Venous Thrombosis and Thrombocyte Activity in Zebrafish Models of Quantitative and Qualitative Fibrinogen Disorders



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Afibrinogenemic Zebrafish Larvae Fail to Support Venous Thrombosis and Have Impaired Thrombocyte Adhesion and Aggregation at Injury Sites
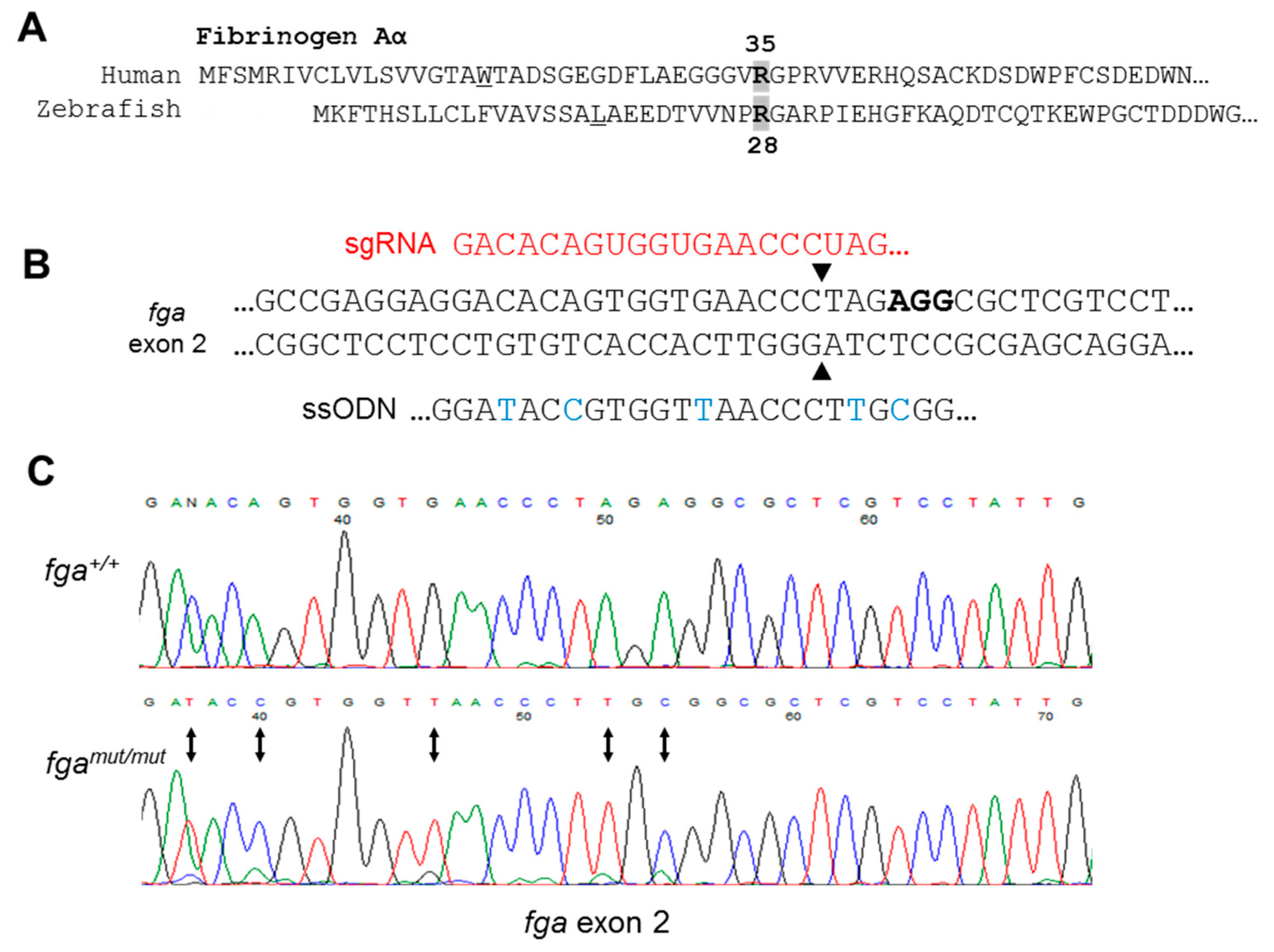
2.2. Genome Editing of fga Exon 2
2.3. Exon 2 Skipping in fga mRNA after Genome Editing
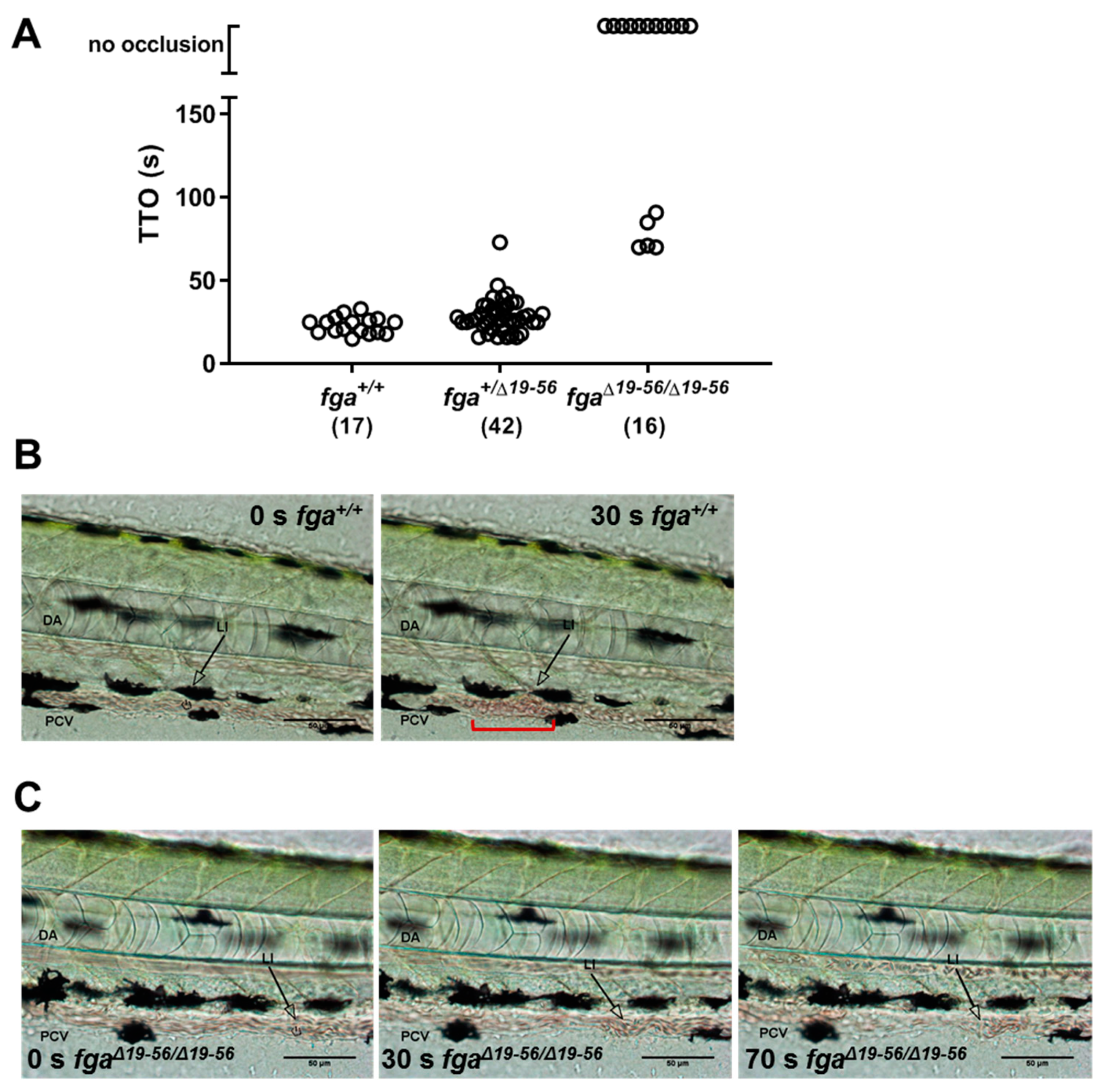
2.4. Venous Thrombosis in Aα Δ19–56-Expressing Zebrafish Larvae
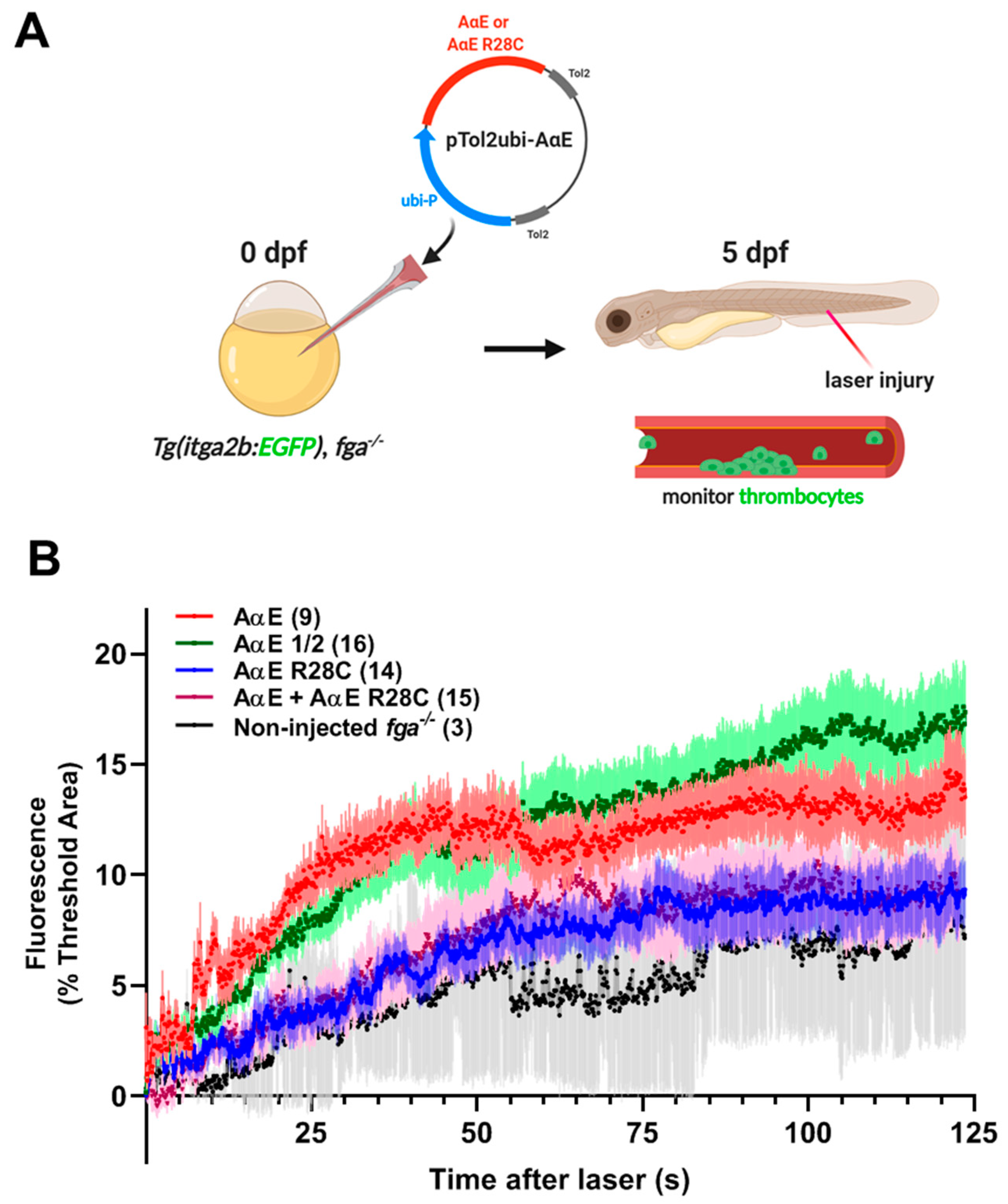
2.5. A Zebrafish Model of Dysfibrinogenemia
3. Discussion
4. Materials and Methods
4.1. Zebrafish
4.2. Production of Zebrafish Fibrinogen in Transfected Cells and Thrombin Cleavage
4.3. Generation of Zebrafish Producing Fibrinogen AαΔ19–56
4.4. Tol2 Plasmid Constructions
4.5. Microinjections for fga mRNA Knockdowns and Fibrinogen Alpha Chain Overexpression
4.6. Genotyping
4.7. Plasma Collection and Western Blots
4.8. Laser-Mediated Thrombosis and Thrombocyte Accumulation in Zebrafish Larvae
4.9. Statistical Analysis and Graphics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DA | Dorsal aorta |
| dpf | Days postfertilization |
| LI | Laser injury |
| MO | Morpholino |
| PCV | Posterior cardinal vein |
| TTO | Time to occlusion |
References
- Neerman-Arbez, M.; de Moerloose, P.; Casini, A. Laboratory and Genetic Investigation of Mutations Accounting for Congenital Fibrinogen Disorders. Semin. Thromb. Hemost. 2016, 42, 356–365. [Google Scholar] [CrossRef]
- Casini, A.; de Moerloose, P. Can the phenotype of inherited fibrinogen disorders be predicted? Haemophilia 2016, 22, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Tajdar, M.; Orlando, C.; Casini, A.; Herpol, M.; De Bisschop, B.; Govaert, P.; Neerman-Arbez, M.; Jochmans, K. Heterozygous FGA p.Asp473Ter (fibrinogen Nieuwegein) presenting as antepartum cerebral thrombosis. Thromb. Res. 2018, 163, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Neerman-Arbez, M. Molecular basis of fibrinogen deficiency. Pathophysiol. Haemost. Thromb. 