Mycobacteriophages as Potential Therapeutic Agents against Drug-Resistant Tuberculosis


Abstract
:1. Introduction
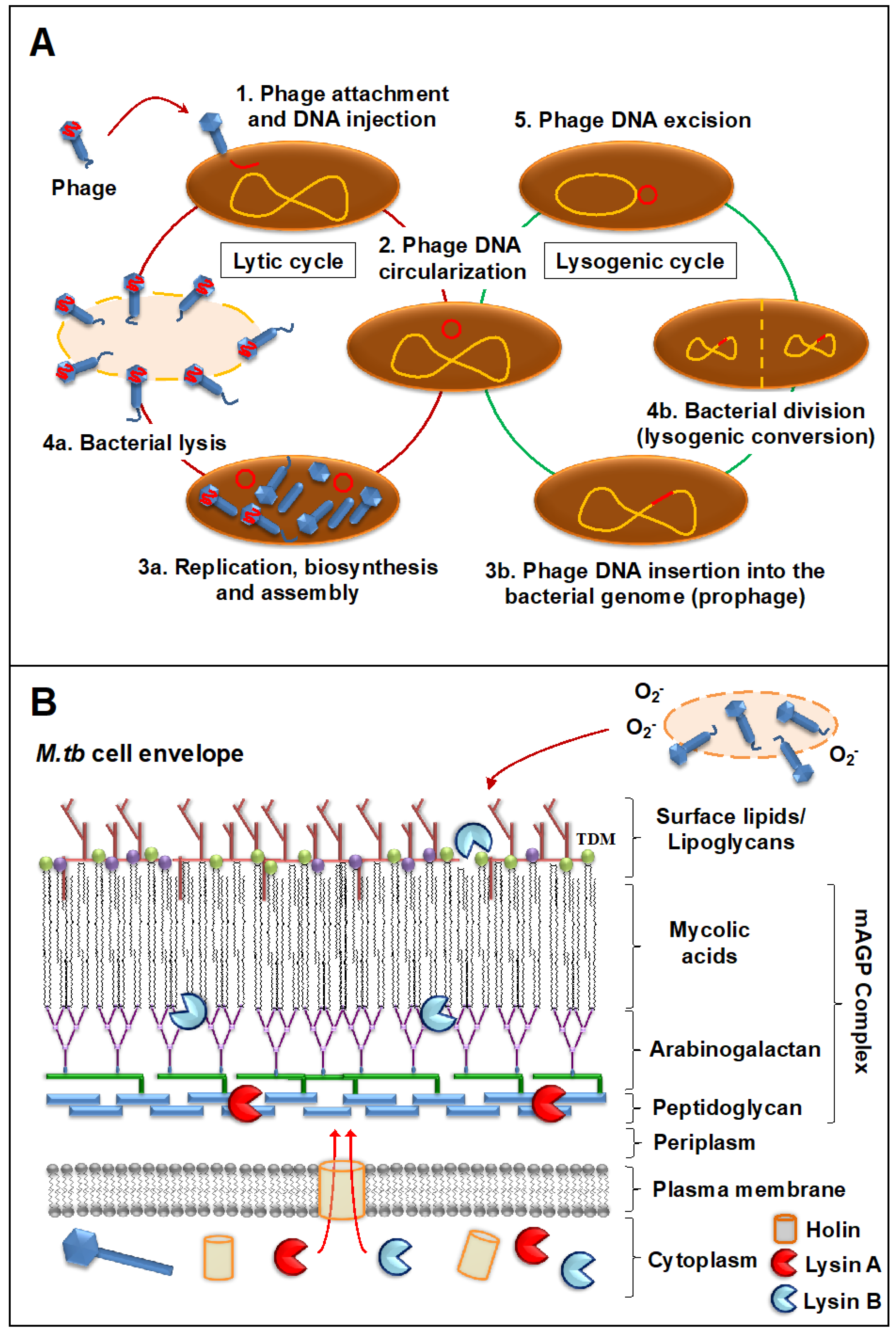
2. Mycobacteriophages
2.1. General Features
2.2. Mode of Action against Mycobacterial Hosts
3. Phage Therapy to Treat Multidrug-Resistant TB
3.1. Mycobacteriophage Host Specificity
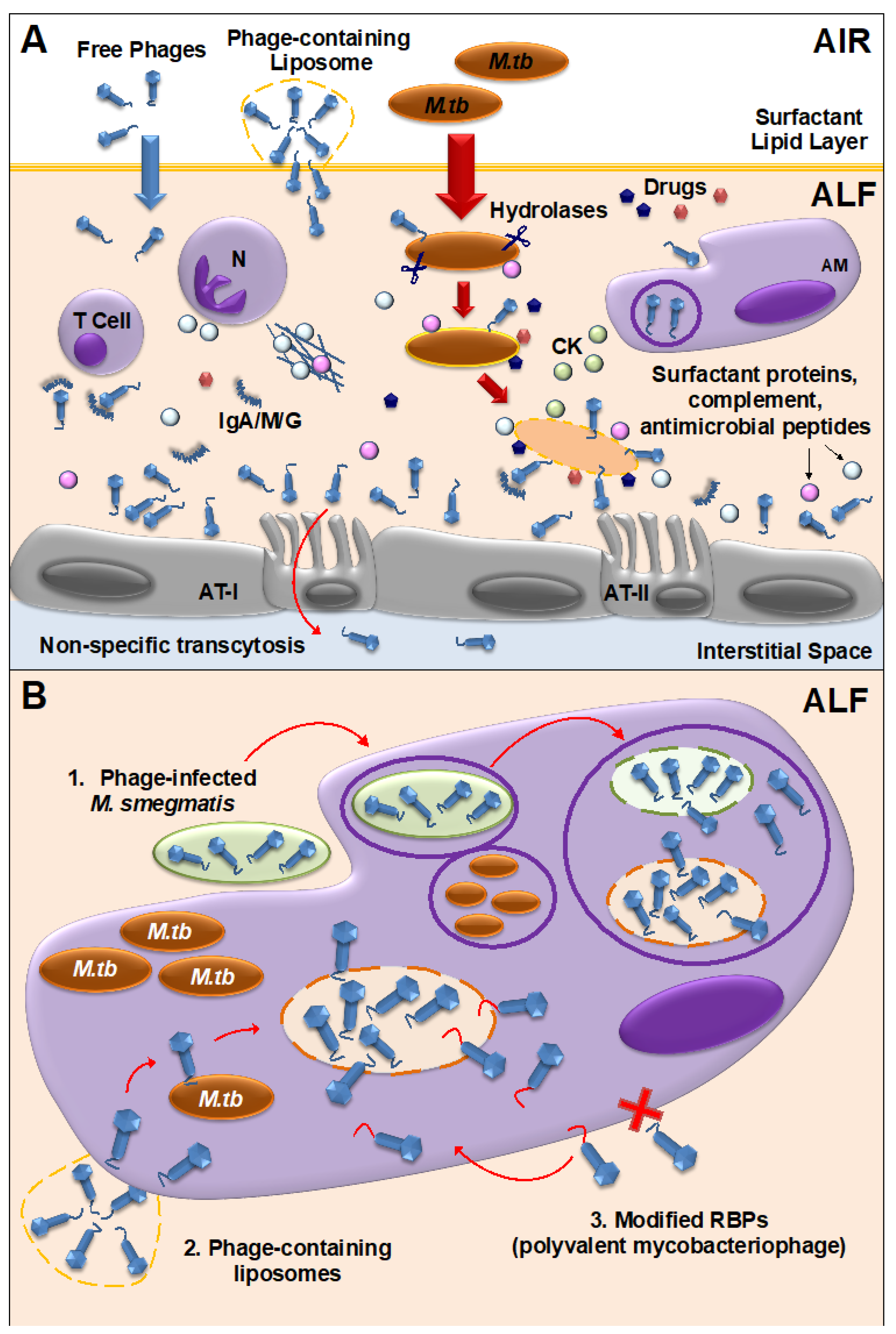
3.2. Mycobacteriophage-M. tuberculosis Interactions within Mammalian Host Cells
3.3. Bacterial Resistance to Mycobacteriophages
3.4. Mycobacteriophage Interactions with the Mammalian Immune System and the Lung Virome
4. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. Global Tuberculosis Report 2020; WHO: Geneva, Switzerland, 2020. [Google Scholar]
- Arcos, J.; Sasindran, S.J.; Fujiwara, N.; Turner, J.; Schlesinger, L.S.; Torrelles, J.B. Human lung hydrolases delineate Mycobacterium tuberculosis-macrophage interactions and the capacity to control infection. J. Immunol. 2011, 187, 372–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torrelles, J.B.; Schlesinger, L.S. Integrating Lung Physiology, Immunology, and Tuberculosis. Trends Microbiol. 2017, 25, 688–697. [Google Scholar] [CrossRef] [PubMed]
- Zaman, K. Tuberculosis: A global health problem. J. Health Popul. Nutr. 2010, 28, 111–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellner, J.J. The emergence of extensively drug-resistant tuberculosis: A global health crisis requiring new interventions: Part I: The origins and nature of the problem. Clin. Transl. Sci. 2008, 1, 249–254. [Google Scholar] [CrossRef] [Green Version]
