An Integrative Transcriptomic Analysis of Systemic Juvenile Idiopathic Arthritis for Identifying Potential Genetic Markers and Drug Candidates


Abstract
:1. Introduction
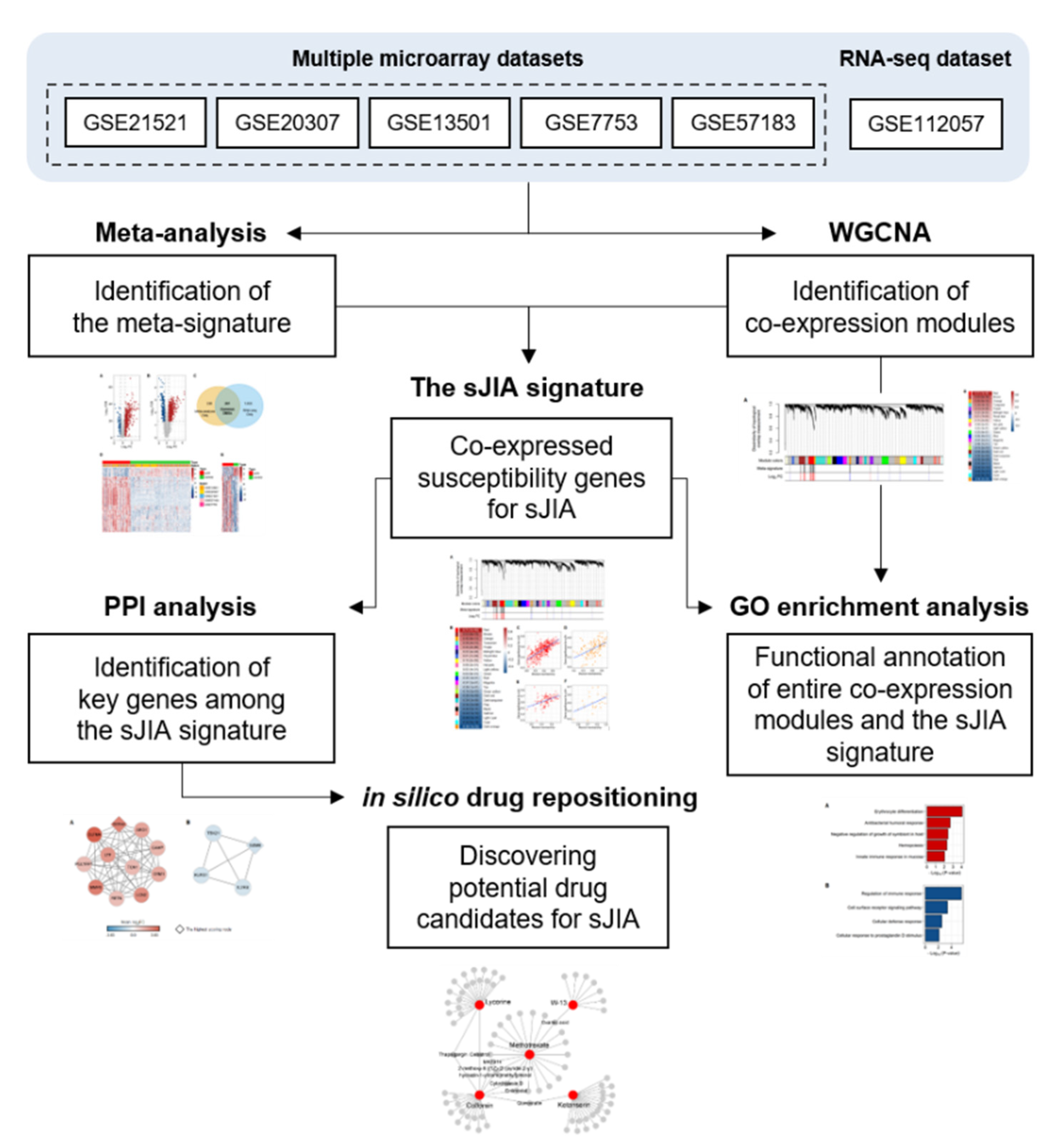
2. Results
2.1. Identification of a Meta-Signature in Human sJIA Datasets

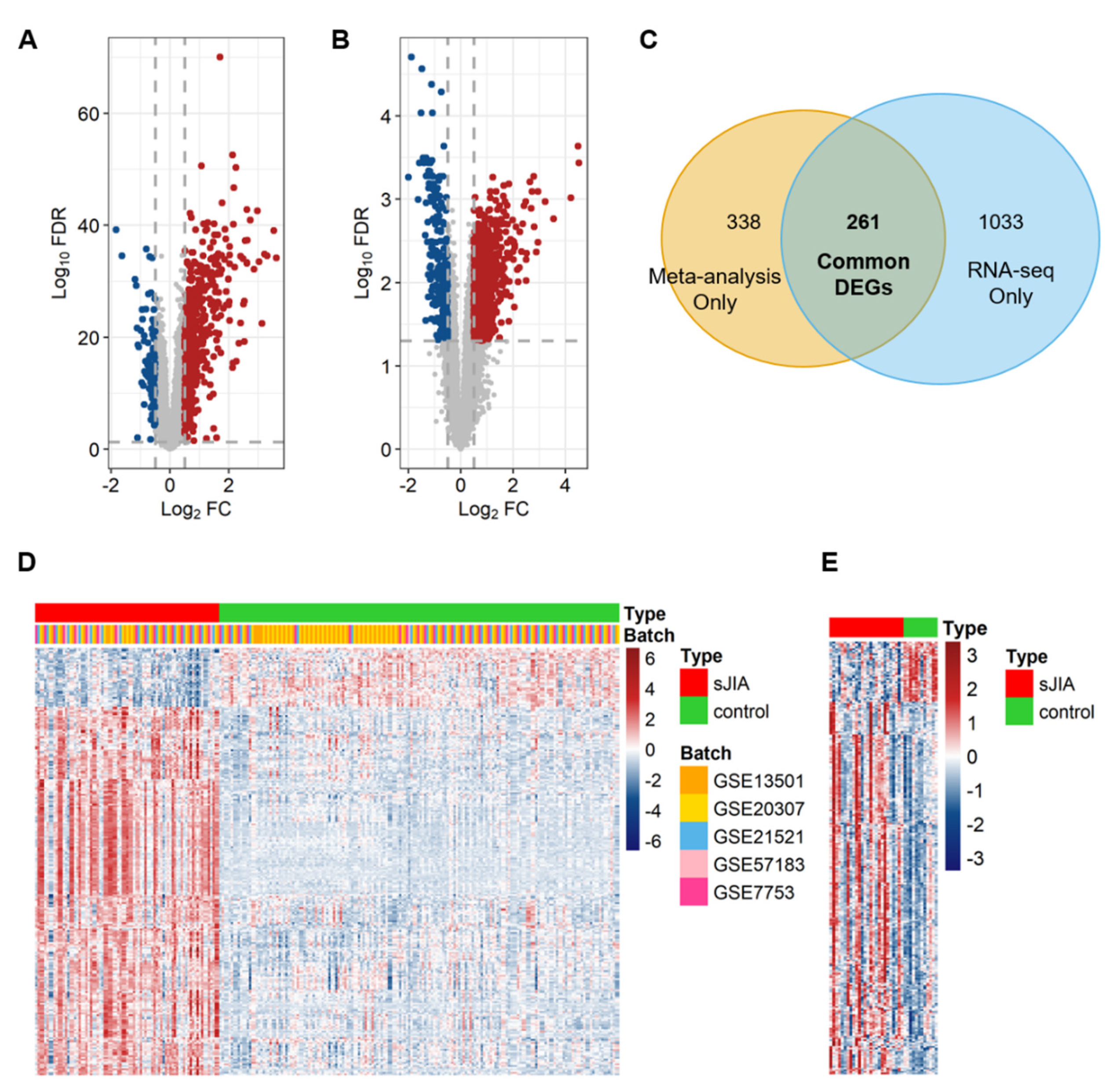{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Type | GEO ID | Tissue Source | Platform | Control/ Disease | Total | PMID |
|---|---|---|---|---|---|---|
| Microarray | GSE21521 | Peripheral blood mononuclear cells (PBMCs) | Affymetrix Human Genome U133 Plus 2.0 | 28/17 | 45 | 20576155 [19] |
| GSE20307 | 56/20 | 76 | 20662067 [20] | |||
| GSE13501 | 59/21 | 80 | 19565513 [21] | |||
| GSE7753 | 30/17 | 47 | 17968951 [22] | |||
| GSE57183 | Whole blood | Illumina HumanHT-12 V4.0 expression beadchip | 3/6 | 9 | 26267155 [23] | |
| RNA-seq | GSE112057 | Illumina HiSeq 2000 | 12/26 | 38 | 29950172 [24] | |
| Total | - | - | - | 188/107 | 295 | - |
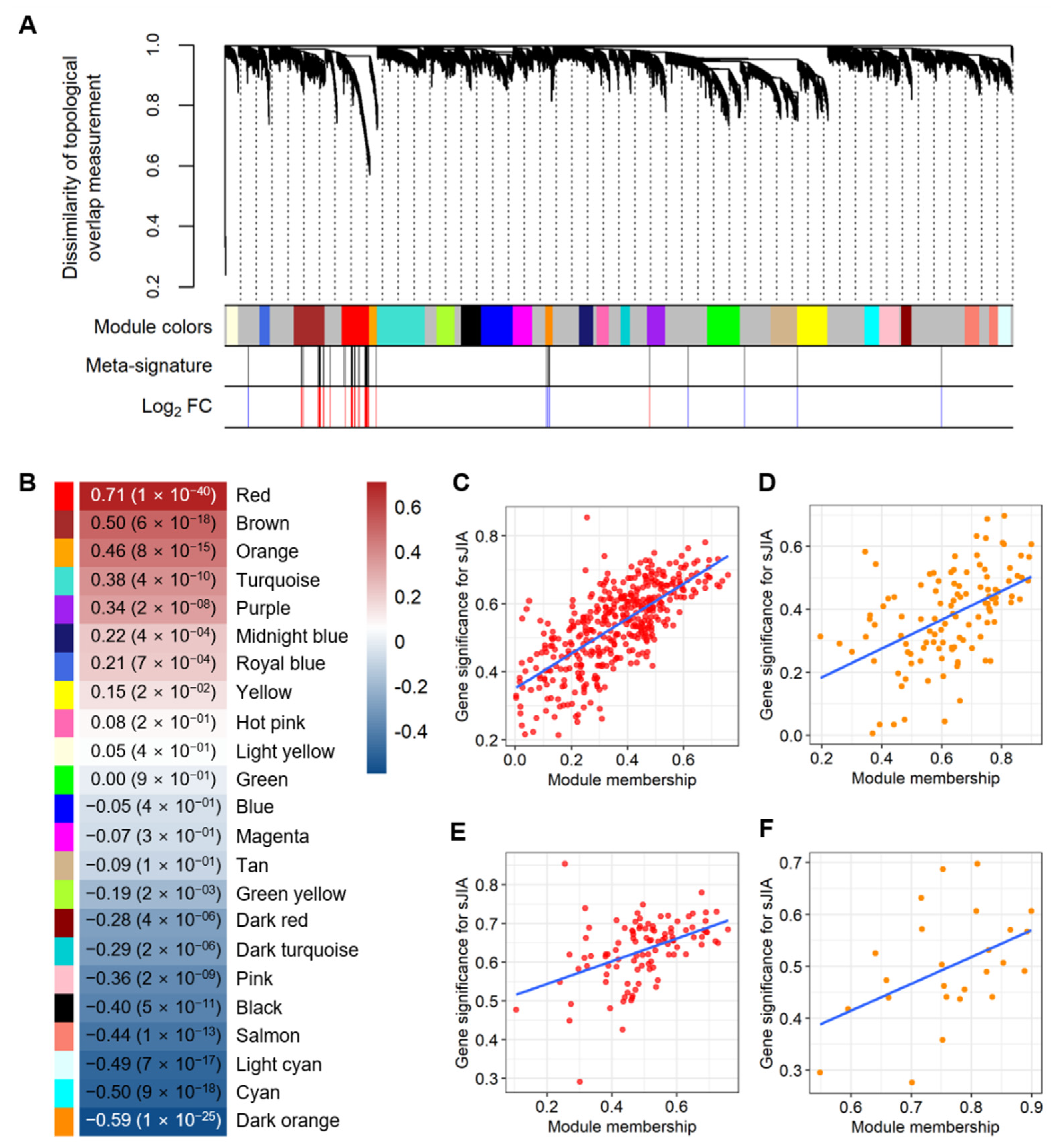
2.2. Selecting Co-Expressed Gene Sets from the Meta-Signature as the sJIA Signature
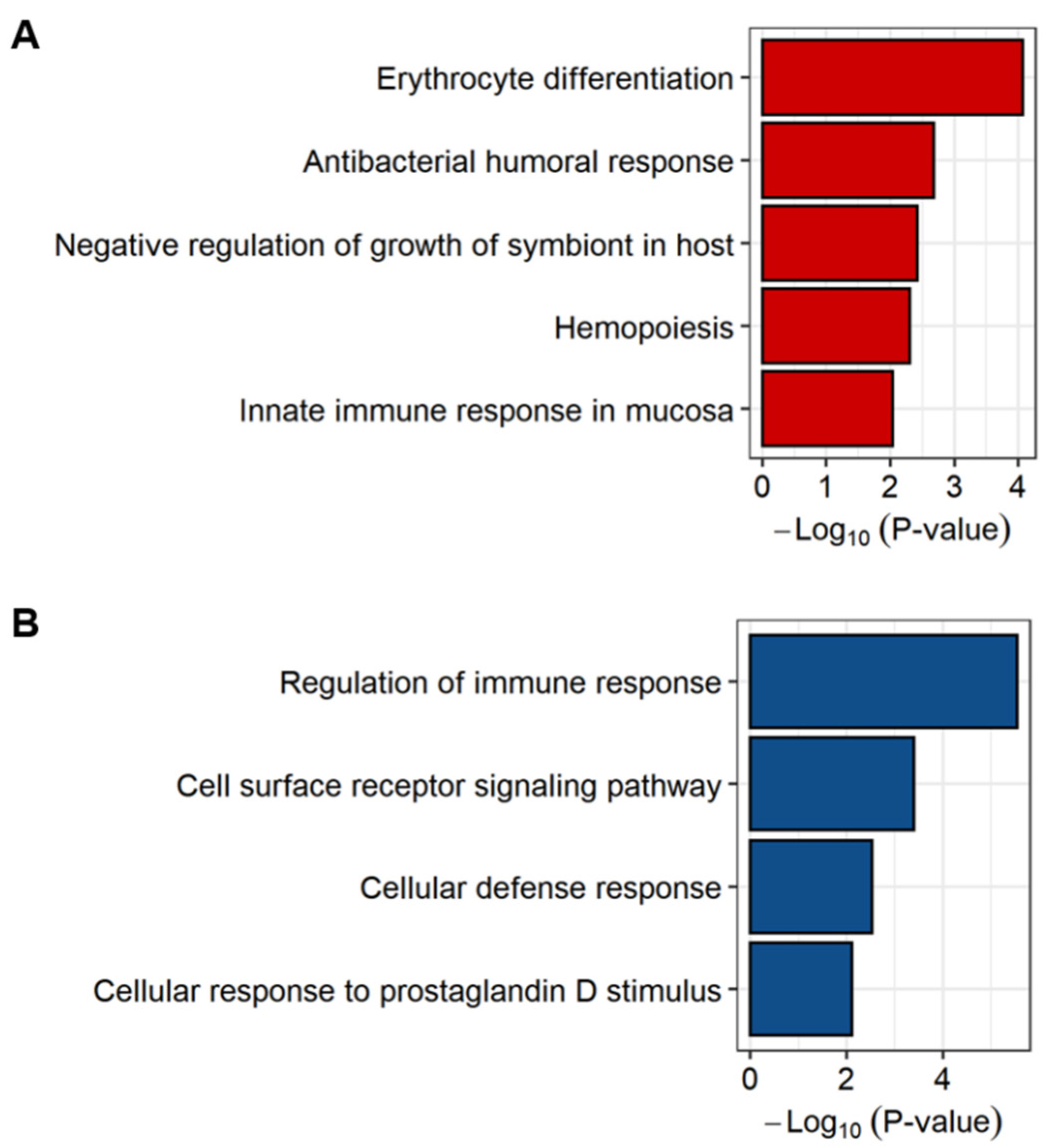
2.3. Exploring the Biological Pathways Associated with the sJIA Signature
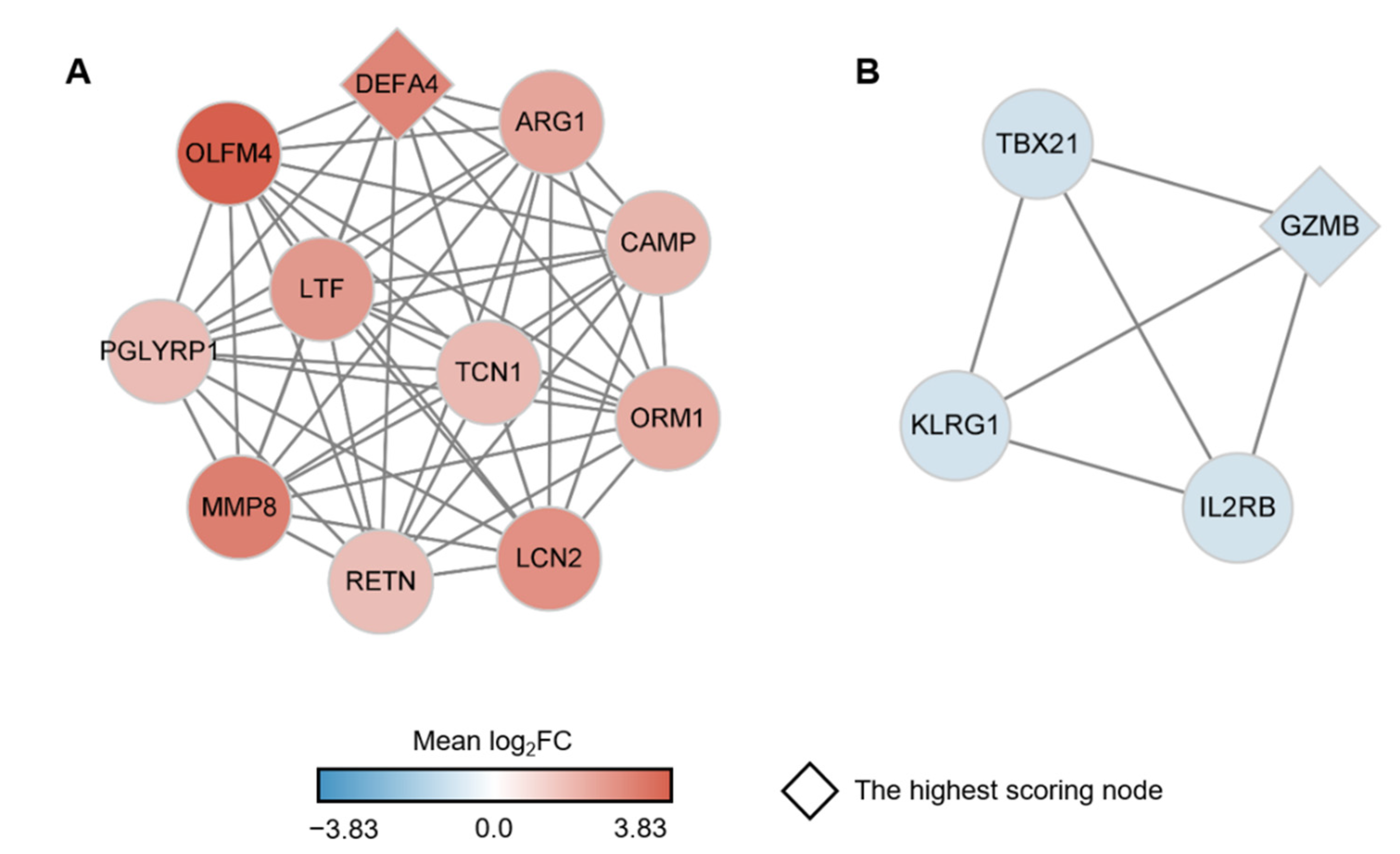
2.4. Identification of Key Genes among the sJIA Signature by Network Analysis
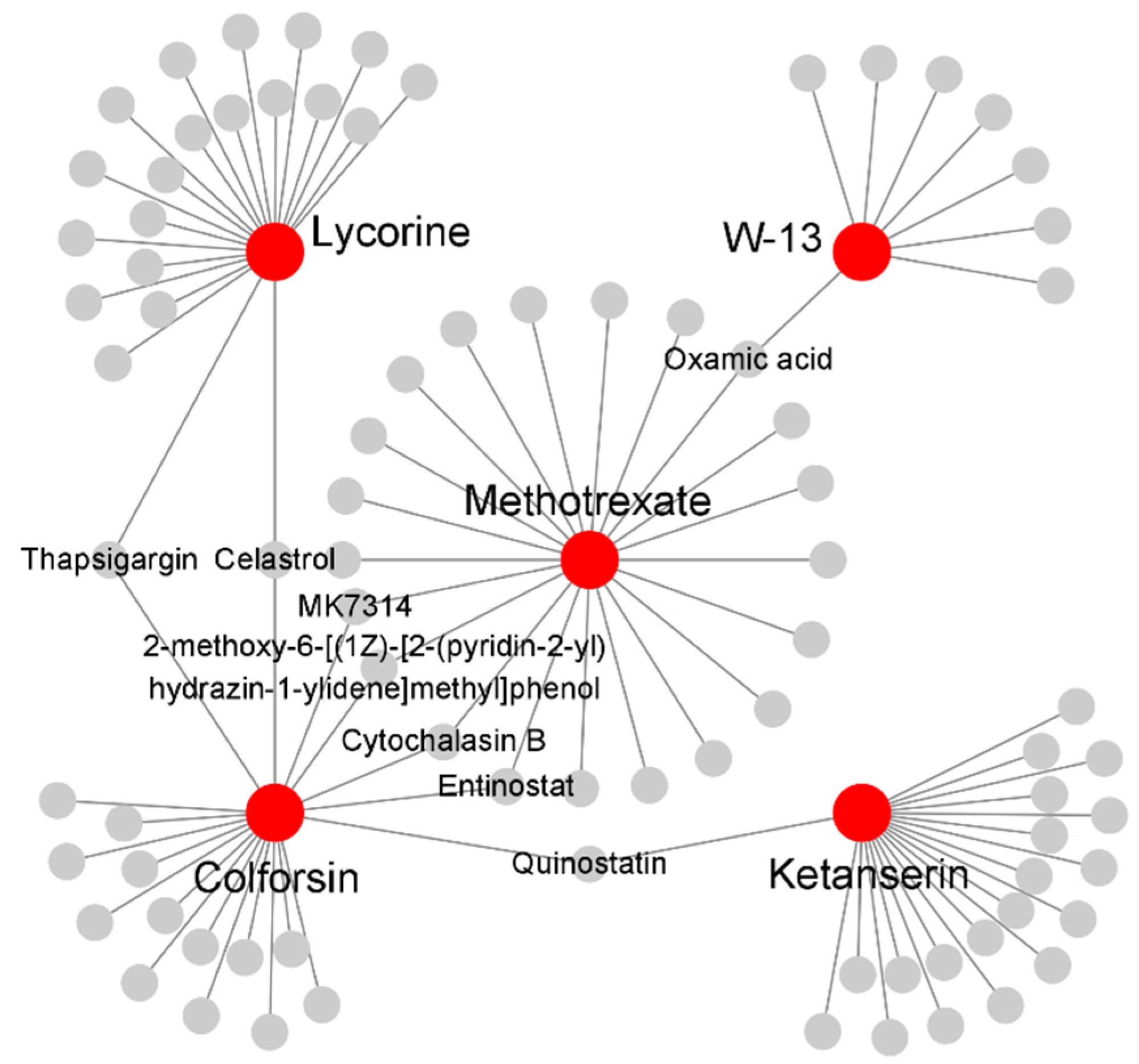
2.5. Identification of in Silico Drug Candidates for sJIA
3. Discussion
4. Materials and Methods
4.1. Data Collection
4.2. Data Preprocessing and Meta-Analysis
4.3. Weighted Gene Co-Expression Network Analysis
4.4. Module Enrichment Analysis
4.5. Functional Annotation of the sJIA Signature
4.6. Network Analysis and Finding Significant Clusters for sJIA
4.7. Identification of Drug Candidates
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| JIA | Juvenile Idiopathic Arthritis |
| sJIA | Systemic Juvenile Idiopathic Arthritis |
| WGCNA | Weighted Gene Co-expression Network Analysis |
| MAS | Macrophage Activation Syndrome |
| DEG | Differentially Expressed Gene |
| IL-1 | Interleukin 1 |
| IL-6 | Interleukin 6 |
| TNF-α | Tumor necrosis factor-α |
| NCBI | National Center for Biotechnology Information |
| GEO | Gene Expression Omnibus |
| PCA | Principal Component Analysis |
| FDR | False Discovery Rate |
| Log2FC | Log2(Fold Change) |
| NGS | Next Generation Sequencing |
| RNA-seq | RNA-sequencing |
| PBMC | Peripheral Blood Mononuclear Cell |
| ME | Module Eigengene |
| GO BP | Gene Ontology Biological Process |
| DAVID | Database for Annotation, Visualization, and Integrated Discovery |
| PPI | Protein–Protein Interaction |
| STRING | Search Tool for the Retrieval of Interacting Genes |
| MCODE | Molecular Complex Detection |
| DEFA4 | Defensin alpha 4 |
| OLFM4 | Olfactomedin 4 |
| ORM1 | Orosomucoid 1 |
| TCN1 | Transcobalamin 1 |
| GZMB | Granzyme B |
| KLR | Killer cell lectin-like receptor |
| KLRG1 | Killer cell lectin-like receptor G1 |
| IL2RB | Interleukin 2 receptor subunit beta |
| TBX21 | T-box transcription factor 21 |
