Some Molecular and Cellular Stress Mechanisms Associated with Neurodegenerative Diseases and Atherosclerosis

 ,
,  , ,
, ,  ,
,  and
and
Abstract
:
1. Introduction
2. Molecular Types of Stress
2.1. Oxidative Stress
The Link of Oxidative Stress with Neurodegenerative Diseases and Mitochondrial Genome Mutations
2.2. Genotoxic Stress
2.2.1. Replicative Stress
2.2.2. Mutation Stress. Genetic Predisposition to Genotoxic Stress
2.3. DNA Methylation Caused by Stress
3. Cellular Types of Stress
3.1. Cellular Stress
3.2. Endoplasmic Reticulum Stress
3.3. Osmotic Stress
3.4. Link between Cellular Senescence, Oxidative Stress and Chronic Stress
4. The Relationship of Chronic Stress with the Occurrence and Development of Atherosclerosis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shafirkin, A.V. A model of ecological hazard and social tension for the description of the health deterioration risk in the Russian population. Aviakosm. Ekol. Med. 2003, 37, 42–49. [Google Scholar]
- Schöner, J.; Heinz, A.; Endres, M.; Gertz, K.; Kronenberg, G. Post-traumatic stress disorder and beyond: An overview of rodent stress models. J. Cell Mol. Med. 2017, 10, 2248–2256. [Google Scholar] [CrossRef] [Green Version]
- Niki, E. Oxidative stress and antioxidants: Distress or eustress? Arch. Biochem. Biophys. 2016, 595, 19–24. [Google Scholar] [CrossRef]
- Parker, K.N.; Ragsdale, J.M. Effects of Distress and Eustress on Changes in Fatigue from Waking to Working. Appl. Psychol. Health Well Being. 2015, 7, 293–315. [Google Scholar] [CrossRef]
- Mesurado, B.; Richaud, M.C.; Mateo, N.J. Engagement, Flow, Self-Efficacy, and Eustress of University Students: A Cross-National Comparison Between the Philippines and Argentina. J. Psychol. 2016, 150, 281–299. [Google Scholar] [CrossRef]
- Vanhooren, S.; Leijssen, M.; Dezutter, J. Loss of Meaning as a Predictor of Distress in Prison. Int. J. Offender Ther. Comp. Criminol. 2017, 61, 1411–1432. [Google Scholar] [CrossRef]
- Tonsing, K.N.; Vungkhanching, M. Assessing psychological distress in cancer patients: The use of distress thermometer in an outpatient cancer/hematology treatment center. Soc. Work Health Care 2018, 57, 126–136. [Google Scholar] [CrossRef]
- Sheth, C.; McGlade, E.; Todd, D.Y. Chronic Stress in Adolescents and Its Neurobiological and Psychopathological Consequences: An RDoC Perspective. Chronic Stress (Thousand Oaks) 2017, 1, 2470547017715645. [Google Scholar] [CrossRef]
- American Psychology Association. Stress in America Press Room. Available online: http://www.apa.org (accessed on 21 October 2020).
- Cohen, M.; Tottenham, N.; Casey, B.J. Translational developmental studies of stress on brain and behavior: Implications for adolescent mental health and illness? Neuroscience 2013, 249, 53–62. [Google Scholar] [CrossRef] [Green Version]
- Shonkoff, J.P.; Boyce, W.T.; McEwen, B.S. Neuroscience, molecular biology, and the childhood roots of health disparities: Building a new framework for health promotion and disease prevention. JAMA 2009, 301, 2252–2259. [Google Scholar] [CrossRef]
- Eiland, L.; Romeo, R.D. Stress and the developing adolescent brain. Neuroscience 2013, 249, 162–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dvir, Y.; Ford, J.; Hill, M.; Frazier, J. Childhood maltreatment, emotional dysregulation, and psychiatric comorbidities. Harv. Rev. Psychiatry 2014, 22, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Yao, B.C.; Meng, L.B.; Hao, M.L.; Zhang, Y.M.; Gong, T.; Guo, Z.G. Chronic stress: A critical risk factor for atherosclerosis. J. Int. Med. Res. 2019, 47, 1429–1440. [Google Scholar] [CrossRef] [PubMed]
- Kershaw, K.N.; Cordova, A.D.L.; Carnethon, M.R.; Tindle, H.A.; Liu, K. Chronic stress and endothelial dysfunction: The Multi-Ethnic Study of Atherosclerosis (MESA). Am. J. Hypertens. 2017, 30, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, S.E.; Baylin, S.B. Stress and the epigenetic landscape: A link to the pathobiology of human diseases? Nat. Rev. Genet. 2010, 11, 806–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jennings, J.R.; Kamarck, T.W.; Rose, S.A.E.; Kaplan, G.A.; Manuck, S.B.; Salonen, J.T. Exaggerated blood pressure responses during mental stress are prospectively related to enhanced carotid atherosclerosis in middle-aged Finnish men. Circulation 2004, 110, 2198–2203. [Google Scholar] [CrossRef] [Green Version]
- Sazonova, M.A.; Sinyov, V.V.; Ryzhkova, A.I.; Galitsyna, E.V.; Khasanova, Z.B.; Postnov, A.Y.; Yarygina, E.I.; Orekhov, A.N.; Sobenin, I.A. Role of Mitochondrial Genome Mutations in Pathogenesis of Carotid Atherosclerosis. Oxid. Med. Cell Longev. 2017, 2017, 6934394. [Google Scholar] [CrossRef]
- Chumaeva, N.; Hintsanen, M.; Hintsa, T.; Ravaja, N.; Juonala, M.; Raitakari, O.T.; Järvinen, L.K. Early atherosclerosis and cardiac autonomic responses to mental stress: A population-based study of the moderating influence of impaired endothelial function. BMC Cardiovas. Disord. 2010, 10, 16–21. [Google Scholar] [CrossRef] [Green Version]
- Werth, D.; Grassi, G.; Konjer, N.; Dapas, B.; Farra, R.; Giansante, C.; Kandolf, R.; Guarnieri, G.; Nordheim, A.; Heidenreich, O. Proliferation of human primary vascular smooth muscle cells depends on serum response factor. Eur. J. Cell Biol. 2010, 89, 216–224. [Google Scholar] [CrossRef]
- Yeung, A.C.; Vekshtein, V.I.; Krantz, D.S.; Vita, J.A.; Ryan, T.J., Jr.; Ganz, P.; Selwyn, A.P. The effect of atherosclerosis on the vasomotor response of coronary arteries to mental stress. N. Engl. J. Med. 1991, 325, 1551–1556. [Google Scholar] [CrossRef]
- Kattoor, A.J.; Pothineni, N.V.K.; Palagiri, D.; Mehta, J.L. Oxidative Stress in Atherosclerosis. Curr. Atheroscler. Rep. 2017, 19, 42. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox. Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Okumura, H.; Guo, R.; Naruse, K. Effect of Oxidative Stress on Cardiovascular System in Response to Gravity. Int. J. Mol. Sci. 2017, 18, 1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sazonova, M.A.; Chicheva, M.M.; Zhelankin, A.V.; Sobenin, I.A.; Bobryshev, Y.V.; Orekhov, A.N. Association of mutations in the mitochondrial genome with the subclinical carotid atherosclerosis in women. Exp. Mol. Pathol. 2015, 99, 25–32. [Google Scholar] [CrossRef]
- Pedzik, A.; Paradowski, M.; Rysz, J. Oxidative stress in nephrology. Pol. Merkur. Lekarski 2010, 28, 56–60. [Google Scholar]
- Kobayashi, C.I.; Suda, T. Regulation of reactive oxygen species in stem cells and cancer stem cells. J. Cell Physiol. 2012, 227, 421–430. [Google Scholar] [CrossRef]
- Cacabelos, R.; Reddy, V.P.; Aliev, G. Editorial: Neurodegeneration, Oxidative Stress, Metabolic Syndrome, Drug Design and Development: Clinical Implications. CNS Neurol. Disord. Drug Targets 2016, 15, 126. [Google Scholar] [CrossRef]
- Ryzhkova, A.I.; Sazonova, M.A.; Sinyov, V.V.; Galitsyna, E.V.; Chicheva, M.M.; Melnichenko, A.A.; Grechko, A.V.; Postnov, A.Y.; Orekhov, A.N.; Shkurat, T.P. Mitochondrial diseases caused by mtDNA mutations: A mini-review. Ther. Clin. Risk Manag. 2018, 14, 1933–1942. [Google Scholar] [CrossRef] [Green Version]
- Griendling, K.K.; Touyz, R.M.; Zweier, J.L.; Dikalov, S.; Chilian, W.; Chen, Y.R.; Harrison, D.G.; Bhatnagar, A. American Heart Association Council on Basic Cardiovascular Sciences. Measurement of Reactive Oxygen Species, Reactive Nitrogen Species, and Redox-Dependent Signaling in the Cardiovascular System: A Scientific Statement from the American Heart Association. Circ. Res. 2016, 119, e39–e75. [Google Scholar] [CrossRef]
- Lacy, F.; O’Connor, D.T.; Schönbein, G.W.S. Plasma Hydrogen Peroxide Production in Hypertensives and Normotensive Subjects at Genetic Risk of Hypertension. J. Hypertens. 1998, 16, 291–303. [Google Scholar] [CrossRef]