- Loewenberg, S. India reports cases of totally drug-resistant tuberculosis. Lancet 2012, 379, 205. [Google Scholar] [CrossRef]
- Klopper, M.; Warren, R.M.; Hayes, C.; Gey van Pittius, N.C.; Streicher, E.M.; Muller, B.; Sirgel, F.A.; Chabula-Nximeni, M.; Hoosain, E.; Coetzee, G.; et al. Emergence and spread of extensively and totally drug-resistant tuberculosis, South Africa. Emerg. Infect. Dis. 2013, 19, 449–455. [Google Scholar] [CrossRef] [Green Version]
- LoBue, P. Extensively drug-resistant tuberculosis. Curr. Opin Infect. Dis. 2009, 22, 167–173. [Google Scholar] [CrossRef] [Green Version]
- Velayati, A.A.; Masjedi, M.R.; Farnia, P.; Tabarsi, P.; Ghanavi, J.; ZiaZarifi, A.H.; Hoffner, S.E. Emergence of new forms of totally drug-resistant tuberculosis bacilli: Super extensively drug-resistant tuberculosis or totally drug-resistant strains in iran. Chest 2009, 136, 420–425. [Google Scholar] [CrossRef]
- Migliori, G.B.; Centis, R.; D’Ambrosio, L.; Spanevello, A.; Borroni, E.; Cirillo, D.M.; Sotgiu, G. Totally drug-resistant and extremely drug-resistant tuberculosis: The same disease? Clin. Infect. Dis 2012, 54, 1379–1380. [Google Scholar] [CrossRef]
- WHO. Drug-Resistant TB: Totally Drug-Resistant TB FAQ. Available online: https://www.who.int/tb/areas-of-work/drug-resistant-tb/totally-drug-resistant-tb-faq/en/ (accessed on 3 March 2020).
