Neurofibromatosis Type 2 (NF2) and the Implications for Vestibular Schwannoma and Meningioma Pathogenesis

 , , ,
, , , 
Abstract
:1. Neurofibromatosis Type 2 (NF2): Introduction and Genetic Overview
2. NF2; Molecular Genetics
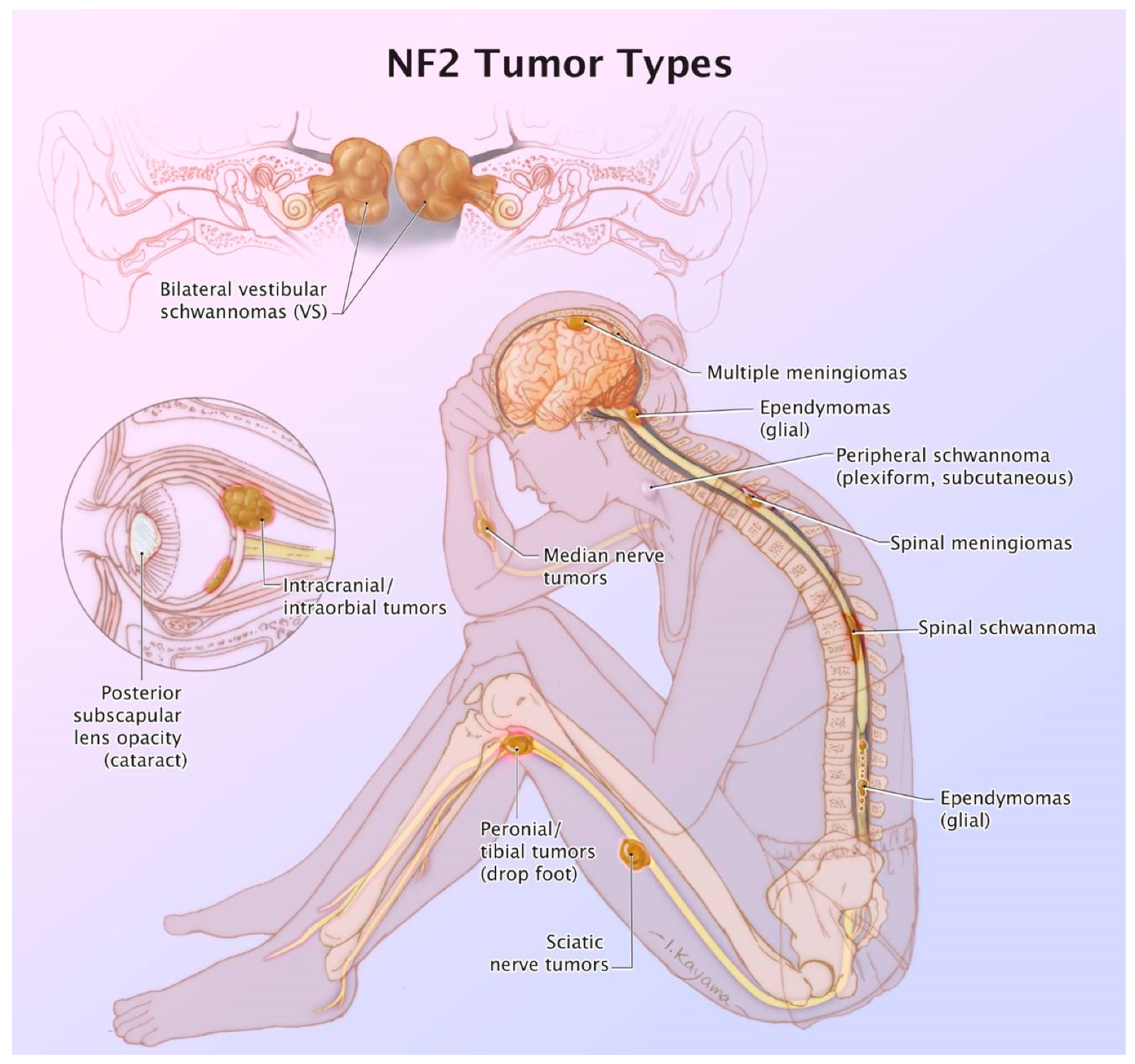
3. NF2: Tumor Types
4. NF2 Meningioma Pathogenesis
5. NF2 Vestibular Schwannoma Pathogenesis
6. The Current State and Future Directions for NF2 Related Meningiomas and Vestibular Schwannomas
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Evans, D.G.R. Neurofibromatosis Type 2 (NF2): A Clinical and Molecular Review. Orphanet J. Rare Dis. 2009, 4, 16. [Google Scholar] [CrossRef] [Green Version]
- Ruggieri, M.; Praticò, A.D.; Evans, D.G. Diagnosis, Management, and New Therapeutic Options in Childhood Neurofibromatosis Type 2 and Related Forms. Semin. Pediatr. Neurol. 2015, 22, 240–258. [Google Scholar] [CrossRef] [PubMed]
- Pećina-Šlaus, N. Merlin, the NF2 gene product. Pathol. Oncol. Res. 2013, 19, 365–373. [Google Scholar] [CrossRef]
- Kresak, J.L.; Walsh, M. Neurofibromatosis: A Review of NF1, NF2, and Schwannomatosis. J. Pediatr. Genet. 2016, 5, 98–104. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, R.; Singh, A.K. Neurofibromatosis Type 2. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Smith, M.J.; Bowers, N.L.; Bulman, M.; Gokhale, C.; Wallace, A.J.; King, A.T.; Lloyd, S.K.; Rutherford, S.A.; Hammerbeck-Ward, C.L.; Freeman, S.R.; et al. Revisiting Neurofibromatosis Type 2 Diagnostic Criteria to Exclude LZTR1-Related Schwannomatosis. Neurology 2017, 88, 87–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, C.; Bedocs, P.M. Neurofibromatosis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Evans, D.G.; King, A.T.; Bowers, N.L.; Tobi, S.; Wallace, A.J.; Perry, M.; Anup, R.; Lloyd, S.K.; Rutherford, S.A.; Hammerbeck-Ward, C.; et al. Identifying the deficiencies of current diagnostic criteria for neurofibromatosis 2 using databases of 2777 individuals with molecular testing. Genet. Med. 2019, 21, 1525–1533. [Google Scholar] [CrossRef]
- Li, W.; You, L.; Cooper, J.; Schiavon, G.; Pepe-Caprio, A.; Zhou, L.; Ishii, R.; Giovannini, M.; Hanemann, C.O.; Long, S.B.; et al. Merlin/NF2 Suppresses Tumorigenesis by Inhibiting the E3 Ubiquitin Ligase CRL4DCAF1 in the Nucleus. Cell 2010, 140, 477–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, G.-H.; Chernoff, J.; Testa, J.R. NF2: The Wizardry of Merlin. Genes Chromosomes Cancer 2003, 38, 389–399. [Google Scholar] [CrossRef]
- Kluwe, L.; Friedrich, R.E.; Hagel, C.; Lindenau, M.; Mautner, V.-F. Mutations and Allelic Loss of the NF2 Gene in Neurofibromatosis 2-Associated Skin Tumors. J. Investig. Dermatol. 2000, 114, 1017–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pemov, A.; Dewan, R.; Hansen, N.F.; Chandrasekharappa, S.C.; Ray-Chaudhury, A.; Jones, K.; Luo, W.; Heiss, J.D.; Mullikin, J.C.; Chittiboina, P.; et al. Comparative clinical and genomic analysis of neurofibromatosis type 2-associated cranial and spinal meningiomas. Sci. Rep. 2020, 10, 12563. [Google Scholar] [CrossRef] [PubMed]
