 5, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciarcia, R.; Damiano, S.; Puzio, M.V.; Montagnaro, S.; Pagnini, F.; Pacilio, C.; Caparrotti, G.; Bellan, C.; Garofano, T.; Polito, M.S.; et al. Comparison of Dasatinib, Nilotinib, and Imatinib in the Treatment of Chronic Myeloid Leukemia. J. Cell Physiol. 2016, 231, 680–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, M.T.; Korfi, K.; Saffrey, P.; Hopcroft, L.E.; Kinstrie, R.; Pellicano, F.; Guenther, C.; Gallipoli, P.; Cruz, M.; Dunn, K.; et al. Epigenetic Reprogramming Sensitizes CML Stem Cells to Combined EZH2 and Tyrosine Kinase Inhibition. Cancer Discov. 2016, 6, 1248–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.Y.; Park, C.-j.; Cho, Y.-U.; You, E.; Jang, S.; Seol, C.A.; Seo, E.-J.; Lee, J.-H. Expression Levels of PD-1 on CD8+ T Cells in Chronic Myeloid Leukemia with and without BCR-ABL1 kinase Mutation. Blood 2017, 130 (Suppl. 1), 4178. [Google Scholar]
- Lee, M.Y.; Park, C.-J.; Cho, Y.-U.; You, E.; Jang, S.; Seol, C.A.; Seo, E.-J.; Choi, E.-J.; Lee, J.-H. Differences in PD-1 expression on CD8+ T-cells in chronic myeloid leukemia patients according to disease phase and TKI medication. Cancer Immunol. Immunother. 2020, 69, 2223–2232. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.; Clarson, J.; Tang, C.; Vidovic, L.; White, D.L.; Hughes, T.P.; Yong, A.S. CML patients with deep molecular responses to TKI have restored immune effectors and decreased PD-1 and immune suppressors. Blood 2017, 129, 1166–1176. [Google Scholar] [CrossRef]
- Molica, M.; Scalzulli, E.; Colafigli, G.; Foà, R.; Breccia, M. Insights into the optimal use of ponatinib in patients with chronic phase chronic myeloid leukaemia. Ther. Adv. Hematol. 2019, 10, 2040620719826444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jabbour, E.; Kantarjian, H.; Cortes, J. Use of second- and third-generation tyrosine kinase inhibitors in the treatment of chronic myeloid leukemia: An evolving treatment paradigm. Clin. Lymphoma Myeloma Leuk. 2015, 15, 323–334. [Google Scholar] [CrossRef] [Green Version]
- Eide, C.A.; Zabriskie, M.S.; Savage Stevens, S.L.; Antelope, O.; Vellore, N.A.; Than, H.; Schultz, A.R.; Clair, P.; Bowler, A.D.; Pomicter, A.D.; et al. Combining the Allosteric Inhibitor Asciminib with Ponatinib Suppresses Emergence of and Restores Efficacy against Highly Resistant BCR-ABL1 Mutants. Cancer Cell 2019, 36, 431–443.e5. [Google Scholar] [CrossRef]
- Jabbour, E.; Kantarjian, H. Chronic myeloid leukemia: 2020 update on diagnosis, therapy and monitoring. Am. J. Hematol. 2020, 95, 691–709. [Google Scholar] [CrossRef]
- Hughes, T.P.; Mauro, M.J.; Cortes, J.E.; Minami, H.; Rea, D.; DeAngelo, D.J.; Breccia, M.; Goh, Y.-T.; Talpaz, M.; Hochhaus, A.; et al. Asciminib in Chronic Myeloid Leukemia after ABL Kinase Inhibitor Failure. N. Engl. J. Med. 2019, 381, 2315–2326. [Google Scholar] [CrossRef] [PubMed]
- Kavanagh, S.; Nee, A.; Lipton, J.H. Emerging alternatives to tyrosine kinase inhibitors for treating chronic myeloid leukemia. Expert Opin. Emerg. Drugs 2018, 23, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Özgür Yurttaş, N.; Eşkazan, A.E. Novel therapeutic approaches in chronic myeloid leukemia. Leuk. Res. 2020, 91, 106337. [Google Scholar] [CrossRef] [PubMed]
- Talpaz, M.; Mercer, J.; Hehlmann, R. The interferon-alpha revival in CML. Ann. Hematol. 2015, 94 (Suppl. 2), S195–S207. [Google Scholar] [CrossRef] [PubMed]
- Talpaz, M.; Hehlmann, R.; Quintás-Cardama, A.; Mercer, J.; Cortes, J. Re-emergence of interferon-α in the treatment of chronic myeloid leukemia. Leukemia 2013, 27, 803–812. [Google Scholar] [CrossRef] [Green Version]
- Chawla-Sarkar, M.; Lindner, D.J.; Liu, Y.F.; Williams, B.R.; Sen, G.C.; Silverman, R.H.; Borden, E.C. Apoptosis and interferons: Role of interferon-stimulated genes as mediators of apoptosis. Apoptosis 2003, 8, 237–249. [Google Scholar] [CrossRef]
- Bhatia, R.; McCarthy, J.B.; Verfaillie, C.M. Interferon-alpha restores normal beta 1 integrin-mediated inhibition of hematopoietic progenitor proliferation by the marrow microenvironment in chronic myelogenous leukemia. Blood 1996, 87, 3883–3891. [Google Scholar] [CrossRef] [Green Version]
- Kunisaki, Y.; Bruns, I.; Scheiermann, C.; Ahmed, J.; Pinho, S.; Zhang, D.; Mizoguchi, T.; Wei, Q.; Lucas, D.; Ito, K.; et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature 2013, 502, 637–643. [Google Scholar] [CrossRef] [Green Version]
- Chronic Myeloid Leukemia Trialists’ Collaborative Group. Interferon Alfa Versus Chemotherapy for Chronic Myeloid Leukemia: A Meta-analysis of Seven Randomized Trials. JNCI J. Natl. Cancer Inst. 1997, 89, 1616–1620. [Google Scholar] [CrossRef] [Green Version]
- Bonifazi, F.; Vivo, A.; Rosti, G.; Guilhot, F.; Guilhot, J.; Trabacchi, E.; Hehlmann, R.; Hochhaus, A.; Shepherd, P.; Steegmann, J.; et al. Chronic myeloid leukemia and interferon-alpha: A study of complete cytogenetic responders. Blood 2001, 98, 3074–3081. [Google Scholar] [CrossRef]
- Tefferi, A.; Barbui, T. Polycythemia vera and essential thrombocythemia: 2019 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2019, 94, 133–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- How, J.; Hobbs, G. Use of Interferon Alfa in the Treatment of Myeloproliferative Neoplasms: Perspectives and Review of the Literature. Cancers 2020, 12, 1954. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Zhang, W.; Li, Y.; Berenzon, D.; Wang, X.; Wang, J.; Mascarenhas, J.; Xu, M.; Hoffman, R. Interferon-α targets JAK2V617F-positive hematopoietic progenitor cells and acts through the p38 MAPK pathway. Exp. Hematol. 2010, 38, 472–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiladjian, J.-J.; Mesa, R.A.; Hoffman, R. The renaissance of interferon therapy for the treatment of myeloid malignancies. Blood 2011, 117, 4706–4715. [Google Scholar] [CrossRef] [Green Version]
- Utke Rank, C.; Weis Bjerrum, O.; Larsen, T.S.; Kjær, L.; De Stricker, K.; Riley, C.H.; Hasselbalch, H.C. Minimal residual disease after long-term interferon-alpha2 treatment: A report on hematological, molecular and histomorphological response patterns in 10 patients with essential thrombocythemia and polycythemia vera. Leuk. Lymphoma 2016, 57, 348–354. [Google Scholar] [CrossRef]
- Skov, V. Next Generation Sequencing in MPNs. Lessons from the Past and Prospects for Use as Predictors of Prognosis and Treatment Responses. Cancers 2020, 12, 2194. [Google Scholar] [CrossRef]
- Hasselbalch, H.C.; Holmström, M.O. Perspectives on interferon-alpha in the treatment of polycythemia vera and related myeloproliferative neoplasms: Minimal residual disease and cure? Semin. Immunopathol. 2019, 41, 5–19. [Google Scholar] [CrossRef] [Green Version]
- Austin, R.J.; Straube, J.; Bruedigam, C.; Pali, G.; Jacquelin, S.; Vu, T.; Green, J.; Gräsel, J.; Lansink, L.; Cooper, L.; et al. Distinct effects of ruxolitinib and interferon-alpha on murine JAK2V617F myeloproliferative neoplasm hematopoietic stem cell populations. Leukemia 2020, 34, 1075–1089. [Google Scholar] [CrossRef]
- Verger, E.; Soret-Dulphy, J.; Maslah, N.; Roy, L.; Rey, J.; Ghrieb, Z.; Kralovics, R.; Gisslinger, H.; Grohmann-Izay, B.; Klade, C.; et al. Ropeginterferon alpha-2b targets JAK2V617F-positive polycythemia vera cells in vitro and in vivo. Blood Cancer J. 2018, 8, 94. [Google Scholar] [CrossRef] [Green Version]
- Czech, J.; Cordua, S.; Weinbergerova, B.; Baumeister, J.; Crepcia, A.; Han, L.; Maié, T.; Costa, I.G.; Denecke, B.; Maurer, A.; et al. JAK2V617F but not CALR mutations confer increased molecular responses to interferon-α via JAK1/STAT1 activation. Leukemia 2019, 33, 995–1010. [Google Scholar] [CrossRef]
- Mosca, M.; Lamrani, L.; Marzac, C.; Tisserand, A.; Edmond, V.; Dagher, T.; El-Khoury, M.; Marty, C.; Campario, H.; Casadevall, N.; et al. Differential Impact of Interferon Alpha on JAK2V617F and Calr Mutated Hematopoietic Stem and Progenitor Cells in Classical MPN. Blood 2018, 132 (Suppl. 1), 4333. [Google Scholar] [CrossRef]
- Kjær, L.; Cordua, S.; Holmström, M.O.; Thomassen, M.; Kruse, T.A.; Pallisgaard, N.; Larsen, T.S.; De Stricker, K.; Skov, V.; Hasselbalch, H.C. Differential Dynamics of CALR Mutant Allele Burden in Myeloproliferative Neoplasms during Interferon Alfa Treatment. PLoS ONE 2016, 11, e0165336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verger, E.; Cassinat, B.; Chauveau, A.; Dosquet, C.; Giraudier, S.; Schlageter, M.-H.; Ianotto, J.-C.; Yassin, M.A.; Al-Dewik, N.; Carillo, S.; et al. Clinical and molecular response to interferon-α therapy in essential thrombocythemia patients with CALR mutations. Blood 2015, 126, 2585–2591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chifotides, H.T.; Bose, P.; Verstovsek, S. Givinostat: An emerging treatment for polycythemia vera. Expert Opin. Investig. Drugs 2020, 29, 525–536. [Google Scholar] [CrossRef] [PubMed]
- Passamonti, F.; Griesshammer, M.; Palandri, F.; Egyed, M.; Benevolo, G.; Devos, T.; Callum, J.; Vannucchi, A.M.; Sivgin, S.; Bensasson, C.; et al. Ruxolitinib for the treatment of inadequately controlled polycythaemia vera without splenomegaly (RESPONSE-2): A randomised, open-label, phase 3b study. Lancet Oncol. 2017, 18, 88–99. [Google Scholar] [CrossRef]
- Verstovsek, S.; Gotlib, J.; Mesa, R.A.; Vannucchi, A.M.; Kiladjian, J.-J.; Cervantes, F.; Harrison, C.N.; Paquette, R.; Sun, W.; Naim, A.; et al. Long-term survival in patients treated with ruxolitinib for myelofibrosis: COMFORT-I and -II pooled analyses. J. Hematol. Oncol. 2017, 10, 156. [Google Scholar] [CrossRef] [Green Version]
- Oh, S.T.; Talpaz, M.; Gerds, A.T.; Gupta, V.; Verstovsek, S.; Mesa, R.; Miller, C.B.; Rivera, C.E.; Fleischman, A.G.; Goel, S.; et al. ACVR1/JAK1/JAK2 inhibitor momelotinib reverses transfusion dependency and suppresses hepcidin in myelofibrosis phase 2 trial. Blood Adv. 2020, 4, 4282–4291. [Google Scholar] [CrossRef]
- Mesa, R.A.; Kiladjian, J.-J.; Catalano, J.V.; Devos, T.; Egyed, M.; Hellmann, A.; Mclornan, D.; Shimoda, K.; Winton, E.F.; Deng, W.; et al. SIMPLIFY-1: A Phase III Randomized Trial of Momelotinib Versus Ruxolitinib in Janus Kinase Inhibitor–Naïve Patients With Myelofibrosis. J. Clin. Oncol. 2017, 35, 3844–3850. [Google Scholar] [CrossRef]
- Talpaz, M.; Kiladjian, J.-J. Fedratinib, a newly approved treatment for patients with myeloproliferative neoplasm-associated myelofibrosis. Leukemia 2020. [Google Scholar] [CrossRef]
- Bose, P.; Verstovsek, S. JAK Inhibition for the Treatment of Myelofibrosis: Limitations and Future Perspectives. HemaSphere 2020, 4, e424. [Google Scholar] [CrossRef]
- Mascarenhas, J.; Hoffman, R.; Talpaz, M.; Gerds, A.T.; Stein, B.; Gupta, V.; Szoke, A.; Drummond, M.; Pristupa, A.; Granston, T.; et al. Pacritinib vs Best Available Therapy, Including Ruxolitinib, in Patients With Myelofibrosis. JAMA Oncol. 2018, 4, 652. [Google Scholar] [CrossRef] [PubMed]
- Harrison, C.N.; Schaap, N.; Mesa, R.A. Management of myelofibrosis after ruxolitinib failure. Ann. Hematol. 2020, 99, 1177–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardanani, A.; Tefferi, A.; Jamieson, C.; Gabrail, N.Y.; Lebedinsky, C.; Gao, G.; Liu, F.; Xu, C.; Cao, H.; Talpaz, M. A phase 2 randomized dose-ranging study of the JAK2-selective inhibitor fedratinib (SAR302503) in patients with myelofibrosis. Blood Cancer J. 2015, 5, e335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardanani, A.; Gotlib, J.R.; Jamieson, C.; Cortes, J.E.; Talpaz, M.; Stone, R.M.; Silverman, M.H.; Gilliland, D.G.; Shorr, J.; Tefferi, A. Safety and Efficacy of TG101348, a Selective JAK2 Inhibitor, in Myelofibrosis. J. Clin. Oncol. 2011, 29, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Pardanani, A.; Harrison, C.; Cortes, J.E.; Cervantes, F.; Mesa, R.A.; Milligan, D.; Masszi, T.; Mishchenko, E.; Jourdan, E.; Vannucchi, A.M.; et al. Safety and Efficacy of Fedratinib in Patients With Primary or Secondary Myelofibrosis. JAMA Oncol. 2015, 1, 643. [Google Scholar] [CrossRef]
