Accelerated Spatial Fibrin Growth and Impaired Contraction of Blood Clots in Patients with Rheumatoid Arthritis


 and
and
Abstract
:1. Introduction
2. Results
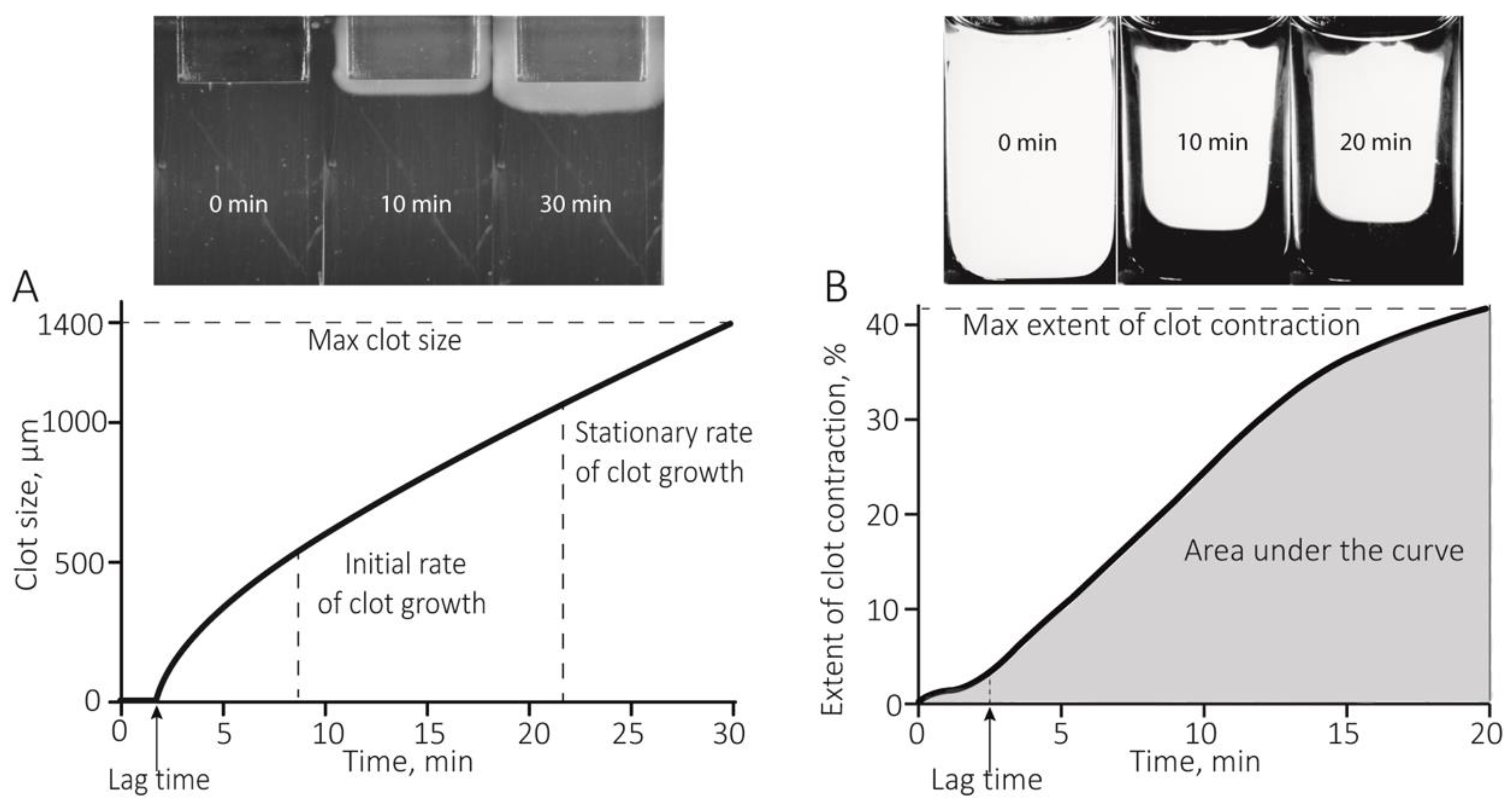
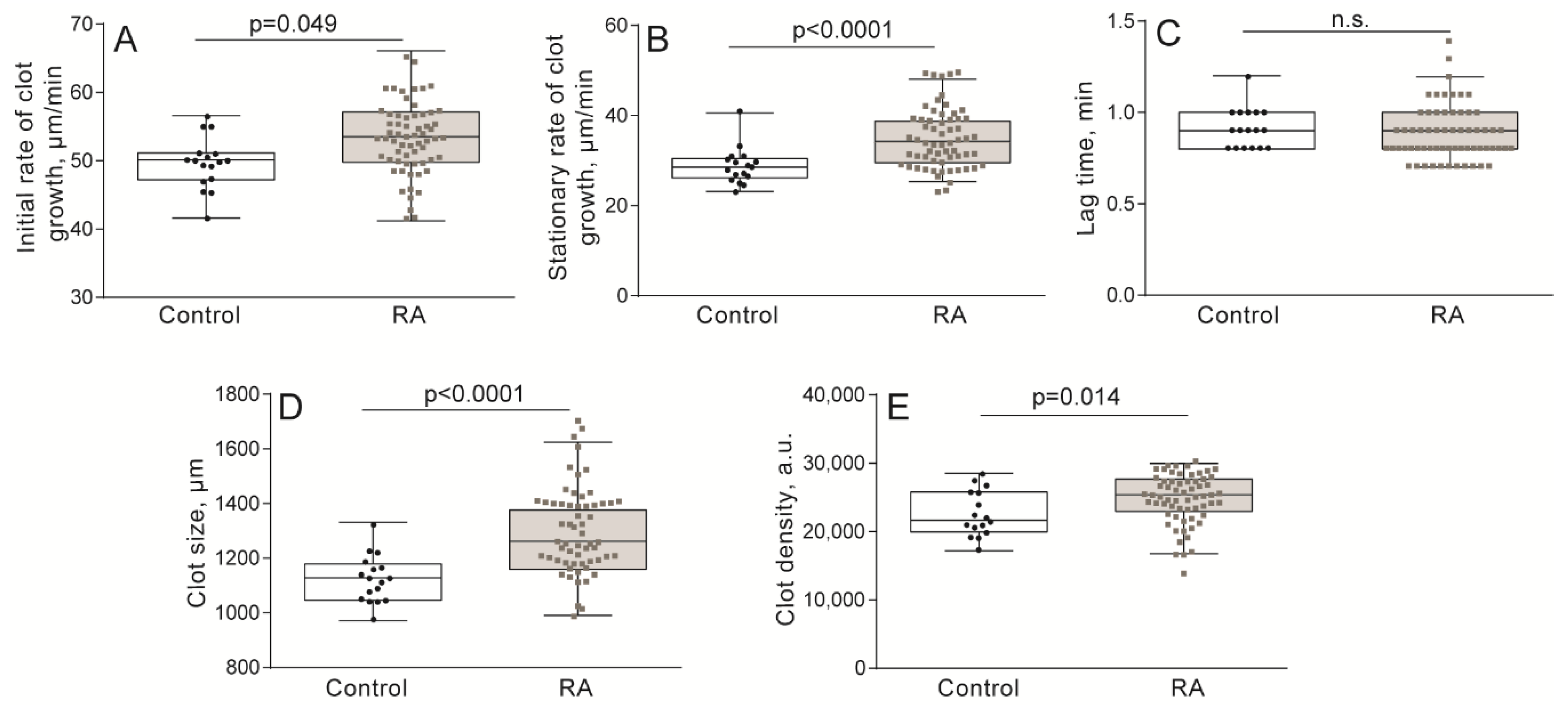
2.1. Hypercoagulability in Patients with RA Revealed by the Thrombodynamics Assay
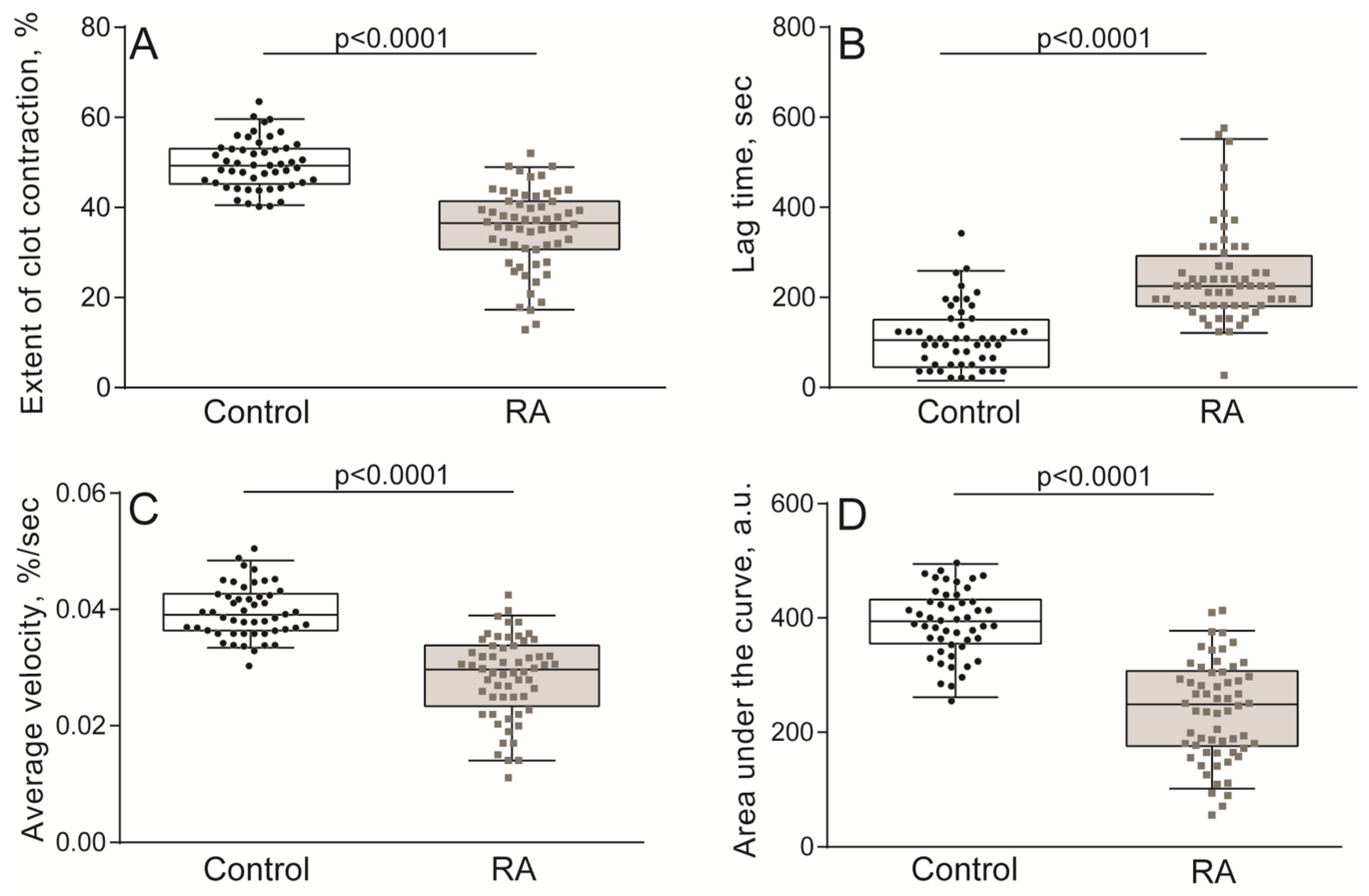
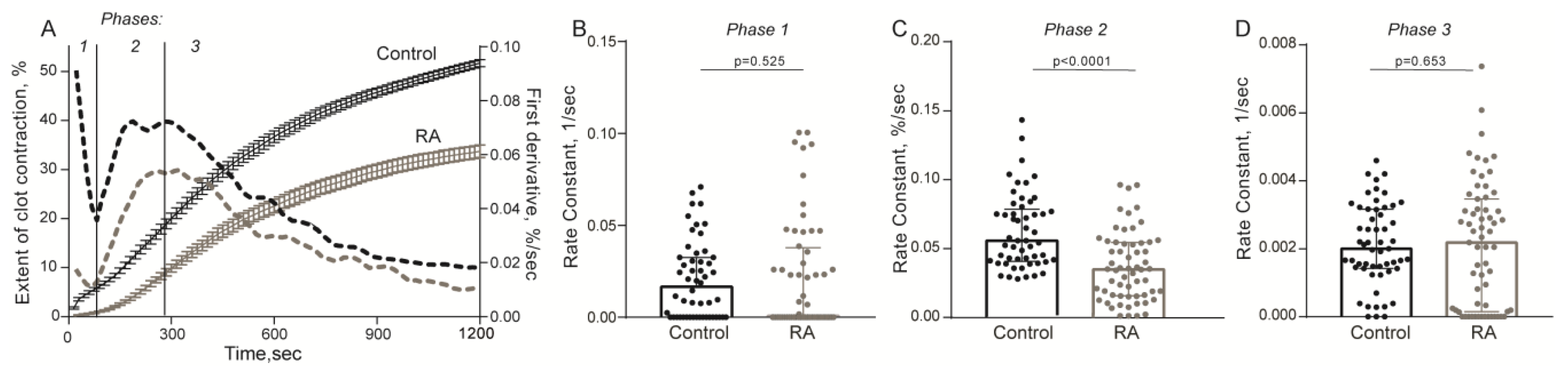
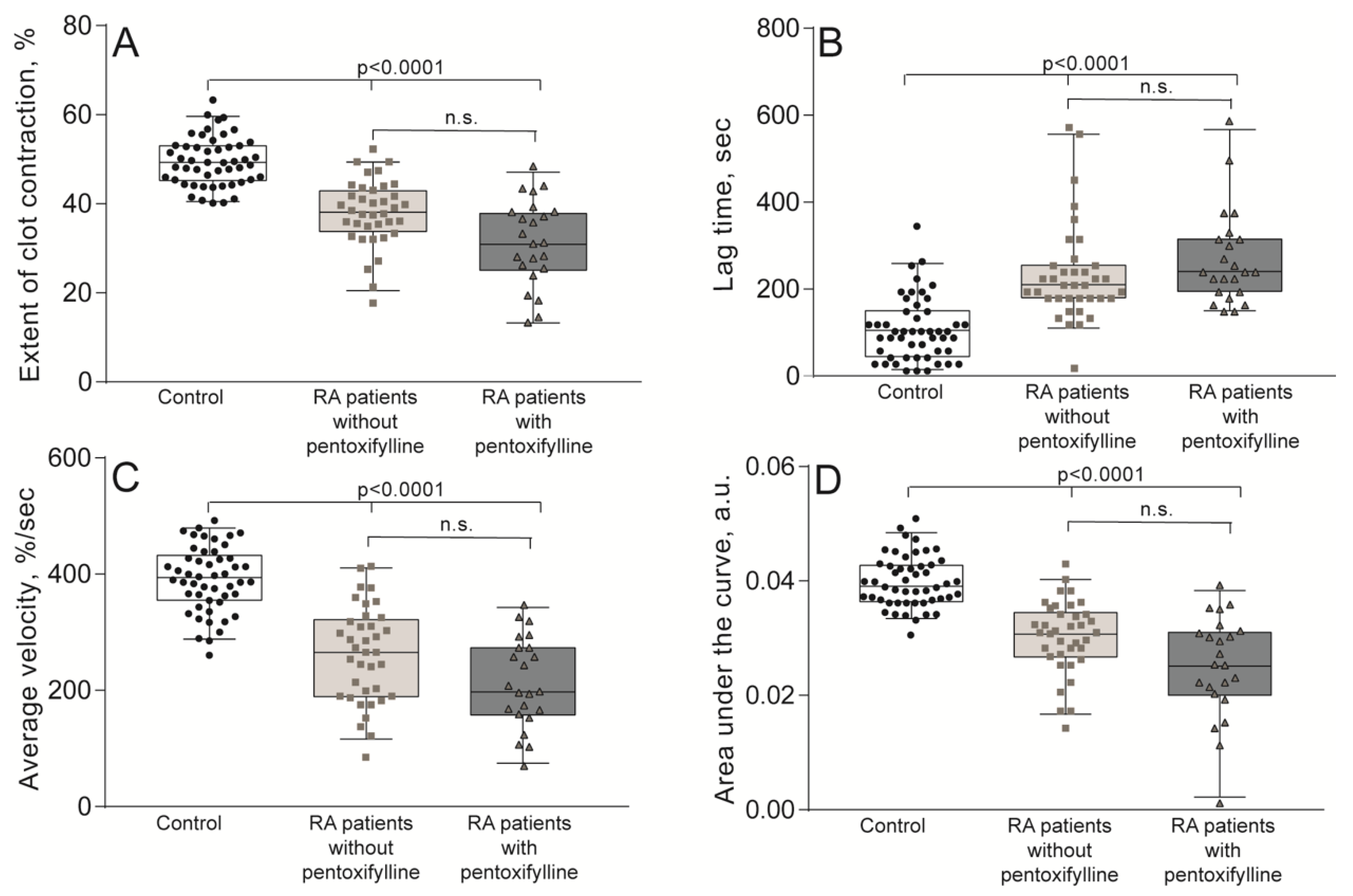
2.2. Impaired Kinetics of Clot Contraction in the Blood of Patients with RA
2.3. Platelet Dysfunction in RA Patients
3. Discussion
4. Materials and Methods
4.1. Patients, Healthy Subjects, and Inclusion and Exclusion Criteria
4.2. Blood Collection and Processing
4.3. Kinetics of Spatially Directed Clot Formation in Blood Plasma (Thrombodynamics Assay)
4.4. Optical Registration of the Blood Clot Contraction Kinetics
4.5. Platelet Isolation
4.6. Flow Cytometry of Resting and Activated Platelets
4.7. Coagulation, Hematological, Biochemical, and Immunological Tests
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| RA | Rheumatoid Arthritis |
| IL-6 | Interleukin-6 |
| Vi | Initial Rate of Clot Growth |
| Vs | Stationary Rate of Clot Growth |
| D | Clot Optical Density |
| RBCs | Red Blood Cells |
| ESR | Erythrocyte Sedimentation Rate |
| IQR | Interquartile Range |
| NSAIDs | Non-Steroidal Anti-Inflammatory Drugs |
| TRAP | Thrombin Receptor-Activating Peptide |
| ACCP | Antibodies Against Cyclic Citrullinated Peptide |
| ACR | American College of Rheumatology |
| EULAR | European League Against Rheumatism |
| DAS | Disease Activity Score |
| EDTA | Ethylenediaminetetraacetic Acid |
| I.V. | Intravenous |
| PAR1 | Protease-Activated Receptor 1 |
References
- McInnes, I.B.; Schett, G. Pathogenetic insights from the treatment of rheumatoid arthritis. Lancet 2017, 389, 2328–2337. [Google Scholar] [CrossRef] [Green Version]
- Alamanos, Y.; Drosos, A.A. Epidemiology of adult rheumatoid arthritis. Autoimmun. Rev. 2005, 4, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Rudan, I.; Sidhu, S.; Papana, A.; Meng, S.-J.; Xin-Wei, Y.; Wang, W.; Campbell-Page, R.M.; Demaio, A.R.; Nair, H.; Sridhar, D.; et al. Prevalence of rheumatoid arthritis in low- and middle-income countries: A systematic review and analysis. J. Glob. Health 2015, 5, 10409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Usenbo, A.; Kramer, V.; Young, T.; Musekiwa, A. Prevalence of arthritis in Africa: A systematic review and meta-analysis. PLoS ONE 2015, 10, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Van Den Oever, I.A.M.; Sattar, N.; Nurmohamed, M.T. Thromboembolic and cardiovascular risk in rheumatoid arthritis: Role of the haemostatic system. Ann. Rheum. Dis. 2014, 73, 954–957. [Google Scholar] [CrossRef]
- Bezuidenhout, J.A.; Pretorius, E. The Central Role of Acute Phase Proteins in Rheumatoid Arthritis: Involvement in Disease Autoimmunity, In fl ammatory Responses, and the Heightened Risk of Cardiovascular Disease. Semin. Thromb. Hemost. 2020, 46, 465–483. [Google Scholar] [CrossRef]