| CMap | Connectivity Map |
| W-13 | N-(4-aminobutyl)-5-chloronaphthalene-2-sulfonamide |
| MANTRA | Mode of Action by Network Analysis |
| MOAs | Mechanisms of Actions |
| GWAS | Genome-Wide Association Study |
| eQTL | Expression Quantitative Trait Loci |
| MSigDB | Molecular Signatures DataBase |
References
- Costello, R.; McDonagh, J.; Dixon, W.; Hyrich, K.; Humphreys, J. Incidence of Juvenile Idiopathic Arthritis in the United Kingdom: Estimates from a National Primary Care Dataset. Arthritis Rheumatol. 2019, 58, 71. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, N.; Yokota, S.; Takei, S.; Okura, Y.; Kubota, T.; Shimizu, M.; Nozawa, T.; Iwata, N.; Umebayashi, H.; Kinjo, N.; et al. Clinical practice guidance for juvenile idiopathic arthritis (JIA) 2018. Mod. Rheumatol. 2019, 29, 41–59. [Google Scholar] [CrossRef] [PubMed]
- Barut, K.; Adrovic, A.; Sahin, S.; Kasapcopur, O. Juvenile Idiopathic Arthritis. Balk. Med. J. 2017, 34, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Mellins, E.D.; Macaubas, C.; Grom, A.A. Pathogenesis of systemic juvenile idiopathic arthritis: Some answers, more questions. Nat. Rev. Rheumatol. 2011, 7, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Ling, X.B.; Park, J.L.; Carroll, T.; Nguyen, K.D.; Lau, K.; Macaubas, C.; Chen, E.; Lee, T.; Sandborg, C.; Milojevic, D.; et al. Plasma profiles in active systemic juvenile idiopathic arthritis: Biomarkers and biological implications. Proteomics 2010, 10, 4415–4430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donn, R.; Ellison, S.; Lamb, R.; Day, T.; Baildam, E.; Ramanan, A.V. Genetic loci contributing to hemophagocytic lymphohistiocytosis do not confer susceptibility to systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 2008, 58, 869–874. [Google Scholar] [CrossRef] [Green Version]
- Put, K.; Vandenhaute, J.; Avau, A.; van Nieuwenhuijze, A.; Brisse, E.; Dierckx, T.; Rutgeerts, O.; Garcia-Perez, J.E.; Toelen, J.; Waer, M.; et al. Inflammatory Gene Expression Profile and Defective Interferon-gamma and Granzyme K in Natural Killer Cells From Systemic Juvenile Idiopathic Arthritis Patients. Arthritis Rheumatol. 2017, 69, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Bracaglia, C.; Prencipe, G.; De Benedetti, F. Macrophage Activation Syndrome: Different mechanisms leading to a one clinical syndrome. Pediatr. Rheumatol. 2017, 15, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Brown, R.A.; Henderlight, M.; Do, T.; Yasin, S.; Grom, A.A.; DeLay, M.; Thornton, S.; Schulert, G.S. Neutrophils From Children With Systemic Juvenile Idiopathic Arthritis Exhibit Persistent Proinflammatory Activation Despite Long-Standing Clinically Inactive Disease. Front. Immunol. 2018, 9, 2995. [Google Scholar] [CrossRef]
- Henderson, L.A.; Hoyt, K.J.; Lee, P.Y.; Rao, D.A.; Jonsson, A.H.; Nguyen, J.P.; Rutherford, K.; Jule, A.M.; Charbonnier, L.M.; Case, S.; et al. Th17 reprogramming of T cells in systemic juvenile idiopathic arthritis. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [Green Version]