- Bankar, A.; Gupta, V. Investigational non-JAK inhibitors for chronic phase myelofibrosis. Expert Opin. Investig. Drugs 2020, 29, 461–474. [Google Scholar] [CrossRef]
- Tamari, R.; Rapaport, F.; Zhang, N.; Mcnamara, C.; Kuykendall, A.; Sallman, D.A.; Komrokji, R.; Arruda, A.; Najfeld, V.; Sandy, L.; et al. Impact of High-Molecular-Risk Mutations on Transplantation Outcomes in Patients with Myelofibrosis. Biol. Blood Marrow Transplant. 2019, 25, 1142–1151. [Google Scholar] [CrossRef]
- Kröger, N.; Panagiota, V.; Badbaran, A.; Zabelina, T.; Triviai, I.; Araujo Cruz, M.M.; Shahswar, R.; Ayuk, F.; Gehlhaar, M.; Wolschke, C.; et al. Impact of Molecular Genetics on Outcome in Myelofibrosis Patients after Allogeneic Stem Cell Transplantation. Biol. Blood Marrow Transplant. 2017, 23, 1095–1101. [Google Scholar] [CrossRef] [Green Version]
- Mannina, D.; Gagelmann, N.; Badbaran, A.; Ditschkowski, M.; Bogdanov, R.; Robin, M.; Cassinat, B.; Heuser, M.; Shahswar, R.; Thol, F.; et al. Allogeneic stem cell transplantation in patients with myelofibrosis harboring the MPL mutation. Eur. J. Haematol. 2019, 103, 552–557. [Google Scholar] [CrossRef]
- Mian, A.A.; Rafiei, A.; Haberbosch, I.; Zeifman, A.; Titov, I.; Stroylov, V.; Metodieva, A.; Stroganov, O.; Novikov, F.; Brill, B.; et al. PF-114, a potent and selective inhibitor of native and mutated BCR/ABL is active against Philadelphia chromosome-positive (Ph+) leukemias harboring the T315I mutation. Leukemia 2015, 29, 1104–1114. [Google Scholar] [CrossRef]
- Ivanova, E.S.; Tatarskiy, V.V.; Yastrebova, M.A.; Khamidullina, A.I.; Shunaev, A.V.; Kalinina, A.A.; Zeifman, A.A.; Novikov, F.N.; Dutikova, Y.V.; Chilov, G.G.; et al. PF-114, a novel selective inhibitor of BCR-ABL tyrosine kinase, is a potent inducer of apoptosis in chronic myelogenous leukemia cells. Int. J. Oncol. 2019, 55, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Turkina, A.G.; Vinogradova, O.; Lomaia, E.; Shatokhina, E.; Shukhov, O.A.; Chelysheva, E.Y.; Shikhbabaeva, D.; Nemchenko, I.; Petrova, A.; Bykova, A.; et al. PF-114: A 4th Generation Tyrosine Kinase-Inhibitor for Chronic Phase Chronic Myeloid Leukaemia Including BCRABL1T315I. Blood 2019, 134 (Suppl. 1), 1638. [Google Scholar] [CrossRef]
- Rossari, F.; Minutolo, F.; Orciuolo, E. Past, present, and future of Bcr-Abl inhibitors: From chemical development to clinical efficacy. J. Hematol. Oncol. 2018, 11, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Stefano, C.; Mirone, G.; Perna, S.; Marfe, G. The roles of microRNAs in the pathogenesis and drug resistance of chronic myelogenous leukemia (Review). Oncol. Rep. 2016, 35, 614–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Litwińska, Z.; Machaliński, B. miRNAs in chronic myeloid leukemia: Small molecules, essential function. Leuk. Lymphoma 2017, 58, 1297–1305. [Google Scholar] [CrossRef] [PubMed]
- Agatheeswaran, S.; Pattnayak, N.C.; Chakraborty, S. Identification and functional characterization of the miRNA-gene regulatory network in chronic myeloid leukemia lineage negative cells. Sci. Rep. 2016, 6, 32493. [Google Scholar] [CrossRef] [Green Version]
- Klümper, T.; Bruckmueller, H.; Diewock, T.; Kaehler, M.; Haenisch, S.; Pott, C.; Bruhn, O.; Cascorbi, I. Expression differences of miR-142-5p between treatment-naïve chronic myeloid leukemia patients responding and non-responding to imatinib therapy suggest a link to oncogenic ABL2, SRI, cKIT and MCL1 signaling pathways critical for development of therapy resistance. Exp. Hematol. Oncol. 2020, 9, 26. [Google Scholar] [PubMed]
- Salati, S.; Salvestrini, V.; Carretta, C.; Genovese, E.; Rontauroli, S.; Zini, R.; Rossi, C.; Ruberti, S.; Bianchi, E.; Barbieri, G.; et al. Deregulated expression of miR-29a-3p, miR-494-3p and miR-660-5p affects sensitivity to tyrosine kinase inhibitors in CML leukemic stem cells. Oncotarget 2017, 8, 49451–49469. [Google Scholar] [CrossRef] [Green Version]
- Fiserova, B.; Kubiczkova, L.; Sedlarikova, L.; Hajek, R.; Sevcikova, S. The miR-29 family in hematological malignancies. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc. Czech Repub. 2015, 159, 184–191. [Google Scholar] [CrossRef] [Green Version]
- Srutova, K.; Curik, N.; Burda, P.; Savvulidi, F.; Silvestri, G.; Trotta, R.; Klamova, H.; Pecherkova, P.; Sovova, Z.; Koblihova, J.; et al. BCR-ABL1 mediated miR-150 downregulation through MYC contributed to myeloid differentiation block and drug resistance in chronic myeloid leukemia. Haematologica 2018, 103, 2016–2025. [Google Scholar] [CrossRef]
- Li, Y.-L.; Tang, J.-M.; Chen, X.-Y.; Luo, B.; Liang, G.-H.; Qu, Q.; Lu, Z.-Y. MicroRNA-153-3p enhances the sensitivity of chronic myeloid leukemia cells to imatinib by inhibiting B-cell lymphoma-2-mediated autophagy. Hum. Cell 2020, 33, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Pan, B.; Yang, J.; Wang, X.; Xu, K.; Ikezoe, T. miR-217 sensitizes chronic myelogenous leukemia cells to tyrosine kinase inhibitors by targeting pro-oncogenic anterior gradient 2. Exp. Hematol. 2018, 68, 80–88.e2. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, C.; Ikezoe, T.; Yang, J.; Nobumoto, A.; Tsuda, M.; Yokoyama, A. Downregulation of miR-217 correlates with resistance of Ph(+) leukemia cells to ABL tyrosine kinase inhibitors. Cancer Sci. 2014, 105, 297–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hershkovitz-Rokah, O.; Modai, S.; Pasmanik-Chor, M.; Toren, A.; Shomron, N.; Raanani, P.; Shpilberg, O.; Granot, G. Restoration of miR-424 suppresses BCR-ABL activity and sensitizes CML cells to imatinib treatment. Cancer Lett. 2015, 360, 245–256. [Google Scholar] [CrossRef]
- Han, B.; Zhao, Y.; Lin, Y.; Fu, S.; Wang, L.; Zhang, M.; Tie, R.; Wang, B.; Luo, Y.; Liu, L.; et al. Hydroxychloroquine sensitizes chronic myeloid leukemia cells to Vγ9Vδ2 T cell-mediated lysis independent of autophagy. Int. J. Oncol. 2017, 50, 1810–1820. [Google Scholar] [CrossRef] [Green Version]
- Lv, M.; Zhang, X.; Jia, H.; Li, D.; Zhang, B.; Zhang, H.; Hong, M.; Jiang, T.; Jiang, Q.; Lu, J.; et al. An oncogenic role of miR-142-3p in human T-cell acute lymphoblastic leukemia (T-ALL) by targeting glucocorticoid receptor-α and cAMP/PKA pathways. Leukemia 2012, 26, 769–777. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.-Z.; Zhang, Y.-Y.; Fu, H.-Y.; Wu, D.-S.; Zhou, H.-R. Overexpression of microRNA-143 inhibits growth and induces apoptosis in human leukemia cells. Oncol. Rep. 2014, 31, 2035–2042. [Google Scholar] [CrossRef] [Green Version]
- Yap, E.; Norziha, Z.A.; Simbun, A.; Tumian, N.R.; Cheong, S.K.; Leong, C.F.; Wong, C.L. Downregulation of Mir-146a-5p, Mir-99b-5p, Mir-143-3p, Mir-10a-5p and Mir-151a-3p Associated with PI3K/AKT, p53, NF-Kb, and Fanconi Anemia/BRCA Signaling Pathways Are Observed in Imatinib-Resistant Chronic Myeloid Leukemia Patients without Detectable BCR-ABL kinase Domain Mutations. Blood 2016, 128, 3060. [Google Scholar]
- Flamant, S.; Ritchie, W.; Guilhot, J.; Holst, J.; Bonnet, M.L.; Chomel, J.C.; Guilhot, F.; Turhan, A.G.; Rasko, J.E. Micro-RNA response to imatinib mesylate in patients with chronic myeloid leukemia. Haematologica 2010, 95, 1325–1333. [Google Scholar] [CrossRef] [Green Version]
- Jurkovicova, D.; Lukackova, R.; Magyerkova, M.; Kulcsar, L.; Krivjanska, M.; Krivjansky, V.; Chovanec, M. microRNA expression profiling as supportive diagnostic and therapy prediction tool in chronic myeloid leukemia. Neoplasma 2015, 62, 949–958. [Google Scholar] [CrossRef]
- Modi, H.; Li, L.; Chu, S.; Rossi, J.; Yee, J.K.; Bhatia, R. Inhibition of Grb2 expression demonstrates an important role in BCR-ABL-mediated MAPK activation and transformation of primary human hematopoietic cells. Leukemia 2011, 25, 305–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Turhan, A.G.; Ding, H.; Lin, Q.; Meng, K.; Jiang, X. Targeting BCR-ABL+ stem/progenitor cells and BCR-ABL-T315I mutant cells by effective inhibition of the BCR-ABL-Tyr177-GRB2 complex. Oncotarget 2017, 8, 43662–43677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tari Ashizawa, A.; Ohanian, M.; Cortes, J.E. BP1001, a Novel Therapeutic for Chronic Myelogenous Leukemia. Blood 2016, 128, 4239. [Google Scholar] [CrossRef]
- Peng, Z.; Luo, H.W.; Yuan, Y.; Shi, J.; Huang, S.F.; Li, C.L.; Cao, W.X.; Huang, Z.G.; Feng, W.L. Growth of chronic myeloid leukemia cells is inhibited by infection with Ad-SH2-HA adenovirus that disrupts Grb2-Bcr-Abl complexes. Oncol. Rep. 2011, 25, 1381–1388. [Google Scholar] [PubMed] [Green Version]
- Ma, L.; Shan, Y.; Bai, R.; Xue, L.; Eide, C.A.; Ou, J.; Zhu, L.J.; Hutchinson, L.; Cerny, J.; Khoury, H.J.; et al. A therapeutically targetable mechanism of BCR-ABL-independent imatinib resistance in chronic myeloid leukemia. Sci. Transl. Med. 2014, 6, 252ra121. [Google Scholar] [CrossRef] [Green Version]
- Chorzalska, A.; Ahsan, N.; Rao, R.S.P.; Roder, K.; Yu, X.; Morgan, J.; Tepper, A.; Hines, S.; Zhang, P.; Treaba, D.O.; et al. Overexpression of Tpl2 is linked to imatinib resistance and activation of MEK-ERK and NF-κB pathways in a model of chronic myeloid leukemia. Mol. Oncol. 2018, 12, 630–647. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.; Saw, T.Y.; Zhang, M.; Janes, M.R.; Nacro, K.; Hill, J.; Lim, A.Q.; Chang, C.T.; Fruman, D.A.; Rizzieri, D.A.; et al. Targeting of the MNK-eIF4E axis in blast crisis chronic myeloid leukemia inhibits leukemia stem cell function. Proc. Natl. Acad. Sci. USA 2013, 110, E2298–E2307. [Google Scholar] [CrossRef] [Green Version]
- Jacquel, A.; Luciano, F.; Robert, G.; Auberger, P. Implication and Regulation of AMPK during Physiological and Pathological Myeloid Differentiation. Int. J. Mol. Sci. 2018, 19, 2991. [Google Scholar] [CrossRef] [Green Version]
- Dinner, S.; Platanias, L.C. Targeting the mTOR Pathway in Leukemia. J. Cell. Biochem. 2016, 117, 1745–1752. [Google Scholar] [CrossRef]
- Frazzi, R.; Guardi, M. Cellular and Molecular Targets of Resveratrol on Lymphoma and Leukemia Cells. Molecules 2017, 22, 885. [Google Scholar] [CrossRef] [Green Version]
- Vakana, E.; Platanias, L.C. AMPK in BCR-ABL expressing leukemias. Regulatory effects and therapeutic implications. Oncotarget 2011, 2, 1322–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shafiei-Irannejad, V.; Samadi, N.; Salehi, R.; Yousefi, B.; Zarghami, N. New insights into antidiabetic drugs: Possible applications in cancer treatment. Chem. Biol. Drug Des. 2017, 90, 1056–1066. [Google Scholar] [CrossRef] [PubMed]
- Biondani, G.; Peyron, J.F. Metformin, an Anti-diabetic Drug to Target Leukemia. Front. Endocrinol 2018, 9, 446. [Google Scholar] [CrossRef] [Green Version]
- Shi, R.; Lin, J.; Gong, Y.; Yan, T.; Shi, F.; Yang, X.; Liu, X.; Naren, D. The antileukemia effect of metformin in the Philadelphia chromosome-positive leukemia cell line and patient primary leukemia cell. Anticancer Drugs 2015, 26, 913–922. [Google Scholar] [CrossRef]
- Wu, X.P.; Xiong, M.; Xu, C.S.; Duan, L.N.; Dong, Y.Q.; Luo, Y.; Niu, T.H.; Lu, C.R. Resveratrol induces apoptosis of human chronic myelogenous leukemia cells in vitro through p38 and JNK-regulated H2AX phosphorylation. Acta Pharmacol. Sin. 2015, 36, 353–361. [Google Scholar] [CrossRef]