2006, 35, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Neerman-Arbez, M.; de Moerloose, P. Mutations in the fibrinogen gene cluster accounting for congenital afibrinogenemia: An update and report of 10 novel mutations. Hum. Mutat. 2007, 28, 540–553. [Google Scholar] [CrossRef]
- Casini, A.; Neerman-Arbez, M.; Ariens, R.A.; de Moerloose, P. Dysfibrinogenemia: From molecular anomalies to clinical manifestations and management. J. Thromb. Haemost. 2015, 13, 909–919. [Google Scholar] [CrossRef]
- Peyvandi, F. Epidemiology and treatment of congenital fibrinogen deficiency. Thromb. Res. 2012, 130 (Suppl. S2), S7–S11. [Google Scholar] [CrossRef]
- Korte, W.; Poon, M.C.; Iorio, A.; Makris, M. Thrombosis in Inherited Fibrinogen Disorders. Transfus. Med. Hemother. 2017, 44, 70–76. [Google Scholar] [CrossRef] [Green Version]
- Vorjohann, S.; Fish, R.J.; Biron-Andreani, C.; Nagaswami, C.; Weisel, J.W.; Boulot, P.; Reyftmann, L.; de Moerloose, P.; Neerman-Arbez, M. Hypodysfibrinogenaemia due to production of mutant fibrinogen alpha-chains lacking fibrinopeptide A and polymerisation knob ‘A’. Thromb. Haemost. 2010, 104, 990–997. [Google Scholar] [CrossRef]
- Ariens, R.A. Fibrin(ogen) and thrombotic disease. J. Thromb. Haemost. 2013, 11 (Suppl. S1), 294–305. [Google Scholar] [CrossRef]
- De Moerloose, P.; Casini, A.; Neerman-Arbez, M. Congenital fibrinogen disorders: An update. Semin. Thromb. Hemost. 2013, 39, 585–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosesson, M.W. Update on antithrombin I (fibrin). Thromb. Haemost. 2007, 98, 105–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bosch, N.B.; Mosesson, M.W.; Ruiz-Saez, A.; Echenagucia, M.; Rodriguez-Lemoin, A. Inhibition of thrombin generation in plasma by fibrin formation (Antithrombin I). Thromb. Haemost. 