- Ungprasert, P.; Srivali, N.; Spanuchart, I.; Thongprayoon, C.; Knight, E.L. Risk of venous thromboembolism in patients with rheumatoid arthritis: A systematic review and meta-analysis. Clin. Rheumatol. 2014, 33, 297–304. [Google Scholar] [CrossRef]
- Bordy, R.; Totoson, P.; Prati, C.; Marie, C.; Wendling, D.; Demougeot, C. Microvascular endothelial dysfunction in rheumatoid arthritis. Nat. Rev. Rheumatol. 2018, 14, 404–420. [Google Scholar] [CrossRef]
- Liang, K.P.; Liang, K.V.; Matteson, E.L.; McClelland, R.L.; Christianson, T.J.H.; Turesson, C. Incidence of noncardiac vascular disease in rheumatoid arthritis and relationship to extraarticular disease manifestations. Arthritis Rheum. 2006, 54, 642–648. [Google Scholar] [CrossRef] [Green Version]
- Zöller, B.; Li, X.; Sundquist, J.; Sundquist, K. Autoimmune diseases and venous thromboembolism: A review of the literature. Am. J. Cardiovasc. Dis. 2012, 2, 171–183. [Google Scholar]
- Beinsberger, J.; Heemskerk, J.W.M.; Cosemans, J.M.E.M. Chronic arthritis and cardiovascular disease: Altered blood parameters give rise to a prothrombotic propensity. Semin. Arthritis Rheum. 2014, 44, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Melki, I.; Tessandier, N.; Zufferey, A.; Boilard, E. Platelet microvesicles in health and disease. Platelets 2017, 28, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Dessein, P.H.; Joffe, B.I.; Singh, S. Biomarkers of endothelial dysfunction, cardiovascular risk factors and atherosclerosis in rheumatoid arthritis. Arthritis Res. Ther. 2005, 7, 634–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wållberg-Jonsson, S.; Cvetkovic, J.T.; Sundqvist, K.G.; Lefvert, A.K.; Rantapää-Dahlqvist, S. Activation of the immune system and inflammatory activity in relation to markers of atherothrombotic disease and atherosclerosis in rheumatoid arthritis. J. Rheumatol. 2002, 29, 875–882. [Google Scholar]
- Ertenli, I.; Haznedaroǧlu, I.C.; Kiraz, S.; Çelik, I.; Çalgüneri, M.; Kirazli, Ş. Cytokines affecting megakaryocytopoiesis in rheumatoid arthritis with thrombocytosis. Rheumatol. Int. 1996, 16, 5–8. [Google Scholar] [CrossRef]
- Boilard, E.; Nigrovic, P.A.; Larabee, K.; Watts, G.F.M.; Coblyn, J.S.; Weinblatt, M.E.; Massarotti, E.M.; Remold-O’Donnell, E.; Farndale, R.W.; Ware, J.; et al. Platelets Amplify Inflammation in Arthritis via Collagen-Dependent Microparticle Production. Science 2010, 327, 580–583. [Google Scholar] [CrossRef] [Green Version]
- Cloutier, N.; Paré, A.; Farndale, R.W.; Schumacher, H.R.; Nigrovic, P.A.; Lacroix, S.; Boilard, E. Platelets can enhance vascular permeability. Blood 2012, 120, 1334–1343. [Google Scholar] [CrossRef]
- Knijff-Dutmer, E.A.J.; Koerts, J.; Nieuwland, R.; Kalsbeek-Batenburg, E.M.; Van De Laar, M.A.F.J. Elevated levels of platelet microparticles are associated with disease activity in rheumatoid arthritis. Arthritis Rheum. 2002, 46, 1498–1503. [Google Scholar] [CrossRef]