- Puissant, A.; Robert, G.; Fenouille, N.; Luciano, F.; Cassuto, J.-P.; Raynaud, S.; Auberger, P. Resveratrol Promotes Autophagic Cell Death in Chronic Myelogenous Leukemia Cells via JNK-Mediated p62/SQSTM1 Expression and AMPK Activation. Cancer Res. 2010, 70, 1042–1052. [Google Scholar] [CrossRef] [Green Version]
- Kirchner, J.; Brüne, B.; Namgaladze, D. AICAR inhibits NFκB DNA binding independently of AMPK to attenuate LPS-triggered inflammatory responses in human macrophages. Sci. Rep. 2018, 8, 7801. [Google Scholar] [CrossRef] [Green Version]
- Carter, B.Z.; Mak, P.Y.; Mu, H.; Zhou, H.; Mak, D.H.; Schober, W.; Leverson, J.D.; Zhang, B.; Bhatia, R.; Huang, X.; et al. Combined targeting of BCL-2 and BCR-ABL tyrosine kinase eradicates chronic myeloid leukemia stem cells. Sci. Transl. Med. 2016, 8, 355ra117. [Google Scholar] [CrossRef] [Green Version]
- Mattes, K.; Gerritsen, M.; Folkerts, H.; Geugien, M.; van den Heuvel, F.A.; Svendsen, A.F.; Yi, G.; Martens, J.H.A.; Vellenga, E. CD34(+) acute myeloid leukemia cells with low levels of reactive oxygen species show increased expression of stemness genes and can be targeted by the BCL2 inhibitor venetoclax. Haematologica 2020, 105, e399–e403. [Google Scholar] [CrossRef]
- Maiti, A.; Franquiz, M.J.; Ravandi, F.; Cortes, J.E.; Jabbour, E.J.; Sasaki, K.; Marx, K.; Daver, N.G.; Kadia, T.M.; Konopleva, M.Y.; et al. Venetoclax and BCR-ABL Tyrosine Kinase Inhibitor Combinations: Outcome in Patients with Philadelphia Chromosome-Positive Advanced Myeloid Leukemias. Acta Haematol. 2020, 143, 567–573. [Google Scholar] [CrossRef]
- Gallipoli, P.; Cook, A.; Rhodes, S.; Hopcroft, L.; Wheadon, H.; Whetton, A.D.; Jørgensen, H.G.; Bhatia, R.; Holyoake, T.L. JAK2/STAT5 inhibition by nilotinib with ruxolitinib contributes to the elimination of CML CD34+ cells in vitro and in vivo. Blood 2014, 124, 1492–1501. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Gallipoli, P.; Degeer, D.; Sloma, I.; Forrest, D.L.; Chan, M.; Lai, D.; Jorgensen, H.; Ringrose, A.; Wang, H.M.; et al. Targeting Primitive Chronic Myeloid Leukemia Cells by Effective Inhibition of a New AHI-1-BCR-ABL-JAK2 Complex. J. Natl. Cancer Inst. 2013, 105, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Sweet, K.; Hazlehurst, L.; Sahakian, E.; Powers, J.; Nodzon, L.; Kayali, F.; Hyland, K.; Nelson, A.; Pinilla-Ibarz, J. A Phase I Study of Ruxolitinib Plus Nilotinib in Chronic Phase CML Patients with Molecular Evidence of Disease. Blood 2016, 128, 1892. [Google Scholar] [CrossRef]
- Sweet, K.; Hazlehurst, L.; Sahakian, E.; Powers, J.; Nodzon, L.; Kayali, F.; Hyland, K.; Nelson, A.; Pinilla-Ibarz, J. A phase I clinical trial of ruxolitinib in combination with nilotinib in chronic myeloid leukemia patients with molecular evidence of disease. Leuk. Res. 2018, 74, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Dao, K.-H.; Collins, R.H.; Cortes, J.E.; Deininger, M.W.; Druker, B.J.; Gotlib, J.R.; Macey, T.A.; Oh, S.T.; Tyner, J.W.; Winton, E.F. Phase 2 Study of Ruxolitinib in Patients with Chronic Neutrophilic Leukemia or Atypical Chronic Myeloid Leukemia. Blood 2018, 132 (Suppl. 1), 350. [Google Scholar] [CrossRef]
- Guerra, V.A.; Kantarjian, H.M.; Borthakur, G.M.; Verstovsek, S.; Pike, A.; Ravandi, F.; Jabbour, E.; Cortes, J.E. A Phase I-II Study of Ruxolitinib (INCB18424) for Patients with Chronic Myeloid Leukemia with Minimal Residual Disease While on Therapy with Imatinib. Blood 2019, 134 (Suppl. 1), 5906. [Google Scholar] [CrossRef]
- Westerweel, P.E.; te Boekhorst, P.A.W.; Levin, M.-D.; Cornelissen, J.J. New Approaches and Treatment Combinations for the Management of Chronic Myeloid Leukemia. Front. Oncol. 2019, 9, 665. [Google Scholar] [CrossRef]
- Rousselot, P.; Prost, S.; Guilhot, J.; Roy, L.; Etienne, G.; Legros, L.; Charbonnier, A.; Coiteux, V.; Cony-Makhoul, P.; Huguet, F.; et al. Pioglitazone together with imatinib in chronic myeloid leukemia: A proof of concept study. Cancer 2017, 123, 1791–1799. [Google Scholar] [CrossRef]
- Glodkowska-Mrowka, E.; Manda-Handzlik, A.; Stelmaszczyk-Emmel, A.; SeferzDska, I.; Stoklosa, T.; Przybylski, J.; Mrowka, P. PPAU≥ ligands increase antileukemic activity of second- and third-generation tyrosine kinase inhibitors in chronic myeloid leukemia cells. Blood Cancer J. 2016, 6, e377. [Google Scholar] [CrossRef] [Green Version]
- Yousefi, B.; Samadi, N.; Baradaran, B.; Shafiei-Irannejad, V.; Zarghami, N. Peroxisome Proliferator-Activated Receptor Ligands and Their Role in Chronic Myeloid Leukemia: Therapeutic Strategies. Chem. Biol. Drug Des. 2016, 88, 17–25. [Google Scholar] [CrossRef]
- Prost, S.; Relouzat, F.; Spentchian, M.; Ouzegdouh, Y.; Saliba, J.; Massonnet, G.; Beressi, J.P.; Verhoeyen, E.; Raggueneau, V.; Maneglier, B.; et al. Erosion of the chronic myeloid leukaemia stem cell pool by PPARγ agonists. Nature 2015, 525, 380–383. [Google Scholar] [CrossRef] [PubMed]
- Eiring, A.M.; Page, B.D.G.; Kraft, I.L.; Mason, C.C.; Vellore, N.A.; Resetca, D.; Zabriskie, M.S.; Zhang, T.Y.; Khorashad, J.S.; Engar, A.J.; et al. Combined STAT3 and BCR-ABL1 inhibition induces synthetic lethality in therapy-resistant chronic myeloid leukemia. Leukemia 2017, 31, 1253–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brachet-Botineau, M.; Polomski, M.; Neubauer, H.A.; Juen, L.; Hédou, D.; Viaud-Massuard, M.-C.; Prié, G.; Gouilleux, F. Pharmacological Inhibition of Oncogenic STAT3 and STAT5 Signaling in Hematopoietic Cancers. Cancers 2020, 12, 240. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; He, B.; Ma, X.; Yu, S.; Bhave, R.R.; Lentz, S.R.; Tan, K.; Guzman, M.L.; Zhao, C.; Xue, H.H. Prostaglandin E1 and Its Analog Misoprostol Inhibit Human CML Stem Cell Self-Renewal via EP4 Receptor Activation and Repression of AP-1. Cell Stem Cell 2017, 21, 359–373.e5. [Google Scholar] [CrossRef] [Green Version]
- El Eit, R.; Itani, A.R.; Nassar, F.; Rasbieh, N.; Jabbour, M.; Santina, A.; Zaatari, G.; Mahon, F.-X.; Bazarbachi, A.; Nasr, R. Antitumor efficacy of arsenic/interferon in preclinical models of chronic myeloid leukemia resistant to tyrosine kinase inhibitors. Cancer 2019, 125, 2818–2828. [Google Scholar] [CrossRef]
- Guarnerio, J.; Mendez, L.M.; Asada, N.; Menon, A.V.; Fung, J.; Berry, K.; Frenette, P.S.; Ito, K.; Pandolfi, P.P. A non-cell-autonomous role for Pml in the maintenance of leukemia from the niche. Nat. Commun. 2018, 9, 66. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Hou, J.; Chan, G.; Sze, D. Arsenic Trioxide for Non Acute Promyelocytic Leukemia Hematological Malignancies: A New Frontier. J. Blood Disord. 2014, 1, 1018. [Google Scholar]
- Du, Y.; Wang, K.; Fang, H.; Li, J.; Xiao, D.; Zheng, P.; Chen, Y.; Fan, H.; Pan, X.; Zhao, C.; et al. Coordination of intrinsic, extrinsic, and endoplasmic reticulum-mediated apoptosis by imatinib mesylate combined with arsenic trioxide in chronic myeloid leukemia. Blood 2006, 107, 1582–1590. [Google Scholar] [CrossRef]
- Wang, W.; Lv, F.-f.; Du, Y.; Li, N.; Chen, Y.; Chen, L. The effect of nilotinib plus arsenic trioxide on the proliferation and differentiation of primary leukemic cells from patients with chronic myoloid leukemia in blast crisis. Cancer Cell Int. 2015, 15, 10. [Google Scholar] [CrossRef] [Green Version]
- El Eit, R.M.; Iskandarani, A.N.; Saliba, J.L.; Jabbour, M.N.; Mahfouz, R.A.; Bitar, N.M.; Ayoubi, H.R.; Zaatari, G.S.; Mahon, F.X.; De Thé, H.B.; et al. Effective targeting of chronic myeloid leukemia initiating activity with the combination of arsenic trioxide and interferon alpha. Int. J. Cancer 2014, 134, 988–996. [Google Scholar] [CrossRef]
- Heibl, S.; Buxhofer-Ausch, V.; Schmidt, S.; Webersinke, G.; Lion, T.; Piringer, G.; Kuehr, T.; Wolf, D.; Melchardt, T.; Greil, R.; et al. A phase 1 study to evaluate the feasibility and efficacy of the addition of ropeginterferon alpha-2b to imatinib treatment in patients with chronic phase chronic myeloid leukemia (CML) not achieving a deep molecular response (molecular remission 4.5)-AGMT_CML 1. Hematol. Oncol. 2020, 28, 792–798. [Google Scholar]
- Mitchell, R.; Copland, M. Defining niche interactions to target chronic myeloid leukemia stem cells. Haematologica 2020, 105, 2–4. [Google Scholar] [CrossRef] [PubMed]
- Murakami, J.L.; Xu, B.; Franco, C.B.; Hu, X.; Galli, S.J.; Weissman, I.L.; Chen, C.C. Evidence that β7 Integrin Regulates Hematopoietic Stem Cell Homing and Engraftment Through Interaction with MAdCAM-1. Stem Cells Dev. 2016, 25, 18–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godavarthy, P.S.; Kumar, R.; Herkt, S.C.; Pereira, R.S.; Hayduk, N.; Weissenberger, E.S.; Aggoune, D.; Manavski, Y.; Lucas, T.; Pan, K.T.; et al. The vascular bone marrow niche influences outcome in chronic myeloid leukemia via the E-selectin—SCL/TAL1—CD44 axis. Haematologica 2020, 105, 136–147. [Google Scholar] [CrossRef] [Green Version]
- Windisch, R.; Pirschtat, N.; Kellner, C.; Chen-Wichmann, L.; Lausen, J.; Humpe, A.; Krause, D.; Wichmann, C. Oncogenic Deregulation of Cell Adhesion Molecules in Leukemia. Cancers 2019, 11, 311. [Google Scholar] [CrossRef] [Green Version]
- Klamer, S.; Voermans, C. The role of novel and known extracellular matrix and adhesion molecules in the homeostatic and regenerative bone marrow microenvironment. Cell Adhes. Migr. 2014, 8, 563–577. [Google Scholar] [CrossRef] [Green Version]
- Villatoro, A.; Konieczny, J.; Cuminetti, V.; Arranz, L. Leukemia Stem Cell Release From the Stem Cell Niche to Treat Acute Myeloid Leukemia. Front. Cell Dev. Biol. 2020, 8, 607. [Google Scholar] [CrossRef]
- Zhou, H.-S.; Carter, B.Z.; Andreeff, M. Bone marrow niche-mediated survival of leukemia stem cells in acute myeloid leukemia: Yin and Yang. Cancer Biol. Med. 2016, 13, 248–259. [Google Scholar] [CrossRef] [Green Version]
- Schepers, K.; Pietras, E.M.; Reynaud, D.; Flach, J.; Binnewies, M.; Garg, T.; Wagers, A.J.; Hsiao, E.C.; Passegué, E. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell 2013, 13, 285–299. [Google Scholar] [CrossRef] [Green Version]
- Schepers, K.; Campbell, T.B.; Passegué, E. Normal and leukemic stem cell niches: Insights and therapeutic opportunities. Cell Stem Cell 2015, 16, 254–267. [Google Scholar] [CrossRef] [Green Version]
- Stramucci, L.; Perrotti, D. Twisting IL-1 signaling to kill CML stem cells. Blood 2016, 128, 2592–2593. [Google Scholar] [CrossRef] [PubMed]
- Krause, D.S.; Lazarides, K.; Lewis, J.B.; von Andrian, U.H.; Van Etten, R.A. Selectins and their ligands are required for homing and engraftment of BCR-ABL1+ leukemic stem cells in the bone marrow niche. Blood 2014, 123, 1361–1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeAngelo, D.J.; Erba, H.P.; Jonas, B.A.; O’Dwyer, M.; Marlton, P.; Huls, G.A.; Liesveld, J.; Cooper, B.W.; Bhatnagar, B.; Armstrong, M.; et al. A phase III trial to evaluate the efficacy of uproleselan (GMI-1271) with chemotherapy in patients with relapsed/refractory acute myeloid leukemia. J. Clin. Oncol. 2019, 37 (Suppl. 15), TPS7066. [Google Scholar] [CrossRef]
- Agarwal, P.; Isringhausen, S.; Li, H.; Paterson, A.J.; He, J.; Gomariz, Á.; Nagasawa, T.; Nombela-Arrieta, C.; Bhatia, R. Mesenchymal Niche-Specific Expression of Cxcl12 Controls Quiescence of Treatment-Resistant Leukemia Stem Cells. Cell Stem Cell 2019, 24, 769–784.e6. [Google Scholar] [CrossRef]
- Weisberg, E.; Azab, A.K.; Manley, P.W.; Kung, A.L.; Christie, A.L.; Bronson, R.; Ghobrial, I.M.; Griffin, J.D. Inhibition of CXCR4 in CML cells disrupts their interaction with the bone marrow microenvironment and sensitizes them to nilotinib. Leukemia 2012, 26, 985–990. [Google Scholar] [CrossRef] [Green Version]