- Kino, T.; Takeshima, H.; Nakao, M.; Nishi, T.; Yamamoto, K.; Kimura, T.; Saito, Y.; Kochi, M.; Kuratsu, J.-I.; Saya, H.; et al. Identification of the Cis-Acting Region in the NF2 Gene Promoter as a Potential Target for Mutation and Methylation-Dependent Silencing in Schwannoma. Genes Cells 2001, 6, 441–454. [Google Scholar] [CrossRef] [PubMed]
- Baser, M.E.; Ragge, N.K.; Riccardi, V.M.; Janus, T.; Gantz, B.; Pulst, S.-M. Phenotypic Variability in Monozygotic Twins with Neurofibromatosis 2. Am. J. Med. Genet. 1996, 64, 563–567. [Google Scholar] [CrossRef]
- Baser, M.E.; Kuramoto, L.; Woods, R.; Joe, H.; Friedman, J.M.; Wallace, A.J.; Ramsden, R.T.; Olschwang, S.; Bijlsma, E.K.; Kalamarides, M.; et al. The Location of Constitutional Neurofibromatosis 2 (NF2) Splice Site Mutations Is Associated with the Severity of NF2. J. Med. Genet. 2005, 42, 540–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruttledge, M.H.; Andermann, A.A.; Phelan, C.M.; Claudio, J.O.; Han, F.Y.; Chretien, N.; Rangaratnam, S.; MacCollin, M.; Short, P.; Parry, D.; et al. Type of Mutation in the Neurofibromatosis Type 2 Gene (NF2) Frequently Determines Severity of Disease. Am. J. Hum. Genet. 1996, 59, 331–342. [Google Scholar]
- Louvrier, C.; Pasmant, E.; Briand-Suleau, A.; Cohen, J.; Nitschké, P.; Nectoux, J.; Orhant, L.; Zordan, C.; Goizet, C.; Goutagny, S.; et al. Targeted next-generation sequencing for differential diagnosis of neurofibromatosis type 2, schwannomatosis, and meningiomatosis. Neuro Oncol. 2018, 20, 917–929. [Google Scholar] [CrossRef] [Green Version]
- Selvanathan, S.K.; Shenton, A.; Ferner, R.; Wallace, A.J.; Huson, S.M.; Ramsden, R.T.; Evans, D.G. Further Genotype—Phenotype Correlations in Neurofibromatosis 2. Clin. Genet. 2010, 77, 163–170. [Google Scholar] [CrossRef]
- Pinto, P.S.; Huisman, T.A.; Ahn, E.; Jordan, L.C.; Burger, P.; Cohen, K.J.; Patay, Z.; Tekes, A. Magnetic Resonance Imaging Features of Meningiomas in Children and Young Adults: A Retrospective Analysis. J. Neuroradiol. 2012, 39, 218–226. [Google Scholar] [CrossRef]
- Coy, S.; Rashid, R.; Stemmer-Rachamimov, A.; Santagata, S. An Update on the CNS Manifestations of Neurofibromatosis Type 2. Acta Neuropathol. 2020, 139, 643–665. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.T.; Lee, C.-H.; Chung, C.K.; Kim, J.H. Is NF2 a Key Player of the Differentially Expressed Gene Between Spinal Cord Ependymoma and Intracranial Ependymoma? World Neurosurg. 2018, 118, e906–e917. [Google Scholar] [CrossRef]
- Smirniotopoulos, J.G.; Murphy, F.M. The phakomatoses. AJNR Am. J. Neuroradiol. 1992, 13, 725–746. [Google Scholar]