- Sellam, J.; Proulle, V.; Jüngel, A.; Ittah, M.; Miceli Richard, C.; Gottenberg, J.E.; Toti, F.; Benessiano, J.; Gay, S.; Freyssinet, J.M.; et al. Increased levels of circulating microparticles in primary Sjögren’s syndrome, systemic lupus erythematosus and rheumatoid arthritis and relation with disease activity. Arthritis Res. Ther. 2009, 11, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Berckmans, R.J.; Nieuwland, R.; Kraan, M.C.; Schaap, M.C.L.; Pots, D.; Smeets, T.J.M.; Sturk, A.; Tak, P.P. Synovial microparticles from arthritic patients modulate chemokine and cytokine release by synoviocytes. Arthritis Res. Ther. 2005, 7. [Google Scholar] [CrossRef] [Green Version]
- Rooney, T.; Scherzer, R.; Shigenaga, J.K.; Graf, J.; Imboden, J.B.; Grunfeld, C. Levels of plasma fibrinogen are elevated in well-controlled rheumatoid arthritis. Rheumatology 2011, 50, 1458–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bunescu, A.; Seiderman, P.; Lenkei, R.; Levin, K.; Egberg, N. Enhanced Fcgamma receptor I, alphaMbeta2 integrin receptor expression by monocytes and neutrophils in rheumatoid arthritis: Interaction with platelets. J. Rheumatol. 2004, 31, 2347–2355. [Google Scholar] [PubMed]
- Undas, A.; Gissel, M.; Kwasny-Krochin, B.; Gluszko, P.; Mann, K.G.; Brummel-Ziedins, K.E. Thrombin generation in rheumatoid arthritis: Dependence on plasma factor composition. Thromb. Haemost. 2010, 104, 224–230. [Google Scholar] [CrossRef] [Green Version]
- Busso, N.; Hamilton, J.A. Extravascular coagulation and the plasminogen activator/plasmin system in rheumatoid arthritis. Arthritis Rheum. 2002, 46, 2268–2279. [Google Scholar] [CrossRef] [PubMed]
- Kopeikina, L.T.; Kamper, E.F.; Koutsoukos, V.; Bassiakos, Y.; Stavridis, I. Imbalance of tissue-type plasminogen activator (t-PA) and its specific inhibitor (PAI-1) in patients with rheumatoid arthritis associated with disease activity. Clin. Rheumatol. 1997, 16, 254–260. [Google Scholar] [CrossRef] [PubMed]
- So, A.K.; Varisco, P.A.; Kemkes-Matthes, B.; Herkenne-Morard, C.; Chobaz-Péclat, V.; Gerster, J.C.; Busso, N. Arthritis is linked to local and systemic activation of coagulation and fibrinolysis pathways. J. Thromb. Haemost. 2003, 1, 2510–2515. [Google Scholar] [CrossRef] [PubMed]
- De Sá, M.C.; Simão, A.N.C.; de Medeiros, F.A.; Iriyoda, T.M.V.; Costa, N.T.; Alfieri, D.F.; Flauzino, T.; Sekiguchi, B.A.; Lozovoy, M.A.B.; Reiche, E.M.V.; et al. Cell adhesion molecules and plasminogen activator inhibitor type-1 (PAI-1) in patients with rheumatoid arthritis: Influence of metabolic syndrome. Clin. Exp. Med. 2018, 18, 495–504. [Google Scholar] [CrossRef]
- McEntegart, A.; Capell, H.A.; Creran, D.; Rumley, A.; Woodward, M.; Lowe, G.D.O. Cardiovascular risk factors, including thrombotic variables, in a population with rheumatoid arthritis. Rheumatology 2001, 40, 640–644. [Google Scholar] [CrossRef] [Green Version]