- Brachat, A.H.; Grom, A.A.; Wulffraat, N.; Brunner, H.I.; Quartier, P.; Brik, R.; McCann, L.; Ozdogan, H.; Rutkowska-Sak, L.; Schneider, R.; et al. Early changes in gene expression and inflammatory proteins in systemic juvenile idiopathic arthritis patients on canakinumab therapy. Arthritis Res. Ther. 2017, 19, 13. [Google Scholar] [CrossRef] [Green Version]
- Frosch, M.; Roth, J. New insights in systemic juvenile idiopathic arthritis--from pathophysiology to treatment. Rheumatology 2008, 47, 121–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, J.; Kim, G.W.; Lee, B.; Joo, J.W.J.; Jang, W. Integrative genomic and transcriptomic analysis of genetic markers in Dupuytren’s disease. BMC Med. Genom. 2019, 12, 98. [Google Scholar] [CrossRef]
- Correll, C.K.; Binstadt, B.A. Advances in the pathogenesis and treatment of systemic juvenile idiopathic arthritis. Pediatr. Res. 2014, 75, 176–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beukelman, T. Treatment advances in systemic juvenile idiopathic arthritis. F1000Prime Rep. 2014, 6, 21. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007, 8, 118–127. [Google Scholar] [CrossRef]
- Hinze, C.H.; Fall, N.; Thornton, S.; Mo, J.Q.; Aronow, B.J.; Layh-Schmitt, G.; Griffin, T.A.; Thompson, S.D.; Colbert, R.A.; Glass, D.N.; et al. Immature cell populations and an erythropoiesis gene-expression signature in systemic juvenile idiopathic arthritis: Implications for pathogenesis. Arthritis Res. Ther. 2010, 12, R123. [Google Scholar] [CrossRef] [Green Version]
- Barnes, M.G.; Grom, A.A.; Thompson, S.D.; Griffin, T.A.; Luyrink, L.K.; Colbert, R.A.; Glass, D.N. Biologic Similarities Based on Age at Onset in Oligoarticular and Polyarticular Subtypes of Juvenile Idiopathic Arthritis. Arthritis Rheum. 2010, 62, 3249–3258. [Google Scholar] [CrossRef] [Green Version]
- Barnes, M.G.; Grom, A.A.; Thompson, S.D.; Griffin, T.A.; Pavlidis, P.; Itert, L.; Fall, N.; Sowders, D.P.; Hinze, C.H.; Aronow, B.J.; et al. Subtype-Specific Peripheral Blood Gene Expression Profiles in Recent-Onset Juvenile Idiopathic Arthritis. Arthritis Rheum. 2009, 60, 2102–2112. [Google Scholar] [CrossRef] [PubMed]
- Fall, N.; Barnes, M.; Thornton, S.; Luyrink, L.; Olson, J.; Ilowite, N.T.; Gottlieb, B.S.; Griffin, T.; Sherry, D.D.; Thompson, S.; et al. Gene expression profiling of peripheral blood from patients with untreated new-onset systemic juvenile idiopathic arthritis reveals molecular heterogeneity that may predict macrophage activation syndrome. Arthritis Rheum. 2007, 56, 3793–3804. [Google Scholar] [CrossRef] [PubMed]