2002, 88, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Dupuy, E.; Soria, C.; Molho, P.; Zini, J.M.; Rosenstingl, S.; Laurian, C.; Bruneval, P.; Tobelem, G. Embolized ischemic lesions of toes in an afibrinogenemic patient: Possible relevance to in vivo circulating thrombin. Thromb. Res. 2001, 102, 211–219. [Google Scholar] [CrossRef]
- Liu, C.Y.; Koehn, J.A.; Morgan, F.J. Characterization of fibrinogen New York 1. A dysfunctional fibrinogen with a deletion of B beta(9-72) corresponding exactly to exon 2 of the gene. J. Biol. Chem. 1985, 260, 4390–4396. [Google Scholar] [CrossRef]
- De Marco, L.; Girolami, A.; Zimmerman, T.S.; Ruggeri, Z.M. von Willebrand factor interaction with the glycoprotein IIb/IIa complex. Its role in platelet function as demonstrated in patients with congenital afibrinogenemia. J. Clin. Investig. 1986, 77, 1272–1277. [Google Scholar] [CrossRef] [Green Version]
- Endenburg, S.C.; Lindeboom-Blokzijl, L.; Zwaginga, J.J.; Sixma, J.J.; de Groot, P.G. Plasma fibrinogen inhibits platelets adhesion in flowing blood to immobilized fibrinogen. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 633–638. [Google Scholar] [CrossRef]
- Koopman, J.; Haverkate, F.; Grimbergen, J.; Lord, S.T.; Mosesson, M.W.; DiOrio, J.P.; Siebenlist, K.S.; Legrand, C.; Soria, J.; Soria, C.; et al. Molecular basis for fibrinogen Dusart (A alpha 554 Arg-->Cys) and its association with abnormal fibrin polymerization and thrombophilia. J. Clin. Investig. 1993, 91, 1637–1643. [Google Scholar] [CrossRef] [Green Version]
- Marchi, R.; Lundberg, U.; Grimbergen, J.; Koopman, J.; Torres, A.; de Bosch, N.B.; Haverkate, F.; Arocha Pinango, C.L. Fibrinogen Caracas V, an abnormal fibrinogen with an Aalpha 532 Ser-->Cys substitution associated with thrombosis. Thromb. Haemost. 2000, 84, 263–270. [Google Scholar] [CrossRef]
- Lozier, J.N.; Nichols, T.C. Animal models of hemophilia and related bleeding disorders. Semin. Hematol. 2013, 50, 175–184. [Google Scholar] [CrossRef] [Green Version]