- Melo, R.; Ferro, K.; Saad, S. CXCR7 participates in CXCL12-mediated migration and homing of leukemic and normal hematopoietic cells. Stem Cell Res. Ther. 2018, 9, 34. [Google Scholar] [CrossRef]
- Kaur, H.; Bruno, J.; Kumar, A.; Sharma, T. Aptamers in the Therapeutics and Diagnostics Pipelines. Theranostics 2018, 8, 4016–4032. [Google Scholar] [CrossRef]
- Weisberg, E.L.; Sattler, M.; Azab, A.K.; Eulberg, D.; Kruschinski, A.; Manley, P.W.; Stone, R.; Griffin, J.D. Inhibition of SDF-1-induced migration of oncogene-driven myeloid leukemia by the L-RNA aptamer (Spiegelmer), NOX-A12, and potentiation of tyrosine kinase inhibition. Oncotarget 2017, 8, 109973–109984. [Google Scholar] [CrossRef] [Green Version]
- Fruehauf, S. Current Clinical Indications for Plerixafor. Transfus. Med. Hemother. 2013, 40, 246–250. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, A.; Fleischman, A.G.; Petersen, C.L.; Mackenzie, R.; Luty, S.; Loriaux, M.; Druker, B.J.; Woltjer, R.L.; Deininger, M.W. Effects of plerixafor in combination with BCR-ABL kinase inhibition in a murine model of CML. Blood 2012, 120, 2658–2668. [Google Scholar] [CrossRef] [Green Version]
- Peng, G.; Liu, Y. Hypoxia-inducible factors in cancer stem cells and inflammation. Trends Pharmacol. Sci. 2015, 36, 374–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Shen, Y.; Gong, F.; Jiang, Y.; Zhang, R. HIF-α Promotes Chronic Myelogenous Leukemia Cell Proliferation by Upregulating p21 Expression. Cell Biochem. Biophys. 2015, 72, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Lin, Y.-C.; Tsai, M.-H.; Lin, C.-S.; Murayama, Y.; Sato, R.; Yokoyama, K.K. Emerging roles of hypoxia-inducible factors and reactive oxygen species in cancer and pluripotent stem cells. Kaohsiung J. Med. Sci. 2015, 31, 279–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheloni, G.; Tanturli, M.; Tusa, I.; Ho DeSouza, N.; Shan, Y.; Gozzini, A.; Mazurier, F.; Rovida, E.; Li, S.; Dello Sbarba, P. Targeting chronic myeloid leukemia stem cells with the hypoxia-inducible factor inhibitor acriflavine. Blood 2017, 130, 655–665. [Google Scholar] [CrossRef] [Green Version]
- Su, W.; Meng, F.; Huang, L.; Zheng, M.; Liu, W.; Sun, H. Sonic hedgehog maintains survival and growth of chronic myeloid leukemia progenitor cells through β-catenin signaling. Exp. Hematol. 2012, 40, 418–427. [Google Scholar] [CrossRef]
- Shah, N.P.; Cortes, J.E.; Martinelli, G.; Smith, B.D.; Clarke, E.; Copland, M.; Strauss, L.; Talpaz, M. Dasatinib Plus Smoothened (SMO) Inhibitor BMS-833923 in Chronic Myeloid Leukemia (CML) with Resistance or Suboptimal Response to a Prior Tyrosine Kinase Inhibitor (TKI): Phase I Study CA180323. Blood 2014, 124, 4539. [Google Scholar] [CrossRef]
- Irvine, D.A.; Zhang, B.; Kinstrie, R.; Tarafdar, A.; Morrison, H.; Campbell, V.L.; Moka, H.A.; Ho, Y.; Nixon, C.; Manley, P.W.; et al. Deregulated hedgehog pathway signaling is inhibited by the smoothened antagonist LDE225 (Sonidegib) in chronic phase chronic myeloid leukaemia. Sci. Rep. 2016, 6, 25476. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, P.; Zhang, B.; Ho, Y.; Cook, A.; Li, L.; Mikhail, F.M.; Wang, Y.; McLaughlin, M.E.; Bhatia, R. Enhanced targeting of CML stem and progenitor cells by inhibition of porcupine acyltransferase in combination with TKI. Blood 2017, 129, 1008–1020. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, P.; Zhang, B.; Ho, Y.; Cook, A.; Li, L.; Wang, Y.; Mclaughlin, M.E.; Bhatia, R. Inhibition of CML Stem Cell Renewal By the Porcupine Inhibitor WNT974. Blood 2015, 126, 54. [Google Scholar] [CrossRef]
- Grassi, S.; Palumbo, S.; Mariotti, V.; Liberati, D.; Guerrini, F.; Ciabatti, E.; Salehzadeh, S.; Baratè, C.; Balducci, S.; Ricci, F.; et al. The WNT Pathway Is Relevant for the BCR-ABL1-Independent Resistance in Chronic Myeloid Leukemia. Front. Oncol. 2019, 9, 532. [Google Scholar] [CrossRef] [Green Version]
- Riether, C.; Schürch, C.M.; Flury, C.; Hinterbrandner, M.; Drück, L.; Huguenin, A.L.; Baerlocher, G.M.; Radpour, R.; Ochsenbein, A.F. Tyrosine kinase inhibitor-induced CD70 expression mediates drug resistance in leukemia stem cells by activating Wnt signaling. Sci. Transl. Med. 2015, 7, 298ra119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pippa, R.; Odero, M.D. The Role of MYC and PP2A in the Initiation and Progression of Myeloid Leukemias. Cells 2020, 9, 544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucas, C.M.; Harris, R.J.; Giannoudis, A.; Clark, R.E. c-Myc inhibition decreases CIP2A and reduces BCR-ABL1 tyrosine kinase activity in chronic myeloid leukemia. Haematologica 2015, 100, e179–e182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiyota, M.; Kuroda, J.; Yamamoto-Sugitani, M.; Shimura, Y.; Nakayama, R.; Nagoshi, H.; Mizutani, S.; Chinen, Y.; Sasaki, N.; Sakamoto, N.; et al. FTY720 induces apoptosis of chronic myelogenous leukemia cells via dual activation of BIM and BID and overcomes various types of resistance to tyrosine kinase inhibitors. Apoptosis 2013, 18, 1437–1446. [Google Scholar] [CrossRef]
- Sharma, N.; Magistroni, V.; Piazza, R.; Citterio, S.; Mezzatesta, C.; Khandelwal, P.; Pirola, A.; Gambacorti-Passerini, C. BCR/ABL1 and BCR are under the transcriptional control of the MYC oncogene. Mol. Cancer 2015, 14, 132. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, A.; Mackenzie, R.; Pippa, R.; Eide, C.; Oddo, J.; Tyner, J.; Sears, R.; Vitek, M.; Christensen, D.; Druker, B. Antagonism of SET Using OP449 Enhances the Efficacy of Tyrosine Kinase Inhibitors and Overcomes Drug Resistance in Myeloid Leukemia. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 2092–2103. [Google Scholar] [CrossRef] [Green Version]
- Albajar, M.; Gomez-Casares, M.T.; Llorca, J.; Mauleon, I.; Vaque, J.P.; Acosta, J.C.; Bermudez, A.; Donato, N.; Delgado, M.D.; Leon, J. MYC in Chronic Myeloid Leukemia: Induction of Aberrant DNA Synthesis and Association with Poor Response to Imatinib. Mol. Cancer Res. 2011, 9, 564–576. [Google Scholar] [CrossRef] [Green Version]
- Neviani, P.; Harb, J.G.; Oaks, J.J.; Santhanam, R.; Walker, C.J.; Ellis, J.J.; Ferenchak, G.; Dorrance, A.M.; Paisie, C.A.; Eiring, A.M.; et al. PP2A-activating drugs selectively eradicate TKI-resistant chronic myeloid leukemic stem cells. J. Clin. Invest. 2013, 123, 4144–4157. [Google Scholar] [CrossRef]
- Keramatinia, A.; Ahadi, A.; Akbari, M.E.; Mohseny, M.; Mosavi Jarahi, A.; Bahadori-Monfared, A.; Hashemi, M.; Moradi, A.; Mehrvar, N.; Kazemi, E.; et al. The roles of DNA epigenetics and clinical significance in Chronic Myeloid Leukemia: A review. Cell Mol. Biol. 2018, 64, 58–63. [Google Scholar] [CrossRef]
- Goldman, J.M.; Gordon, M.; Bazeos, A.; Marin, D. Biology of CML stem cells: The basis for clinical heterogeneity? Leuk. Suppl. 2012, 1, S43–S45. [Google Scholar] [CrossRef]
- Biray Avci, C.; Goker Bagca, B.; Tetik Vardarli, A.; Saydam, G.; Gunduz, C. Epigenetic modifications in chronic myeloid leukemia cells through ruxolitinib treatment. J. Cell Biochem. 2019, 120, 4555–4563. [Google Scholar] [CrossRef] [PubMed]
- Olszewski, A.J.; Chorzalska, A.D.; Kim, A.S.; Quesenberry, P.J.; Lopresti, M.L.; Fenton, M.A.; Reagan, J.L.; Butera, J.N.; Sahin, I.; Hamel, C.; et al. Clonal haematopoiesis of indeterminate potential among cancer survivors exposed to myelotoxic chemotherapy. Br. J. Haematol. 2019, 186, e31–e35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steensma, D.P. Myelodysplastic syndromes current treatment algorithm 2018. Blood Cancer J. 2018, 8, 47. [Google Scholar] [CrossRef] [PubMed]
- Busque, L.; Buscarlet, M.; Mollica, L.; Levine, R.L. Concise Review: Age-Related Clonal Hematopoiesis: Stem Cells Tempting the Devil. Stem Cells 2018, 36, 1287–1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valent, P.; Kern, W.; Hoermann, G.; Milosevic Feenstra, J.; Sotlar, K.; Pfeilstöcker, M.; Germing, U.; Sperr, W.; Reiter, A.; Wolf, D.; et al. Clonal Hematopoiesis with Oncogenic Potential (CHOP): Separation from CHIP and Roads to AML. Int. J. Mol. Sci. 2019, 20, 789. [Google Scholar] [CrossRef] [Green Version]
- Park, S.J.; Bejar, R. Clonal Hematopoiesis in Aging. Curr. Stem Cell Rep. 2018, 4, 209–219. [Google Scholar] [CrossRef]
- Abelson, S.; Wang, J.C.Y. Age-related clonal hematopoiesis: Implications for hematopoietic stem cell transplantation. Curr. Opin. Hematol. 2018, 25, 441–445. [Google Scholar] [CrossRef]
- Jaiswal, S.; Ebert, B.L. Clonal hematopoiesis in human aging and disease. Science 2019, 366, eaan4673. [Google Scholar] [CrossRef]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [Green Version]
- Doroshow, D.B.; Eder, J.P.; LoRusso, P.M. BET inhibitors: A novel epigenetic approach. Ann. Oncol. 2017, 28, 1776–1787. [Google Scholar] [CrossRef]
- White, M.E.; Fenger, J.M.; Carson, W.E. 3rd, Emerging roles of and therapeutic strategies targeting BRD4 in cancer. Cell Immunol. 2019, 337, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Peter, B.; Eisenwort, G.; Keller, A.; Bauer, K.; Berger, D.; Sadovnik, I.; Stefanzl, G.; Hoermann, G.; Wolf, D.; Racil, Z.; et al. BRD4 Degradation Is a Potent Approach to Block MYC Expression and to Overcome Multiple Forms of Stem Cell Resistance in Ph+ CML. Blood 2018, 132 (Suppl. 1), 1722. [Google Scholar] [CrossRef]
- White, E. The role for autophagy in cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Bengsch, F.; Svoronos, N.; Rutkowski, M.R.; Bitler, B.G.; Allegrezza, M.J.; Yokoyama, Y.; Kossenkov, A.V.; Bradner, J.E.; Conejo-Garcia, J.R.; et al. BET Bromodomain Inhibition Promotes Anti-tumor Immunity by Suppressing PD-L1 Expression. Cell Rep. 2016, 16, 2829–2837. [Google Scholar] [CrossRef] [Green Version]
- Hogg, S.J.; Vervoort, S.J.; Deswal, S.; Ott, C.J.; Li, J.; Cluse, L.A.; Beavis, P.A.; Darcy, P.K.; Martin, B.P.; Spencer, A.; et al. BET-Bromodomain Inhibitors Engage the Host Immune System and Regulate Expression of the Immune Checkpoint Ligand PD-L1. Cell Rep. 2017, 18, 2162–2174. [Google Scholar] [CrossRef] [Green Version]
- Jones, B.A.; Varambally, S.; Arend, R.C. Histone Methyltransferase EZH2: A Therapeutic Target for Ovarian Cancer. Mol. Cancer Ther. 2018, 17, 591–602. [Google Scholar] [CrossRef] [Green Version]
- Ueda, K.; Yoshimi, A.; Kagoya, Y.; Nishikawa, S.; Marquez, V.E.; Nakagawa, M.; Kurokawa, M. Inhibition of histone methyltransferase EZH2 depletes leukemia stem cell of mixed lineage leukemia fusion leukemia through upregulation of p16. Cancer Sci. 2014, 105, 512–519. [Google Scholar] [CrossRef]
- Rinke, J.; Chase, A.; Cross, N.C.P.; Hochhaus, A.; Ernst, T. EZH2 in Myeloid Malignancies. Cells 2020, 9, 1639. [Google Scholar] [CrossRef]
- Wen, Y.; Cai, J.; Hou, Y.; Huang, Z.; Wang, Z. Role of EZH2 in cancer stem cells: From biological insight to a therapeutic target. Oncotarget 2017, 8, 37974–37990. [Google Scholar] [CrossRef] [Green Version]
- Xie, H.; Peng, C.; Huang, J.; Li, B.E.; Kim, W.; Smith, E.C.; Fujiwara, Y.; Qi, J.; Cheloni, G.; Das, P.P.; et al. Chronic Myelogenous Leukemia- Initiating Cells Require Polycomb Group Protein EZH2. Cancer Discov. 2016, 6, 1237–1247. [Google Scholar] [CrossRef] [Green Version]