- Shenoi, S.; Ou, J.N.; Ni, C.; Macaubass, C.; Gersuk, V.H.; Wallace, C.A.; Mellins, E.D.; Stevens, A.M. Comparison of biomarkers for systemic juvenile idiopathic arthritis. Pediatr. Res. 2015, 78, 554–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mo, A.; Marigorta, U.M.; Arafat, D.; Chan, L.H.K.; Ponder, L.; Jang, S.R.; Prince, J.; Kugathasan, S.; Prahalad, S.; Gibson, G. Disease-specific regulation of gene expression in a comparative analysis of juvenile idiopathic arthritis and inflammatory bowel disease. Genome Med. 2018, 10, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Luo, R.; Oldham, M.C.; Horvath, S. Is my network module preserved and reproducible? PLoS Comput. Biol. 2011, 7, e1001057. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- The Gene Ontology, C. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar] [CrossRef] [Green Version]
- Nandakumar, S.K.; Ulirsch, J.C.; Sankaran, V.G. Advances in understanding erythropoiesis: Evolving perspectives. Br. J. Haematol. 2016, 173, 206–218. [Google Scholar] [CrossRef] [Green Version]
- Barreda, D.R.; Belosevic, M. Transcriptional regulation of hemopoiesis. Dev. Comp. Immunol. 2001, 25, 763–789. [Google Scholar] [CrossRef]
- Pretorius, E. The adaptability of red blood cells. Cardiovasc. Diabetol. 2013, 12, 63. [Google Scholar] [CrossRef] [Green Version]
- Von Mering, C.; Jensen, L.J.; Snel, B.; Hooper, S.D.; Krupp, M.; Foglierini, M.; Jouffre, N.; Huynen, M.A.; Bork, P. STRING: Known and predicted protein-protein associations, integrated and transferred across organisms. Nucleic Acids Res. 2005, 33, D433–D437. [Google Scholar] [CrossRef] [PubMed]
- Bader, G.D.; Hogue, C.W. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003, 4, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilde, C.G.; Griffith, J.E.; Marra, M.N.; Snable, J.L.; Scott, R.W. Purification and characterization of human neutrophil peptide 4, a novel member of the defensin family. J. Biol. Chem. 1989, 264, 11200–11203. [Google Scholar] [CrossRef]
- Park, K.S.; Kim, K.K.; Piao, Z.H.; Kim, M.K.; Lee, H.J.; Kim, Y.C.; Lee, K.S.; Lee, J.H.; Kim, K.E. Olfactomedin 4 suppresses tumor growth and metastasis of mouse melanoma cells through downregulation of integrin and MMP genes. Mol. Cells 2012, 34, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Prada-Medina, C.A.; Peron, J.P.S.; Nakaya, H.I. Immature neutrophil signature associated with the sexual dimorphism of systemic juvenile idiopathic arthritis. J. Leukoc. Biol. 2020, 108, 1319–1327. [Google Scholar] [CrossRef]
- Ramanathan, K.; Glaser, A.; Lythgoe, H.; Ong, J.; Beresford, M.W.; Midgley, A.; Wright, H.L. Neutrophil activation signature in juvenile idiopathic arthritis indicates the presence of low-density granulocytes. Rheumatology 2018, 57, 488–498. [Google Scholar] [CrossRef] [Green Version]
- Hu, N.; Mora-Jensen, H.; Theilgaard-Monch, K.; der Meer, B.D.-V.; Huitema, M.G.; Stegeman, C.A.; Heeringa, P.; Kallenberg, C.G.M.; Westra, J. Differential Expression of Granulopoiesis Related Genes in Neutrophil Subsets Distinguished by Membrane Expression of CD177. PLoS ONE 2014, 9, e99671. [Google Scholar] [CrossRef] [Green Version]
- El Mesallamy, H.O.; Hamdy, N.M.; Mostafa, D.M.; Amin, A.I. The Serine Protease Granzyme B as an Inflammatory Marker, in Relation to the Insulin Receptor Cleavage in Human Obesity and Type 2 Diabetes Mellitus. J. Interf. Cytokine Res. 2014, 34, 179–186. [Google Scholar] [CrossRef]