- Hamilton, C.A.; Brosnan, M.J.; McIntyre, M.; Graham, D.; Dominiczak, A.F. Superoxide excess in hypertension and aging: A common cause of endothelial dysfunction. Hypertension 2001, 37, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Case, A.J.; Tian, J.; Zimmerman, M.C. Increased mitochondrial superoxide in the brain, but not periphery, sensitizes mice to angiotensin II-mediated hypertension. Redox Biol. 2017, 11, 82–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hackl, C.Z.; von Känel, R.; Thomas, L.; Hauser, M.; Kuebler, U.; Widmer, H.R.; Wirtz, P.H. Macrophage Superoxide Anion Production in Essential Hypertension: Associations With Biological and Psychological Cardiovascular Risk Factors. Psychosom. Med. 2016, 78, 750–757. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, M.M.; Borodachev, E.N.; Sazonova, M.A. Human pathologies associated with mutations of mitochondrial genome. Patol. Fiziol. Eksp. Ter. 2012, 3, 115–122. [Google Scholar]
- Kirichenko, T.V.; Ryzhkova, A.I.; Sinyov, V.V.; Sazonova, M.D.; Orekhova, V.A.; Karagodin, V.P.; Gerasimova, E.V.; Voevoda, M.I.; Orekhov, A.N.; Sobenin, I.A. Impact of mitochondrial DNA mutations of carotid intima-media thickness in the Novosibersk region. Life 2020, 10, 160. [Google Scholar] [CrossRef]
- Sazonova, M.A.; Zhelankin, A.V.; Barinova, V.A.; Sinyov, V.V.; Khasanova, Z.B.; Postnov, A.Y.; Orekhov, A.N.; Bobryshev, Y.V.; Sobenin, I.A. Mutations of mitochondrial genome in patients with carotid atherosclerosis. Front. Genet. 2015, 6, 111. [Google Scholar] [CrossRef] [Green Version]
- Hrycay, E.G.; Bandiera, S.M. Involvement of Cytochrome P450 in Reactive Oxygen Species Formation and Cancer. Adv. Pharm. 2015, 74, 35–84. [Google Scholar] [CrossRef]
- Takeshima, T.; Yumura, Y.; Yasuda, K.; Sanjo, H.; Kuroda, S.; Yamanaka, H.; Iwasaki, A. Inverse correlation between reactive oxygen species in unwashed semen and sperm motion parameters as measured by a computer-assisted semen analyzer. Asian J. Androl. 2017, 19, 350–354. [Google Scholar] [CrossRef]
- Keng, C.L.; Lin, Y.C.; Tseng, W.L.; Lu, C.Y. Design of Peptide-Based Probes for the Microscale Detection of Reactive Oxygen Species. Anal. Chem. 2017, 89, 10883–10888. [Google Scholar] [CrossRef]
- Sobenin, I.A.; Sazonova, M.A.; Postnov, A.Y.; Salonen, J.T.; Bobryshev, Y.V.; Orekhov, A.N. Association of mitochondrial genetic variation with carotid atherosclerosis. PLoS ONE 2013, 8, e68070. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.T.; Simon, D.K.; Ahn, C.H.; Kim, L.M.; Beal, M.F. High aggregate burden of somatic mtDNA point mutations in aging and Alzheimer’s disease brain. Hum. Mol. Genet. 2002, 11, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.; Krishnan, K.J.; Morris, C.M.; Taylor, G.A.; Reeve, A.K.; Perry, R.H.; Jaros, E.; Hersheson, J.S.; Betts, J.; Klopstock, T.; et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 2006, 38, 515–517. [Google Scholar] [CrossRef] [PubMed]
- Saffari, A.; Kölker, S.; Hoffmann, G.F.; Fakhari, D.E. Linking mitochondrial dysfunction to neurodegeneration in lysosomal storage diseases. J. Inherit. Metab. Dis. 2017, 40, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, J.; Zhao, A.; Li, J. SIRT1 activation inhibits hyperglycemia-induced apoptosis by reducing oxidative stress and mitochondrial dysfunction in human endothelial cells. Mol. Med. Rep. 2017, 16, 3331–3338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karam, B.S.; Moreno, A.C.; Koh, W.; Akar, J.G.; Akar, F.G. Oxidative Stress and Inflammation as Central Mediators of Atrial Fibrillation in Obesity and Diabetes. Cardiovasc. Diabetol. 2017, 16, 120. [Google Scholar] [CrossRef] [PubMed]
- Klaunig, J.E. Oxidative Stress and Cancer. Curr. Pharm. Des. 2018, 24, 4771–4778. [Google Scholar] [CrossRef]
- Jenner, P. Oxidative stress in Parkinson’s disease. Ann. Neurol. 2003, 53 (Suppl. 3), S26–S36, discussion S36–S38. [Google Scholar] [CrossRef]
- Çubukçu, H.C.; Yurtdaş, M.; Durak, Z.E.; Aytaç, B.; Güneş, H.N.; Çokal, B.G.; Yoldaş, T.K.; Durak, İ. Oxidative and nitrosative stress in serum of patients with Parkinson’s disease. Neurol. Sci. 2016, 37, 1793–1798. [Google Scholar] [CrossRef]
- Paul, R.; Choudhury, A.; Kumar, S.; Giri, A.; Sandhir, R.; Borah, A. Cholesterol contributes to dopamine-neuronal loss in MPTP mouse model of Parkinson’s disease: Involvement of mitochondrial dysfunctions and oxidative stress. PLoS ONE 2017, 12, e0171285. [Google Scholar] [CrossRef] [Green Version]
- Dawson, T.M.; Dawson, V.L. Molecular pathways of neurodegeneration in Parkinson’s disease. Science 2003, 302, 819–822. [Google Scholar] [CrossRef]
- Kish, S.J.; Shannak, K.; Rajput, A.; Deck, J.H.; Hornykiewicz, O. Aging produces a specific pattern of striatal dopamine loss: Implications for the etiology of idiopathic Parkinson’s disease. J. Neurochem. 1992, 58, 642–648. [Google Scholar] [CrossRef] [PubMed]
- Oh, M.; Kim, J.S.; Kim, J.Y.; Shin, K.H.; Park, S.H.; Kim, H.O.; Moon, D.H.; Oh, S.J.; Chung, S.J.; Lee, C.S. Subregional patterns of preferential striatal dopamine transporter loss differ in Parkinson disease, progressive supranuclear palsy, and multiple-system atrophy. J. Nucl. Med. 2012, 53, 399–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fredenburg, R.A.; Rospigliosi, C.; Meray, R.K.; Kessler, J.C.; Lashuel, H.A.; Eliezer, D.; Lansbury, P.T., Jr. The impact of the E46K mutation on the properties of alpha-synuclein in its monomeric and oligomeric states. Biochemistry 2007, 46, 7107–7118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.H.; Kuo, C.W.; Lin, T.K.; Tsai, M.H.; Liou, C.W. Dopamine Therapy and the Regulation of Oxidative Stress and Mitochondrial DNA Copy Number in Patients with Parkinson’s Disease. Antioxidants 2020, 9, 1159. [Google Scholar] [CrossRef]
- Nido, G.S.; Dölle, C.; Flønes, I.; Tuppen, H.A.; Alves, G.; Tysnes, O.B.; Haugarvoll, K.; Tzoulis, C. Ultradeep mapping of neuronal mitochondrial deletions in Parkinson’s disease. Neurobiol. Aging 2018, 63, 120–127. [Google Scholar] [CrossRef]
- Bose, A.; Beal, M.F. Mitochondrial dysfunction in Parkinson’s disease. Neurochem 2016, 139 (Suppl. 1), 216–231. [Google Scholar] [CrossRef]
- Xu, W.; Tan, L.; Yu, J.T. Link between the SNCA gene and parkinsonism. Neurobiol. Aging 2015, 36, 1505–1518. [Google Scholar] [CrossRef]
- Chen, C.M.; Chen, I.C.; Huang, Y.C.; Juan, H.F.; Chen, Y.L.; Chen, Y.C.; Lin, C.H.; Lee, L.C.; Lee, C.M.; Chen, G.J.L. FBXO7 Y52C polymorphism as a potential protective factor in Parkinson’s disease. PLoS ONE 2014, 9, e101392. [Google Scholar] [CrossRef]
- Tsika, E.; Glauser, L.; Moser, R.; Fiser, A.; Daniel, G.; Sheerin, U.M.; Lees, A.; Troncoso, J.C.; Lewis, P.A.; Bandopadhyay, R.; et al. Parkinson’s disease-linked mutations in VPS35 induce dopaminergic neurodegeneration. Hum. Mol. Genet. 2014, 23, 4621–4638. [Google Scholar] [CrossRef] [Green Version]
- Bonifati, V. Genetics of Parkinson’s disease—State of the art. Parkinsonism Relat. Disord. 2014, 20 (Suppl. 1), S23–S28. [Google Scholar] [CrossRef]
- Güell, C.V.; Rajput, A.; Milnerwood, A.J.; Shah, B.; Tu, C.S.; Trinh, J.; Yu, I.; Encarnacion, M.; Munsie, L.N.; Tapia, L.; et al. DNAJC13 mutations in Parkinson disease. Hum. Mol. Genet. 2014, 23, 1794–1801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keck, S.; Nitsch, R.; Grune, T.; Ullrich, O. Proteasome inhibition by paired helical filament-tau in brains of patients with Alzheimer’s disease. J. Neurochem. 2003, 8, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Agholme, L.; Nath, S.; Domert, J.; Marcusson, J.; Kågedal, K.; Hallbeck, M. Proteasome inhibition induces stress kinase dependent transport deficits—Implications for Alzheimer’s disease. Mol. Cell Neurosci. 2014, 58, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Adel, C.A.; Chen, S.; Chohan, M.O.; Akkad, E.E.; Gong, C.X.; Khatoon, S.; Li, B.; Liu, F.; Rahman, A.; et al. Tau pathology in Alzheimer disease and other tauopathies. Biochim. Biophys. Acta 2005, 1739, 198–210. [Google Scholar] [CrossRef] [Green Version]
- Fitzpatrick, A.W.P.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H.W. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef] [Green Version]
- Cras, P.; Smith, M.A.; Richey, P.L.; Siedlak, S.L.; Mulvihill, P.; Perry, G. Extracellular neurofibrillary tangles reflect neuronal loss and provide further evidence of extensive protein cross-linking in Alzheimer disease. Acta Neuropathol. 1995, 89, 291–295. [Google Scholar] [CrossRef]