- Proctor, D.T.; Ramachandran, S.; Lama, S.; Sutherland, G.R. Towards Molecular Classification of Meningioma: Evolving Treatment and Diagnostic Paradigms. World Neurosurg. 2018, 119, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Karsy, M.; Azab, M.A.; Abou-Al-Shaar, H.; Guan, J.; Eli, I.; Jensen, R.L.; Ormond, D.R. Clinical potential of meningioma genomic insights: A practical review forneurosurgeons. Neurosurg. Focus 2018, 44, E10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrom, Q.T.; Gittleman, H.; Fulop, J.; Liu, M.; Blanda, R.; Kromer, C.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS statistical Report: Primary brain and central nervous system tumors diagnosed in the United States in 2008-2012. Neuro Oncol. 2015, 17, iv1–iv62. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; Von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harter, P.N.; Braun, Y.; Plate, K.H. Classification of meningiomas-advances and controversies. Chin. Clin. Oncol. 2017, 6, S2. [Google Scholar] [CrossRef] [PubMed]
- Mirian, C.; Duun-Henriksen, A.K.; Juratli, T.; Sahm, F.; Spiegl-Kreinecker, S.; Peyre, M.; Biczok, A.; Tonn, J.-C.; Goutagny, S.; Bertero, L.; et al. Poor prognosis associated with TERT gene alterations in meningioma is independent of the WHO classification: An individual patient data meta-analysis. J. Neurol. Neurosurg. Psychiatry 2020, 91, 378–387. [Google Scholar] [CrossRef]
- Sahm, F.; Schrimpf, D.; Stichel, D.; Jones, D.T.W.; Hielscher, T.; Schefzyk, S.; Okonechnikov, K.; Koelsche, C.; Reuss, D.E.; Capper, D.; et al. DNA methylation-based classification and grading system for meningioma: A multicentre, retrospective analysis. Lancet Oncol. 2017, 18, 682–694. [Google Scholar] [CrossRef] [Green Version]
- Goutagny, S.; Bah, A.B.; Henin, D.; Parfait, B.; Grayeli, A.B.; Sterkers, O.; Kalamarides, M. Long-term follow-up of 287 meningiomas in neurofibromatosis type 2 patients: Clinical, radiological, and molecular features. Neuro Oncol. 2012, 14, 1090–1096. [Google Scholar] [CrossRef] [Green Version]
- Clark, V.E.; Erson-Omay, E.Z.; Serin, A.; Yin, J.; Cotney, J.; Özduman, K.; Avşar, T.; Li, J.; Murray, P.B.; Henegariu, O.; et al. Genomic Analysis of Non-NF2 Meningiomas Reveals Mutations in TRAF7, KLF4, AKT1, and SMO. Science 2013, 339, 1077–1080. [Google Scholar] [CrossRef] [Green Version]
- Bi, W.L.; Greenwald, N.F.; Abedalthagafi, M.; Wala, J.; Gibson, W.J.; Agarwalla, P.K.; Horowitz, P.; Schumacher, S.E.; Esaulova, E.; Mei, Y.; et al. Genomic landscape of high-grade meningiomas. NPJ Genom. Med. 2017, 2, 1–14. [Google Scholar] [CrossRef]