- Zhou, J.; Tang, X.M.; Ding, Y.; An, Y.F.; Zhao, X.D. Natural killer cell activity and frequency of killer cell immunoglobulin-like receptors in children with different forms of juvenile idiopathic arthritis. Pediatr. Allergy Immunol. 2013, 24, 691–696. [Google Scholar] [CrossRef]
- Vandenhaute, J.; Avau, A.; Filtjens, J.; Malengier-Devlies, B.; Imbrechts, M.; Van den Berghe, N.; Ahmadzadeh, K.; Mitera, T.; Boon, L.; Leclercq, G.; et al. Regulatory Role for NK Cells in a Mouse Model of Systemic Juvenile Idiopathic Arthritis. J. Immunol. 2019, 203, 3339–3348. [Google Scholar] [CrossRef]
- Suzuki, H.; Kundig, T.M.; Furlonger, C.; Wakeham, A.; Timms, E.; Matsuyama, T.; Schmits, R.; Simard, J.J.L.; Ohashi, P.S.; Griesser, H.; et al. Deregulated T-Cell Activation and Autoimmunity in Mice Lacking Interleukin-2 Receptor-Beta. Science 1995, 268, 1472–1476. [Google Scholar] [CrossRef] [PubMed]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.P.; Subramanian, A.; Ross, K.N.; et al. The connectivity map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ido, M.; Kato, S.; Ogawa, H.; Hayashi, K.; Komada, Y.; Zhang, X.L.; Sakurai, M.; Suzuki, K. Ca2+-dependent activation of the 33-kDa protein kinase transmits thrombin receptor signals in human platelets. Thromb. Haemost. 1996, 76, 439–443. [Google Scholar] [CrossRef] [PubMed]
- Audran, E.; Dagher, R.; Gioria, S.; Tsvetkov, P.O.; Kulikova, A.A.; Didier, B.; Villa, P.; Makarov, A.A.; Kilhoffer, M.C.; Haiech, J. A general framework to characterize inhibitors of calmodulin: Use of calmodulin inhibitors to study the interaction between calmodulin and its calmodulin binding domains. Biochim. Biophys. Acta BBA Bioenerg. 2013, 1833, 1720–1731. [Google Scholar] [CrossRef] [Green Version]
- Hong, Y.G.; Ji, H.M.; Wei, H. Topical ketanserin attenuates hyperalgesia and inflammation in arthritis in rats. Pain 2006, 124, 27–33. [Google Scholar] [CrossRef]
- Lin, L.; Zheng, J.; Zhu, W.P.; Jia, N. Nephroprotective Effect of Gelsemine Against Cisplatin-Induced Toxicity is Mediated Via Attenuation of Oxidative Stress. Cell Biochem. Biophys. 2015, 71, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Li, K.C.; Ho, Y.L.; Chen, C.Y.; Hsieh, W.T.; Chang, Y.S.; Huang, G.J. Lobeline improves acute lung injury via nuclear factor-kappaB-signaling pathway and oxidative stress. Respir. Physiol. Neurobiol. 2016, 225, 19–30. [Google Scholar] [CrossRef]
- Hayashida, N.; Chihara, S.; Tayama, E.; Takaseya, T.; Enomoto, N.; Kawara, T.; Aoyagi, S. Antiinflammatory effects of colforsin daropate hydrochloride, a novel water-soluble forskolin derivative. Ann. Thorac. Surg. 2001, 71, 1931–1938. [Google Scholar] [CrossRef]
- Awouters, F.; Niemegeers, C.J.; Lenaerts, F.M.; Janssen, P.A. The effects of suprofen in rats with Mycobacterium butyricum-induced arthritis. Arzneimittelforschung 1975, 25, 1526–1537. [Google Scholar]