- Santos, M.J.; Carmona-Fernandes, D.; Canhão, H.; Canas da Silva, J.; Fonseca, J.E.; Gil, V. Early vascular alterations in SLE and RA patients- a step towards understanding the associated cardiovascular risk. PLoS ONE 2012, 7, 7–12. [Google Scholar] [CrossRef]
- Tuktamyshov, R.; Zhdanov, R. The method of in vivo evaluation of hemostasis: Spatial thrombodynamics. Hematology 2015, 20, 584–586. [Google Scholar] [CrossRef]
- Balandina, A.N.; Koltsova, E.M.; Teterina, T.A.; Yakovenko, A.G.; Simonenko, E.U.; Poletaev, A.V.; Zorina, I.V.; Shibeko, A.M.; Vuimo, T.A.; Yakovenko, S.A.; et al. An enhanced clot growth rate before in vitro fertilization decreases the probability of pregnancy. PLoS ONE 2019, 14, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Sinauridze, E.I.; Vuimo, T.A.; Tarandovskiy, I.D.; Ovsepyan, R.A.; Surov, S.S.; Korotina, N.G.; Serebriyskiy, I.I.; Lutsenko, M.M.; Sokolov, A.L.; Ataullakhanov, F.I. Thrombodynamics, a new global coagulation test: Measurement of heparin efficiency. Talanta 2018, 180, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Tutwiler, V.; Litvinov, R.I.; Lozhkin, A.P.; Peshkova, A.D.; Lebedeva, T.; Ataullakhanov, F.I.; Spiller, K.L.; Cines, D.B.; Weisel, J.W. Kinetics and mechanics of clot contraction are governed by the molecular and cellular composition of the blood. Blood 2016, 127. [Google Scholar] [CrossRef] [Green Version]
- Tutwiler, V.; Peshkova, A.D.; Andrianova, I.A.; Khasanova, D.R.; Weisel, J.W.; Litvinov, R.I. Contraction of blood clots is impaired in acute ischemic stroke. Arterioscler. Thromb. Vasc. Biol. 2017, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peshkova, A.D.; Malyasev, D.V.; Bredikhin, R.A.; Giang, L.M.; Litvinov, R.I. Contraction of Blood Clots Is Impaired in Deep Vein Thrombosis. Bionanoscience 2016, 6. [Google Scholar] [CrossRef]
- Le Minh, G.; Peshkova, A.D.; Andrianova, I.A.; Sibgatullin, T.B.; Maksudova, A.N.; Weisel, J.W.; Litvinov, R.I. Impaired contraction of blood clots as a novel prothrombotic mechanism in systemic lupus erythematosus. Clin. Sci. 2018, 132. [Google Scholar] [CrossRef] [Green Version]
- Peshkova, A.D.; Safiullina, S.I.; Evtugina, N.G.; Baras, Y.S.; Ataullakhanov, F.I.; Weisel, J.W.; Litvinov, R.I. Premorbid Hemostasis in Women with a History of Pregnancy Loss. Thromb. Haemost. 2019, 119. [Google Scholar] [CrossRef]
- Peshkova, A.; Malyasyov, D.; Bredikhin, R.; Le Minh, G.; Andrianova, I.; Tutwiler, V.; Nagaswami, C.; Weisel, J.; Litvinov, R. Reduced Contraction of Blood Clots in Venous Thromboembolism Is a Potential Thrombogenic and Embologenic Mechanism. TH Open 2018, 2, e104–e115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanly, J.G.; Smith, S.A. Anti-beta2-glycoprotein I autoantibodies, in vitro thrombin generation, and the antiphospholipid syndrome. J. Rheumatol. 2000, 27, 2152–2159. [Google Scholar]