- Wallesch, M.; Pachow, D.; Blücher, C.; Firsching, R.; Warnke, J.-P.; Braunsdorf, W.E.; Kirches, E.; Mawrin, C. Altered expression of E-Cadherin-related transcription factors indicates partial epithelial-mesenchymal transition in aggressive meningiomas. J. Neurol. Sci. 2017, 380, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Petrilli, A.M.; Fernández-Valle, C. Role of Merlin/NF2 inactivation in tumor biology. Oncogene 2016, 35, 537–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angus, S.P.; Oblinger, J.L.; Stuhlmiller, T.J.; DeSouza, P.A.; Beauchamp, R.L.; Witt, L.; Chen, X.; Jordan, J.T.; Gilbert, T.S.K.; Stemmer-Rachamimov, A.; et al. EPH receptor signaling as a novel therapeutic target in NF2-deficient meningioma. Neuro Oncol. 2018, 20, 1185–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reuss, D.E.; Piro, R.M.; Jones, D.T.W.; Simon, M.; Ketter, R.; Kool, M.; Becker, A.; Sahm, F.; Pusch, S.; Meyer, J.; et al. Secretory meningiomas are defined by combined KLF4 K409Q and TRAF7 mutations. Acta Neuropathol. 2013, 125, 351–358. [Google Scholar] [CrossRef]
- Sahm, F.; Bissel, J.; Koelsche, C.; Schweizer, L.; Capper, D.; Reuss, D.; Böhmer, K.; Lass, U.; Göck, T.; Kalis, K.; et al. AKT1E17K mutations cluster with meningothelial and transitional meningiomas and can be detected by SFRP1 immunohistochemistry. Acta Neuropathol. 2013, 126, 757–762. [Google Scholar] [CrossRef]
- Abedalthagafi, M.; Bi, W.L.; Aizer, A.A.; Merrill, P.H.; Brewster, R.; Agarwalla, P.K.; Listewnik, M.L.; Dias-Santagata, D.; Thorner, A.R.; Van Hummelen, P.; et al. Oncogenic PI3K mutations are as common asAKT1andSMOmutations in meningioma. Neuro Oncol. 2016, 18, 649–655. [Google Scholar] [CrossRef] [Green Version]
- Bi, W.L.; Abedalthagafi, M.; Horowitz, P.; Agarwalla, P.K.; Mei, Y.; Aizer, A.A.; Brewster, R.; Dunn, G.P.; Al-Mefty, O.; Alexander, B.M.; et al. Genomic landscape of intracranial meningiomas. J. Neurosurg. 2016, 125, 525–535. [Google Scholar] [CrossRef] [Green Version]
- Brastianos, P.K.; Horowitz, P.M.; Santagata, S.; Jones, R.T.; McKenna, A.; Getz, G.; Ligon, K.L.; Palescandolo, E.; Van Hummelen, P.; Ducar, M.D.; et al. Genomic sequencing of meningiomas identifies oncogenic SMO and AKT1 mutations. Nat. Genet. 2013, 45, 285–289. [Google Scholar] [CrossRef]
- Goutagny, S.; Kalamarides, M. Meningiomas and neurofibromatosis. J. Neuro Oncol. 2010, 99, 341–347. [Google Scholar] [CrossRef]
- Lee, Y.S.; Lee, Y.-S. Molecular characteristics of meningiomas. J. Pathol. Transl. Med. 2020, 54, 45–63. [Google Scholar] [CrossRef] [Green Version]
- Al Hinai, Q.; Zeitouni, A.; Sirhan, D.; Sinclair, D.; Melancon, D.; Richardson, J.; Leblanc, R. Communicating Hydrocephalus and Vestibular Schwannomas: Etiology, Treatment, and Long-Term Follow-Up. J. Neurol. Surg. Part B Skull Base 2013, 74, 068–074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howitz, M.F.; Johansen, C.; Tos, M.; Charabi, S.; Olsen, J.H. Incidence of vestibular schwannoma in Denmark, 1977–1995. Am. J. Otol. 2000, 21, 690–694. [Google Scholar] [PubMed]