- Li, S.; Liu, X.; Chen, X.; Bi, L. Research Progress on Anti-Inflammatory Effects and Mechanisms of Alkaloids from Chinese Medical Herbs. Evid. Based Complement. Altern. Med. 2020, 2020, 1303524. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Ye, M.; Zhang, J. Vincamine prevents lipopolysaccharide induced inflammation and oxidative stress via thioredoxin reductase activation in human corneal epithelial cells. Am. J. Transl. Res. 2018, 10, 2195–2204. [Google Scholar] [PubMed]
- Ali, A.; El-Zaitony, A.; Abd Al Haleem, E. Evaluation of Therapeutic Efficacy of Vinpocetine in Adjuvant Induced Arthritis Model in Rats. J. Pain Manag. Med. 2016, 2. [Google Scholar] [CrossRef]
- Eberlin, M.; Chen, M.; Mueck, T.; Dabritz, J. Racecadotril in the treatment of acute diarrhea in children: A systematic, comprehensive review and meta-analysis of randomized controlled trials. BMC Pediatr. 2018, 18, 124. [Google Scholar] [CrossRef] [PubMed]
- Kantele, A.M.; Takanen, R.; Arvilommi, H. Immune response to acute diarrhea seen as circulating antibody-secreting cells. J. Infect. Dis. 1988, 158, 1011–1016. [Google Scholar] [CrossRef]
- Yeo, W.S.; Arya, R.; Kim, K.K.; Jeong, H.; Cho, K.H.; Bae, T. The FDA-approved anti-cancer drugs, streptozotocin and floxuridine, reduce the virulence of Staphylococcus aureus. Sci. Rep. 2018, 8, 2521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vavra, J.J.; Deboer, C.; Dietz, A.; Hanka, L.J.; Sokolski, W.T. Streptozotocin, a new antibacterial antibiotic. Antibiot. Annu. 1959, 7, 230–235. [Google Scholar] [PubMed]
- Carrella, D.; Napolitano, F.; Rispoli, R.; Miglietta, M.; Carissimo, A.; Cutillo, L.; Sirci, F.; Gregoretti, F.; Di Bernardo, D. Mantra 2.0: An online collaborative resource for drug mode of action and repurposing by network analysis. Bioinformatics 2014, 30, 1787–1788. [Google Scholar] [CrossRef] [Green Version]
- Grevich, S.; Shenoi, S. Update on the management of systemic juvenile idiopathic arthritis and role of IL-1 and IL-6 inhibition. Adolesc. Health Med. Ther. 2017, 8, 125–135. [Google Scholar] [CrossRef]
- Ivliev, A.E.; Hoen, P.A.C.; Borisevich, D.; Nikolsky, Y.; Sergeeva, M.G. Drug Repositioning through Systematic Mining of Gene Coexpression Networks in Cancer. PLoS ONE 2016, 11, e0165059. [Google Scholar] [CrossRef]
- Wang, C.; Gong, B.S.; Bushel, P.R.; Thierry-Mieg, J.; Thierry-Mieg, D.; Xu, J.S.; Fang, H.; Hong, H.X.; Shen, J.; Su, Z.Q.; et al. The concordance between RNA-seq and microarray data depends on chemical treatment and transcript abundance. Nat. Biotechnol. 2014, 32, 926–932. [Google Scholar] [CrossRef]
- Van Dam, S.; Vosa, U.; van der Graaf, A.; Franke, L.; de Magalhaes, J.P. Gene co-expression analysis for functional classification and gene-disease predictions. Brief. Bioinform. 2018, 19, 575–592. [Google Scholar] [CrossRef]
- Long, T.H.; Lynn, D.J.; Henn, M.; Birren, B.W.; Lennon, N.J.; Phuong, T.L.; Kien, T.H.D.; Tham, T.H.N.; Lanh, N.M.; Farrar, J.J.; et al. The Early Whole-Blood Transcriptional Signature of Dengue Virus and Features Associated with Progression to Dengue Shock Syndrome in Vietnamese Children and Young Adults. J. Virol. 2010, 84, 12982–12994. [Google Scholar] [CrossRef] [Green Version]