- Jagadeeswaran, P.; Cooley, B.C.; Gross, P.L.; Mackman, N. Animal Models of Thrombosis From Zebrafish to Nonhuman Primates: Use in the Elucidation of New Pathologic Pathways and the Development of Antithrombotic Drugs. Circ. Res. 2016, 118, 1363–1379. [Google Scholar] [CrossRef]
- Jagadeeswaran, P.; Sheehan, J.P. Analysis of blood coagulation in the zebrafish. Blood Cells Mol. Dis. 1999, 25, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Jagadeeswaran, P.; Liu, Y.C. A hemophilia model in zebrafish: Analysis of hemostasis. Blood Cells Mol. Dis. 1997, 23, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Weyand, A.C.; Shavit, J.A. Zebrafish as a model system for the study of hemostasis and thrombosis. Curr. Opin. Hematol. 2014, 21, 418–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fish, R.J.; Di Sanza, C.; Neerman-Arbez, M. Targeted mutation of zebrafish fga models human congenital afibrinogenemia. Blood 2014, 123, 2278–2281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grzegorski, S.J.; Hu, Z.; Liu, Y.; Yu, X.; Ferguson, A.C.; Madarati, H.; Friedmann, A.P.; Reyon, D.; Kim, P.Y.; Kretz, C.A.; et al. Disruption of the kringle 1 domain of prothrombin leads to late onset mortality in zebrafish. Sci. Rep. 2020, 10, 4049. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Lavik, K.I.; Liu, Y.; Vo, A.H.; Richter, C.E.; Di Paola, J.; Shavit, J.A. Loss of fibrinogen in zebrafish results in an asymptomatic embryonic hemostatic defect and synthetic lethality with thrombocytopenia. J. Thromb. Haemost. 2019, 17, 607–617. [Google Scholar] [CrossRef]
- Hu, Z.; Liu, Y.; Huarng, M.C.; Menegatti, M.; Reyon, D.; Rost, M.S.; Norris, Z.G.; Richter, C.E.; Stapleton, A.N.; Chi, N.C.; et al. Genome editing of factor X in zebrafish reveals unexpected tolerance of severe defects in the common pathway. Blood 2017, 130, 666–676. [Google Scholar] [CrossRef] [Green Version]