- Bavaro, L.; Martelli, M.; Cavo, M.; Soverini, S. Mechanisms of Disease Progression and Resistance to Tyrosine Kinase Inhibitor Therapy in Chronic Myeloid Leukemia: An Update. Int. J. Mol. Sci. 2019, 20, 6141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuda, Y.; Yamauchi, T.; Hosono, N.; Uzui, K.; Negoro, E.; Morinaga, K.; Nishi, R.; Yoshida, A.; Kimura, S.; Maekawa, T.; et al. Combination of panobinostat with ponatinib synergistically overcomes imatinib-resistant CML cells. Cancer Sci. 2016, 107, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Strauss, A.C.; Chu, S.; Li, M.; Ho, Y.; Shiang, K.D.; Snyder, D.S.; Huettner, C.S.; Shultz, L.; Holyoake, T.; et al. Effective targeting of quiescent chronic myelogenous leukemia stem cells by histone deacetylase inhibitors in combination with imatinib mesylate. Cancer Cell 2010, 17, 427–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lernoux, M.; Schnekenburger, M.; Losson, H.; Vermeulen, K.; Hahn, H.; Gérard, D.; Lee, J.-Y.; Mazumder, A.; Ahamed, M.; Christov, C.; et al. Novel HDAC inhibitor MAKV-8 and imatinib synergistically kill chronic myeloid leukemia cells via inhibition of BCR-ABL/MYC-signaling: Effect on imatinib resistance and stem cells. Clin. Epigenet. 2020, 12, 69. [Google Scholar] [CrossRef]
- He, B.; Wang, Q.; Xu, N.; Lu, Z.; Pan, C.; Sun, J.; Zhou, H.; Xiaoli, L. Chidamide, a Novel Histone Deacetylase Inhibitor, Combined with Imatinib Induces Synthetic Lethality in Drug-Resistant Chronic Myeloid Leukemia. Blood 2017, 130 (Suppl. 1), 4990. [Google Scholar]
- Zhu, F.; Rui, L. PRMT5 in gene regulation and hematologic malignancies. Genes Dis. 2019, 6, 247–257. [Google Scholar] [CrossRef]
- Xiao, W.; Chen, X.; Liu, L.; Shu, Y.; Zhang, M.; Zhong, Y. Role of protein arginine methyltransferase 5 in human cancers. Biomed. Pharmacother. 2019, 114, 108790. [Google Scholar] [CrossRef]
- Jin, Y.; Zhou, J.; Xu, F.; Jin, B.; Cui, L.; Wang, Y.; Du, X.; Li, J.; Li, P.; Ren, R.; et al. Targeting methyltransferase PRMT5 eliminates leukemia stem cells in chronic myelogenous leukemia. J. Clin. Invest. 2016, 126, 3961–3980. [Google Scholar] [CrossRef] [Green Version]
- Abraham, S.A.; Hopcroft, L.E.; Carrick, E.; Drotar, M.E.; Dunn, K.; Williamson, A.J.; Korfi, K.; Baquero, P.; Park, L.E.; Scott, M.T.; et al. Dual targeting of p53 and c-MYC selectively eliminates leukaemic stem cells. Nature 2016, 534, 341–346. [Google Scholar] [CrossRef]
- Crivellaro, S.; Panuzzo, C.; Carrà, G.; Volpengo, A.; Crasto, F.; Gottardi, E.; Familiari, U.; Papotti, M.; Torti, D.; Piazza, R.; et al. Non genomic loss of function of tumor suppressors in CML: BCR-ABL promotes IκBα mediated p53 nuclear exclusion. Oncotarget 2015, 6, 25217–25225. [Google Scholar] [CrossRef]
- Li, L.; Wang, L.; Li, L.; Wang, Z.; Ho, Y.; McDonald, T.; Holyoake, T.L.; Chen, W.; Bhatia, R. Activation of p53 by SIRT1 inhibition enhances elimination of CML leukemia stem cells in combination with imatinib. Cancer Cell 2012, 21, 266–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mobaraki, R.N.; Karimi, M.; Alikarami, F.; Farhadi, E.; Amini, A.; Bashash, D.; Paridar, M.; Kokhaei, P.; Rezvani, M.R.; Kazemi, A.; et al. RITA induces apoptosis in p53-null K562 leukemia cells by inhibiting STAT5, Akt, and NF-κB signaling pathways. Anticancer Drugs 2018, 29, 847–853. [Google Scholar] [CrossRef] [PubMed]
- Issaeva, N.; Bozko, P.; Enge, M.; Protopopova, M.; Verhoef, L.G.; Masucci, M.; Pramanik, A.; Selivanova, G. Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nat. Med. 2004, 10, 1321–1328. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Yamada, S.; Ichwan, S.J.; Iseki, S.; Ohtani, K.; Otsu, M.; Ikeda, M.A. Inability of p53-reactivating compounds Nutlin-3 and RITA to overcome p53 resistance in tumor cells deficient in p53Ser46 phosphorylation. Biochem. Biophys. Res. Commun. 2012, 417, 931–937. [Google Scholar] [CrossRef]
- Hientz, K.; Mohr, A.; Bhakta-Guha, D.; Efferth, T. The role of p53 in cancer drug resistance and targeted chemotherapy. Oncotarget 2017, 8, 8921–8946. [Google Scholar] [CrossRef] [Green Version]
- Abraham, A.; Qiu, S.; Chacko, B.K.; Li, H.; Paterson, A.J.; He, J.; Agarwal, P.; Shah, M.; Welner, R.S.; Darley-Usmar, V.M.; et al. SIRT1 Mediates Enhanced Mitochondrial Oxidative Phosphorylation in Chronic Myelogenous Leukemia Stem Cells. Blood 2018, 132 (Suppl. 1), 932. [Google Scholar] [CrossRef]
- Peterson, L.F.; Lo, M.-C.; Liu, Y.; Giannola, D.; Mitrikeska, E.; Donato, N.J.; Johnson, C.N.; Wang, S.; Mercer, J.; Talpaz, M. Induction of p53 suppresses chronic myeloid leukemia. Leuk. Lymphoma 2017, 58, 2165–2175. [Google Scholar] [CrossRef]
- Sanz, G.; Singh, M.; Peuget, S.; Selivanova, G. Inhibition of p53 inhibitors: Progress, challenges and perspectives. J. Mol. Cell Biol. 2019, 11, 586–599. [Google Scholar] [CrossRef] [Green Version]
- Grant, S. Recruiting TP53 to target chronic myeloid leukemia stem cells. Haematologica 2020, 105, 1172–1174. [Google Scholar] [CrossRef]
- Yun, C.W.; Lee, S.H. The Roles of Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Li, Y.; Shou, L.; Li, L.; Chen, Z.; Ye, X.; Qian, W. The molecular mechanisms underlying BCR/ABL degradation in chronic myeloid leukemia cells promoted by Beclin1-mediated autophagy. Cancer Manag. Res. 2019, 11, 5197–5208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Z.; Xu, N.; He, B.; Pan, C.; Lan, Y.; Zhou, H.; Liu, X. Inhibition of autophagy enhances the selective anti-cancer activity of tigecycline to overcome drug resistance in the treatment of chronic myeloid leukemia. J. Exp. Clin. Cancer Res. 2017, 36, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuntz, E.M.; Baquero, P.; Michie, A.M.; Dunn, K.; Tardito, S.; Holyoake, T.L.; Helgason, G.V.; Gottlieb, E. Targeting mitochondrial oxidative phosphorylation eradicates therapy-resistant chronic myeloid leukemia stem cells. Nat. Med. 2017, 23, 1234–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greer, N.D. Tigecycline (Tygacil): The First in the Glycylcycline Class of Antibiotics. Bayl. Univ. Med. Cent. Proc. 2006, 19, 155–161. [Google Scholar] [CrossRef]
- Santana-Codina, N.; Mancias, J.D.; Kimmelman, A.C. The Role of Autophagy in Cancer. Annu. Rev. Cancer Biol. 2017, 1, 19–39. [Google Scholar] [CrossRef]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.-J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Calabretta, B.; Salomoni, P. Inhibition of autophagy: A new strategy to enhance sensitivity of chronic myeloid leukemia stem cells to tyrosine kinase inhibitors. Leuk. Lymphoma 2011, 52 (Suppl. 1), 54–59. [Google Scholar] [CrossRef]
- Horne, G.A.; Stobo, J.; Kelly, C.; Mukhopadhyay, A.; Latif, A.L.; Dixon-Hughes, J.; McMahon, L.; Cony-Makhoul, P.; Byrne, J.; Smith, G.; et al. A randomised phase II trial of hydroxychloroquine and imatinib versus imatinib alone for patients with chronic myeloid leukaemia in major cytogenetic response with residual disease. Leukemia 2020, 34, 1775–1786. [Google Scholar] [CrossRef]
- Riether, C.; Gschwend, T.; Huguenin, A.L.; Schürch, C.M.; Ochsenbein, A.F. Blocking programmed cell death 1 in combination with adoptive cytotoxic T-cell transfer eradicates chronic myelogenous leukemia stem cells. Leukemia 2015, 29, 1781–1785. [Google Scholar] [CrossRef] [Green Version]
- Rousselot, P.; Renard, P.; de Buyer, A.; Finet, A.; Spentchian, M.; Saiag, P. Nivolumab to control molecular response in chronic myeloid leukemia. Leuk. Res. 2018, 72, 5–6. [Google Scholar] [CrossRef]
- Collins, J.M.; Gulley, J.L. Product review: Avelumab, an anti-PD-L1 antibody. Hum. Vaccin. Immunother. 2019, 15, 891–908. [Google Scholar] [CrossRef] [PubMed]
- Mosoyan, G.; Kraus, T.; Ye, F.; Eng, K.; Crispino, J.D.; Hoffman, R.; Iancu-Rubin, C. Imetelstat, a telomerase inhibitor, differentially affects normal and malignant megakaryopoiesis. Leukemia 2017, 31, 2458–2467. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hu, C.S.; Petersen, B.; Qiu, J.; Ye, F.; Houldsworth, J.; Eng, K.; Huang, F.; Hoffman, R. Imetelstat, a telomerase inhibitor, is capable of depleting myelofibrosis stem and progenitor cells. Blood Adv. 2018, 2, 2378–2388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tefferi, A.; Lasho, T.L.; Begna, K.H.; Patnaik, M.M.; Zblewski, D.L.; Finke, C.M.; Laborde, R.R.; Wassie, E.; Schimek, L.; Hanson, C.A.; et al. A Pilot Study of the Telomerase Inhibitor Imetelstat for Myelofibrosis. N. Engl. J. Med. 2015, 373, 908–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mascarenhas, J.; Komrokji, R.S.; Cavo, M.; Martino, B.; Niederwieser, D.; Reiter, A.; Scott, B.L.; Baer, M.R.; Hoffman, R.; Odenike, O.; et al. Imetelstat Is Effective Treatment for Patients with Intermediate-2 or High-Risk Myelofibrosis Who Have Relapsed on or Are Refractory to Janus Kinase Inhibitor Therapy: Results of a Phase 2 Randomized Study of Two Dose Levels. Blood 2018, 132 (Suppl. 1), 685. [Google Scholar] [CrossRef]
- Baerlocher, G.M.; Oppliger Leibundgut, E.; Ottmann, O.G.; Spitzer, G.; Odenike, O.; Mcdevitt, M.A.; Röth, A.; Daskalakis, M.; Burington, B.; Stuart, M.; et al. Telomerase Inhibitor Imetelstat in Patients with Essential Thrombocythemia. N. Engl. J. Med. 2015, 373, 920–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mascarenhas, J.; Lu, M.; Kosiorek, H.; Virtgaym, E.; Xia, L.; Sandy, L.; Mesa, R.; Petersen, B.; Farnoud, N.; Najfeld, V.; et al. Oral idasanutlin in patients with polycythemia vera. Blood 2019, 134, 525–533. [Google Scholar] [CrossRef] [Green Version]
- Hobbs, G.S.; Hanasoge Somasundara, A.V.; Kleppe, M.; Litvin, R.; Arcila, M.; Ahn, J.; Mckenney, A.S.; Knapp, K.; Ptashkin, R.; Weinstein, H.; et al. Hsp90 inhibition disrupts JAK-STAT signaling and leads to reductions in splenomegaly in patients with myeloproliferative neoplasms. Haematologica 2018, 103, e5–e9. [Google Scholar] [CrossRef] [Green Version]
- Pemmaraju, N.; Gupta, V.; Ali, H.; Yacoub, A.; Wang, E.S.; Lee, S.; Schiller, G.J.; Sardone, M.; Wysowskyj, H.; Chen, J.; et al. Results from a Phase 1/2 Clinical Trial of Tagraxofusp (SL-401) in Patients with Intermediate, or High Risk, Relapsed/Refractory Myelofibrosis. Blood 2019, 134 (Suppl. 1), 558. [Google Scholar] [CrossRef]
- Pettit, K.; Gerds, A.T.; Yacoub, A.; Watts, J.M.; Tartaczuch, M.; Bradley, T.J.; Shortt, J.; Stevenson, W.S.; Curtin, N.J.; Rossetti, J.M.; et al. A Phase 2a Study of the LSD1 Inhibitor Img-7289 (bomedemstat) for the Treatment of Myelofibrosis. Blood 2019, 134 (Suppl. 1), 556. [Google Scholar] [CrossRef]
- Hergert, J. First-in-Class LSD1 Inhibitor Elicits Clinical Activity in Advanced Myelofibrosis. Available online: onclive.com/view/first-in-class-lsd1-inhibitor-elicits-clinical-activity-in-advanced-myelofibrosis (accessed on 30 November 2019).
- Hugh Rienhoff, M. IMG-7289 in Patients With Essential Thrombocythemia, 2020-02-05 ed.; U.S. National Library of Medicine: Bethesda, MD, USA, 2020.
- Rambaldi, A.; Iurlo, A.; Vannucchi, A.M.; Noble, R.; Von Bubnoff, N.; Guarini, A.; Martino, B.; Pezzutto, A.; Carli, G.; De Muro, M.; et al. Safety and efficacy of the maximum tolerated dose of givinostat in polycythemia vera: A two-part Phase Ib/II study. Leukemia 2020, 34, 2234–2237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deangelo, D.J.; Mesa, R.A.; Fiskus, W.; Tefferi, A.; Paley, C.; Wadleigh, M.; Ritchie, E.K.; Snyder, D.S.; Begna, K.; Ganguly, S.; et al. Phase II trial of panobinostat, an oral pan-deacetylase inhibitor in patients with primary myelofibrosis, post-essential thrombocythaemia, and post-polycythaemia vera myelofibrosis. Br. J. Haematol. 2013, 162, 326–335. [Google Scholar] [CrossRef] [PubMed]
- A Study of PRT543 in Participants With Advanced Solid Tumors and Hematologic Malignancies, 2019-03-22 ed.; U.S. National Library of Medicine: Bethesda, MD, USA, 2019.