- Evtugina, N.G.; Peshkova, A.D.; Pichugin, A.A.; Weisel, J.W.; Litvinov, R.I. Impaired contraction of blood clots precedes and predicts postoperative venous thromboembolism. Sci. Rep. 2020, 10, 18261. [Google Scholar] [CrossRef]
- Habets, K.L.L.; Trouw, L.A.; Levarht, E.W.N.; Korporaal, S.J.A.; Habets, P.A.M.; de Groot, P.; Huizinga, T.W.J.; Toes, R.E.M. Anti-citrullinated protein antibodies contribute to platelet activation in rheumatoid arthritis. Arthritis Res. Ther. 2015, 17, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Joseph, J.E.; Harrison, P.; Mackie, I.J.; Isenberg, D.A.; Machin, S.J. Increased circulating platelet-leucocyte complexes and platelet activation in patients with antiphospholipid syndrome, systemic lupus erythematosus and rheumatoid arthritis. Br. J. Haematol. 2001, 115, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.S.; Peng, C.L.; Lin, C.L.; Chang, Y.J.; Chen, Y.F.; Chiang, J.Y.; Sung, F.C.; Kao, C.H. Rheumatoid arthritis increases the risk of deep vein thrombosis and pulmonary thromboembolism: A nationwide cohort study. Ann. Rheum. Dis. 2014, 73, 1774–1780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solomon, D.H.; Karlson, E.W.; Rimm, E.B.; Cannuscio, C.C.; Mandl, L.A.; Manson, J.A.E.; Stampfer, M.J.; Curhan, G.C. Cardiovascular morbidity and mortality in women diagnosed with rheumatoid arthritis. Circulation 2003, 107, 1303–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Türk, S.M.; Cansu, D.Ü.; Teke, H.Ü.; Kaşifoğlu, T.; Meltem Akay, O.; Bilgin, M.; Korkmaz, C. Can we predict thrombotic tendency in rheumatoid arthritis? A thromboelastographic analysis (with ROTEM). Clin. Rheumatol. 2018, 37, 2341–2349. [Google Scholar] [CrossRef]
- Göbel, K.; Eichler, S.; Wiendl, H.; Chavakis, T.; Kleinschnitz, C.; Meuth, S.G. The coagulation factors fibrinogen, thrombin, and factor XII in inflammatory disorders-a systematic review. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Tomasiak-Lozowska, M.M.; Rusak, T.; Misztal, T.; Bodzenta-Lukaszyk, A.; Tomasiak, M. Reduced clot retraction rate and altered platelet energy production in patients with asthma. J. Asthma 2016, 53, 589–598. [Google Scholar] [CrossRef]
- Olumuyiwa-Akeredolu, O.O.; Pretorius, E. Platelet and red blood cell interactions and their role in rheumatoid arthritis. Rheumatol. Int. 2015, 35, 1955–1964. [Google Scholar] [CrossRef] [Green Version]
- Yazici, S.; Yazici, M.; Erer, B.; Erer, B.; Calik, Y.; Ozhan, H.; Ataoglu, S. The platelet indices in patients with rheumatoid arthritis: Mean platelet volume reflects disease activity. Platelets 2010, 21, 122–125. [Google Scholar] [CrossRef]
- Vasina, E.M.; Cauwenberghs, S.; Staudt, M.; Feijge, M.A.; Weber, C.; Koenen, R.R.; Heemskerk, J.W. Aging- and activation-induced platelet microparticles suppress apoptosis in monocytic cells and differentially signal to proinflammatory mediator release. Am. J. Blood Res. 2013, 3, 107–123. [Google Scholar]