- Iyer, N.; Tcheuyap, V.T.; Schneider, S.; Marshall, V.; Jagadeeswaran, P. Knockout of von Willebrand factor in Zebrafish by CRISPR/Cas9 mutagenesis. Br. J. Haematol. 2019, 186, e76–e80. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Kretz, C.A.; Maeder, M.L.; Richter, C.E.; Tsao, P.; Vo, A.H.; Huarng, M.C.; Rode, T.; Hu, Z.; Mehra, R.; et al. Targeted mutagenesis of zebrafish antithrombin III triggers disseminated intravascular coagulation and thrombosis, revealing insight into function. Blood 2014, 124, 142–150. [Google Scholar] [CrossRef] [Green Version]
- Weyand, A.C.; Grzegorski, S.J.; Rost, M.S.; Lavik, K.I.; Ferguson, A.C.; Menegatti, M.; Richter, C.E.; Asselta, R.; Duga, S.; Peyvandi, F.; et al. Analysis of factor V in zebrafish demonstrates minimal levels needed for early hemostasis. Blood Adv. 2019, 3, 1670–1680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagadeeswaran, P.; Sheehan, J.P.; Craig, F.E.; Troyer, D. Identification and characterization of zebrafish thrombocytes. Br. J. Haematol. 1999, 107, 731–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khandekar, G.; Kim, S.; Jagadeeswaran, P. Zebrafish thrombocytes: Functions and origins. Adv. Hematol 2012, 2012, 857058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregory, M.; Hanumanthaiah, R.; Jagadeeswaran, P. Genetic analysis of hemostasis and thrombosis using vascular occlusion. Blood Cells Mol. Dis. 2002, 29, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Freire, C.; Fish, R.J.; Vilar, R.; Di Sanza, C.; Grzegorski, S.J.; Richter, C.E.; Shavit, J.A.; Neerman-Arbez, M. A genetic modifier of venous thrombosis in zebrafish reveals a functional role for fibrinogen AalphaE in early hemostasis. Blood Adv. 2020, 4, 5480–5491. [Google Scholar] [CrossRef]
- Hanss, M.; Biot, F. A database for human fibrinogen variants. Ann. N. Y. Acad. Sci. 2001, 936, 89–90. [Google Scholar] [CrossRef]