- Colombo, S.; Buclin, T.; Decosterd, L.A.; Telenti, A.; Furrer, H.; Lee, B.L.; Biollaz, J.; Eap, C.B.; Swiss, H.I.V.C.S. Orosomucoid (alpha1-acid glycoprotein) plasma concentration and genetic variants: Effects on human immunodeficiency virus protease inhibitor clearance and cellular accumulation. Clin. Pharmacol. Ther. 2006, 80, 307–318. [Google Scholar] [CrossRef]
- Ye, X.D.; Zhang, N.; Jin, Y.X.; Xu, B.; Guo, C.Y.; Wang, X.Q.; Su, Y.T.; Yang, Q.; Song, J.Q.; Yu, W.H.; et al. Dramatically changed immune-related molecules as early diagnostic biomarkers of non-small cell lung cancer. FEBS J. 2020, 287, 783–799. [Google Scholar] [CrossRef] [PubMed]
- Ter Haar, N.M.; Tak, T.; Mokry, M.; Scholman, R.C.; Meerding, J.M.; de Jager, W.; Verwoerd, A.; Foell, D.; Vogl, T.; Roth, J.; et al. Reversal of Sepsis-Like Features of Neutrophils by Interleukin-1 Blockade in Patients with Systemic-Onset Juvenile Idiopathic Arthritis. Arthritis Rheumatol. 2018, 70, 943–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rousalova, I.; Krepela, E. Granzyme B-induced apoptosis in cancer cells and its regulation (Review). Int. J. Oncol. 2010, 37, 1361–1378. [Google Scholar] [CrossRef] [Green Version]
- Abel, A.M.; Yang, C.; Thakar, M.S.; Malarkannan, S. Natural Killer Cells: Development, Maturation, and Clinical Utilization. Front. Immunol. 2018, 9, 1869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, I.Z.; Baxter, R.M.; Garcia-Perez, J.E.; Vendrame, E.; Ranganath, T.; Kong, D.S.; Lundquist, K.; Nguyen, T.; Ogolla, S.; Black, J.; et al. A novel human IL2RB mutation results in T and NK cell-driven immune dysregulation. J. Exp. Med. 2019, 216, 1465. [Google Scholar] [CrossRef] [Green Version]
- Gordon, S.M.; Chaix, J.; Rupp, L.J.; Wu, J.M.; Madera, S.; Sun, J.C.; Lindsten, T.; Reiner, S.L. The Transcription Factors T-bet and Eomes Control Key Checkpoints of Natural Killer Cell Maturation. Immunity 2012, 36, 55–67. [Google Scholar] [CrossRef] [Green Version]
- Leavy, O. Natural Killer Cells Maturation and function of NK cells. Nat. Rev. Immunol. 2012, 12, 151. [Google Scholar] [CrossRef]
- Avau, A.; Put, K.; Wouters, C.H.; Matthys, P. Cytokine balance and cytokine-driven natural killer cell dysfunction in systemic juvenile idiopathic arthritis. Cytokine Growth Factor Rev. 2015, 26, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Naccache, P.H.; Molski, T.F.; Alobaidi, T.; Becker, E.L.; Showell, H.J.; Sha’Afi, R.I. Calmodulin inhibitors block neutrophil degranulation at a step distal from the mobilization of calcium. Biochem. Biophys. Res. Commun. 1980, 97, 62–68. [Google Scholar] [CrossRef]
- Alobaidi, T.; Naccache, P.H.; Sha’Afi, R.I. Calmodulin antagonists modulate rabbit neutrophil degranulation, aggregation and stimulated oxygen consumption. Biochim. Biophys. Acta 1981, 675, 316–321. [Google Scholar] [CrossRef]