- Gasparyan, A.Y.; Stavropoulos-Kalinoglou, A.; Mikhailidis, D.P.; Douglas, K.M.J.; Kitas, G.D. Platelet function in rheumatoid arthritis: Arthritic and cardiovascular implications. Rheumatol. Int. 2011, 31, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Stack, J.R.; Madigan, A.; Helbert, L.; Dunne, E.; Gardiner, E.E.; Andrews, R.K.; Finan, R.; Smyth, E.; Kenny, D.; McCarthy, G.M. Soluble glycoprotein VI, a specific marker of platelet activation is increased in the plasma of subjects with seropositive rheumatoid arthritis. PLoS ONE 2017, 12, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceruso, M.A.; McComsey, D.F.; Leo, G.C.; Andrade-Gordon, P.; Addo, M.F.; Scarborough, R.M.; Oksenberg, D.; Maryanoff, B.E. Thrombin receptor-activating peptides (TRAPs): Investigation of bioactive conformations via structure-activity, spectroscopic, and computational studies. Bioorg. Med. Chem. 1999, 7, 2353–2371. [Google Scholar] [CrossRef]
- Cloutier, N.; Tan, S.; Boudreau, L.H.; Cramb, C.; Subbaiah, R.; Lahey, L.; Albert, A.; Shnayder, R.; Gobezie, R.; Nigrovic, P.A.; et al. The exposure of autoantigens by microparticles underlies the formation of potent inflammatory components: The microparticle-associated immune complexes. EMBO Mol. Med. 2013, 5, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Maino, A.; Rosendaal, F.R.; Algra, A.; Peyvandi, F.; Siegerink, B. Hypercoagulability is a stronger risk factor for ischaemic stroke than for myocardial infarction: A systematic review. PLoS ONE 2015, 10, 1–12. [Google Scholar] [CrossRef]
- Mcdonagh, J.; Lee, M.H. How does hyperfibrinogenemia lead to thrombosis? Fibrinolysis Proteolysis 1997, 11, 13–17. [Google Scholar] [CrossRef]
- Machlus, K.R.; Cardenas, J.C.; Church, F.C.; Wolberg, A.S. Causal relationship between hyperfibrinogenemia, thrombosis, and resistance to thrombolysis in mice. Blood 2011, 117, 4953–4963. [Google Scholar] [CrossRef] [Green Version]
- Litvinov, R.I.; Nabiullina, R.M.; Zubairova, L.D.; Shakurova, M.A.; Andrianova, I.A.; Weisel, J.W. Lytic susceptibility, structure, and mechanical properties of fibrin in systemic lupus erythematosus. Front. Immunol. 2019, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Aletaha, D.; Neogi, T.; Silman, A.J.; Funovits, J.; Felson, D.T.; Bingham, C.O.; Birnbaum, N.S.; Burmester, G.R.; Bykerk, V.P.; Cohen, M.D.; et al. Rheumatoid arthritis classification criteria: An American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010, 62, 2569–2581. [Google Scholar] [CrossRef]
- Costabile, M. Measuring the 50% haemolytic complement (CH50) activity of serum. J. Vis. Exp. 2010, 8–10. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Groups | P-Selectin Expression | Fibrinogen-Binding Capacity | ||||
|---|---|---|---|---|---|---|
| Unstimulated Platelets | TRAP-Activated (3-min Activation) | TRAP-Activated (10-min Activation) | Unstimulated Platelets | TRAP-Activated (3-min Activation) | TRAP-Activated (10-min Activation) | |