- Velayati, A.A.; Farnia, P.; Masjedi, M.R. The totally drug resistant tuberculosis (TDR-TB). Int. J. Clin. Exp. Med. 2013, 6, 307–309. [Google Scholar] [PubMed]
- Burki, T.K. The uphill battle to find new TB treatments. Lancet Respir Med. 2017. [Google Scholar] [CrossRef]
- Lange, C.; Chesov, D.; Heyckendorf, J.; Leung, C.C.; Udwadia, Z.; Dheda, K. Drug-resistant tuberculosis: An update on disease burden, diagnosis and treatment. Respirology 2018, 23, 656–673. [Google Scholar] [CrossRef]
- Velayati, A.A.; Farnia, P.; Masjedi, M.R.; Ibrahim, T.A.; Tabarsi, P.; Haroun, R.Z.; Kuan, H.O.; Ghanavi, J.; Farnia, P.; Varahram, M. Totally drug-resistant tuberculosis strains: Evidence of adaptation at the cellular level. Eur. Respir. J. 2009, 34, 1202–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkinson, L. Felix d’Herelle and the origins of molecular biology. Med. Hist. 2001, 45, 294–295. [Google Scholar] [CrossRef] [Green Version]
- Chanishvili, N. Phage Therapy-History from Twort and d’Herelle Through Soviet Experience to Current Approaches. Adv. Virus Res. 2012, 83, 3–40. [Google Scholar] [CrossRef] [PubMed]
- Sulakvelidze, A.; Alavidze, Z.; Morris, J.G., Jr. Bacteriophage therapy. Antimicrob. Agents Chemother. 2001, 45, 649–659. [Google Scholar] [CrossRef] [Green Version]
- Dublanchet, A.; Bourne, S. The epic of phage therapy. Can. J. Infect. Dis. Med. Microbiol. 2007, 18, 15–18. [Google Scholar] [CrossRef] [Green Version]
- Kokin, G.A. Phage therapy and phage prophylaxis of gas gangrene. In Experience of the Soviet Military Medicine during the Great Patriotic War 1941–1945; Medgiz: Moscow, Russia, 1946; Volume 3. [Google Scholar]
- Lin, D.M.; Koskella, B.; Lin, H.C. Phage therapy: An alternative to antibiotics in the age of multi-drug resistance. World J. Gastrointest. Pharmacol. Ther. 2017, 8, 162–173. [Google Scholar] [CrossRef]
- Merabishvili, M.; Pirnay, J.P.; Verbeken, G.; Chanishvili, N.; Tediashvili, M.; Lashkhi, N.; Glonti, T.; Krylov, V.; Mast, J.; Van Parys, L.; et al. Quality-Controlled Small-Scale Production of a Well-Defined Bacteriophage Cocktail for Use in Human Clinical Trials. PLoS ONE 2009, 4, e4944. [Google Scholar] [CrossRef]
- Markoishvili, K.; Tsitlanadze, G.; Katsarava, R.; Morris, J.G.; Sulakvelidze, A. A novel sustained-release matrix based on biodegradable poly(ester amide)s and impregnated with bacteriophages and an antibiotic shows promise in management of infected venous stasis ulcers and other poorly healing wounds. Int. J. Dermatol. 2002, 41, 453–458. [Google Scholar] [CrossRef]
- McCallin, S.; Sarker, S.A.; Sultana, S.; Oechslin, F.; Brussow, H. Metagenome analysis of Russian and Georgian Pyophage cocktails and a placebo-controlled safety trial of single phage versus phage cocktail in healthy Staphylococcus aureus carriers. Environ. Microbiol. 2018, 20, 3278–3293. [Google Scholar] [CrossRef] [PubMed]
- Villarroel, J.; Larsen, M.V.; Kilstrup, M.; Nielsen, M. Metagenomic Analysis of Therapeutic PYO Phage Cocktails from 1997 to 2014. Viruses 2017, 9, 328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kutateladze, M. Experience of the Eliava Institute in bacteriophage therapy. Virol. Sin. 2015, 30, 80–81. [Google Scholar] [CrossRef]
- Gorski, A.; Miedzybrodzki, R.; Borysowski, J.; Weber-Dabrowska, B.; Lobocka, M.; Fortuna, W.; Letkiewicz, S.; Zimecki, M.; Filby, G. Bacteriophage therapy for the treatment of infections. Curr. Opin Investig. Drugs 2009, 10, 766–774. [Google Scholar]
- Furfaro, L.L.; Payne, M.S.; Chang, B.J. Bacteriophage Therapy: Clinical Trials and Regulatory Hurdles. Front. Cell Infect. Microbiol. 2018, 8, 376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wienhold, S.M.; Lienau, J.; Witzenrath, M. Towards Inhaled Phage Therapy in Western Europe. Viruses 2019, 11, 295. [Google Scholar] [CrossRef] [Green Version]