- MacCollin, M.; Chiocca, E.A.; Evans, D.G.; Friedman, J.M.; Horvitz, R.; Jaramillo, D.; Lev, M.; Mautner, V.F.; Niimura, M.; Plotkin, S.R.; et al. Diagnostic criteria for schwannomatosis. Neurology 2005, 64, 1838–1845. [Google Scholar] [CrossRef] [PubMed]
- Neff, B.A.; Welling, D.B.; Akhmametyeva, E.; Chang, L.-S. The Molecular Biology of Vestibular Schwannomas: Dissecting the Pathogenic Process at the Molecular Level. Otol. Neurotol. 2006, 27, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Rouleau, G.A.; Merel, P.; Lutchman, M.; Sanson, M.; Zucman, J.; Marineau, C.; Hoang-Xuan, K.; Demczuk, S.; Desmaze, C.; Plougastel, B.; et al. Alteration in a new gene encoding a putative membrane-organizing protein causes neuro-fibromatosis type 2. Nat. Cell Biol. 1993, 363, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Trofatter, J.A.; MacCollin, M.M.; Rutter, J.L.; Murrell, J.R.; Duyao, M.P.; Parry, D.M.; Eldridge, R.; Kley, N.; Menon, A.G.; Pulaski, K.; et al. A novel Moesin-, Exrin-, Radixin-like gene is a candidate for the neurofibromatosis 2 tumor-suppressor. Cell 1993, 72, 791–800. [Google Scholar] [CrossRef]
- Jacoby, L.B.; MacCollin, M.; Louls, D.N.; Mohney, T.; Rublo, M.-P.; Pulaski, K.; Trofatter, J.A.; Kley, N.; Seizinger, B.; Ramesh, V.; et al. Exon scanning for mutation of the NF2 gene in schwannomas. Hum. Mol. Genet. 1994, 3, 413–419. [Google Scholar] [CrossRef]
- Welling, D.B.; Guida, M.; Goll, F.; Pearl, D.K.; Glasscock, M.E.; Pappas, D.G.; Linthicum, F.H.; Rogers, D.; Prior, T.W. Mutational spectrum in the neurofibromatosis type 2 gene in sporadic and familial schwannomas. Qual. Life Res. 1996, 98, 189–193. [Google Scholar] [CrossRef]
- Welling, D.B.; Lasak, J.M.; Akhmametyeva, E.; Ghaheri, B.; Chang, L.-S. cDNA Microarray Analysis of Vestibular Schwannomas. Otol. Neurotol. 2002, 23, 736–748. [Google Scholar] [CrossRef]
- Baser, M.E. The distribution of constitutional and somatic mutations in the neurofibromatosis 2 gene. Hum. Mutat. 2006, 27, 297–306. [Google Scholar] [CrossRef]
- Martuza, R.L.; Eldridge, R. Neurofibromatosis 2 (bilateral acoustic neurofibromatosis). N. Engl. J. Med. 1988, 318, 684–688. [Google Scholar] [CrossRef] [PubMed]
- Kluwe, L.; Mautner, V.F. Mosaicism in sporadic neurofibromatosis 2 patients. Hum. Mol. Genet. 1998, 7, 2035–2051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlson, M.L.; Smadbeck, J.B.; Link, M.J.; Klee, E.W.; Vasmatzis, G.; Schimmenti, L.A. Next Generation Sequencing of Sporadic Vestibular Schwannoma: Necessity of Biallelic NF2 Inactivation and Implications of Accessory Non-NF2 Variants. Otol. Neurotol. 2018, 39, e860–e871. [Google Scholar] [CrossRef] [PubMed]
- Parry, D.M.; MacCollin, M.M.; Kaiser-Kupfer, M.I.; Pulaski, K.; Nicholson, H.S.; Bolesta, M.; Eldridge, R.; Gusella, J.F. Germ-line mutations in the neurofibromatosis 2 gene: Correlations with disease severity and retinal abnormalities. Am. J. Hum. Genet. 1996, 59, 529–539. [Google Scholar]