| Healthy subjects (n = 9) | 1.4 ± 0.2 1 | 68.4 ± 3.5 1 | 87.3 ± 4.7 1 | 0.8 ± 0.2 1 | 74.9 ± 5.5 1 | 75.1 ± 4.7 1 |
| RA patients (n = 12) | 3.4 ± 0.6 1 | 38.8 ± 5.9 1 *** | 67.8 ± 5.8 1 ** | 2.6 ± 1.1 1 | 53.2 ± 6.2 1 ** | 59.7 ± 6.2 1 * |
| Characteristics | RA Patients (n = 60) | |
|---|---|---|
| Age, years (mean ± SEM) | 57 ± 1 | |
| Gender | Men | 14 (23%) |
| Women | 46 (77%) | |
| Duration of the disease, years (mean ± SEM) | 9 ± 1 | |
| Index of disease activity DAS28 | Low (2.6–3.2) | 18 (30%) |
| Medium (3.2–5.1) | 21 (35%) | |
| High (>5.1) | 21 (35%) | |
| Main clinical manifestations | Polyarthritis | 49 (82%) |
| Extra-articular manifestations | 8 (13%) | |
| Associated musculoskeletal disorders | Osteoarthrosis | 48 (80%) |
| Osteoporosis | 11 (18%) | |
| Cardiovascular disorders | Hypertonic disease | 30 (50%) |
| Heart lesions (hypertrophy, coronary heart disease, mitral valve prolapse, dilatation, extrasystole, hydropericardium) | 23 (38%) | |
| Chronic heart failure | 26 (43%) | |
| Hematologic disorders | Anemia (Hb < 110 g/L) | 23 (38%) |
| Thrombocytosis (>420 × 109/L) | 8 (13%) | |
| Hyperfibrinogenemia (>4.5 g/L) | 34 (57%) | |
| Leukocytosis (>9 × 109/L) | 4 (6%) | |
| Leukopenia (<4 × 109/L) | 2 (3%) | |
| Renal disorders | Kidney cyst | 16 (27%) |
| Chronic renal failure | 7 (12%) | |
| Gastrointestinal disorders | Liver steatosis | 9 (15%) |
| Chronic cholecystitis | 22 (37%) | |
| Chronic gastroduodenitis | 12 (19%) | |
| Chronic gastritis | 22 (37%) | |
| Chronic pancreatitis | 10 (17%) | |
| Parameters (in Parentheses—Reference Values) | RA Patients (n = 60) | Control Group (n = 50) |
|---|---|---|
| Hemostatic parameters | ||
| aPTT (25–36), s | 34.3 (31.2; 37.3) ** | 32.2 (29.7; 34,1) |
| Prothrombin time (9.4–12.5), s | 10.9 (10.4; 11.4) * | 11.3 (10.8; 11.9) |
| Prothrombin index (77–137), % | 109 (99; 117) * | 99 (92; 110) |
| Fibrinogen (1.8–4.0), g/L | 4.6 (4.1; 5.0) *** | 3.8 (3.1; 5.0) |
| Hematological parameters | ||
| Platelet count (180–320) × 109/L | 281 (230; 338) *** | 231 (174; 275) |
| Red blood cells (3.7–4.7) × 1012/L | 4.3 (3.9; 4.6) *** | 4.5 (4.3; 4.9) |
| Leukocytes (4–9) × 109/L | 6.1 (5.0; 8.2) *** | 4.9 (4.3; 6.1) |
| Neutrophils (47–72), % | 58 (51; 65) | 57 (52; 61) |
| Lymphocytes (17–48), % | 28 (22; 34) * | 34 (26; 36) |
| Monocytes (4–10), % | 8.5 (6.4; 10.8) *** | 6.2 (5.2; 7.1) |
| Eosinophils (1–5), % | 2.2 (1.3; 3.7) | 2.0 (2.0; 4.0) |
| Erythrocyte sedimentation rate (ESR) (2–20), mm/h | 34 (25; 45) *** | 7.0 (5.5; 11.5) |
| Hemoglobin (11.0–16.5), g/dL | 11.3 (10.5;12.9) *** | 14.3 (12.8; 15.2) |
| Mean corpuscular volume (MCV) (80–97), fL | 84 (74; 90) | 86 (83; 89) |
| Mean corpuscular hemoglobin (MCH) (26.5–33.5), pg | 28 (24; 30) *** | 32 (31; 34) |
| Mean corpuscular hemoglobin concentration (MCHC) (31.5–36.0), g/dL | 32.5 (31.4; 34.1) *** | 35.6 (35.0; 36.2) |
| Red cell distribution width (RDW) (10–15), % | 14.8 (13.4; 16) ** | 12.6 (12.1; 13.6) |