- Hoopes, J.T.; Stark, C.J.; Kim, H.A.; Sussman, D.J.; Donovan, D.M.; Nelson, D.C. Use of a bacteriophage lysin, PlyC, as an enzyme disinfectant against Streptococcus equi. Appl. Environ. Microbiol. 2009, 75, 1388–1394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brovko, L.Y.; Anany, H.; Griffiths, M.W. Bacteriophages for detection and control of bacterial pathogens in food and food-processing environment. Adv. Food Nutr. Res. 2012, 67, 241–288. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, B.; Biosca, E.G. Bacteriophage-Based Bacterial Wilt Biocontrol for an Environmentally Sustainable Agriculture. Front. Plant. Sci. 2017, 8, 1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, C.C.; Chang, D.C.; Chang, K.C. Development of a Biocontrol Method Applying Bacteriophage-Containing Aerosol against Mycobacterium tuberculosis Using the Bacteriophage BTCU-1 and M. smegmatis as Models. Microorganisms 2019, 7, 237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, E.; Campbell, K.; Grant, I.; McAuliffe, O. Understanding and Exploiting Phage-Host Interactions. Viruses 2019, 11, 567. [Google Scholar] [CrossRef] [Green Version]
- Lohrasbi, V.; Talebi, M.; Bialvaei, A.Z.; Fattorini, L.; Drancourt, M.; Heidary, M.; Darban-Sarokhalil, D. Trends in the discovery of new drugs for Mycobacterium tuberculosis therapy with a glance at resistance. Tuberculosis 2018, 109, 17–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaVergne, S.; Hamilton, T.; Biswas, B.; Kumaraswamy, M.; Schooley, R.T.; Wooten, D. Phage Therapy for a Multidrug-Resistant Acinetobacter baumannii Craniectomy Site Infection. Open Forum. Infect. Dis. 2018, 5, ofy064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dedrick, R.M.; Guerrero-Bustamante, C.A.; Garlena, R.A.; Russell, D.A.; Ford, K.; Harris, K.; Gilmour, K.C.; Soothill, J.; Jacobs-Sera, D.; Schooley, R.T.; et al. Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat. Med. 2019, 25, 730–733. [Google Scholar] [CrossRef]
- Jeon, J.; Park, J.H.; Yong, D. Efficacy of bacteriophage treatment against carbapenem-resistant Acinetobacter baumannii in Galleria mellonella larvae and a mouse model of acute pneumonia. BMC Microbiol. 2019, 19, 70. [Google Scholar] [CrossRef] [PubMed]
- Tkhilaishvili, T.; Winkler, T.; Muller, M.; Perka, C.; Trampuz, A. Bacteriophages as adjuvant to antibiotics for the treatment of periprosthetic joint infection caused by multidrug-resistant Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2019, 64, 1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kortright, K.E.; Chan, B.K.; Koff, J.L.; Turner, P.E. Phage Therapy: A Renewed Approach to Combat Antibiotic-Resistant Bacteria. Cell Host Microbe 2019, 25, 219–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardner, G.M.; Weiser, R.S. A bacteriophage for Mycobacterium smegmatis. Proc. Soc. Exp. Biol Med. 1947, 66, 205. [Google Scholar] [CrossRef] [PubMed]
- Sula, L.; Sulova, J.; Stolcpartova, M. Phage typing of South Indian M. tuberculosis strains by “surface” and “overlay” method. Czech. Med. 1981, 4, 215–223. [Google Scholar]
- Goode, D. Bacteriophage typing of strains of Mycobacterium tuberculosis from Nepal. Tubercle 1983, 64, 15–21. [Google Scholar] [CrossRef]
- Raj, C.V.; Ramakrishnan, T. Transduction in Mycobacterium smegmatis. Nature 1970, 228, 280–281. [Google Scholar] [CrossRef]
- Imaeda, T. Ultrastructure of L-phase variants isolated from a culture of Mycobacterium phlei. J. Med. Microbiol. 1975, 8, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Fay, D.; Bowman, B.U. Structure of native and chloroform-methanol-treated mycobacteriophage R1. J. Virol. 1978, 27, 432–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, W.; White, A. Lysogeny in mycobacteria. I. Conversion of colony morphology, nitrate reductase activity, and tween 80 hydrolysis of Mycobacterium sp. ATCC 607 associated with lysogeny. Can. J. Microbiol. 1968, 14, 551–555. [Google Scholar] [CrossRef]
- Schafer, R.; Huber, U.; Franklin, R.M. Chemical and physical properties of mycobacteriophage D29. Eur. J. Biochem. 1977, 73, 239–246. [Google Scholar] [CrossRef]