- Curto, M.; Cole, B.K.; Lallemand, D.; Liu, C.-H.; McClatchey, A.I. Contact-dependent inhibition of EGFR signaling by Nf2/Merlin. J. Cell Biol. 2007, 177, 893–903. [Google Scholar] [CrossRef] [Green Version]
- Scoles, D.R.; Yong, W.H.; Qin, Y.; Wawrowsky, K.; Pulst, S.M. Schwannomin inhibits tumorigenesis through direct interaction with the eukaryotic initiation factor subunit c (eIF3c). Hum. Mol. Genet. 2006, 15, 1059–1070. [Google Scholar] [CrossRef] [Green Version]
- Ryu, C.H.; Kim, S.-W.; Lee, K.H.; Lee, J.Y.; Kim, H.; Lee, W.K.; Choi, B.H.; Lim, Y.; Kim, Y.H.; Lee, K.-H.; et al. The merlin tumor suppressor interacts with Ral guanine nucleotide dissociation stimulator and inhibits its activity. Oncogene 2005, 24, 5355–5364. [Google Scholar] [CrossRef] [Green Version]
- Rong, R.; Tang, X.; Gutmann, D.H.; Ye, K. Neurofibromatosis 2 (NF2) tumor suppressor merlin inhibits phosphatidylinositol 3-kinase through binding to PIKE-L. Proc. Natl. Acad. Sci. USA 2004, 101, 18200–18205. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-Y.; Kim, H.; Ryu, C.H.; Kim, J.-Y.; Choi, B.H.; Lim, Y.; Huh, P.-W.; Kim, Y.-H.; Lee, K.-H.; Jun, T.-Y.; et al. Merlin, a Tumor Suppressor, Interacts with Transactivation-responsive RNA-binding Protein and Inhibits Its Oncogenic Activity. J. Biol. Chem. 2004, 279, 30265–30273. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.Y.; Kim, H.; Jeun, S.-S.; Kang, S.; Lee, K.J. Merlin inhibits growth hormone-regulated Raf–ERKs pathways by binding to Grb2 protein. Biochem. Biophys. Res. Commun. 2006, 340, 1151–1157. [Google Scholar] [CrossRef]
- Morrison, H.; Sperka, T.; Manent, J.; Giovannini, M.; Ponta, H.; Herrlich, P. Merlin/Neurofibromatosis Type 2 Suppresses Growth by Inhibiting the Activation of Ras and Rac. Cancer Res. 2007, 67, 520–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiederhold, T.; Lee, M.-F.; James, M.; Neujahr, R.; Smith, N.; Murthy, A.; Hartwig, J.; Gusella, J.F.; Ramesh, V. Magicin, a novel cytoskeletal protein associates with the NF2 tumor suppressor merlin and Grb2. Oncogene 2004, 23, 8815–8825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, N.; Kerl, K. Preclinical Evaluation of Combined Targeted Approaches in Malignant Rhabdoid Tumors. Anticancer Res. 2016, 36, 3883–3887. [Google Scholar] [PubMed]
- Plotkin, S.R.; Stemmer-Rachamimov, A.O.; Ii, F.G.B.; Halpin, C.; Padera, T.P.; Tyrrell, A.; Sorensen, A.G.; Jain, R.K.; Di Tomaso, E. Hearing Improvement after Bevacizumab in Patients with Neurofibromatosis Type 2. N. Engl. J. Med. 2009, 361, 358–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitte, J.; Gao, F.; Coppola, G.; Judkins, A.R.; Giovannini, M. Timing of Smarcb1 and Nf2 inactivation determines schwannoma versus rhabdoid tumor development. Nat. Commun. 