- Jeong, S. Molecular and Cellular Basis of Neurodegeneration in Alzheimer’s Disease. Mol. Cells 2017, 40, 613–620. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhou, X.; Li, G.; Zhang, Y.; Wu, Y.; Song, W. Modifications and Trafficking of APP in the Pathogenesis of Alzheimer’s Disease. Front. Mol. Neurosci. 2017, 10, 294. [Google Scholar] [CrossRef]
- Alikhani, N.; Guo, L.; Yan, S.; Du, H.; Pinho, C.M.; Chen, J.X.; Glaser, E.; Yan, S.S. Decreased proteolytic activity of the mitochondrial amyloid-β degrading enzyme, PrePpeptidasome, in Alzheimer’s disease brain mitochondria. J. Alzheimers Dis. 2011, 27, 75–87. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, P.F.; Pinho, C.M.; Branca, R.M.; Lehtiö, J.; Levine, R.L.; Glaser, E. In vitro oxidative inactivation of human presequence protease (hPreP). Free Radic. Biol. Med. 2012, 53, 2188–2195. [Google Scholar] [CrossRef] [Green Version]
- Alikhani, N.; Ankarcrona, M.; Glaser, E. Mitochondria and Alzheimer’s disease: Amyloid-beta peptide uptake and degradation by the presequence protease, hPreP. J. Bioenerg. Biomembr. 2009, 41, 447–451. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Du, H.; Yan, S.; Fang, F.; Wang, C.; Lue, L.F.; Guo, L.; Chen, D.; Stern, D.M.; Moore, F.J.G.; et al. Inhibition of amyloid-beta (Abeta) peptide-binding alcohol dehydrogenase-Abeta interaction reduces Abeta accumulation and improves mitochondrial function in a mouse model of Alzheimer’s disease. J. Neurosci. 2011, 31, 2313–2320. [Google Scholar] [CrossRef] [PubMed]
- Fang, D.; Wang, Y.; Zhang, Z.; Du, H.; Yan, S.; Sun, Q.; Zhong, C.; Wu, L.; Vangavaragu, J.R.; Yan, S.; et al. Increased neuronal PreP activity reduces Aβ accumulation, attenuates neuroinflammation and improves mitochondrial and synaptic function in Alzheimer disease’s mouse model. Hum. Mol. Genet. 2015, 24, 5198–5210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langer, Y.; Aran, A.; Gulsuner, S.; Libdeh, B.A.; Renbaum, P.; Brunetti, D.; Teixeira, P.F.; Walsh, T.; Zeligson, S.; Ruotolo, R.; et al. Mitochondrial PITRM1 peptidase loss-of-function in childhood cerebellar atrophy. J. Med. Genet. 2018, 55, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Pérez, M.J.; Ivanyuk, D.; Panagiotakopoulou, V.; di Napoli, G.; Kalb, S.; Brunetti, D.; Shaana, R.A.; Kaeser, S.A.; Fraschka, S.A.; Jucker, M.; et al. Loss of function of the mitochondrial peptidase PITRM1 induces proteotoxic stress and Alzheimer’s disease-like pathology in human cerebral organoids. Mol. Psychiatry 2020. [Google Scholar] [CrossRef]
- Phillips, N.R.; Simpkins, J.W.; Roby, R.K. Mitochondrial DNA deletions in Alzheimer’s brains: A review. Alzheimers Dement. 2014, 10, 393–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lezza, A.M.; Mecocci, P.; Cormio, A.; Beal, M.F.; Cherubini, A.; Cantatore, P.; Senin, U.; Gadaleta, M.N. Mitochondrial DNA 4977 bp deletion and OH8dG levels correlate in the brain of aged subjects but not Alzheimer’s disease patients. FASEB J. 1999, 13, 1083–1088. [Google Scholar] [CrossRef]
- Hou, Y.; Song, H.; Croteau, D.L.; Akbari, M.; Bohr, V.A. Genome instability in Alzheimer disease. Mech. Ageing Dev. 2017, 161, 83–94. [Google Scholar] [CrossRef] [Green Version]
- Karch, C.M.; Cruchaga, C.; Goate, A.M. Alzheimer’s disease genetics: From the bench to the clinic. Neuron 2014, 83, 11–26. [Google Scholar] [CrossRef] [Green Version]
- Arun, S.; Liu, L.; Donmez, G. Mitochondrial Biology and Neurological Diseases. Curr. Neuropharmacol. 2016, 14, 143–154. [Google Scholar] [CrossRef]
- Iwata, A.; Nagata, K.; Hatsuta, H.; Takuma, H.; Bundo, M.; Iwamoto, K.; Tamaoka, A.; Murayama, S.; Saido, T.; Tsuji, S. Altered CpG methylation in sporadic Alzheimer’s disease is associated with APP and MAPT dysregulation. Hum. Mol. Genet. 2014, 23, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Benitez, B.A.; Karch, C.M.; Cai, Y.; Jin, S.C.; Cooper, B.; Carrell, D.; Bertelsen, S.; Chibnik, L.; Schneider, J.A.; Bennett, D.A.; et al. The PSEN1, p.E318G variant increases the risk of Alzheimer’s disease in APOE-ε4 carriers. PLoS Genet. 2013, 9, e1003685. [Google Scholar] [CrossRef] [PubMed]
- Bekris, L.M.; Tsuang, D.W.; Peskind, E.R.; Yu, C.E.; Montine, T.J.; Zhang, J.; Zabetian, C.P.; Leverenz, J.B. Cerebrospinal fluid Aβ42 levels and APP processing pathway genes in Parkinson’s disease. Mov. Disord. 2015, 30, 936–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Border, R.; Johnson, E.C.; Evans, L.M.; Smolen, A.; Berley, N.; Sullivan, P.F.; Keller, M.C. No Support for Historical Candidate Gene or Candidate Gene-by-Interaction Hypotheses for Major Depression Across Multiple Large Samples. Am. J. Psychiatry 2019, 176, 376–387. [Google Scholar] [CrossRef]
- Igata, N.; Kakeda, S.; Watanabe, K.; Ide, S.; Kishi, T.; Abe, O.; Igata, R.; Katsuki, A.; Iwata, N.; Yoshimura, R.; et al. Voxel-based morphometric brain comparison between healthy subjects and major depressive disorder patients in Japanese with the s/s genotype of 5-HTTLPR. Sci. Rep. 2017, 21, 3931. [Google Scholar] [CrossRef]
- Ming, Q.; Zhang, Y.; Yi, J.; Wang, X.; Zhu, X.; Yao, S. Serotonin transporter gene polymorphism (5-HTTLPR) L allele interacts with stress to increase anxiety symptoms in Chinese adolescents: A multiwave longitudinal study. BMC Psychiatry 2015, 15, 248. [Google Scholar] [CrossRef] [Green Version]
- Sazonova, M.A.; Shkurat, T.P.; Demakova, N.A.; Zhelankin, A.V.; Barinova, V.A.; Sobenin, I.A.; Orekhov, A.N. Mitochondrial genome sequencing in atherosclerosis: What’s next? Curr. Pharm. Des. 2016, 22, 390–396. [Google Scholar] [CrossRef] [Green Version]
- Zaragoza, M.V.; Fass, J.; Diegoli, M.; Lin, D.; Arbustini, E. Mitochondrial DNA variant discovery and evaluation in human Cardiomyopathies through next-generation sequencing. PLoS ONE 2010, 5, e12295. [Google Scholar] [CrossRef] [Green Version]
- Prentice, H.; Modi, J.P.; Wu, J.Y. Mechanisms of Neuronal Protection against Excitotoxicity, Endoplasmic Reticulum Stress, and Mitochondrial Dysfunction in Stroke and Neurodegenerative Diseases. Oxid. Med. Cell Longev. 2015, 2015, 964518. [Google Scholar] [CrossRef] [Green Version]
- Gallina, I.; Colding, C.; Henriksen, P.; Beli, P.; Nakamura, K.; Offman, J.; Mathiasen, D.P.; Silva, S.; Hoffmann, E.; Growth, A.; et al. Cmr1/WDR76 defines a nuclear genotoxic stress body linking genome integrity and protein quality control. Nat. Commun. 2015, 6, 6533. [Google Scholar] [CrossRef]
- Rué, L.; Coronel, M.B.; Muncunill, J.C.; Giralt, A.; Vida, R.A.; Mentxaka, G.; Kagerbauer, B.; Abellán, M.T.Z.; Aranda, Z.; Venturi, V.; et al. Targeting CAG repeat RNAs reduces Huntington’s disease phenotype independently of huntingtin levels. J. Clin. Investig. 2016, 126, 4319–4330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz, P.J.G.; Caldentey, J.G.; Feliz, C.; del Val, J.; Herranz, A.; Castrillo, J.C.M. Late onset Huntington’s disease with 29 CAG repeat expansion. J. Neurol. Sci. 2016, 363, 114–115. [Google Scholar] [CrossRef] [PubMed]
- Faye, C.; Mcgowan, J.C.; Denny, C.A.; David, D.J. Neurobiological Mechanisms of Stress Resilience and Implications for the Aged Population. Curr. Neuropharmacol. 2018, 16, 234–270. [Google Scholar] [CrossRef]
- de Magalhães, J.P.; Passos, J.F. Stress, cell senescence and organismal ageing. Mech. Ageing Dev. 2018, 170, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Sazonova, M.A.; Ryzhkova, A.I.; Sinyov, V.V.; Sazonova, M.D.; Kirichenko, T.V.; Doroschuk, N.A.; Karagodin, V.P.; Orekhov, A.N.; Sobenin, I.A. Mutations of mtDNA in some vascular and metabolic diseases. Curr. Pharm. Des. 2020, 26, 1–10. [Google Scholar] [CrossRef]
- Bersani, F.S.; Morley, C.; Lindqvist, D.; Epel, E.S.; Picard, M.; Yehuda, R.; Flory, J.; Bierer, L.M.; Makotkine, I.; Amara, D.A.; et al. Mitochondrial DNA copy number is reduced in male combat veterans with PTSD. Prog. Neuropsychopharmacol. Biol. Psychiatry 2016, 64, 10–17. [Google Scholar] [CrossRef] [Green Version]