- Bardarov, S.; Kriakov, J.; Carriere, C.; Yu, S.; Vaamonde, C.; McAdam, R.A.; Bloom, B.R.; Hatfull, G.F.; Jacobs, W.R., Jr. Conditionally replicating mycobacteriophages: A system for transposon delivery to Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 1997, 94, 10961–10966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatfull, G.F.; Sarkis, G.J. DNA sequence, structure and gene expression of mycobacteriophage L5: A phage system for mycobacterial genetics. Mol. Microbiol. 1993, 7, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, W.R., Jr.; Tuckman, M.; Bloom, B.R. Introduction of foreign DNA into mycobacteria using a shuttle phasmid. Nature 1987, 327, 532–535. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, W.R., Jr. Gene Transfer in Mycobacterium tuberculosis: Shuttle Phasmids to Enlightenment. Microbiol. Spectr. 2014, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Kriakov, J.; Vilcheze, C.; Dai, Z.; Hatfull, G.F.; Jacobs, W.R., Jr. Bxz1, a new generalized transducing phage for mycobacteria. FEMS Microbiol. Lett. 2004, 241, 271–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Kessel, J.C.; Hatfull, G.F. Efficient point mutagenesis in mycobacteria using single-stranded DNA recombineering: Characterization of antimycobacterial drug targets. Mol. Microbiol. 2008, 67, 1094–1107. [Google Scholar] [CrossRef] [PubMed]
- Hatfull, G.F. Molecular Genetics of Mycobacteriophages. Microbiol. Spectr. 2014, 2, 1–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arora, G.; Chaudhary, D.; Kidwai, S.; Sharma, D.; Singh, R. CitE Enzymes Are Essential for Mycobacterium tuberculosis to Establish Infection in Macrophages and Guinea Pigs. Front. Cell Infect. Microbiol. 2018, 8, 385. [Google Scholar] [CrossRef] [PubMed]
- Marinelli, L.J.; Piuri, M.; Hatfull, G.F. Genetic Manipulation of Lytic Bacteriophages with BRED: Bacteriophage Recombineering of Electroporated DNA. Methods Mol. Biol. 2019, 1898, 69–80. [Google Scholar] [CrossRef]
- Petrova, Z.O.; Broussard, G.W.; Hatfull, G.F. Mycobacteriophage-repressor-mediated immunity as a selectable genetic marker: Adephagia and BPs repressor selection. Microbiology 2015, 161, 1539–1551. [Google Scholar] [CrossRef] [Green Version]
- McNerney, R.; Kiepiela, P.; Bishop, K.S.; Nye, P.M.; Stoker, N.G. Rapid screening of Mycobacterium tuberculosis for susceptibility to rifampicin and streptomycin. Int. J. Tuberc Lung Dis. 2000, 4, 69–75. [Google Scholar]
- Swift, B.M.; Convery, T.W.; Rees, C.E. Evidence of Mycobacterium tuberculosis complex bacteraemia in intradermal skin test positive cattle detected using phage-RPA. Virulence 2016, 7, 779–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs, W.R., Jr.; Barletta, R.G.; Udani, R.; Chan, J.; Kalkut, G.; Sosne, G.; Kieser, T.; Sarkis, G.J.; Hatfull, G.F.; Bloom, B.R. Rapid assessment of drug susceptibilities of Mycobacterium tuberculosis by means of luciferase reporter phages. Science 1993, 260, 819–822. [Google Scholar] [CrossRef] [Green Version]
- Carriere, C.; Riska, P.F.; Zimhony, O.; Kriakov, J.; Bardarov, S.; Burns, J.; Chan, J.; Jacobs, W.R., Jr. Conditionally replicating luciferase reporter phages: Improved sensitivity for rapid detection and assessment of drug susceptibility of Mycobacterium tuberculosis. J. Clin. Microbiol. 1997, 35, 3232–3239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riska, P.F.; Jacobs, W.R., Jr.; Bloom, B.R.; McKitrick, J.; Chan, J. Specific identification of Mycobacterium tuberculosis with the luciferase reporter mycobacteriophage: Use of p-nitro-alpha-acetylamino-beta-hydroxy propiophenone. J. Clin. Microbiol. 1997, 35, 3225–3231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, P.; Garing, S.; Verma, D.; Saranathan, R.; Clute-Reinig, N.; Gadwa, J.; Peterson, C.; Hermansky, G.; Astashkina Fernandez, A.; Asare, E.; et al. Nanoluciferase Reporter Mycobacteriophage for Sensitive and Rapid Detection of Mycobacterium tuberculosis Drug Susceptibility. J. Bacteriol. 2020, 202. [Google Scholar] [CrossRef]
- Piuri, M.; Jacobs, W.R., Jr.; Hatfull, G.F. Fluoromycobacteriophages for rapid, specific, and sensitive antibiotic susceptibility testing of Mycobacterium tuberculosis. PLoS ONE 2009, 4, e4870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crowley, G.C.; O’Mahony, J.; Coffey, A.; Sayers, R.; Cotter, P. A rapid viability and drug-susceptibility assay utilizing mycobacteriophage as an indicator of drug susceptibilities of Anti-TB drugs against Mycobacterium smegmatis mc(2) 155. Int. J. Mycobacteriol. 2019, 8, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Rondon, L.; Urdaniz, E.; Latini, C.; Payaslian, F.; Matteo, M.; Sosa, E.J.; Do Porto, D.F.; Turjanski, A.G.; Nemirovsky, S.; Hatfull, G.F.; et al. Fluoromycobacteriophages Can Detect Viable Mycobacterium tuberculosis and Determine Phenotypic Rifampicin Resistance in 3-5 Days From Sputum Collection. Front. Microbiol. 2018, 9, 1471. [Google Scholar] [CrossRef] [PubMed]