2017, 8, 300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuse, M.A.; Dinh, C.T.; Vitte, J.; Kirkpatrick, J.; Mindos, T.; Plati, S.K.; Young, J.I.; Huang, J.; Carlstedt, A.; Franco, M.C.; et al. Preclinical assessment of MEK1/2 inhibitors for neurofibromatosis type 2–associated schwannomas reveals differences in efficacy and drug resistance development. Neuro Oncol. 2019, 21, 486–497. [Google Scholar] [CrossRef]
- Pećina-Šlaus, N.; Kafka, A.; Lechpammer, M. Molecular Genetics of Intracranial Meningiomas with Emphasis on Canonical Wnt Signalling. Cancers 2016, 8, 67. [Google Scholar] [CrossRef] [Green Version]
- Pećina-Šlaus, N.; Kafka, A.; Bukovac, A.; Vladušić, T.; Tomas, D.; Hrašćan, R. Genetic changes of MLH1 and MSH2 genes could explain constant findings on microsatellite instability in intracranial meningioma. Tumor Biol. 2017, 39, 1010428317705791. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1. Bilateral vestibular schwannomas < 70 years of age. |
| 2. Unilateral vestibular schwannoma < 70 years and a first-degree relative with NF2. |
| 3. Any two of the following: meningioma, schwannoma (non-vestibular), ependymoma, cerebral calcification, cataract AND first-degree relative to NF2 OR unilateral vestibular schwannoma and negative LZTR1 testing. Note: recent data have excluded glioma in the criteria. |
| 4. Multiple meningiomas and unilateral vestibular schwannoma or any two of the following: schwannoma (non-vestibular), neurofibroma, glioma, cerebral calcification, cataract. |
| 5. Constitutional or mosaic pathogenic NF2 gene mutation from the blood or by the identification of an identical mutation from two separate tumors in the same individual. |
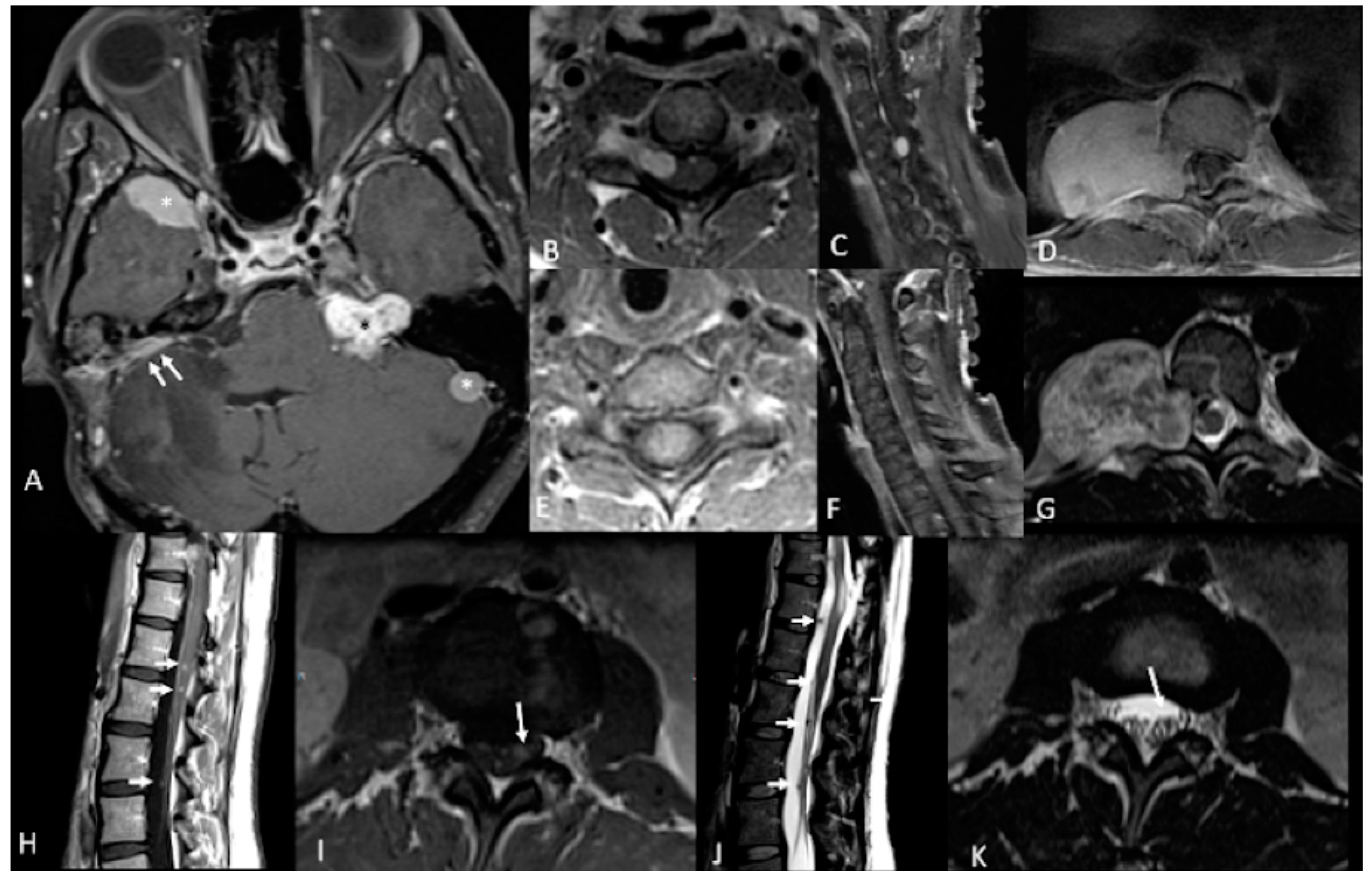
| NF2 Tumor Types | % | Clinical Presentation | Histology | Imaging | Treatment | Complications |
|---|---|---|---|---|---|---|
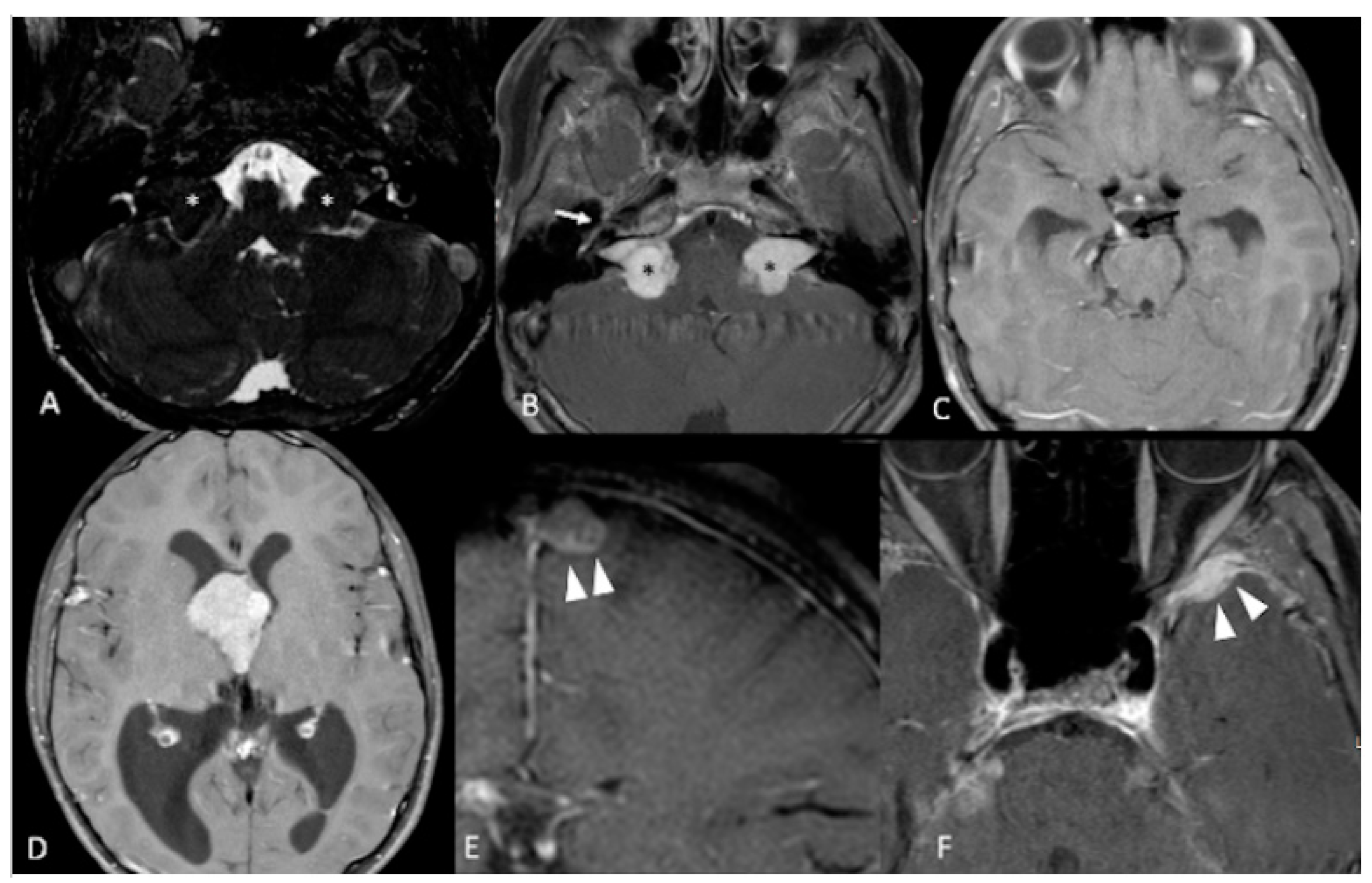
| Vestibular Schwannomas | ~90% | Tinnitus Hearing loss Ataxia | Antoni A, B regions Verocay bodies Hyalinzed vessels | Often bilateral. Slightly T1 hypointense (63%) or isointense (37%). Heterogeneously T2 hyperintense (Antoni A: relatively low, Antoni B: high), cystic degenerative areas may be present if large tumor. Intense contrast enhancement on T1 C+ (Gd) | Radiosurgery Chemotherapy; Bevacizumab | Facial nerve injury Malignant transformation |
| Peripheral Schwannomas -Tumorlets -Plexiform | ~70% | Neuropathic pain Loss of sensation Weakness Tumors on skin, head and neck region (Plexiform) | Antoni A, B regions Verocay bodies Hyalinzed vessels Infiltration of nerve | T1: 75% are isointense, 25% are hypointense. T2: more than 95% are hyperintense, often with mixed signal. Intense contrast enhancement on T1 C+ (Gd) | Intraneural dissection Excision | Rarely undergo malignant transformation although high risk of nerve infiltration |