- Brunst, K.J.; Guerra, M.S.; Gennings, C.; Hacker, M.; Jara, C.; Enlow, M.B.; Wright, R.O.; Baccarelli, A.; Wright, R.J. Maternal Lifetime Stress and Prenatal Psychological Functioning and Decreased Placental Mitochondrial DNA Copy Number in the PRISM Study. Am. J. Epidemiol. 2017, 186, 1227–1236. [Google Scholar] [CrossRef]
- Kranenburg, M.J.B.; VanIjzendoorn, M.H. Gene-environment interaction of the dopamine D4 receptor (DRD4) and observed maternal insensitivity predicting externalizing behavior in preschoolers. Dev. Psychobiol. 2006, 48, 406–409. [Google Scholar] [CrossRef]
- Kirichenko, T.V.; Ragino, Y.I.; Voevoda, M.I.; Urazalina, S.J.; Khasanova, Z.B.; Orekhova, V.A.; Sinyov, V.V.; Sazonova, M.A.; Orekhov, A.N.; Sobenin, I.A. Data on association of mitochondrial heteroplasmy with carotid intima-media thickness in subjects from Russian and Kazakh populations. Data Brief. 2020, 29, 105136. [Google Scholar] [CrossRef]
- Novoselov, V.V.; Sazonova, M.A.; Ivanova, E.A.; Orekhov, A.N. Study of the activated macrophage transcriptome. Exp. Mol. Pathol. 2015, 99, 575–580. [Google Scholar] [CrossRef]
- Rodrigues, L.T.; Jacques, S.M.C.; Erler, M.L.P.; Tsuneto, L.; Salzano, F.M.; Hutz, M.H. Dopamine receptor D4 allele distribution in Amerindians: A reflection of past behavior differences? Am. J. Phys. Anthropol. 2010, 143, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Hollerbach, P.; Johansson, A.; Ventus, D.; Jern, P.; Neuman, C.S.; Westberg, L.; Santtila, P.; Habermeyer, E.; Mokros, A. Main and interaction effects of childhood trauma and the MAOA uVNTR polymorphism on psychopathy. Psychoneuroendocrinology 2018, 95, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Youdim, M.B.H. Monoamine oxidase inhibitors, and iron chelators in depressive illness and neurodegenerative diseases. J. Neural Transm. (Vienna) 2018, 125, 1719–1733. [Google Scholar] [CrossRef] [PubMed]
- Eusebi, P.G.; Sevane, N.; Cortes, O.; Contreras, E.; Canon, J.; Dunner, S. Aggressive behavior in cattle is associated with polymorphism in the MAOA gene promoter. Anim. Genet. 2020, 51, 14–21. [Google Scholar] [CrossRef]
- Caspi, A.; Sugden, K.; Moffitt, T.E.; Taylor, A.; Craig, I.W.; Harrington, H.; McClay, J.; Mill, J.; Martin, J.; Braithwaite, A.; et al. Influence of life stress on depression: Moderation by a polymorphism in the 5-HTT gene. Science 2003, 301, 386–389. [Google Scholar] [CrossRef]
- Zammit, S.; Owen, M.J. Stressful life events, 5-HTT genotype and risk of depression. Br. J. Psychiatry 2006, 188, 199–201. [Google Scholar] [CrossRef] [Green Version]
- Karg, K.; Burmeister, M.; Shedden, K.; Sen, S. The serotonin transporter promoter variant (5-httlpr), stress, and depression meta-analysis revisited. Arch. Gen. Psychiatry 2011, 68, 444–454. [Google Scholar] [CrossRef] [Green Version]
- Sazonova, M.D.; Ryzhkova, A.I.; Sinyov, V.V.; Khasanova, Z.B.; Doroschuk, N.A.; Nikitina, N.A.; Sobenin, I.A.; Orekhov, A.N.; Sazonova, M.A. Analysis of mtDNA mutations in atherosclerotic plaques in individuals from Novosibirsk. Atherosclerosis 2019, 287, e166–e167. [Google Scholar] [CrossRef]
- McGuffin, P.; Alsabban, S.; Uher, R. The truth about genetic variation in the serotonin transporter gene and response to stress and medication. Br. J. Psychiatry 2011, 198, 424–427. [Google Scholar] [CrossRef]
- Munafò, M.R.; Brown, S.M.; Hariri, A.R. Serotonin transporter (5-HTTLPR) genotype and amygdala activation: A meta-analysis. Biol. Psychiatry 2008, 63, 852–857. [Google Scholar] [CrossRef] [Green Version]
- Murphy, S.E.; Norbury, R.; Godlewska, B.R.; Cowen, P.J.; Mannie, Z.M.; Harmer, C.J.; Munafò, M.R. The effect of the serotonin transporter polymorphism (5-HTTLPR) on amygdala function: A meta-analysis. Mol. Psychiatry 2013, 18, 512–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seeger, G.; Schloss, P.; Schmidt, M.H.; Jungfleisch, A.R.; Henn, F.A. Gene-environment interaction in hyperkinetic conduct disorder (HD + CD) as indicated by season of birth variations in dopamine receptor (DRD4) gene polymorphism. Neurosci. Lett. 2004, 366, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Belsky, J.; Jonassaint, C.; Pluess, M.; Stanton, M.; Brummett, B.; Williams, R. Vulnerability genes or plasticity genes? Mol. Psychiatry 2009, 14, 746–754. [Google Scholar] [CrossRef] [PubMed]
- Alieva, A.; Rudenok, M.; Filatova, E.; Karabanov, A.; Doronina, O.; Doronina, K.; Kolacheva, A.A.; Ugryumov, M.V.; Illarioshkin, S.; Slominsky, P.; et al. VCP expression decrease as a biomarker of preclinical and early clinical stages of Parkinson’s disease. Sci. Rep. 2020, 10, 827. [Google Scholar] [CrossRef]
- Shulskaya, M.V.; Alieva, A.K.; Vlasov, I.N.; Zyrin, V.V.; Fedotova, E.Y.; Abramycheva, N.Y.; Usenko, T.S.; Yakimovsky, A.F.; Emelyanov, A.K.; Pchelina, S.N.; et al. Whole-exome sequencing in searching for new variants associated with the development of Parkinson’s disease. Front. Aging Neurosci. 2018, 10, 136. [Google Scholar] [CrossRef]
- Rudenok, M.M.; Alieva, A.K.; Nikolaev, M.A.; Kolacheva, A.A.; Ugryumov, M.V.; Pchelina, S.N.; Slominsky, P.A.; Shadrina, M.I. Possible Involvement of Genes Related to Lysosomal Storage Disorders in the Pathogenesis of Parkinson’s Disease. Mol. Biol. (Mosk.) 2019, 53, 28–36. [Google Scholar] [CrossRef]
- Yang, X.; Gao, L.; Zhang, S. Comparative pan-cancer DNA methylation analysis reveals cancer common and specific patterns. Brief. Bioinform. 2017, 18, 761–773. [Google Scholar] [CrossRef]
- Trimarchi, M.P.; Yan, P.; Groden, J.; Bundschuh, R.; Goodfellow, P.J. Identification of endometrial cancer methylation features using combined methylation analysis methods. PLoS ONE 2017, 12, e0173242. [Google Scholar] [CrossRef]
- Champagne, D.L.; Bagot, R.C.; van Hasselt, F.; Ramakers, G.; Meaney, M.J.; de Kloet, E.R.; Joëls, M.; Krugers, H. Maternal care and hippocampal plasticity: Evidence for experience-dependent structural plasticity, altered synaptic functioning, and differential responsiveness to glucocorticoids and stress. J. Neurosci. 2008, 28, 6037–6045. [Google Scholar] [CrossRef] [Green Version]
- Tyrka, A.R.; Price, L.H.; Marsit, C.; Walters, O.C.; Carpenter, L.L. Childhood adversity and epigenetic modulation of the leukocyte glucocorticoid receptor: Preliminary findings in healthy adults. PLoS ONE 2012, 7, e30148. [Google Scholar] [CrossRef] [Green Version]
- Manneville, S.E. Cytoplasmic Intermediate Filaments in Cell Biology. Annu. Rev. Cell Dev. Biol. 2018, 34, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Pockley, A.G.; Henderson, B.; Multhoff, G. Extracellular cell stress proteins as biomarkers of human disease. Biochem. Soc. Trans. 2014, 42, 1744–1751. [Google Scholar] [CrossRef] [PubMed]
- Waltenberger, J. Stress testing at the cellular and molecular level to unravel cellular dysfunction and growth factor signal transduction defects: What Molecular Cell Biology can learn from Cardiology. Thromb. Haemost. 2007, 98, 975–979. [Google Scholar] [CrossRef] [PubMed]
- Gundamaraju, R.; Vemuri, R.; Chong, W.C.; Geraghty, D.P.; Eri, R. Cell Stress Signaling Cascades Regulating Cell Fate. Curr. Pharm. Des. 2018, 24, 3176–3183. [Google Scholar] [CrossRef]
- Wojsiat, J.; Prandelli, C.; Laskowska-Kaszub, K.; Requero, A.M.; Wojda, U. Oxidative Stress and Aberrant Cell Cycle in Alzheimer’s Disease Lymphocytes: Diagnostic Prospects. J. Alzheimers Dis. 2015, 46, 329–350. [Google Scholar] [CrossRef] [PubMed]
- Dada, G.O.L. Ferroptosis: Role of lipid peroxidation, iron and ferritinophagy. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 1893–1900. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Liu, C.; Zhao, Y.; Gao, G. Mitochondria regulation in ferroptosis. Eur. J. Cell Biol. 2020, 99, 151058. [Google Scholar] [CrossRef]
- Galluzzi, L.; Pedro, J.M.B.S.; Kroemer, G. Ferroptosis in p53-dependent oncosuppression and organismal homeostasis. Cell Death Differ. 2015, 22, 1237–1238. [Google Scholar] [CrossRef]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53—Mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Xiang, D.M.; Sun, W.; Zhou, T.; Zhang, C.; Cheng, Z.; Li, S.C.; Jiang, W.; Wang, R.; Fu, G.; Cui, X.; et al. Oncofetal HLF transactivates c-Jun to promote hepatocellular carcinoma development and sorafenib resistance. Gut 2019, 68, 1858–1871. [Google Scholar] [CrossRef]