- Piuri, M.; Hatfull, G.F. Fluoromycobacteriophages for Drug Susceptibility Testing (DST) of Mycobacteria. Methods Mol. Biol. 2019, 1898, 27–36. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, M.R.; Larsen, M.H.; Brown, T.S.; Jain, P.; Munsamy, V.; Wolf, A.; Uccellini, L.; Karim, F.; de Oliveira, T.; Mathema, B.; et al. Early Detection of Emergent Extensively Drug-Resistant Tuberculosis by Flow Cytometry-Based Phenotyping and Whole-Genome Sequencing. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef] [Green Version]
- Jain, P.; Weinrick, B.C.; Kalivoda, E.J.; Yang, H.; Munsamy, V.; Vilcheze, C.; Weisbrod, T.R.; Larsen, M.H.; O’Donnell, M.R.; Pym, A.; et al. Dual-Reporter Mycobacteriophages (Phi2DRMs) Reveal Preexisting Mycobacterium tuberculosis Persistent Cells in Human Sputum. MBio 2016, 7, e01023-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatfull, G.F.; Russell, D.; Jacobs-Sera, D.; Pope, W.H.; Sivanathan, V. The Actinobacteriophage Database. Available online: https://phagesdb.org/hosts/genera/1/ (accessed on 3 March 2020).
- Russell, D.A.; Hatfull, G.F. PhagesDB: The actinobacteriophage database. Bioinformatics 2017, 33, 784–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajpai, U.; Mehta, A.K.; Eniyan, K.; Sinha, A.; Ray, A.; Virdi, S.; Ahmad, S.; Shah, A.; Arora, D.; Marwaha, D.; et al. Isolation and characterization of bacteriophages from India, with lytic activity against Mycobacterium tuberculosis. Can. J. Microbiol. 2018, 64, 483–491. [Google Scholar] [CrossRef] [Green Version]
- Satish, R.; Desouza, A. Study of characteristics of mycobacteriophage—A novel tool to treat Mycobacterium spp. Int. J. Mycobacteriol. 2019, 8, 170–174. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, H.W. Tailed bacteriophages: The order caudovirales. Adv. Virus Res. 1998, 51, 135–201. [Google Scholar] [CrossRef] [PubMed]
- Dion, M.B.; Oechslin, F.; Moineau, S. Phage diversity, genomics and phylogeny. Nat. Rev. Microbiol. 2020, 18, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Hatfull, G.F. Mycobacteriophages. Microbiol. Spectr. 2018, 6. [Google Scholar] [CrossRef] [PubMed]
- Pope, W.H.; Bowman, C.A.; Russell, D.A.; Jacobs-Sera, D.; Asai, D.J.; Cresawn, S.G.; Jacobs, W.R.; Hendrix, R.W.; Lawrence, J.G.; Hatfull, G.F.; et al. Whole genome comparison of a large collection of mycobacteriophages reveals a continuum of phage genetic diversity. Elife 2015, 4, e06416. [Google Scholar] [CrossRef] [PubMed]
- Pedulla, M.L.; Ford, M.E.; Houtz, J.M.; Karthikeyan, T.; Wadsworth, C.; Lewis, J.A.; Jacobs-Sera, D.; Falbo, J.; Gross, J.; Pannunzio, N.R.; et al. Origins of highly mosaic mycobacteriophage genomes. Cell 2003, 113, 171–182. [Google Scholar] [CrossRef] [Green Version]
- Hatfull, G.F.; Pedulla, M.L.; Jacobs-Sera, D.; Cichon, P.M.; Foley, A.; Ford, M.E.; Gonda, R.M.; Houtz, J.M.; Hryckowian, A.J.; Kelchner, V.A.; et al. Exploring the mycobacteriophage metaproteome: Phage genomics as an educational platform. PLoS Genet. 2006, 2, e92. [Google Scholar] [CrossRef] [Green Version]
- Hatfull, G.F. Mycobacteriophages: Genes and genomes. Annu. Rev. Microbiol. 2010, 64, 331–356. [Google Scholar] [CrossRef] [PubMed]
- Dedrick, R.M.; Mavrich, T.N.; Ng, W.L.; Cervantes Reyes, J.C.; Olm, M.R.; Rush, R.E.; Jacobs-Sera, D.; Russell, D.A.; Hatfull, G.F. Function, expression, specificity, diversity and incompatibility of actinobacteriophage parABS systems. Mol. Microbiol. 2016, 101, 625–644. [Google Scholar] [CrossRef] [Green Version]
- Morgado, S.M.; Vicente, A.C.P. Beyond the Limits: tRNA Array Units in Mycobacterium Genomes. Front. Microbiol. 2018, 9, 1042. [Google Scholar] [CrossRef] [PubMed]
- Kelley, D.S.; Lennon, C.W.; Sea, P.; Belfort, M.; Novikova, O. Mycobacteriophages as Incubators for Intein Dissemination and Evolution. MBio 2016, 7, e01537-16. [Google Scholar] [CrossRef] [Green Version]