- Flood, V.H.; Nagaswami, C.; Chernysh, I.N.; Al-Mondhiry, H.A.; Weisel, J.W.; Farrell, D.H. Incorporation of fibrin molecules containing fibrinopeptide A alters clot ultrastructure and decreases permeability. Br. J. Haematol. 2007, 138, 117–124. [Google Scholar] [CrossRef]
- Berget, S.M. Exon recognition in vertebrate splicing. J. Biol. Chem. 1995, 270, 2411–2414. [Google Scholar] [CrossRef] [Green Version]
- Vilar, R.; Lukowski, S.W.; Garieri, M.; Di Sanza, C.; Neerman-Arbez, M.; Fish, R.J. Chemical Modulators of Fibrinogen Production and Their Impact on Venous Thrombosis. Thromb. Haemost. 2020. [Google Scholar] [CrossRef]
- Ni, H.; Denis, C.V.; Subbarao, S.; Degen, J.L.; Sato, T.N.; Hynes, R.O.; Wagner, D.D. Persistence of platelet thrombus formation in arterioles of mice lacking both von Willebrand factor and fibrinogen. J. Clin. Investig. 2000, 106, 385–392. [Google Scholar] [CrossRef] [Green Version]
- Okumura, N.; Terasawa, F.; Haneishi, A.; Fujihara, N.; Hirota-Kawadobora, M.; Yamauchi, K.; Ota, H.; Lord, S.T. B:b interactions are essential for polymerization of variant fibrinogens with impaired holes ‘a’. J. Thromb. Haemost. 2007, 5, 2352–2359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flood, V.H.; Al-Mondhiry, H.A.; Farrell, D.H. The fibrinogen Aalpha R16C mutation results in fibrinolytic resistance. Br. J. Haematol. 2006, 134, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.Y.; Fu, Y.; Reyon, D.; Maeder, M.L.; Tsai, S.Q.; Sander, J.D.; Peterson, R.T.; Yeh, J.R.; Joung, J.K. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 2013, 31, 227–229. [Google Scholar] [CrossRef] [PubMed]
- Mosimann, C.; Kaufman, C.K.; Li, P.; Pugach, E.K.; Tamplin, O.J.; Zon, L.I. Ubiquitous transgene expression and Cre-based recombination driven by the ubiquitin promoter in zebrafish. Development 2011, 138, 169–177. [Google Scholar] [CrossRef] [Green Version]
- Ablain, J.; Durand, E.M.; Yang, S.; Zhou, Y.; Zon, L.I. A CRISPR/Cas9 vector system for tissue-specific gene disruption in zebrafish. Dev. Cell 2015, 32, 756–764. [Google Scholar] [CrossRef] [Green Version]