- Schwartz, L.M.; Milligan, C.E. Cold thoughts of death: The role of ICE proteases in neuronal cell death. Trends Neurosci. 1996, 19, 555–562. [Google Scholar] [CrossRef]
- Saghiri, M.A.; Asatourian, A.; Gurel, Z.; Sorenson, C.M.; Sheibani, N. Bcl-2 expression is essential for development and normal physiological properties of tooth hard tissue and saliva production. Exp. Cell Res. 2017, 358, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Parsi, S.; Soltani, B.M.; Hosseini, E.; Tousi, S.E.; Mowla, S.J. Experimental verification of a predicted intronic microRNA in human NGFR gene with a potential pro-apoptotic function. PLoS ONE 2012, 7, e35561. [Google Scholar] [CrossRef] [PubMed]
- Fricker, M.; Tolkovsky, A.M.; Borutaite, V.; Coleman, M.; Brown, G.C. Neuronal Cell Death. Physiol. Rev. 2018, 98, 813–880. [Google Scholar] [CrossRef]
- Shimizu, S.; Eguchi, Y.; Kamiike, W.; Waguri, S.; Uchiyama, Y.; Matsuda, H.; Tsujimoto, Y. Retardation of chemical hypoxia-induced necrotic cell death by Bcl-2 and ICE inhibitors: Possible involvement of common mediators in apoptotic and necrotic signal transductions. Oncogene 1996, 12, 2045–2050. [Google Scholar]
- Lavoie, C.; Paiement, J. Topology of molecular machines of the endoplasmic reticulum: A compilation of proteomics and cytological data. Histochem. Cell Biol. 2008, 129, 117–128. [Google Scholar] [CrossRef] [Green Version]
- Song, S.; Tan, J.; Miao, Y.; Zhang, Q. Crosstalk of ER stress-mediated autophagy and ER-phagy: Involvement of UPR and the core autophagy machinery. J. Cell Physiol. 2018, 233, 3867–3874. [Google Scholar] [CrossRef]
- Wang, H.F.; Wang, Z.Q.; Ding, Y.; Piao, M.H.; Feng, C.S.; Chi, G.F.; Luo, Y.N.; Ge, P.F. Endoplasmic reticulum stress regulates oxygen-glucose deprivation-induced parthanatos in human SH-SY5Y cells via improvement of intracellular ROS. CNS Neurosci. Ther. 2018, 24, 29–38. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Guo, X.; Ge, Q.; Zhao, Y.; Mu, H.; Zhang, J. ER Stress Activates the NLRP3 Inflammasome: A Novel Mechanism of Atherosclerosis. Oxid. Med. Cell Longev. 2019, 2019, 3462530. [Google Scholar] [CrossRef]
- Petkova, A.T.; Ishii, Y.; Balbach, J.J.; Antzutkin, O.N.; Leapman, R.D.; Delaglio, F.; Tycko, R.A. A structural model for Alzheimer’s beta-amyloid fibrils based on experimental constraints from solid state NMR. Proc. Natl. Acad. Sci. USA 2002, 99, 16742–16747. [Google Scholar] [CrossRef] [Green Version]
- Mota, S.I.; Costa, R.O.; Ferreira, I.L.; Santana, I.; Caldeira, G.L.; Padovano, C.; Fonseca, A.C.; Baldeiras, I.; Cunha, C.; Letra, L.; et al. Oxidative stress involving changes in Nrf2 and ER stress in early stages of Alzheimer’s disease. Biochim. Biophys. Acta 2015, 1852, 1428–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saliba, R.S.; Munro, P.M.; Luthert, P.J.; Cheetham, M.E. The cellular fate of mutant rhodopsin: Quality control, degradation and aggresome formation. J. Cell Sci. 2002, 115, 2907–2918. [Google Scholar] [PubMed]
- Sobenin, I.A.; Sazonova, M.A.; Postnov, A.Y.; Bobryshev, Y.V.; Orekhov, A.N. Changes of mitochondria in atherosclerosis: Possible determinant in the pathogenesis of the disease. Atherosclerosis 2013, 227, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Xiong, S.; Xia, X. Retinitis pigmentosa associated rhodopsin mutant T17M induces endoplasmic reticulum (ER) stress and sensitizes cells to ER stress-induced cell death. Mol. Med. Rep. 2014, 9, 1737–17421. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.J.; Chung, J.; Ryoo, H.D. CDK5 and MEKK1 mediate pro-apoptotic signalling following endoplasmic reticulum stress in an autosomal dominant retinitis pigmentosa model. Nat. Cell. Biol. 2012, 14, 409–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sazonova, M.A.; Sinyov, V.V.; Ryzhkova, A.I.; Sazonova, M.D.; Zhelankin, A.V.; Khasanova, Z.B.; Postnov, A.Y.; Orekhov, A.N.; Sobenin, I.A. Detection of homoplasmic MTDNA mutations by NGS method in patients with carotid atherosclerosis. Atherosclerosis 2018, 275, e128. [Google Scholar] [CrossRef]
- Bals, R. Alpha 1-antitrypsin deficiency. Best Pract. Res. Clin. Gastroenterol. 2010, 24, 629–633. [Google Scholar] [CrossRef] [PubMed]
- Greene, C.M.; McElvaney, N.G. Z α-1 antitrypsin deficiency and the endoplasmic reticulum stress response. World J. Gastrointest. Pharm. Ther. 2010, 1, 94–101. [Google Scholar] [CrossRef] [Green Version]
- Teckman, J.H.; Blomenkamp, K.S. Pathophysiology of Alpha-1 Antitrypsin Deficiency Liver Disease. Methods Mol. Biol. 2017, 1639, 1–8. [Google Scholar] [CrossRef]
- Sobenin, I.A.; Orekhova, V.A.; Grechko, A.V.; Orekhov, A.N. Is insulin pro-atherogenic at the cellular level? Vessel Plus 2017, 2017, 174–181. [Google Scholar] [CrossRef] [Green Version]
- Schleicher, S.M.; Moretti, L.; Varki, V.; Lu, B. Progress in the unraveling of the endoplasmic reticulum stress/autophagy pathway and cancer: Implications for future therapeutic approaches. Drug Res. Update 2010, 13, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Sopha, P.; Ren, H.Y.; Grove, D.E.; Cyr, D.M. Endoplasmic reticulum stress-induced degradation of DNAJB12 stimulates BOK accumulation and primes cancer cells for apoptosis. J. Biol. Chem. 2017, 292, 11792–11803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.; Liang, Y.; Ma, X. Alpha-mangostin induces endoplasmic reticulum stress and autophagy which count against fatty acid synthase inhibition mediated apoptosis in human breast cancer cells. Cancer Cell Int. 2019, 19, 151. [Google Scholar] [CrossRef] [PubMed]
- Sinyov, V.V.; Ryzhkova, A.I.; Sazonova, M.D.; Doroschuk, A.D.; Karagodin, V.P.; Sobenin, I.A.; Orekhov, A.N.; Sazonova, M.A. The influence of the mutational burden of mitochondrial genome on cellular respiration. Atherosclerosis 2020, 315. [Google Scholar] [CrossRef]
- Anfinsen, C.B. Principles that govern the folding of protein chains. Science 1973, 181, 223–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdullahi, A.; Stanojcic, M.; Parousis, A.; Patsouris, D.; Jeschke, M.G. Modeling Acute ER Stress In Vivo and In Vitro. Shock 2017, 47, 506–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhelankin, A.V.; Sazonova, M.A. Association of the mutations in the human mitochondrial genome with chronic non-inflammatory diseases: Type 2 diabetes, hypertension and different types of cardiomyopathy. Patol. Fiziol. Eksp. Ter. 2012, 3, 123–128. [Google Scholar]
- Enyedi, B.; Várnai, P.; Geiszt, M. Redox state of the endoplasmic reticulum is controlled by Ero1L-alpha and intraluminal calcium. Antioxid. Redox Signal. 2010, 13, 721–729. [Google Scholar] [CrossRef]
- Gross, E.; Sevier, C.S.; Heldman, N.; Vitu, E.; Bentzur, M.; Kaiser, C.A.; Thorpe, C.; Fass, D. Generating disulfides enzymatically: Reaction products and electron acceptors of the endoplasmic reticulum thiol oxidase Ero1p. Proc. Natl. Acad. Sci. USA 2006, 103, 299–304. [Google Scholar] [CrossRef] [Green Version]
- Haynes, C.M.; Titus, E.A.; Cooper, A.A. Degradation of misfolded proteins prevents ER-derived oxidative stress and cell death. Mol. Cell 2004, 15, 767–776. [Google Scholar] [CrossRef]
- Sobenin, I.A.; Sazonova, M.A.; Ivanova, M.M.; Zhelankin, A.V.; Myasoedova, V.A.; Postnov, A.Y.; Nurbaev, S.D.; Bobryshev, Y.V.; Orekhov, A.N. Mutation C3256T of Mitochondrial Genome in White Blood Cells: Novel Genetic Marker of Atherosclerosis and Coronary Heart Disease. PLoS ONE 2012, 7, e46573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheper, W.; Hoozemans, J.J. The unfolded protein response in neurodegenerative diseases: A neuropathological perspective. Acta Neuropathol. 2015, 130, 315–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakae, N.; Liu, C.C.; Shinohara, M.; Daiello, J.F.; Ma, L.; Yamazaki, Y.; Tachibana, M.; Younkin, L.; Kurti, A.; Carrasquillo, M.M.; et al. ABCA7 Deficiency Accelerates Amyloid-β Generation and Alzheimer’s Neuronal Pathology. J. Neurosci. 2016, 36, 3848–3859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bharathi, M.D.; Thenmozhi, A.J.; Manivasagam, T.; Rather, M.A.; Babu, C.S.; Essa, M.M.; Guillemin, G.J. Amelioration of Aluminum Maltolate-Induced Inflammation and Endoplasmic Reticulum Stress-Mediated Apoptosis by Tannoid Principles of Emblica officinalis in Neuronal Cellular Model. Neurotox. Res. 2019, 35, 318–330. [Google Scholar] [CrossRef]