- Curran, P.J.; Hussong, A.M. Integrative data analysis: The simultaneous analysis of multiple data sets. Psychol. Methods 2009, 14, 81–100. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, B.S.; Irizarry, R.A. A framework for oligonucleotide microarray preprocessing. Bioinformatics 2010, 26, 2363–2367. [Google Scholar] [CrossRef] [PubMed]
- Irizarry, R.A.; Bolstad, B.M.; Collin, F.; Cope, L.M.; Hobbs, B.; Speed, T.P. Summaries of affymetrix GeneChip probe level data. Nucleic Acids Res. 2003, 31, e15. [Google Scholar] [CrossRef]
- Bolstad, B.M.; Irizarry, R.A.; Astrand, M.; Speed, T.P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irizarry, R.A.; Hobbs, B.; Collin, F.; Beazer-Barclay, Y.D.; Antonellis, K.J.; Scherf, U.; Speed, T.P. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003, 4, 249–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.F.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Jiao, X.L.; Sherman, B.T.; Huang, D.W.; Stephens, R.; Baseler, M.W.; Lane, H.C.; Lempicki, R.A. DAVID-WS: A stateful web service to facilitate gene/protein list analysis. Bioinformatics 2012, 28, 1805–1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdottir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]






| CMap Name | PubChem Name | PubChem CID | Enrichment Score | p-Value |
|---|---|---|---|---|
| W-13 | N-(4-Aminobutyl)-5-chloronaphthalene-2-sulfonamide | 4299 | 0.95 | 5.77 × 10−03 |
| Ketanserin | Ketanserin | 3822 | 0.79 | 3.94 × 10−03 |
| Gelsemine | Gelsemine | 279057 | 0.77 | 5.61 × 10−03 |
| Lobeline | Lobeline | 101616 | 0.74 | 8.55 × 10−03 |
| Suprofen | Suprofen | 5359 | 0.69 | 1.99 × 10−02 |
| Racecadotril | Racecadotril | 107751 | 0.67 | 2.47 × 10−02 |
| Streptozocin | Streptozocin | 29327 | 0.65 | 3.34 × 10−02 |
| Lycorine | Lycorine | 72378 | 0.65 | 1.40 × 10−02 |
| Colforsin | Forskolin | 47936 | 0.62 | 2.33 × 10−02 |
| Vincamine | Vincamine | 15376 | 0.53 | 4.21 × 10−02 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, D.; Song, J.; Lee, S.; Jung, J.; Jang, W. An Integrative Transcriptomic Analysis of Systemic Juvenile Idiopathic Arthritis for Identifying Potential Genetic Markers and Drug Candidates. Int. J. Mol. Sci. 2021, 22, 712. https://doi.org/10.3390/ijms22020712
Kim D, Song J, Lee S, Jung J, Jang W. An Integrative Transcriptomic Analysis of Systemic Juvenile Idiopathic Arthritis for Identifying Potential Genetic Markers and Drug Candidates. International Journal of Molecular Sciences. 2021; 22(2):712. https://doi.org/10.3390/ijms22020712
Chicago/Turabian StyleKim, Daeun, Jaeseung Song, Sora Lee, Junghyun Jung, and Wonhee Jang. 2021. "An Integrative Transcriptomic Analysis of Systemic Juvenile Idiopathic Arthritis for Identifying Potential Genetic Markers and Drug Candidates" International Journal of Molecular Sciences 22, no. 2: 712. https://doi.org/10.3390/ijms22020712
APA StyleKim, D., Song, J., Lee, S., Jung, J., & Jang, W. (2021). An Integrative Transcriptomic Analysis of Systemic Juvenile Idiopathic Arthritis for Identifying Potential Genetic Markers and Drug Candidates. International Journal of Molecular Sciences, 22(2), 712. https://doi.org/10.3390/ijms22020712