- Kwan, K.M.; Fujimoto, E.; Grabher, C.; Mangum, B.D.; Hardy, M.E.; Campbell, D.S.; Parant, J.M.; Yost, H.J.; Kanki, J.P.; Chien, C.B. The Tol2kit: A multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Dev. Dyn 2007, 236, 3088–3099. [Google Scholar] [CrossRef]
- Babaei, F.; Ramalingam, R.; Tavendale, A.; Liang, Y.; Yan, L.S.; Ajuh, P.; Cheng, S.H.; Lam, Y.W. Novel blood collection method allows plasma proteome analysis from single zebrafish. J. Proteome Res. 2013, 12, 1580–1590. [Google Scholar] [CrossRef]
- Jagadeeswaran, P.; Carrillo, M.; Radhakrishnan, U.P.; Rajpurohit, S.K.; Kim, S. Laser-induced thrombosis in zebrafish. Methods Cell Biol. 2011, 101, 197–203. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fish, R.J.; Freire, C.; Di Sanza, C.; Neerman-Arbez, M. Venous Thrombosis and Thrombocyte Activity in Zebrafish Models of Quantitative and Qualitative Fibrinogen Disorders. Int. J. Mol. Sci. 2021, 22, 655. https://doi.org/10.3390/ijms22020655
Fish RJ, Freire C, Di Sanza C, Neerman-Arbez M. Venous Thrombosis and Thrombocyte Activity in Zebrafish Models of Quantitative and Qualitative Fibrinogen Disorders. International Journal of Molecular Sciences. 2021; 22(2):655. https://doi.org/10.3390/ijms22020655
Chicago/Turabian StyleFish, Richard J., Cristina Freire, Corinne Di Sanza, and Marguerite Neerman-Arbez. 2021. "Venous Thrombosis and Thrombocyte Activity in Zebrafish Models of Quantitative and Qualitative Fibrinogen Disorders" International Journal of Molecular Sciences 22, no. 2: 655. https://doi.org/10.3390/ijms22020655
APA StyleFish, R. J., Freire, C., Di Sanza, C., & Neerman-Arbez, M. (2021). Venous Thrombosis and Thrombocyte Activity in Zebrafish Models of Quantitative and Qualitative Fibrinogen Disorders. International Journal of Molecular Sciences, 22(2), 655. https://doi.org/10.3390/ijms22020655