- Hatfull, G.F.; Jacobs-Sera, D.; Lawrence, J.G.; Pope, W.H.; Russell, D.A.; Ko, C.C.; Weber, R.J.; Patel, M.C.; Germane, K.L.; Edgar, R.H.; et al. Comparative genomic analysis of 60 Mycobacteriophage genomes: Genome clustering, gene acquisition, and gene size. J. Mol. Biol. 2010, 397, 119–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, C.C.; Hatfull, G.F. Mycobacteriophage Fruitloop gp52 inactivates Wag31 (DivIVA) to prevent heterotypic superinfection. Mol. Microbiol. 2018, 108, 443–460. [Google Scholar] [CrossRef] [Green Version]
- Ko, C.C.; Hatfull, G.F. Identification of mycobacteriophage toxic genes reveals new features of mycobacterial physiology and morphology. Sci. Rep. 2020, 10, 14670. [Google Scholar] [CrossRef] [PubMed]
- Dedrick, R.M.; Jacobs-Sera, D.; Bustamante, C.A.; Garlena, R.A.; Mavrich, T.N.; Pope, W.H.; Reyes, J.C.; Russell, D.A.; Adair, T.; Alvey, R.; et al. Prophage-mediated defence against viral attack and viral counter-defence. Nat. Microbiol. 2017, 2, 16251. [Google Scholar] [CrossRef] [PubMed]
- Gentile, G.M.; Wetzel, K.S.; Dedrick, R.M.; Montgomery, M.T.; Garlena, R.A.; Jacobs-Sera, D.; Hatfull, G.F. More Evidence of Collusion: A New Prophage-Mediated Viral Defense System Encoded by Mycobacteriophage Sbash. MBio 2019, 10, e00196-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mavrich, T.N.; Hatfull, G.F. Evolution of Superinfection Immunity in Cluster A Mycobacteriophages. MBio 2019, 10, e00971-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lima-Junior, J.D.; Viana-Niero, C.; Conde Oliveira, D.V.; Machado, G.E.; Rabello, M.C.; Martins-Junior, J.; Martins, L.F.; Digiampietri, L.A.; da Silva, A.M.; Setubal, J.C.; et al. Characterization of mycobacteria and mycobacteriophages isolated from compost at the Sao Paulo Zoo Park Foundation in Brazil and creation of the new mycobacteriophage Cluster U. BMC Microbiol. 2016, 16, 111. [Google Scholar] [CrossRef]
- Suarez, C.A.; Franceschelli, J.J.; Morbidoni, H.R. Mycobacteriophage CRB2 defines a new subcluster in mycobacteriophage classification. PLoS ONE 2019, 14, e0212365. [Google Scholar] [CrossRef] [PubMed]
- Fast, K.M.; Keener, T.; Ali, R.; Butcher, B.M.; Millwood, J.D.; Odom, T.; Schellhammer, P.K.; Ufomadu, E.; Sandel, M.W. Genome Sequence of a Newly Isolated F2 Subcluster Mycobacteriophage from the Black Belt Geological Region of Western Alabama. Genome Announc. 2018, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saadhali, S.A.; Hassan, S.; Hanna, L.E.; Ranganathan, U.D.; Kumar, V. Homology modeling, substrate docking, and molecular simulation studies of mycobacteriophage Che12 lysin A. J. Mol. Model. 2016, 22, 180. [Google Scholar] [CrossRef]
- Pimentel, M. Genetics of Phage Lysis. Microbiol. Spectr. 2014, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, H.; Nair, G.; Gangakhedkar, R.; Jain, V. Understanding the role of the lysozyme-like domain of D29 mycobacteriophage-encoded endolysin in host cell lysis and phage propagation. Microbiology 2019, 165, 1013–1023. [Google Scholar] [CrossRef] [PubMed]
- Catalao, M.J.; Pimentel, M. Mycobacteriophage Lysis Enzymes: Targeting the Mycobacterial Cell Envelope. Viruses 2018, 10, 428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, H.; Seniya, S.P.; Suryanarayanan, V.; Patidar, N.D.; Singh, S.K.; Jain, V. Dissecting the structure-function relationship in lysozyme domain of mycobacteriophage D29-encoded peptidoglycan hydrolase. FEBS Lett. 2017, 591, 3276–3287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bavda, V.R.; Jain, V. Deciphering the Role of Holin in Mycobacteriophage D29 Physiology. Front. Microbiol. 2020, 11, 883. [Google Scholar] [CrossRef] [PubMed]
- Catalao, M.J.; Gil, F.; Moniz-Pereira, J.; Pimentel, M. The mycobacteriophage Ms6 encodes a chaperone-like protein involved in the endolysin delivery to the peptidoglycan. Mol. Microbiol. 2010, 77, 672–686. [Google Scholar] [CrossRef] [PubMed]
- Dulberger, C.L.; Rubin, E.J.; Boutte, C.C. The mycobacterial cell envelope—A moving target. Nat. Rev. Microbiol. 2020, 18, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Gigante, A.M.; Hampton, C.M.; Dillard, R.S.; Gil, F.; Catalao, M.J.; Moniz-Pereira, J.; Wright, E.R.; Pimentel, M. The Ms6 Mycolyl-Arabinogalactan Esterase LysB is Essential for an Efficient Mycobacteriophage-Induced Lysis. Viruses 2017, 9, 343. [Google Scholar] [CrossRef] [Green Version]