| Mean platelet volume (MPV) (6.5–11.0), fL | 10.3 (9.2; 10.8) *** | 8.2 (7.4; 8.9) |
| Platelet distribution width (PDW) (10–18), % | 12.3 (10.2; 12.9) | 12.7 (11.9; 18.4) |
| Thrombocrit (PCT) (0.12–0.36), % | 0.27 (0.23; 0.33) ** | 0.20 (0.16; 0.24) |
| Immunological parameters | ||
| IgA (0.8–4.0), mg/mL | 1.9 (1.7; 2.6) | 2.1 (1.4; 3.3) |
| IgM (0.5–2.0), mg/mL | 2.2 (1.6; 3.3) *** | 1.4 (1.3; 1.5) |
| IgG (5.3–16.5), mg/mL | 12.5 (10.5; 17.6) ** | 9.8 (9.2; 11.6) |
| Circulating immune complexes (CICs) (0–120), a.u. | 99 (84; 146) | 95 (86; 101) |
| Complement CH50 (50–80), hemolytic units | 39 (33; 44) *** | 56 (52; 61) |
| Anti-DNA antibody (≤25), IU/mL | 4.8 (2.9; 7.5) | 2.8 (2.2; 8.1) |
| Anti-cardiolipin antibody (≤10), IU/mL | 2.2 (1.4; 4.0) ** | 1.2 (0.9; 1.2) |
| Antibodies to citrullinated peptide (0–5), IU/mL | 200 (40; 954) *** | 2.0 (1.2; 3.6) |
| Biochemical parameters | ||
| ALT (5–40), IU/L | 13 (9; 20) ** | 17 (13; 28) |
| AST (10–36), IU/L | 16 (13; 23) | 18 (14; 20) |
| Protein (64–84), g/L | 69 (66; 73) *** | 73 (70; 76) |
| Glucose (3.8–6.1), mmol/L | 4.9 (4.6; 5.1) * | 5.2 (4.7; 5.5) |
| Creatinine (62–110), μmol/L | 53 (44; 65) *** | 89 (80; 98) |
| Bilirubin (3.5–25), μmol/L | 6.4 (5.5; 9.0) | 8.0 (5.8; 10.0) |
| Cholesterol (2.2–5.7), μmol/L | 4.6 (4.0; 5.4) | 4.7 (4.1; 5.0) |
| Uric acid (202–420), μmol/L | 251 (186; 310) *** | 355 (284; 399) |
| Serum iron (10–34.5), μmol/L | 9.1 (7.0; 11.9) *** | 19 (11; 27) |
| Alkaline phosphatase (40–130), IU/L | 81 (59; 105) | 92 (76; 104) |
| GGT (8–61), IU/L | 23 (15; 45) | 20 (11; 27) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peshkova, A.D.; Evdokimova, T.A.; Sibgatullin, T.B.; Ataullakhanov, F.I.; Litvinov, R.I.; Weisel, J.W. Accelerated Spatial Fibrin Growth and Impaired Contraction of Blood Clots in Patients with Rheumatoid Arthritis. Int. J. Mol. Sci. 2020, 21, 9434. https://doi.org/10.3390/ijms21249434
Peshkova AD, Evdokimova TA, Sibgatullin TB, Ataullakhanov FI, Litvinov RI, Weisel JW. Accelerated Spatial Fibrin Growth and Impaired Contraction of Blood Clots in Patients with Rheumatoid Arthritis. International Journal of Molecular Sciences. 2020; 21(24):9434. https://doi.org/10.3390/ijms21249434
Chicago/Turabian StylePeshkova, Alina D., Tatiana A. Evdokimova, Timur B. Sibgatullin, Fazoil I. Ataullakhanov, Rustem I. Litvinov, and John W. Weisel. 2020. "Accelerated Spatial Fibrin Growth and Impaired Contraction of Blood Clots in Patients with Rheumatoid Arthritis" International Journal of Molecular Sciences 21, no. 24: 9434. https://doi.org/10.3390/ijms21249434
APA StylePeshkova, A. D., Evdokimova, T. A., Sibgatullin, T. B., Ataullakhanov, F. I., Litvinov, R. I., & Weisel, J. W. (2020). Accelerated Spatial Fibrin Growth and Impaired Contraction of Blood Clots in Patients with Rheumatoid Arthritis. International Journal of Molecular Sciences, 21(24), 9434. https://doi.org/10.3390/ijms21249434