- Drexler, B.; Passweg, J.R.; Tzankov, A.; Bigler, M.; Theocharides, A.P.; Cantoni, N.; Keller, P.; Stussi, G.; Ruefer, A.; Benz, R.; et al. The sympathomimetic agonist mirabegron did not lower JAK2-V617F allele burden, but restored nestin-positive cells and reduced reticulin fibrosis in patients with myeloproliferative neoplasms: Results of phase II study SAKK 33/14. Haematologica 2019, 104, 710–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerds, A.T.; Vannucchi, A.M.; Passamonti, F.; Kremyanskaya, M.; Gotlib, J.R.; Palmer, J.M.; McCaul, K.; Ribrag, V.; Mead, A.J.; Harrison, C.N.; et al. A Phase 2 Study of Luspatercept in Patients with Myelofibrosis-Associated Anemia. Blood 2019, 134 (Suppl. 1), 557. [Google Scholar] [CrossRef]
- Bose, P.; Pemmaraju, N.; Masarova, L.; Bledsoe, S.D.; Daver, N.; Jabbour, E.; Kadia, T.M.; Estrov, Z.E.; Kornblau, S.M.; Andreeff, M.; et al. Sotatercept (ACE-011) for Anemia of Myelofibrosis: A Phase 2 Study. Blood 2020, 136 (Suppl. 1), 10–11. [Google Scholar] [CrossRef]
- Piszczatowski, R.T.; Steidl, U. Aurora Kinase A Inhibition: A Mega-Hit for Myelofibrosis Therapy? Clin. Cancer Res. 2019, 25, 4868–4870. [Google Scholar] [CrossRef]
- Gangat, N.; Marinaccio, C.; Swords, R.; Watts, J.M.; Gurbuxani, S.; Rademaker, A.; Fought, A.J.; Frankfurt, O.; Altman, J.K.; Wen, Q.J.; et al. Aurora Kinase A Inhibition Provides Clinical Benefit, Normalizes Megakaryocytes, and Reduces Bone Marrow Fibrosis in Patients with Myelofibrosis: A Phase I Trial. Clin. Cancer Res. 2019, 25, 4898–4906. [Google Scholar] [CrossRef] [Green Version]
- Durrant, S.T.; Nagler, A.; Guglielmelli, P.; Lavie, D.; Le Coutre, P.; Gisslinger, H.; Chuah, C.; Maffioli, M.; Bharathy, S.; Dong, T.; et al. Results from HARMONY: An open-label, multicenter, 2-arm, phase 1b, dose-finding study assessing the safety and efficacy of the oral combination of ruxolitinib and buparlisib in patients with myelofibrosis. Haematologica 2019, 104, e551–e554. [Google Scholar] [CrossRef] [Green Version]
- Saenz, D.T.; Fiskus, W.; Manshouri, T.; Rajapakshe, K.; Krieger, S.; Sun, B.; Mill, C.P.; Dinardo, C.; Pemmaraju, N.; Kadia, T.; et al. BET protein bromodomain inhibitor-based combinations are highly active against post-myeloproliferative neoplasm secondary AML cells. Leukemia 2017, 31, 678–687. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Salvia, M.; Esteller, M. Bromodomain inhibitors and cancer therapy: From structures to applications. Epigenetics 2017, 12, 323–339. [Google Scholar] [CrossRef]
- Jiang, Q.; Jamieson, C. BET’ing on Dual JAK/BET Inhibition as a Therapeutic Strategy for Myeloproliferative Neoplasms. Cancer Cell 2018, 33, 3–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleppe, M.; Koche, R.; Zou, L.; Van Galen, P.; Hill, C.E.; Dong, L.; De Groote, S.; Papalexi, E.; Hanasoge Somasundara, A.V.; Cordner, K.; et al. Dual Targeting of Oncogenic Activation and Inflammatory Signaling Increases Therapeutic Efficacy in Myeloproliferative Neoplasms. Cancer Cell 2018, 33, 29–43.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sashida, G.; Wang, C.; Tomioka, T.; Oshima, M.; Aoyama, K.; Kanai, A.; Mochizuki-Kashio, M.; Harada, H.; Shimoda, K.; Iwama, A. The loss of Ezh2 drives the pathogenesis of myelofibrosis and sensitizes tumor-initiating cells to bromodomain inhibition. J. Exp. Med. 2016, 213, 1459–1477. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Xia, L.; Li, Y.; Wang, X.; Hoffman, R. The orally bioavailable MDM2 antagonist RG7112 and pegylated interferon α 2a target JAK2V617F-positive progenitor and stem cells. Blood 2014, 124, 771–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eran, Z.; Zingariello, M.; Bochicchio, M.T.; Bardelli, C.; Migliaccio, A.R. Novel strategies for the treatment of myelofibrosis driven by recent advances in understanding the role of the microenvironment in its etiology. F1000Research 2019, 8, 1662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcellino, B.K.; Hoffman, R.; Tripodi, J.; Lu, M.; Kosiorek, H.; Mascarenhas, J.; Rampal, R.K.; Dueck, A.; Najfeld, V. Advanced forms of MPNs are accompanied by chromosomal abnormalities that lead to dysregulation of TP53. Blood Adv. 2018, 2, 3581–3589. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Delgado, R.; McLornan, D.P.; Rejtő, L.; Jourdan, E.; Al-Ali, H.K.; Pluta, A.; Hus, M.; Ewing, J.; Khan, I.; Hebart, H.; et al. An Open-Label, Phase 2 Study of KRT-232, a First-in-Class, Oral Small Molecule Inhibitor of MDM2, for the Treatment of Patients with Myelofibrosis (MF) Who Have Previously Received Treatment with a JAK Inhibitor. Blood 2019, 134 (Suppl. 1), 2945. [Google Scholar] [CrossRef]
- Gotlib, J.; Gabrail, N.; O’Connell, C.L.; Garcia-Delgado, R.; Sbardellati, T.; Rothbaum, W.M.; McGreivy, J.; Harrison, C.N.; Kiladjian, J.-J. A Randomized, Open-Label, Multicenter, Phase 2 Study to Evaluate the Efficacy, Safety, and Pharmacokinetics of KRT-232 Compared with Ruxolitinib in Patients with Phlebotomy-Dependent Polycythemia Vera. Blood 2019, 134 (Suppl. 1), 4168. [Google Scholar] [CrossRef]
- Kuykendall, A.T.; Horvat, N.P.; Pandey, G.; Komrokji, R.; Reuther, G.W. Finding a Jill for JAK: Assessing Past, Present, and Future JAK Inhibitor Combination Approaches in Myelofibrosis. Cancers 2020, 12, 2278. [Google Scholar] [CrossRef]
- De Almeida, S.; Regimbeau, M.; Jego, G.; Garrido, C.; Girodon, F.; Hermetet, F. Heat Shock Proteins and PD-1/PD-L1 as Potential Therapeutic Targets in Myeloproliferative Neoplasms. Cancers 2020, 12, 2592. [Google Scholar] [CrossRef]
- Gilani, J.A.; Ashfaq, M.A.; Mansoor, A.-E.-R.; Abdul Jabbar, A.; Siddiqui, T.; Khan, M. Overview of the Mutational Landscape in Primary Myelofibrosis and Advances in Novel Therapeutics. Asian Pac. J. Cancer Prev. 2019, 20, 1691–1699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiskus, W.; Verstovsek, S.; Manshouri, T.; Rao, R.; Balusu, R.; Venkannagari, S.; Rao, N.N.; Ha, K.; Smith, J.E.; Hembruff, S.L.; et al. Heat Shock Protein 90 Inhibitor Is Synergistic with JAK2 Inhibitor and Overcomes Resistance to JAK2-TKI in Human Myeloproliferative Neoplasm Cells. Clin. Cancer Res. 2011, 17, 7347–7358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marubayashi, S.; Koppikar, P.; Taldone, T.; Abdel-Wahab, O.; West, N.; Bhagwat, N.; Caldas-Lopes, E.; Ross, K.N.; Gönen, M.; Gozman, A.; et al. HSP90 is a therapeutic target in JAK2-dependent myeloproliferative neoplasms in mice and humans. J. Clin. Investig. 2010, 120, 3578–3593. [Google Scholar] [CrossRef] [PubMed]
- Nieborowska-Skorska, M.; Maifrede, S.; Dasgupta, Y.; Sullivan, K.; Flis, S.; Le, B.V.; Solecka, M.; Belyaeva, E.A.; Kubovcakova, L.; Nawrocki, M.; et al. Ruxolitinib-induced defects in DNA repair cause sensitivity to PARP inhibitors in myeloproliferative neoplasms. Blood 2017, 130, 2848–2859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyner, J.W. DNA distress creates lethal opportunity in MPN. Blood 2017, 130, 2814–2816. [Google Scholar] [CrossRef] [PubMed]
- Rose, M.; Burgess, J.T.; O’Byrne, K.; Richard, D.J.; Bolderson, E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front. Cell Dev. Biol. 2020, 8, 564601. [Google Scholar] [CrossRef]
- Braun, L.M.; Zeiser, R. Immunotherapy in Myeloproliferative Diseases. Cells 2020, 9, 1559. [Google Scholar] [CrossRef]
- Alkharabsheh, O.; Frankel, A.E. Clinical Activity and Tolerability of SL-401 (Tagraxofusp): Recombinant Diphtheria Toxin and Interleukin-3 in Hematologic Malignancies. Biomedicines 2019, 7, 6. [Google Scholar] [CrossRef] [Green Version]
- Testa; Pelosi; Castelli, CD123 as a Therapeutic Target in the Treatment of Hematological Malignancies. Cancers 2019, 11, 1358. [CrossRef] [Green Version]
- Krishnan, A.; Pagane, M.; Roshal, M.; McGovern, E.; Stone-Molloy, Z.; Chen, J.; Brooks, C.; Levine, R.L.; Rampal, R.K. Evaluation of Tagraxofusp (SL-401) Alone and in Combination with Ruxolitinib for the Treatment of Myeloproliferative Neoplasms. Blood 2019, 134 (Suppl. 1), 2967. [Google Scholar] [CrossRef]
- Bose, P.; Masarova, L.; Verstovsek, S. Novel Concepts of Treatment for Patients with Myelofibrosis and Related Neoplasms. Cancers 2020, 12, 2891. [Google Scholar] [CrossRef] [PubMed]
- Mazzacurati, L.; Lambert, Q.T.; Pradhan, A.; Griner, L.N.; Huszar, D.; Reuther, G.W. The PIM inhibitor AZD1208 synergizes with ruxolitinib to induce apoptosis of ruxolitinib sensitive and resistant JAK2-V617F-driven cells and inhibit colony formation of primary MPN cells. Oncotarget 2015, 6, 40141–40157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzacurati, L.; Collins, R.J.; Pandey, G.; Lambert-Showers, Q.T.; Amin, N.E.; Zhang, L.; Stubbs, M.C.; Epling-Burnette, P.K.; Koblish, H.K.; Reuther, G.W. The pan-PIM inhibitor INCB053914 displays potent synergy in combination with ruxolitinib in models of MPN. Blood Adv. 2019, 3, 3503–3514. [Google Scholar] [CrossRef] [PubMed]
- Bartalucci, N.; Calabresi, L.; Balliu, M.; Martinelli, S.; Rossi, M.C.; Villeval, J.L.; Annunziato, F.; Guglielmelli, P.; Vannucchi, A.M. Inhibitors of the PI3K/mTOR pathway prevent STAT5 phosphorylation in JAK2V617F mutated cells through PP2A/CIP2A axis. Oncotarget 2017, 8, 96710–96724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiskus, W.; Verstovsek, S.; Manshouri, T.; Smith, J.E.; Peth, K.; Abhyankar, S.; McGuirk, J.; Bhalla, K.N. Dual PI3K/AKT/mTOR Inhibitor BEZ235 Synergistically Enhances the Activity of JAK2 Inhibitor against Cultured and Primary Human Myeloproliferative Neoplasm Cells. Mol. Cancer Ther. 2013, 12, 577–588. [Google Scholar] [CrossRef] [Green Version]
- Moyo, T.K.; Sochacki, A.; Ayers, G.D.; Byrne, M.T.; Strickland, S.A.; Mohan, S.R.; Harrison, J.; Berry, L.D.; Dudley, C.V.; Severs, R.; et al. Preliminary Results from a Phase I Dose Escalation Trial of Ruxolitinib and the PI3Kδ Inhibitor TGR-1202 in Myelofibrosis. Blood 2016, 128, 1125. [Google Scholar] [CrossRef]
- Petiti, J.; Lo Iacono, M.; Rosso, V.; Andreani, G.; Jovanovski, A.; Podestà, M.; Lame, D.; Gobbi, M.D.; Fava, C.; Saglio, G.; et al. Bcl-xL represents a therapeutic target in Philadelphia negative myeloproliferative neoplasms. J. Cell. Mol. Med. 2020, 24, 10978–10986. [Google Scholar] [CrossRef]
- Winter, P.S.; Sarosiek, K.A.; Lin, K.H.; Meggendorfer, M.; Schnittger, S.; Letai, A.; Wood, K.C. RAS signaling promotes resistance to JAK inhibitors by suppressing BAD-mediated apoptosis. Sci. Signal. 2014, 7, ra122. [Google Scholar] [CrossRef] [Green Version]
- Waibel, M.; Solomon, V.S.; Knight, D.A.; Ralli, R.A.; Kim, S.K.; Banks, K.M.; Vidacs, E.; Virely, C.; Sia, K.C.; Bracken, L.S.; et al. Combined Targeting of JAK2 and Bcl-2/Bcl-xL to Cure Mutant JAK2-Driven Malignancies and Overcome Acquired Resistance to JAK2 Inhibitors. Cell Rep. 2013, 5, 1047–1059. [Google Scholar] [CrossRef] [Green Version]
- Harrison, C.N.; Garcia, J.S.; Mesa, R.A.; Somervaille, T.C.; Komrokji, R.S.; Pemmaraju, N.; Jamieson, C.; Papadantonakis, N.; Foran, J.M.; O’Connell, C.L.; et al. Results from a Phase 2 Study of Navitoclax in Combination with Ruxolitinib in Patients with Primary or Secondary Myelofibrosis. Blood 2019, 134 (Suppl. 1), 671. [Google Scholar] [CrossRef]
- Jutzi, J.S.; Kleppe, M.; Dias, J.; Staehle, H.F.; Shank, K.; Teruya-Feldstein, J.; Gambheer, S.M.M.; Dierks, C.; Rienhoff, H.Y.J.; Levine, R.L.; et al. LSD1 Inhibition Prolongs Survival in Mouse Models of MPN by Selectively Targeting the Disease Clone. HemaSphere 2018, 2, e54. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zhao, X.; Perna, F.; Wang, L.; Koppikar, P.; Abdel-Wahab, O.; Harr, M.W.; Levine, R.L.; Xu, H.; Tefferi, A.; et al. JAK2V617F-Mediated Phosphorylation of PRMT5 Downregulates Its Methyltransferase Activity and Promotes Myeloproliferation. Cancer Cell 2011, 19, 283–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastore, F.; Bhagwat, N.; Krishnan, A.; Pastore, A.; Li, B.; Bowman, R.L.; Xiao, W.; Viny, A.D.; Karzai, A.; Zouak, A.; et al. PRMT5 Inhibition Modulates E2F1 Methylation and Gene Regulatory Networks Leading to Therapeutic Efficacy in JAK2VF Mutant MPN. Blood 2019, 134 (Suppl. 1), 473. [Google Scholar] [CrossRef]