- Sobenin, I.A.; Chistiakov, D.A.; Sazonova, M.A.; Ivanova, M.M.; Bobryshev, Y.V.; Orekhov, A.N.; Postnov, A.Y. Association of the level of heteroplasmy of the 15059G>A mutation in the MT-CYB mitochondrial gene with essential hypertension. World J. Cardiol. 2013, 5, 132–140. [Google Scholar] [CrossRef]
- Wnag, H.; Takahashi, R. Expanding insights on the involvement of endoplasmic reticulum stress in Parkinson’s disease. Antioxid. Redox Signal. 2007, 9, 553–561. [Google Scholar] [CrossRef]
- LaVoie, M.J.; Ostaszewski, B.L.; Weihofen, A.; Schlossmacher, M.G.; Selkoe, D.J. Dopamine Covalently Modifies and Functionally Inactivates Parkin. Nat. Med. 2005, 11, 1214–1221. [Google Scholar] [CrossRef]
- Tsai, Y.; Weissman, A. The Unfolded Protein Response, Degradation from the Endoplasmic Reticulum, and Cancer. Genes Cancer 2010, 1, 764–778. [Google Scholar] [CrossRef]
- Eizirik, D.; Cardozo, A.; Cnop, M. The Role for Endoplasmic Reticulum Stress in Diabetes Mellitus. Endocr. Rev. 2008, 29, 42–61. [Google Scholar] [CrossRef] [Green Version]
- Marciniak, S.; Ron, D. Endoplasmic Reticulum Stress Signaling in Disease. Physiol. Rev. 2006, 86, 1133–1149. [Google Scholar] [CrossRef]
- Back, S.H.; Kang, S.W.; Han, J.; Chung, H.T. Endoplasmic reticulum stress in the β-cell pathogenesis of type 2 diabetes. Exp. Diabet. Res. 2012, 2012, 618396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobenin, I.A.; Mitrofanov, K.Y.; Zhelankin, A.V.; Sazonova, M.A.; Postnov, A.Y.; Revin, V.V.; Bobryshev, Y.V.; Orekhov, A.N. Quantitative assessment of heteroplasmy of mitochondrial genome: Perspectives in diagnostics and methodological pitfalls. BioMed Res. Int. 2014, 2014, 292017. [Google Scholar] [CrossRef] [PubMed]
- van Raalte, D.H.; Diamant, M. Glucolipotoxicity and beta cells in type 2 diabetes mellitus: Target for durable therapy? Diabet. Res. Clin. Pract. 2011, 93 (Suppl. 1), S37–S46. [Google Scholar] [CrossRef]
- Karunakaran, U.; Kim, H.J.; Kim, J.Y.; Lee, I.K. Guards and culprits in the endoplasmic reticulum: Glucolipotoxicity and β-cell failure in type II diabetes. Exp. Diabet. Res. 2012, 2012, 639762. [Google Scholar] [CrossRef] [Green Version]
- Leem, J.; Koh, E.H. Interaction between mitochondria and the endoplasmic reticulum: Implications for the pathogenesis of type 2 diabetes mellitus. Exp. Diabet. Res. 2012, 2012, 242984. [Google Scholar] [CrossRef] [Green Version]
- Chávez, A.C.; Argueta, S.E.; Reyes, G.G.; Pedroza, J.I.L. Pathophysiological implications between chronic inflammation and the development of diabetes and obesity. Cir. Cir. 2011, 79, 209–216. [Google Scholar]
- Karuppagounder, S.S.; Alim, I.; Khim, S.J.; Bourassa, M.W.; Sleiman, S.F.; John, R.; Thinnes, C.C.; Yeh, T.L.; Demetriades, M.; Neitemeier, S.; et al. Therapeutic targeting of oxygen-sensing prolyl hydroxylases abrogates ATF4-dependent neuronal death and improves outcomes after brain hemorrhage in several rodent models. Sci. Transl. Med. 2016, 8, 328ra29. [Google Scholar] [CrossRef] [Green Version]
- Lange, P.S.; Chavez, J.C.; Pinto, J.T.; Coppola, G.; Sun, C.W.; Townes, T.M.; Geschwind, D.H.; Ratan, R.R. ATF4 is an oxidative stress-inducible, prodeath transcription factor in neurons in vitro and in vivo. J. Exp. Med. 2008, 205, 1227–1242. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Naguro, I.; Ichijo, H.; Watanabe, K. Mitogen-activated protein kinases as key players in osmotic stress signaling. Biochim. Biophys. Acta 2016, 1860, 2037–2052. [Google Scholar] [CrossRef]
- Zhang, D.; Chen, X.; Wang, Q.; Wu, S.; Zheng, Y.; Liu, X. Role of the MAPKs/TGF-β1/TRAF6 signaling pathway in postoperative atrial fibrillation. PLoS ONE 2017, 12, e0173759. [Google Scholar] [CrossRef]
- Andersen, O.S. Perspectives on: The response to osmotic challenges. J. Gen. Physiol. 2015, 145, 371–372. [Google Scholar] [CrossRef] [PubMed]
- Sinyov, V.V.; Sazonova, M.A.; Ryzhkova, A.I.; Postnov, A.Y.; Orekhov, A.N.; Sobenin, I.A. Heteroplasmy level of five mitochondrial mutations in different types of leucocytes in human blood. Atherosclerosis 2014, 23, e220. [Google Scholar] [CrossRef]
- Finan, J.D.; Guilak, F. The effects of osmotic stress on the structure and function of the cell nucleus. J. Cell Biochem. 2010, 109, 460–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jadiya, P.; Mir, S.S.; Nazir, A. Osmotic stress induced toxicity exacerbates Parkinson’s associated effects via dysregulation of autophagy in transgenic C. elegans model. Cell Signal. 2018, 45, 71–80. [Google Scholar] [CrossRef]
- Postnov, A.Y.; Sazonova, M.A.; Budnikov, Y.Y.; Khazanova, Z.B.; Sobenin, I.A.; Orekhov, A.N. Association of somatic mitochondrial mutations with atherosclerosis. Atheroscler. Suppl. 2007, 8, 46. [Google Scholar] [CrossRef]
- Velichko, A.K.; Petrova, N.V.; Luzhin, A.V.; Strelkova, O.S.; Ovsyannikova, N.; Kireev, I.I.; Petrova, N.V.; Razin, S.V.; Kantidze, O.L. Hypoosmotic stress induces R loop formation in nucleoli and ATR/ATM-dependent silencing of nucleolar transcription. Nucleic. Acids Res. 2019, 47, 6811–6825. [Google Scholar] [CrossRef] [Green Version]
- Kabeche, L.; Nguyen, H.D.; Buisson, R.; Zou, L. A mitosis-specific and R loop-driven ATR pathway promotes faithful chromosome segregation. Science 2018, 359, 108–114. [Google Scholar] [CrossRef] [Green Version]
- Subhi, N.A.; Ali, R.; Fatah, T.A.; Moseley, P.M.; Chan, S.Y.T.; Green, A.R.; Ellis, I.O.; Rakha, E.A.; Madhusudan, S. Targeting ataxia telangiectasia-mutated- and Rad3-related kinase (ATR) in PTEN-deficient breast cancers for personalized therapy. Breast. Cancer Res. Treat. 2018, 169, 277–286. [Google Scholar] [CrossRef] [Green Version]
- Ryzhkova, A.; Sazonova, M.; Sazonova, M.; Nikitina, N.; Galitsyna, E.; Melnichenko, A.; Demakova, N.; Shkurat, T.; Sobenin, I. Investigation of mutations m.3256C>T and m.12315G>A in women with asymptomatic atherosclerosis. Atherosclerosis 2018, 275, e254. [Google Scholar] [CrossRef]
- Kim, E.K.; Choi, E.J. Compromised MAPK signaling in human diseases: An update. Arch. Toxicol. 2015, 89, 867–882. [Google Scholar] [CrossRef]
- Kim, E.K.; Choi, E.J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta 2010, 1802, 396–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossiello, F.; Aguado, J.; Sepe, S.; Iannelli, F.; Nguyen, Q.; Pitchiaya, S.; Carninci, P.; d’Adda di Fagagna, F. DNA damage response inhibition at dysfunctional telomeres by modulation of telomeric DNA damage response RNAs. Nat. Commun. 2017, 8, 13980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regulski, M.J. Cellular Senescence: What, Why, and How. Wounds 2017, 29, 168–174. [Google Scholar] [PubMed]
- Sazonova, M.A.; Sinyov, V.V.; Barinova, V.A.; Ryzhkova, A.I.; Bobryshev, Y.V.; Orekhov, A.N.; Sobenin, I.A. Association of mitochondrial mutations with the age of patients having atherosclerotic lesions. Exp. Mol. Pathol. 2015, 99, 717–719. [Google Scholar] [CrossRef]
- Brand, T. Length doesn’t matter—Telomere damage triggers cellular senescence in the ageing heart. EMBO J. 2019, 38, e101571. [Google Scholar] [CrossRef]
- Stein, J.Y.; Levin, Y.; Uziel, O.; Abumock, H.; Solomon, Z. Traumatic stress and cellular senescence: The role of war-captivity and homecoming stressors in later life telomere length. J. Affect. Disord. 2018, 238, 129–135. [Google Scholar] [CrossRef]
- Doroshchuk, N.A.; Tihaze, A.K.; Lankin, V.Z.; Konovalova, G.G.; Mednikova, T.K.; Postnov, A.Y.; Kuharchuk, V.V. The effect of oxidative stress on the length of telomeric repeats in chromosomes of blood leukocytes in persons with different risk of cardiovascular death and coronary heart disease. Kardiol. Vestn. 2017, 12, 32–37. (In Russian) [Google Scholar]