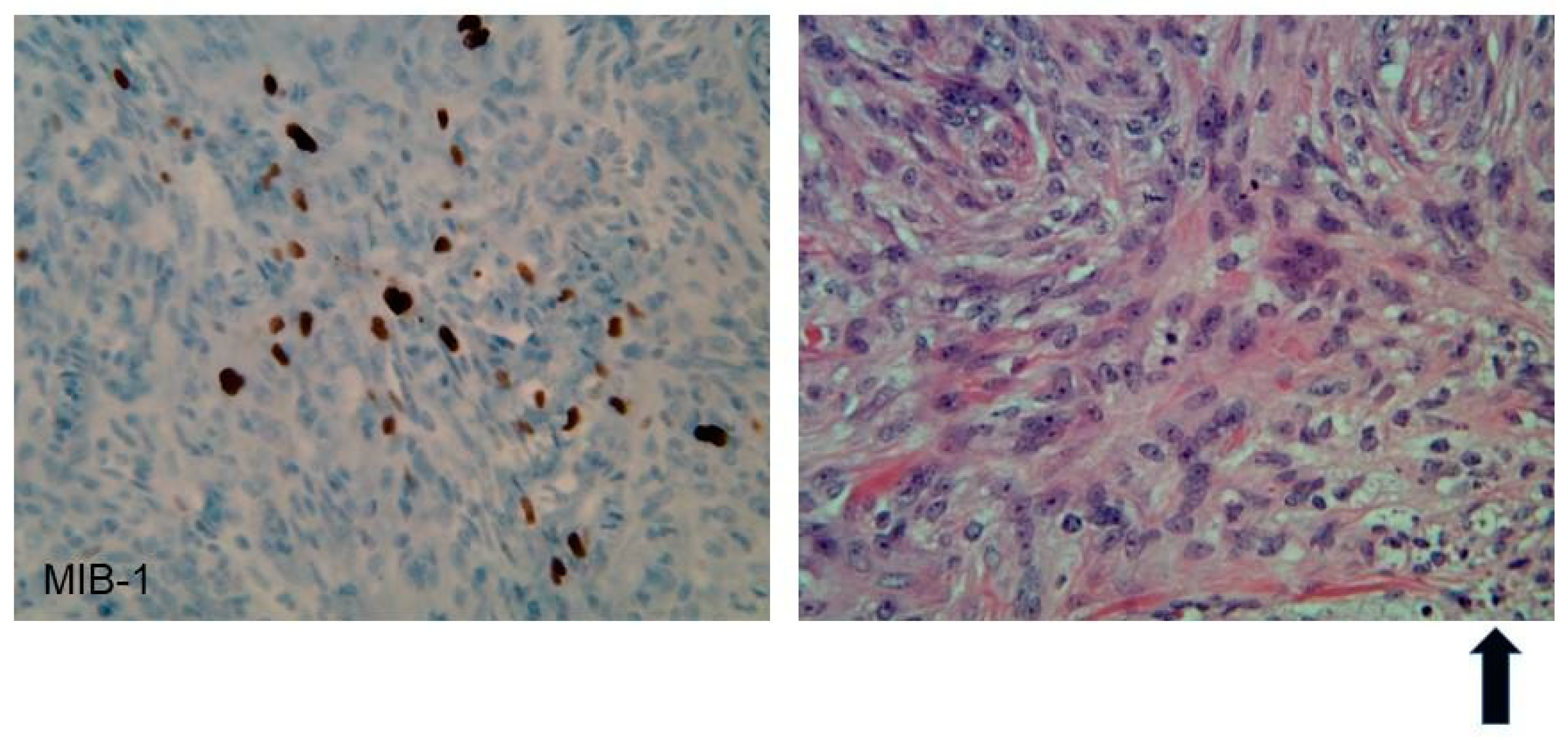
| Meningiomas | ~50% (20% are in kids) | Headache Seizure | Fibrous morphology Psamomma Bodies High mitotic index | Intense and homogeneous enhancement. Frequent cystic components Can be multiple Present in unusual locations: craniocervical junction. | Surgical excision Radiosurgery Current clinical trial: mTORC1/2 inhibitor AZD2014 (NCT02831257, NCT03071874) | Malignant transformation Invasion to vascular brain structures Compression effect |
| Ependymoma Glial | ~30% | Asymptomatic | Perivascular pseudorosettes Ependymal rosettes | Usually spinal intramedullary (not intracranial/intraventricular). “String of pearls” appearance along the spinal cord and cauda equina. | Monitoring/surveillance Surgical resection if symptomatic | Malignant transformation is rare |
| Menigioangiomatosis | rare | Headache Seizures Behavioral changes Cortical blindness Paresis | Plaque like leptomeningeal and perivascular proliferation Fibroblastic and meningothelial appearing cells | Cortical/subcortical white matter mass characterized by Ca⁺⁺, enhancing meningovascular proliferation. Most common in temporal and frontal lobes. | Surgical excision | Intracerebral hemorrhage |
| Glial micro hamartomas | Common | Asymptomatic | Atypical pleomorphic nuclei, Occasional multi-nucleation, Eosinophilic cytoplasm | Cortical hyperintense T2/FLAIR lesions “Transmantle sign” | Surveillance and monitoring | None |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bachir, S.; Shah, S.; Shapiro, S.; Koehler, A.; Mahammedi, A.; Samy, R.N.; Zuccarello, M.; Schorry, E.; Sengupta, S. Neurofibromatosis Type 2 (NF2) and the Implications for Vestibular Schwannoma and Meningioma Pathogenesis. Int. J. Mol. Sci. 2021, 22, 690. https://doi.org/10.3390/ijms22020690
Bachir S, Shah S, Shapiro S, Koehler A, Mahammedi A, Samy RN, Zuccarello M, Schorry E, Sengupta S. Neurofibromatosis Type 2 (NF2) and the Implications for Vestibular Schwannoma and Meningioma Pathogenesis. International Journal of Molecular Sciences. 2021; 22(2):690. https://doi.org/10.3390/ijms22020690
Chicago/Turabian StyleBachir, Suha, Sanjit Shah, Scott Shapiro, Abigail Koehler, Abdelkader Mahammedi, Ravi N. Samy, Mario Zuccarello, Elizabeth Schorry, and Soma Sengupta. 2021. "Neurofibromatosis Type 2 (NF2) and the Implications for Vestibular Schwannoma and Meningioma Pathogenesis" International Journal of Molecular Sciences 22, no. 2: 690. https://doi.org/10.3390/ijms22020690
APA StyleBachir, S., Shah, S., Shapiro, S., Koehler, A., Mahammedi, A., Samy, R. N., Zuccarello, M., Schorry, E., & Sengupta, S. (2021). Neurofibromatosis Type 2 (NF2) and the Implications for Vestibular Schwannoma and Meningioma Pathogenesis. International Journal of Molecular Sciences, 22(2), 690. https://doi.org/10.3390/ijms22020690