- Sonderegger, S.; Cerruti, L.; Tremblay, C.; Toulmin, E.; Saw, J.; Nebl, T.; Hannan, K.M.; Lane, S.W.; Falk, H.; Unnikrishnan, A.; et al. Small-Molecule Inhibition of PRMT5 Induces Translational Stress and p53 in JAK2V617F Mutant Myeloproliferative Neoplasms. Blood 2018, 132 (Suppl. 1), 53. [Google Scholar] [CrossRef]
- Bose, P.; Verstovsek, S. Investigational histone deacetylase inhibitors (HDACi) in myeloproliferative neoplasms. Expert Opin. Investig. Drugs 2016, 25, 1393–1403. [Google Scholar] [CrossRef] [PubMed]
- Amaru Calzada, A.; Todoerti, K.; Donadoni, L.; Pellicioli, A.; Tuana, G.; Gatta, R.; Neri, A.; Finazzi, G.; Mantovani, R.; Rambaldi, A.; et al. The HDAC inhibitor Givinostat modulates the hematopoietic transcription factors NFE2 and C-MYB in JAK2V617F myeloproliferative neoplasm cells. Exp. Hematol. 2012, 40, 634–645.e10. [Google Scholar] [CrossRef]
- Wang, Y.; Fiskus, W.; Chong, D.G.; Buckley, K.M.; Natarajan, K.; Rao, R.; Joshi, A.; Balusu, R.; Koul, S.; Chen, J.; et al. Cotreatment with panobinostat and JAK2 inhibitor TG101209 attenuates JAK2V617F levels and signaling and exerts synergistic cytotoxic effects against human myeloproliferative neoplastic cells. Blood 2009, 114, 5024–5033. [Google Scholar] [CrossRef]
- Harrison, C.N.; Kiladjian, J.-J.; Heidel, F.H.; Vannucchi, A.M.; Passamonti, F.; Hayat, A.; Conneally, E.; Martino, B.; Kindler, T.; Lipka, D.B.; et al. Efficacy, Safety, and Confirmation of the Recommended Phase 2 Starting Dose of the Combination of Ruxolitinib (RUX) and Panobinostat (PAN) in Patients (Pts) with Myelofibrosis (MF). Blood 2015, 126, 4060. [Google Scholar] [CrossRef]
- Gleitz, H.F.; Kramann, R.; Schneider, R.K. Understanding deregulated cellular and molecular dynamics in the haematopoietic stem cell niche to develop novel therapeutics for bone marrow fibrosis. J. Pathol. 2018, 245, 138–146. [Google Scholar] [CrossRef] [Green Version]
- Arranz, L.; Sánchez-Aguilera, A.; Martín-Pérez, D.; Isern, J.; Langa, X.; Tzankov, A.; Lundberg, P.; Muntión, S.; Tzeng, Y.-S.; Lai, D.-M.; et al. Neuropathy of haematopoietic stem cell niche is essential for myeloproliferative neoplasms. Nature 2014, 512, 78–81. [Google Scholar] [CrossRef]
- Fielding, C.; Méndez-Ferrer, S. Neuronal regulation of bone marrow stem cell niches. F1000Research 2020, 9, 614. [Google Scholar] [CrossRef] [PubMed]
- Herlihy, N.; Harrison, C.N.; McLornan, D.P. Exploitation of the neural-hematopoietic stem cell niche axis to treat myeloproliferative neoplasms. Haematologica 2019, 104, 639–641. [Google Scholar] [CrossRef] [PubMed]
- Archana, A.; Morrone, K.; Bartenstein, M.; Zhao, Z.J.; Verma, A.; Goel, S. Bone marrow fibrosis in primary myelofibrosis: Pathogenic mechanisms and the role of TGF-β. Stem Cell Investig. 2016, 3, 5. [Google Scholar]
- Teodorescu, P.; Pasca, S.; Jurj, A.; Gafencu, G.; Joelsson, J.P.; Selicean, S.; Moldovan, C.; Munteanu, R.; Onaciu, A.; Tigu, A.B.; et al. Transforming growth factor β-mediated micromechanics modulates disease progression in primary myelofibrosis. J. Cell. Mol. Med. 2020, 24, 11100. [Google Scholar] [CrossRef]
- Yue, L.; Bartenstein, M.; Zhao, W.; Ho, W.T.; Han, Y.; Murdun, C.; Mailloux, A.W.; Zhang, L.; Wang, X.; Budhathoki, A.; et al. Efficacy of ALK5 inhibition in myelofibrosis. JCI Insight 2017, 2, e90932. [Google Scholar] [CrossRef] [Green Version]
- Ceglia, I.; Dueck, A.C.; Masiello, F.; Martelli, F.; He, W.; Federici, G.; Petricoin, E.F.; Zeuner, A.; Iancu-Rubin, C.; Weinberg, R.; et al. Preclinical rationale for TGF-β inhibition as a therapeutic target for the treatment of myelofibrosis. Exp. Hematol. 2016, 44, 1138–1155.e4. [Google Scholar] [CrossRef] [Green Version]
- Jeremy Wen, Q.; Yang, Q.; Goldenson, B.; Malinge, S.; Lasho, T.; Schneider, R.K.; Breyfogle, L.J.; Schultz, R.; Gilles, L.; Koppikar, P.; et al. Targeting megakaryocytic-induced fibrosis in myeloproliferative neoplasms by AURKA inhibition. Nat. Med. 2015, 21, 1473–1480. [Google Scholar] [CrossRef]
- Pilling, D.; Gomer, R.H. The Development of Serum Amyloid P as a Possible Therapeutic. Front. Immunol. 2018, 9, 2328. [Google Scholar] [CrossRef] [Green Version]
- Verstovsek, S.; Manshouri, T.; Pilling, D.; Bueso-Ramos, C.E.; Newberry, K.J.; Prijic, S.; Knez, L.; Bozinovic, K.; Harris, D.M.; Spaeth, E.L.; et al. Role of neoplastic monocyte-derived fibrocytes in primary myelofibrosis. J. Exp. Med. 2016, 213, 1723–1740. [Google Scholar] [CrossRef]
- Verstovsek, S.; Hasserjian, R.P.; Pozdnyakova, O.; Veletic, I.; Mesa, R.A.; Foltz, L.; Mascarenhas, J.; Ritchie, E.K.; Palmer, J.; Silver, R.T.; et al. PRM-151 in Myelofibrosis: Efficacy and Safety in an Open Label Extension Study. Blood 2018, 132 (Suppl. 1), 686. [Google Scholar] [CrossRef]
- Verstovsek, S.; Mesa, R.A.; Foltz, L.M.; Gupta, V.; Mascarenhas, J.O.; Ritchie, E.K.; Hoffman, R.; Silver, R.T.; Kremyanskaya, M.; Pozdnyakova, O.; et al. PRM-151 in Myelofibrosis: Durable Efficacy and Safety at 72 Weeks. Blood 2015, 126, 56. [Google Scholar] [CrossRef]
- Wang, J.-C.; Chen, C.; Kundra, A.; Kodali, S.; Pandey, A.; Wong, C.; Cheung, T.; Gotlieb, V.; Joseph, G.; Tribie, S. Programmed Cell Death Receptor (PD-1) Ligand (PD-L1) expression in Philadelphia chromosome-negative myeloproliferative neoplasms. Leuk. Res. 2019, 79, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Holmström, M.O.; Riley, C.H.; Skov, V.; Svane, I.M.; Hasselbalch, H.C.; Andersen, M.H. Spontaneous T-cell responses against the immune check point programmed-death-ligand 1 (PD-L1) in patients with chronic myeloproliferative neoplasms correlate with disease stage and clinical response. OncoImmunology 2018, 7, e1433521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prestipino, A.; Emhardt, A.J.; Aumann, K.; O’Sullivan, D.; Gorantla, S.P.; Duquesne, S.; Melchinger, W.; Braun, L.; Vuckovic, S.; Boerries, M.; et al. Oncogenic JAK2V617Fcauses PD-L1 expression, mediating immune escape in myeloproliferative neoplasms. Sci. Transl. Med. 2018, 10, eaam7729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinton, L.; Solito, S.; Damuzzo, V.; Francescato, S.; Pozzuoli, A.; Berizzi, A.; Mocellin, S.; Rossi, C.R.; Bronte, V.; Mandruzzato, S. Activated T cells sustain myeloid-derived suppressor cell-mediated immune suppression. Oncotarget 2016, 7, 1168–1184. [Google Scholar] [CrossRef] [Green Version]
- John Mascarenhas, M.; Gabriela Hobbs, M.D. PD-1 Inhibition in Advanced Myeloproliferative Neoplasms, 2017-02-27 ed.; U.S. National Library of Medicine: Bethesda, MD, USA, 2017.
- Verstovsek, S. Nivolumab in Treating Patients With Primary Myelofibrosis, Post-Essential Thrombocythemia Myelofibrosis, or Post-Polycythemia Vera Myelofibrosis, 2019-05-15 ed.; U.S. National Library of Medicine: Bethesda, MD, USA, 2019.
- Brady Stein, M. MHS, Durvalumab in Treating Patients With Primary, Post-Polycythemia Vera, or Post-Essential Thrombocythemia Myelofibrosis, 2019-08-20 ed.; U.S. National Library of Medicine: Bethesda, MD, USA, 2016.
- Holmström, M.O.; Martinenaite, E.; Ahmad, S.M.; Met, Ö.; Friese, C.; Kjær, L.; Riley, C.H.; Thor Straten, P.; Svane, I.M.; Hasselbalch, H.C.; et al. The calreticulin (CALR) exon 9 mutations are promising targets for cancer immune therapy. Leukemia 2018, 32, 429–437. [Google Scholar] [CrossRef]
- How, J.; Hobbs, G.S.; Mullally, A. Mutant calreticulin in myeloproliferative neoplasms. Blood 2019, 134, 2242–2248. [Google Scholar] [CrossRef] [Green Version]
- Jacob, H.; Grauslund, M. CALR Exon 9 Mutant Peptide Vaccine to Patients With CALR-mutant Myeloproliferative Neoplasms, 2018-06-25 ed.; U.S. National Library of Medicine: Bethesda, MD, USA, 2018.
- Sørensen, A.L.; Mikkelsen, S.U.; Knudsen, T.A.; Bjørn, M.E.; Andersen, C.L.; Bjerrum, O.W.; Brochmann, N.; Patel, D.A.; Gjerdrum, L.M.R.; El Fassi, D.; et al. Ruxolitinib and interferon-α2 combination therapy for patients with polycythemia vera or myelofibrosis: A phase II study. Haematologica 2020, 105, 2262–2272. [Google Scholar] [CrossRef] [Green Version]
- Mascarenhas, J.; Kremyanskaya, M.; Hoffman, R.; Bose, P.; Talpaz, M.; Harrison, C.N.; Gupta, V.; Leber, B.; Sirhan, S.; Kabir, S.; et al. MANIFEST, a Phase 2 Study of CPI-0610, a Bromodomain and Extraterminal Domain Inhibitor (BETi), As Monotherapy or "Add-on" to Ruxolitinib, in Patients with Refractory or Intolerant Advanced Myelofibrosis. Blood 2019, 134 (Suppl. 1), 670. [Google Scholar] [CrossRef]
- Study of Oral Navitoclax Tablet in Combination with Oral Ruxolitinib Tablet when Compared with Oral Ruxolitinib Tablet to Assess Change in Spleen Volume in Adult Participants with Myelofibrosis (TRANSFORM-1), 2020-07-15 ed.; U.S. National Library of Medicine: Bethesda, MD, USA, 2020.
- Safety, Efficacy, and Assessment of Change in Spleen Volume Study of Oral Navitoclax Tablet in Combination with Oral Ruxolitinib Tablet in Adult Participants with Relapsed/Refractory Myelofibrosis (TRANSFORM-2), 2020-07-13 ed.; U.S. National Library of Medicine: Bethesda, MD, USA, 2020.
- Dagher, T.; Maslah, N.; Edmond, V.; Cassinat, B.; Vainchenker, W.; Giraudier, S.; Pasquier, F.; Verger, E.; Niwa-Kawakita, M.; Lallemand-Breitenbach, V.; et al. JAK2V617F myeloproliferative neoplasm eradication by a novel interferon/arsenic therapy involves PML. J. Exp. Med. 2021, 218, e20201268. [Google Scholar] [CrossRef]
- Maslah1, N.; Dagher, T.; Edmond, V.; Cassinat, B.; Lallemand-Breitenbach, V.; Verger, E.; Plo, I.; Kiladjian, J.-J.; Villeval, J.-L.; de Thé, H. Specific and Synergistic Targeting of JAK2V617F Cells By Interferon Alpha and Arsenic. Blood 2018, 132 (Suppl. 1), 52. [Google Scholar] [CrossRef]
- Ichiro Kawashima, K.K. Metformin inhibits JAK2V617F activity in MPN cells by activating AMPK and PP2A complexes containing the B56α subunit. Exp. Hematol. 2016, 44, 1156–1165. [Google Scholar] [CrossRef] [PubMed]
- Machado-Neto, J.A.; Fenerich, B.A.; Scopim-Ribeiro, R.; Eide, C.A.; Coelho-Silva, J.L.; Dechandt, C.R.P.; Fernandes, J.C.; Rodrigues Alves, A.P.N.; Scheucher, P.S.; Simões, B.P.; et al. Metformin exerts multitarget antileukemia activity in JAK2V617F-positive myeloproliferative neoplasms. Cell Death Dis. 2018, 9, 311. [Google Scholar] [CrossRef] [PubMed]
- Campos, P.D.M.; Pagnano, K.B.; Mancuso, R.; Teófilo, F.B.S.; Coelho-Silva, J.L.; Della Via, F.I.; Freitas, L.L.L.; Carvalho, H.F.; Costa, F.F.; Traina, F.; et al. Analysis of Metformin Effects on Bone Marrow Fibrosis and Disease Progression in Primary Myelofibrosis Patients: Preliminary Results of an Open Label Phase II Trial (FIBROMET). Blood 2019, 134 (Suppl. 1), 554. [Google Scholar] [CrossRef]
- Azizian, N.G.; Li, Y. XPO1-dependent nuclear export as a target for cancer therapy. J. Hematol. Oncol. 2020, 13. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Pomicter, A.D.; Tantravahi, S.; Mason, C.C.; Senina, A.V.; Ahmann, J.M.; Wang, Q.; Than, H.; Patel, A.B.; Heaton, W.L.; et al. Nuclear–Cytoplasmic Transport Is a Therapeutic Target in Myelofibrosis. Clin. Cancer Res. 2019, 25, 2323–2335. [Google Scholar] [CrossRef] [PubMed]
- Srinivas Tantravahi, M. Selinexor in Myelofibrosis Refractory or Intolerant to JAK1/2 Inhibitors (ESSENTIAL), 2018-08-13 ed.; U.S. National Library of Medicine: Bethesda, MD, USA, 2018.