- Doroshchuk, N.A.; Lankin, V.Z.; Tihaze, A.K.; Odinokova, O.A.; Konovalova, G.G.; Postnov, A.Y. Oxidative stress and shortening of telomeres in blood leukocytes of patients with newly diagnosed type 2 diabetes mellitus. Kardiol. Vestn. 2016, 11, 56–60. (In Russian) [Google Scholar]
- Doroshchuk, N.A.; Doroshchuk, A.D.; Rodnenkov, O.V.; Osyaeva, M.K.; Hasanova, Z.B.; Hesuani, Y.D.; Postnov, A.Y.; Chazova, I.E. The change of length of telomeres in chromosomes under the influence of environmental conditions, imitating the heat in summer 2010, in Moscow. Kardiol. Vestn. 2013, 8, 32–35. (In Russian) [Google Scholar]
- Doroshchuk, N.A.; Postnov, A.Y.; Doroshchuk, A.D.; Khasanova, Z.B.; Konovalova, N.V.; Hesuani, Y.D.; Osyaeva, M.K.; Rodnenkov, O.V.; Chazova, I.E. Direct deleterious effect on human DNA of adverse environmental and climatic factors. Ther. Arch. 2014, 86, 72–77. (In Russian) [Google Scholar]
- Sager, R. Senescence as a mode of tumor suppression. Env. Health Persp. 1991, 93, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Baskara, I.; Kerbat, S.; Dagouassat, M.; Nguyen, H.Q.; Delost, M.G.; Surenud, M.; Baillou, C.; Lemoine, F.M.; Morin, D.; Boczkowski, J.; et al. Cigarette smoking induces human CCR6 (+) Th17 lymphocytes senescence and VEGF-A secretion. Sci. Rep. 2020, 10, 6488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.P. Aging mechanisms and intervention targets. Clin. Exp. Pharmacol. Physiol. 2017, 44, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Armstrong, J.L.; Tchkonia, T.; Kirkland, J.L. Cellular senescence and the senescence secretory phenotype and age-related chronic diseases. Curr. Opin. Clin. Nutr. Metab. Care 2014, 17, 324–328. [Google Scholar] [CrossRef]
- Wang, L.; Xie, R.; Fan, Z.; Yang, J.; Liang, W.; Wu, Q.; Wu, M.X.; Wang, Z.; Lu, Y. The contribution of oxidative stress to platelet senescence during storage. Transfusion 2019, 59, 2389–2402. [Google Scholar] [CrossRef]
- d’Adda di Fagagna, F. Cellular senescence and cellulal longevity: Nearly 50 years on and still working on it. Exp. Cell Res. 2008, 314, 1907–1908. [Google Scholar] [CrossRef]
- Lee, B.Y.; Han, J.A.; Im, J.S.; Morrone, A.; Johung, K.; Goodwin, E.C.; Kleijer, W.J.; di Maio, D.; Hwang, E.S. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell 2006, 5, 187–195. [Google Scholar] [CrossRef]
- di Micco, R.; Krizhanovsky, V.; Baker, D.; d’Adda di Fagagna, F. Cellular senescence in ageing from mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 2020. [Google Scholar] [CrossRef]
- Campisi, J.; Andersen, J.K.; Kapahi, P.; Melov, S. Cellular senescence: A link between cancer and age-related degenerative disease? Semin. Cancer Biol. 2011, 21, 354–359. [Google Scholar] [CrossRef] [Green Version]
- Coppé, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [Green Version]
- Tchkonia, T.; Morbeck, D.E.; von Zglinicki, T.; van Deursen, J.; Lustgarten, J.; Scrable, H.; Khosla, S.; Jensen, M.D.; Kirkland, J.L. Fat tissue, aging, and cellular senescence. Aging Cell 2010, 9, 667–684. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Exosomal vesicles enhance immunosuppression in chronic inflammation: Impact in cellular senescence and the aging process. Cell Signal. 2020, 75, 109771. [Google Scholar] [CrossRef] [PubMed]
- Chinta, S.J.; Woods, G.; Demaria, M.; Rene, A.; Zou, Y.; McQuade, A.; Rajagopalan, S.; Limbad, C.; Madden, D.T.; Campisi, J.; et al. Cellular senescence is induced by the environmental neurotoxin paraquat and contributes to neuropathology linked to Parkinson’s disease. Cell Rep. 2018, 22, 930–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, B.; Choi, Y.; Kim, H.S.; Im, H.I. Methyl-VpG Binding protein 2 in Alzheimer dementia. Int. Neurourol. J. 2019, 23 (Suppl. 2), S72–S81. [Google Scholar] [CrossRef]
- Prattichizzo, F.; Giuliani, A.; Recchioni, R.; Bonafe, M.; Marcheselli, F.; De Carolis, S.; Campanati, A.; Giuliodori, K.; Rippo, M.R.; Bruge, F.; et al. Anti TNF-alpha treatment modulates SASP and SASP-related microRNAs in endothelial cells and in circulating angiogenic cells. Oncotarget 2016, 7, 11945–11958. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Zhang, R.X.; Xie, X.J.; Gu, H.F. Autophagy in chronic stress induced atherosclerosis. Clin. Chim. Acta 2020, 503, 70–75. [Google Scholar] [CrossRef]
- Heidt, T.; Sager, H.B.; Courties, G.; Dutta, P.; Iwamoto, Y.; Zaltsman, A.; von ZurMuhlen, C.; Bode, C.; Fricchione, G.L.; Denninger, J.; et al. Chronic variable stress activates hematopoietic stem cells. Nat. Med. 2014, 20, 754–758. [Google Scholar] [CrossRef] [Green Version]
- Arnold, S.V.; Smolderen, K.G.; Buchanan, D.M.; Spertus, J.A. Perceived stress in myocardial infarction: Long-term mortality and health status outcomes. J. Am. Coll. Cardiol. 2012, 60, 1756–1763. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Ong, L.K.; Johnson, S.; Nilsson, M.; Walker, F.R. Chronic stress induced disruption of the peri-infarct neurovascular unit following experimentally induced photothrombotic stroke. J. Cereb. Blood Flow Metab. 2017, 37, 3709–3724. [Google Scholar] [CrossRef] [Green Version]
- Scally, C.; Abbas, H.; Ahearn, T.; Srinivasan, J.; Mezincescu, A.; Rudd, A.; Spath, N.; Finn, A.Y.; Yuecel, R.; Oldroyd, K.; et al. Myocardial and systemic inflammation in acute stress-induced (Takotsubo) cardiomyopathy. Circulation 2019, 139, 1581–1592. [Google Scholar] [CrossRef]
- Civelek, M.; Manduchi, E.; Riley, R.J.; Stoeckert, C.J., Jr.; Davies, P.F. Chronic endoplasmic reticulum stress activates unfolded protein response in arterial endothelium in regions of susceptibility to atherosclerosis. Circ. Res. 2009, 105, 453–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanova, E.A.; Orekhov, A.N. The role of endoplasmic reticulum stress and unfolded protein response in atherosclerosis. Int. J. Mol. Sci. 2016, 17, 193. [Google Scholar] [CrossRef] [PubMed]
- Grechowa, I.; Horke, S.; Wallrath, A.; Vahl, C.F.; Dorweiler, B. Human neutrophil elastase induces endothelial cell apoptosis by activating the PERK-CHOP branch of the unfolded protein response. FASEB J. 2017, 31, 3868–3881. [Google Scholar] [CrossRef] [Green Version]
- Hamczyk, M.R.; Bellosta, R.V.; Quesada, V.; Gonzalo, P.; Vidak, S.; Nevado, R.M.; Manzano, M.J.A.; Misteli, T.; Otín, C.L.; Andrés, V. Progerin accelerates atherosclerosis by inducing endoplasmic reticulum stress in vascular smooth muscle cells. EMBO Mol. Med. 2019, 11, e9736. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhang, D.; Liu, X.; Li, X.; Liu, F.; Yu, Y.; Jia, S.; Zhou, Y.; Zhao, Y. Endoplasmic reticulum stress affects lipid metabolism in atherosclerosis via CHOP activation and over-expression of miR-33. Cell Physiol. Biochem. 2018, 48, 1995–2010. [Google Scholar] [CrossRef] [PubMed]
- di Pasquale, E.; Condorelli, G. Endoplasmic reticulum stress at the crossroads of progeria and atherosclerosis. EMBO Mol. Med. 2019, 11, e10360. [Google Scholar] [CrossRef] [PubMed]
- Abe, J.; Berk, B.C. Atheroprone flow activation of the sterol regulatory element binding protein 2 and nod-like receptor protein 3 inflammasome mediates focal atherosclerosis. Circulation 2013, 128, 579–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chistiakov, D.A.; Sobenin, I.A.; Orekhov, A.N.; Bobryshev, Y.V. Role of endoplasmatic reticulum stress in atherosclerosis and diabetic macrovascular complications. BioMed Res. Int. 2014, 2014, 610140. [Google Scholar] [CrossRef] [Green Version]
- Zheng, F.; Xing, S.; Gong, Z.; Mu, W.; Xing, Q. Silence of NLRP3 suppresses atherosclerosis and stabilizes plaques in apolipoprotein E-deficient mice. Mediat. Inflamm. 2014, 2014, 507208. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Syed, T.W.; Liu, R.; Yu, J. Role of endoplasmic reticulum stress, autophagy, and inflammation in cardiovascular disease. Front. Cardiovasc. Med. 2017, 4, 29. [Google Scholar] [CrossRef]
- Tabas, I. Macrophage apoptosis in atherosclerosis: Consequences on plaque progression and the role of endoplasmic reticulum stress. Antioxid. Redox. Signal. 2009, 11, 2333–2339. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, Y.; Ren, X.; Zhang, X.; Hu, D.; Gao, Y.; Xing, Y.; Shang, H. Oxidative Stress-Mediated Atherosclerosis: Mechanisms and Therapies. Front. Physiol. 2017, 8, 600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boudjeltia, K.Z.; Moguilevsky, N.; Legssyer, I.; Babar, S.; Guillaume, M.; Delree, P.; Vanhaeverbeek, M.; Brohee, D.; Ducobu, J.; Remacle, C. Oxidation of low density lipoproteins by myeloperoxidase at the surface of endothelial cells: An additional mechanism to subendothelium oxidation. Biochem. Biophys. Res. Commun. 2004, 325, 434–438. [Google Scholar] [CrossRef] [PubMed]
- Sazonova, M.; Budnikov, E.; Khasanova, Z.; Sobenin, I.; Postnov, A.; Orekhov, A. Studies of the human aortic intima by a direct quantitative assay of mutant alleles in the mitochondrial genome. Atherosclerosis 2009, 204, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Sazonova, M.A.; Ryzhkova, A.I.; Sinyov, V.V.; Galitsyna, E.V.; Orekhova, V.A.; Melnichenko, A.A.; Orekhov, A.N.; Ravani, A.L.; Sobenin, I.A. New markers of atherosclerosis: A threshold level of heteroplasmy in mtDNA mutations. Vessel Plus 2017, 1, 182–191. [Google Scholar] [CrossRef]
- Sazonova, M.A.; Postnov, A.I.; Orekhov, A.N.; Sobenin, I.A. A new method of quantitative estimation of mutant allele in mitochondrial genome. Patol. Fiziol. Eksp. Ter. 2011, 4, 81–84. [Google Scholar]
- Sazonova, M.; Andrianova, I.; Khasanova, Z.; Sobenin, I.; Postnov, A. Quantitative mitochondrial genome mutation investigation and possible role of the somatic mutations in development of atherosclerotic lesion of human aorta. Atheroscler. Suppl. 2008, 9, 113. [Google Scholar] [CrossRef]
- Sazonova, M.A.; Budnikov, Y.Y.; Khazanova, Z.B.; Postnov, A.Y.; Sobenin, I.A.; Orekhov, A.N. Direct quantitative assessment of mutant allele in mitochondrial genome in atherosclerotic lesion of human aorta. Atheroscler. Suppl. 2007, 8, 45–46. [Google Scholar] [CrossRef]
- Sazonova, M.A.; Sinyov, V.V.; Ryzhkova, A.I.; Sazonova, M.D.; Khasanova, Z.B.; Shkurat, T.P.; Karagodin, V.P.; Orekhov, A.N.; Sobenin, I.A. Creation of Cybrid Cultures Containing mtDNA Mutations m.12315G>A and m.1555G>A, Associated with Atherosclerosis. Biomolecules 2019, 9, 499. [Google Scholar] [CrossRef] [Green Version]
- Sazonova, M.A.; Ryzhkova, A.I.; Sinyov, V.V.; Sazonova, M.D.; Khasanova, Z.B.; Nikitina, N.A.; Karagodin, V.P.; Orekhov, A.N.; Sobenin, I.A. Creation of Cultures Containing Mutations Linked with Cardiovascular Diseases using Transfection and Genome Editing. Curr. Pharm. Des. 2019, 25, 693–699. [Google Scholar] [CrossRef]
- Sazonova, M.A.; Sinyov, V.V.; Ryzhkova, A.I.; Galitsyna, E.V.; Melnichenko, A.A.; Postnov, A.Y.; Orekhov, A.N.; Sobenin, I.A. Cybrid Models of Pathological Cell Processes in Different Diseases. Oxid. Med. Cell Longev. 2018, 2018, 4647214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinyov, V.V.; Sazonova, M.A.; Ryzhkova, A.I.; Galitsyna, E.V.; Zhelankin, A.V.; Mitrofanov, K.Y.; Bobryshev, Y.V.; Orekhov, A.N.; Sobenin, I.A. Creation of cybrid cultures containing mitochondrial genome mutation m.12315G>A, associated with atherosclerosis. Atherosclerosis 2017, 263, e201. [Google Scholar] [CrossRef]
- Sinyov, V.; Sazonova, M.A.; Ryzhkova, A.I.; Doroshchuk, A.D.; Kuzmin, A.V.; Sazonova, M.; Khasanova, Z.B.; Orekhov, A.N.; Sobenin, I.A. Cellular respiration in cytoplasmic hybrids with different heteroplasmy levels of mitochondrial genome mutations. Atherosclerosis 2018, 275, e254. [Google Scholar] [CrossRef]
- Sazonova, M.A.; Ryzhkova, A.I.; Sinyov, V.V.; Galitsyna, E.V.; Melnichenko, A.A.; Demakova, N.A.; Sobenin, I.A.; Shkurat, T.P.; Orekhov, A.N. Mitochondrial Genome Mutations Associated with Myocardial Infarction. Dis. Mark. 2018, 2018, 9749457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryzhkova, A.I.; Sinyov, V.V.; Sazonova, M.D.; Khasanova, Z.B.; Doroschuk, N.A.; Nikitina, N.A.; Sobenin, I.A.; Orekhov, A.N.; Sazonova, M.A. Heteroplasmy level analysis of mtDNA mutations in intima-medial layer of Novosibirsk region patients. Atherosclerosis 2019, 287, e163. [Google Scholar] [CrossRef]
- Sazonova, M.A.; Sinyov, V.V.; Ryzhkova, A.I.; Sazonova, M.D.; Khasanova, Z.B.; Sobenin, I.A. MtDNA mutations linked with left ventricular hypertrophy. Vessel Plus 2019, 3, 5. [Google Scholar] [CrossRef]
- Sazonova, M.A.; Ryzhkova, A.I.; Sinyov, V.V.; Sazonova, M.D.; Nikitina, N.A.; Shkurat, T.P.; Sobenin, I.A.; Orekhov, A.N. Mitochondrial mutations associated with cardiac angina. Vessel Plus 2019, 3, 8. [Google Scholar] [CrossRef]
- Sazonova, M.A.; Sinyov, V.V.; Ryzhkova, A.I.; Barinova, V.A.; Zhelankin, A.V.; Mitrofanov, K.Y.; Postnov, A.Y.; Sobenin, I.A.; Orekhov, A.N. Atherosclerosis and ageing: Common mutations of mitochondrial genome. Atherosclerosis 2015, 241, e228. [Google Scholar] [CrossRef]



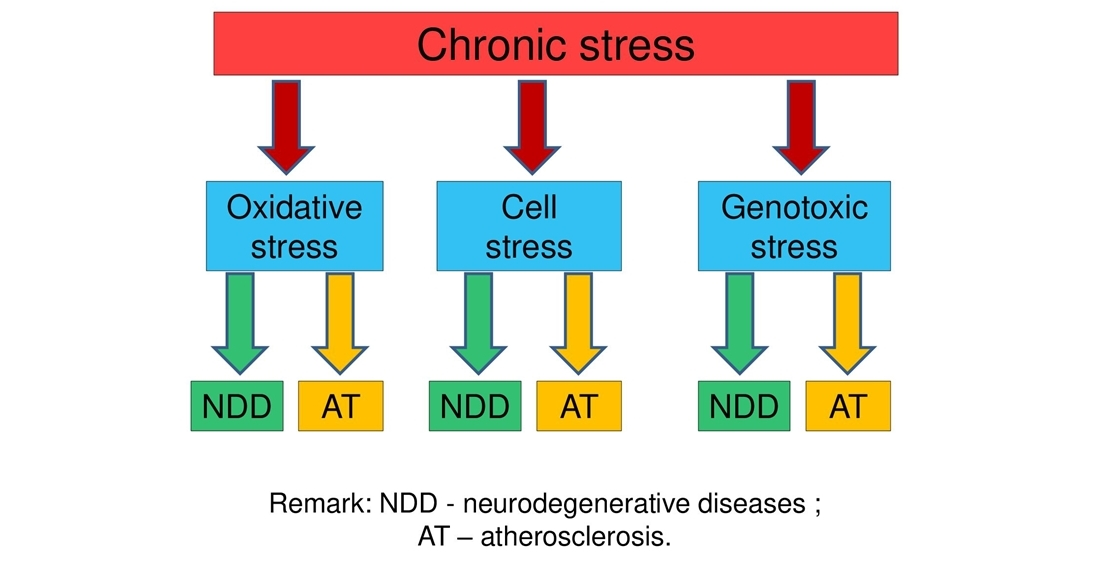{kind=link}
{kind=link}
{kind=link}
| Number | Reactive Oxygen Species | Chemical Formula |
|---|---|---|
| 1 | Superoxide anion | O2− |
| 2 | Hydroxyl radical | OH- |
| 3 | Nitrogen oxide anion | NO- |
| 4 | Hydrogen peroxide | H2O2 |
| 5 | Peroxynitrite anion | ONOO− |
| 6 | Hydroperoxide radical | HO2- |
| 7 | Singlet oxygen | O12 |
| 8 | Hypochlorite anion | OCl− |
| 9 | Hypobromite anion | OBr− |
| 10 | Hypoiodic anion | OI− |
| 11 | Alkyldioxyl radical | ROO- |
| 12 | Alkoxyl radical | RO- |
| 13 | Organic radical | R- |
| Genes | Mutations | Threshold Value of Heteroplasmy Level (%) |
|---|---|---|
| MT-TL2 | m.12315G>A | 7.6 |
| MT-RNR1 | m.652delG | 20.4 |
| MT-ND1 | m.3336T>C | 6.6 |
| MT-TL1 | m.3256C>T | 15.4 |
| MT-CYTB | m.14846G>A * | 17.6 |
| MT-RNR1 | m.652insG | 20.1 |
| MT-RNR1 | m.1555A>G * | 17.5 |
| MT-ND6 | m.14459G>A | 4.5 |
| MT-ND2 | m.5178C>A | 6.5 |
| MT-ND5 | m.13513G>A * | 32.4 |
| MT-CYTB | m.15059G>A | 24.6 |
| Genes | Mutations | Threshold Value of Heteroplasmy Level (%) |
|---|---|---|
| MT-TL2 | m.12315G>A | 10.4 |
| MT-RNR1 | m.652delG | 21.6 |
| MT-ND1 | m.3336T>C | 7.5 |
| MT-TL1 | m.3256C>T | 16.4 |
| MT-CYTB | m.14846G>A * | 17.4 |
| MT-RNR1 | m.652insG | 20.2 |
| MT-RNR1 | m.1555A>G * | 19.6 |
| MT-ND6 | m.14459G>A | 4.6 |
| MT-ND2 | m.5178C>A | 6.4 |
| MT-ND5 | m.13513G>A * | 33.6 |
| MT-CYTB | m.15059G>A | 26.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sazonova, M.A.; Sinyov, V.V.; Ryzhkova, A.I.; Sazonova, M.D.; Kirichenko, T.V.; Khotina, V.A.; Khasanova, Z.B.; Doroschuk, N.A.; Karagodin, V.P.; Orekhov, A.N.; et al. Some Molecular and Cellular Stress Mechanisms Associated with Neurodegenerative Diseases and Atherosclerosis. Int. J. Mol. Sci. 2021, 22, 699. https://doi.org/10.3390/ijms22020699
Sazonova MA, Sinyov VV, Ryzhkova AI, Sazonova MD, Kirichenko TV, Khotina VA, Khasanova ZB, Doroschuk NA, Karagodin VP, Orekhov AN, et al. Some Molecular and Cellular Stress Mechanisms Associated with Neurodegenerative Diseases and Atherosclerosis. International Journal of Molecular Sciences. 2021; 22(2):699. https://doi.org/10.3390/ijms22020699
Chicago/Turabian StyleSazonova, Margarita A., Vasily V. Sinyov, Anastasia I. Ryzhkova, Marina D. Sazonova, Tatiana V. Kirichenko, Victoria A. Khotina, Zukhra B. Khasanova, Natalya A. Doroschuk, Vasily P. Karagodin, Alexander N. Orekhov, and et al. 2021. "Some Molecular and Cellular Stress Mechanisms Associated with Neurodegenerative Diseases and Atherosclerosis" International Journal of Molecular Sciences 22, no. 2: 699. https://doi.org/10.3390/ijms22020699
APA StyleSazonova, M. A., Sinyov, V. V., Ryzhkova, A. I., Sazonova, M. D., Kirichenko, T. V., Khotina, V. A., Khasanova, Z. B., Doroschuk, N. A., Karagodin, V. P., Orekhov, A. N., & Sobenin, I. A. (2021). Some Molecular and Cellular Stress Mechanisms Associated with Neurodegenerative Diseases and Atherosclerosis. International Journal of Molecular Sciences, 22(2), 699. https://doi.org/10.3390/ijms22020699