- Uras, I.Z.; Maurer, B.; Nivarthi, H.; Jodl, P.; Kollmann, K.; Prchal-Murphy, M.; Milosevic Feenstra, J.D.; Zojer, M.; Lagger, S.; Grausenburger, R.; et al. CDK6 coordinates JAK2V617F mutant MPN via NF-κB and apoptotic networks. Blood 2019, 133, 1677–1690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uras, I.Z.; Sexl, V.; Kollmann, K. CDK6 Inhibition: A Novel Approach in AML Management. Int. J. Mol. Sci. 2020, 21, 2528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohi, G.; Dutta, A.; Nath, D.; Yang, Y. The CDK6 Inhibitor Palbociclib in Combination with Ruxolitinib Remarkably Improves Myelofibrosis in Murine Models. Blood 2019, 134 (Suppl. 1), 4200. [Google Scholar] [CrossRef]
- Lu, M.; Xia, L.; Salama, M.E.; Hoffman, R. Splenic Micro Environmental Cells from Patients with Myelofibrosis Elaborate a Cascade of Cytokines and Serve As a Niche for Malignant Hematopoiesis. Blood 2016, 128, 953. [Google Scholar] [CrossRef]



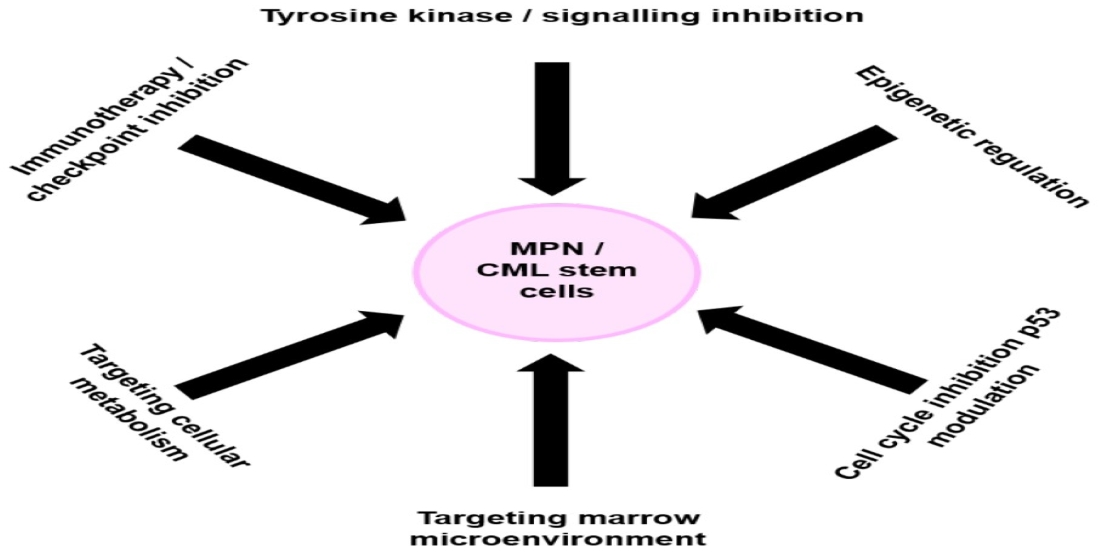{kind=link}
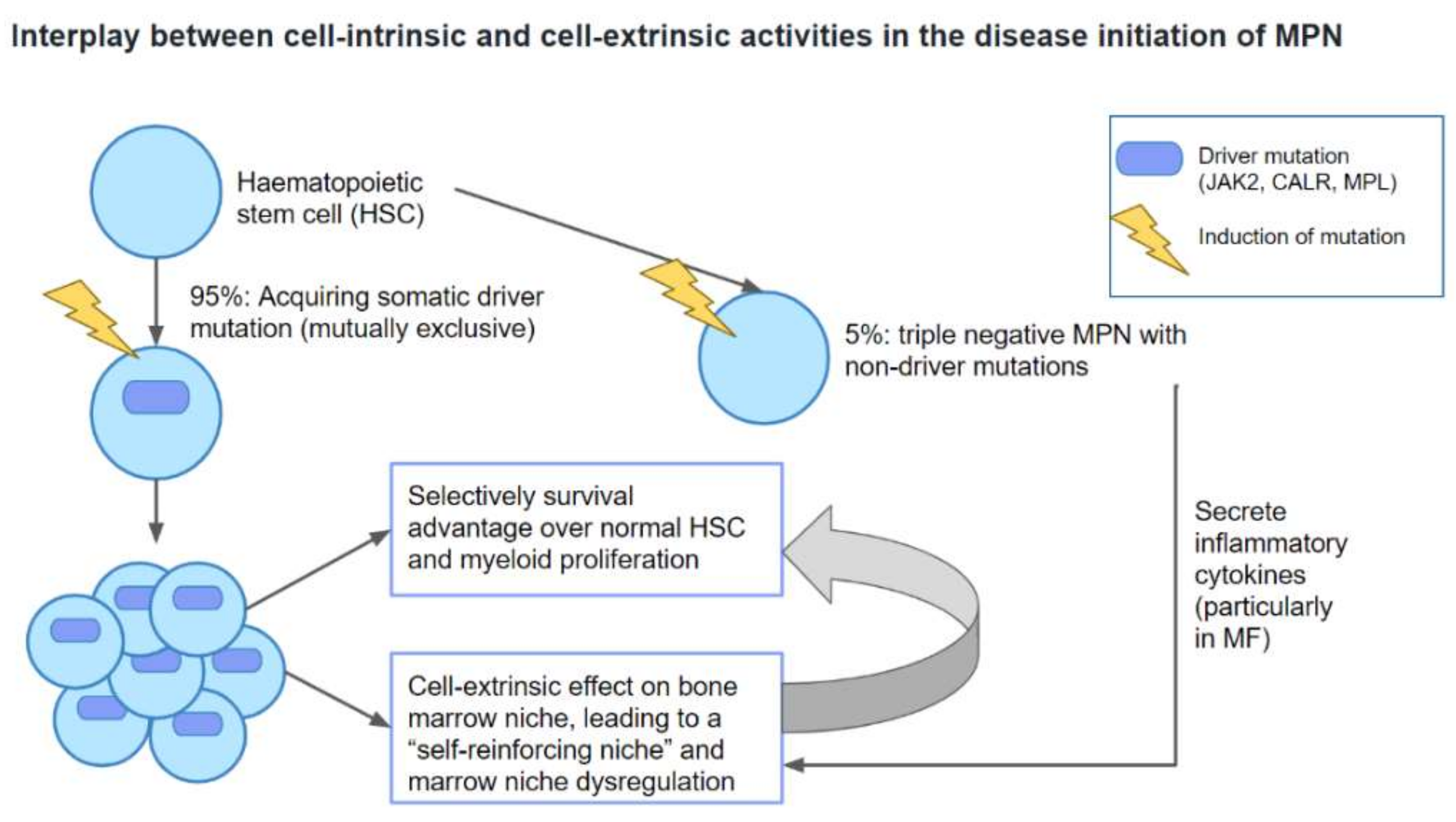{kind=link}
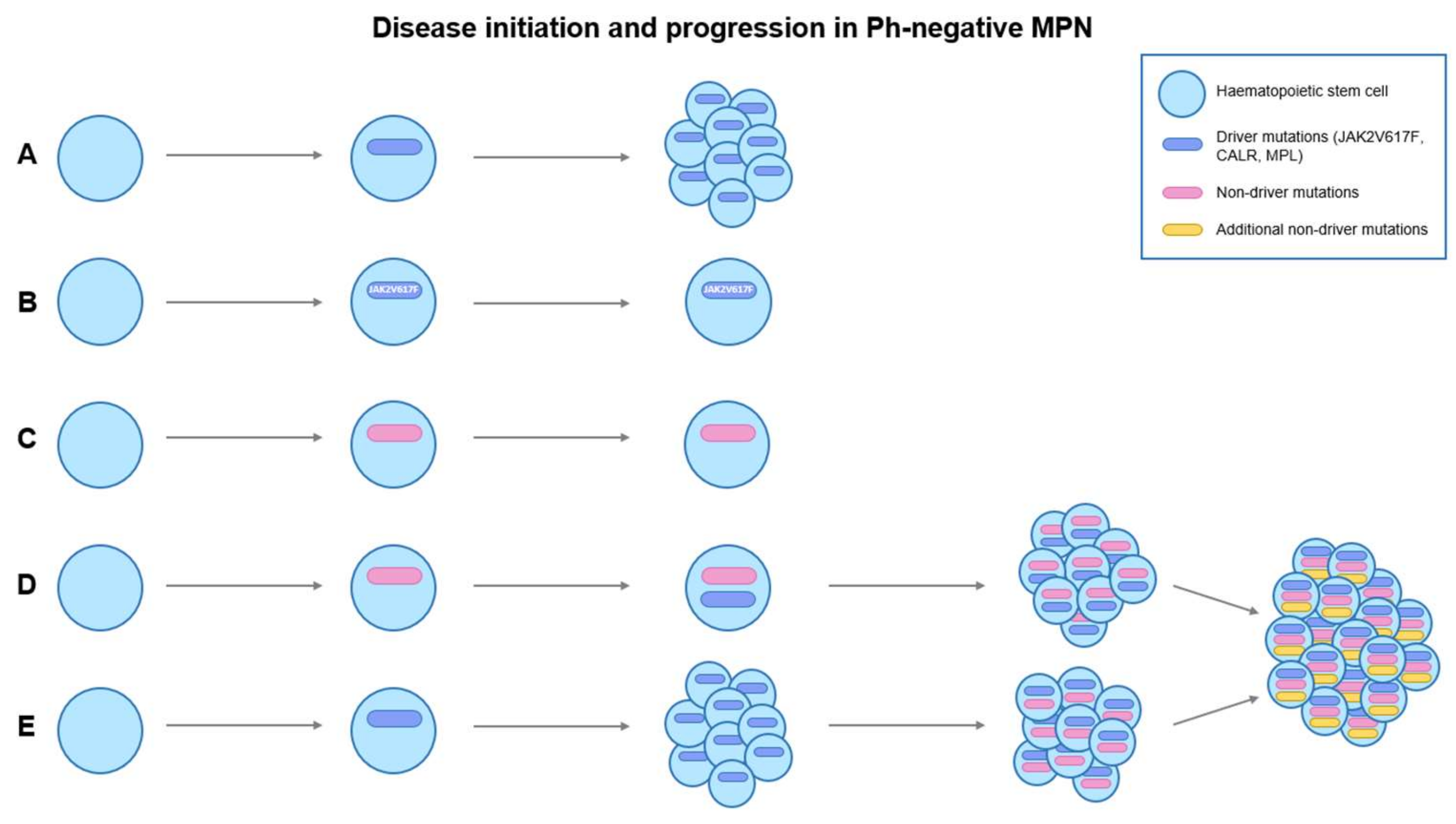{kind=link}
| TKI | Observations | References |
|---|---|---|
| Imatinib |
| [16,34] |
| Dasatinib |
| [34] |
| Nilotinib |
| [36,37] |
| Bosutinib |
| [31,38] |
| Ponatinib |
| [22] |
| IFNα Preparation | MPN Subtype | Observations | References |
|---|---|---|---|
| IFNα | PV, ET, MF |
| [71] |
| CALR-mutated ET, MF |
| [62,72] | |
| Peg-IFNα-2a | CALR-mutated ET |
| [73] |
| Ropeginterferon α-2b | PV |
| [69] |
|
| Novel Therapy | MPN Subtype | Observations | References |
|---|---|---|---|
| Telomerase inhibitor (imetelstat) | DIPSS-plus Int-2/high risk MF |
| [244] |
| DIPSS Int-2/high risk MF |
| [245] | |
| ET |
| [246] | |
| MDM2 inhibitor (idasanutlin) | PV/ET (only 1 ET patient was enrolled) |
| [247] |
| HSP90 inhibitor (AUY922) | PV, ET, MF (IPSS score: ≥2) |
| [248] |
| CD123 (tagraxofusp) | DIPSS-plus int-1, int-2, high risk MF |
| [249] |
| LSD-1 inhibitor (IMG-7289) | IPSS Int-2, high risk MF |
| [250,251] |
| ET | Ongoing | [252] | |
| HDAC inhibitor (givinostat) | PV |
| [74,253] |
| HDAC inhibitor (panobinostat) | PMF, SMF |
| [254] |
| PRMT5 inhibitor (PRT-543) | R/R MF | Ongoing | [255] |
| β-3 sympathomimetic (mirabegron) | 39 JAK2 V617F MPN patients (7 ET, 21 PV, 5 PMF, 3 post-PV MF, 3 post-ET MF) |
| [256] |
| ActRIIA (luspatercept) | DIPSS int-1, int-2, high risk MF |
| [257] |
| ActRIIA (sotatercept) | DIPSS int-1, int-2, high risk MF |
| [258] |
| AURKA inhibitor (alisertib) | DIPSS int-1, int-2, high risk MF |
| [259,260] |
| Anti-PD-1 (nivolumab) | PMF, post-PV, ET MF, hepatomegaly, splenomegaly |
| [261] |
| Combination | MPN Subtype | Study | Observations | References |
|---|---|---|---|---|
| Combination with IFNα | ||||
| MDM2 inhibitor (RG-7112) ± Peg-IFNα-2a | PV, PMF | Preclinical |
| [267] |
| IFNα | PV, MF | Phase II |
| [325] |
| Combinations with Ruxolitinib | ||||
| BET inhibitor (CPI-0610) ± ruxolitinib | MF | Phase II |
| [326] |
| CD123 (tagraxofusp) ± ruxolitinib | MF-AP, BP | Preclinical |
| [283] |
| Bcl-2 family protein inhibitor (navitoclax) + ruxolitinib | PMF, SMF | Phase II |
| [293] |
| MF | Phase III | Ongoing | [327] | |
| R/R MF | Phase III | Ongoing | [328] | |
| HDAC inhibitor (panobinostat) + ruxolitinib | IPSS int-1, int-2, high risk PMF, SMF | Phase Ib |
| [301] |
| PI3K-delta inhibitor (umbralisib; TGR-1202) + ruxolitinib | MF | Phase I |
| [289] |
| Pan-PI3K inhibitor (buparlisib; BKM120) + ruxolitinib | IPSS int, high risk | Phase Ib |
| [261] |
| SAP (PRM-151) ± ruxolitinib | DIPSS int-1, int-2 PMF, SMF | Phase II |
| [314] |
| PMF, SMF | Phase II |
| [313] | |
| Novel Therapy | Combination | Mechanisms | Observations | References |
|---|---|---|---|---|
| Arsenic trioxide | Peg-IFNα-2a | In MPN, expression of JAK2V617F increases PML expression ATO and IFNα enhanced the formation of PML-NB, amplifying activation of p53 and the eradication of JAK2V617F MPN stem cells |
| [329,330] |
| Metformin | N/A | Activation of AMPK pathway suppresses mTOR and reduces synthesis of anti-apoptotic Mcl-1 | Apoptosis of JAK2V617F cell lines | [331,332] |
| Attenuation of JAK2V617F downstream signaling by activating B56α subunit of PP2A | Reduced JAK2 and STAT5 phosphorylation in MPN cells | [331] | ||
| Ruxolitinib | Delayed transition from G1 to S phase by downregulation of cyclin D1 and upregulation of p27 | Combination treatment showed significant downregulation of cyclin D1 and upregulation of p27 in PCR and qPCR | [332] | |
| N/A | FIBROMET (phase II clinical trial): metformin is safe and well-tolerated. Reduction in BM fibrosis was seen but the results were not statistically significant due to small sample size | [333] | ||
| XPO1 inhibitor/SINE compounds | N/A | XPO1 exports tumour suppressor proteins (e.g., p53, NPM, p27) from the nucleus to cytosol XPO1 inhibitor (KPT-330, KPT-8602) decreases NCT | Nuclear retention of p53 in MF CD34+ cells, leading to apoptosis and inhibition of colony formation in MF CD34+ cells | [334,335] |
| Ruxolitinib | Co-treatment showed greater diminution of JAK2V617F-expressing cells in blood and spleen in mouse model | |||
| N/A | ESSENTIAL (phase II clinical trial): ongoing; Assessment of safety and efficacy of selinexor in MF patients who are refractory and intolerant to JAK1/2 inhibitors | [336] | ||
| CDK6 inhibitor | N/A | Overexpressed CDK6 in MPN increases chronic inflammation
| Absence of CDK6 decreased spleen size and induced symptom relief in murine modelsReduced pro-inflammatory cytokines in plasma of JAK2V617F/CDK6−/− murine modelsPalbociclib is a CDK4/6 inhibitor that decreased splenomegaly in MPL mouse models, synergistic effect was seen when co-treated with ruxolitinib | [337,338,339] |
| Promotion of quiescence and deactivation of JAK2V617F MPN stem cells | JAK2V617F/CDK6−/− murine models had elevated short term LT-HSCs | |||
| Apoptosis | Palbociclib drove apoptosis in JAK2V617F haematopoietic cells in combination with ruxolitinib | |||
| Reparixin | N/A | Reparixin inhibits IL-8 receptor and reduces proliferation of splenic endothelial cells, leading to a disrupted niche that cannot sustain MF HSC growth | MF spleen showed elevated LCN2 levels that increased MF haematopoiesis Reparixin led to the reduction of MF CD34+ cells by 35% | [14,268,340] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yung, Y.; Lee, E.; Chu, H.-T.; Yip, P.-K.; Gill, H. Targeting Abnormal Hematopoietic Stem Cells in Chronic Myeloid Leukemia and Philadelphia Chromosome-Negative Classical Myeloproliferative Neoplasms. Int. J. Mol. Sci. 2021, 22, 659. https://doi.org/10.3390/ijms22020659
Yung Y, Lee E, Chu H-T, Yip P-K, Gill H. Targeting Abnormal Hematopoietic Stem Cells in Chronic Myeloid Leukemia and Philadelphia Chromosome-Negative Classical Myeloproliferative Neoplasms. International Journal of Molecular Sciences. 2021; 22(2):659. https://doi.org/10.3390/ijms22020659
Chicago/Turabian StyleYung, Yammy, Emily Lee, Hiu-Tung Chu, Pui-Kwan Yip, and Harinder Gill. 2021. "Targeting Abnormal Hematopoietic Stem Cells in Chronic Myeloid Leukemia and Philadelphia Chromosome-Negative Classical Myeloproliferative Neoplasms" International Journal of Molecular Sciences 22, no. 2: 659. https://doi.org/10.3390/ijms22020659
APA StyleYung, Y., Lee, E., Chu, H. -T., Yip, P. -K., & Gill, H. (2021). Targeting Abnormal Hematopoietic Stem Cells in Chronic Myeloid Leukemia and Philadelphia Chromosome-Negative Classical Myeloproliferative Neoplasms. International Journal of Molecular Sciences, 22(2), 659. https://doi.org/10.3390/ijms22020659