- Payne, K.; Sun, Q.; Sacchettini, J.; Hatfull, G.F. Mycobacteriophage Lysin B is a novel mycolylarabinogalactan esterase. Mol. Microbiol. 2009, 73, 367–381. [Google Scholar] [CrossRef] [Green Version]
- Gil, F.; Grzegorzewicz, A.E.; Catalao, M.J.; Vital, J.; McNeil, M.R.; Pimentel, M. Mycobacteriophage Ms6 LysB specifically targets the outer membrane of Mycobacterium smegmatis. Microbiology 2010, 156, 1497–1504. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Vilanova, A.; Chan, J.; Torrelles, J.B. Underestimated Manipulative Roles of Mycobacterium tuberculosis Cell Envelope Glycolipids During Infection. Front. Immunol. 2019, 10, 2909. [Google Scholar] [CrossRef] [PubMed]
- Fraga, A.G.; Trigo, G.; Murthy, R.K.; Akhtar, S.; Hebbur, M.; Pacheco, A.R.; Dominguez, J.; Silva-Gomes, R.; Goncalves, C.M.; Oliveira, H.; et al. Antimicrobial activity of Mycobacteriophage D29 Lysin B during Mycobacterium ulcerans infection. PLoS Negl. Trop Dis. 2019, 13, e0007113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samaddar, S.; Grewal, R.K.; Sinha, S.; Ghosh, S.; Roy, S.; Das Gupta, S.K. Dynamics of Mycobacteriophage-Mycobacterial Host Interaction: Evidence for Secondary Mechanisms for Host Lethality. Appl. Environ. Microbiol. 2016, 82, 124–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Belleghem, J.D.; Dabrowska, K.; Vaneechoutte, M.; Barr, J.J.; Bollyky, P.L. Interactions between Bacteriophage, Bacteria, and the Mammalian Immune System. Viruses 2018, 11, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lood, R.; Winer, B.Y.; Pelzek, A.J.; Diez-Martinez, R.; Thandar, M.; Euler, C.W.; Schuch, R.; Fischetti, V.A. Novel phage lysin capable of killing the multidrug-resistant gram-negative bacterium Acinetobacter baumannii in a mouse bacteremia model. Antimicrob. Agents Chemother. 2015, 59, 1983–1991. [Google Scholar] [CrossRef] [Green Version]
- Diez-Martinez, R.; De Paz, H.D.; Garcia-Fernandez, E.; Bustamante, N.; Euler, C.W.; Fischetti, V.A.; Menendez, M.; Garcia, P. A novel chimeric phage lysin with high in vitro and in vivo bactericidal activity against Streptococcus pneumoniae. J. Antimicrob. Chemother. 2015, 70, 1763–1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, G.; Yang, R.; Fan, K.; Dong, H.; Gao, C.; Wang, S.; Yu, L.; Cheng, Z.; Lei, L. External lysis of Escherichia coli by a bacteriophage endolysin modified with hydrophobic amino acids. AMB Express 2019, 9, 106. [Google Scholar] [CrossRef] [Green Version]
- Zampara, A.; Sorensen, M.C.H.; Grimon, D.; Antenucci, F.; Vitt, A.R.; Bortolaia, V.; Briers, Y.; Brondsted, L. Exploiting phage receptor binding proteins to enable endolysins to kill Gram-negative bacteria. Sci. Rep. 2020, 10, 12087. [Google Scholar] [CrossRef]
- Dams, D.; Briers, Y. Enzybiotics: Enzyme-Based Antibacterials as Therapeutics. Adv. Exp. Med. Biol. 2019, 1148, 233–253. [Google Scholar] [CrossRef] [PubMed]
- Catalao, M.J.; Filipe, S.R.; Pimentel, M. Revisiting Anti-tuberculosis Therapeutic Strategies That Target the Peptidoglycan Structure and Synthesis. Front. Microbiol. 2019, 10, 190. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhou, M.; Fan, X.; Yan, J.; Li, W.; Xie, J. Mycobacteriophage SWU1 gp39 can potentiate multiple antibiotics against Mycobacterium via altering the cell wall permeability. Sci. Rep. 2016, 6, 28701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puiu, M.; Julius, C. Bacteriophage gene products as potential antimicrobials against tuberculosis. Biochem. Soc. Trans. 2019, 47, 847–860. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Godavarthi, S.; Kumar, A.; Sen, R. A mycobacteriophage genomics approach to identify novel mycobacteriophage proteins with mycobactericidal properties. Microbiology 2019, 165, 722–736. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, Z.; He, X.; Yang, J.; Wu, J.; Yang, H.; Li, M.; Qian, Q.; Lai, R.; Xu, W.; et al. A small mycobacteriophage-derived peptide and its improved isomer restrict mycobacterial infection via dual mycobactericidal-immunoregulatory activities. J. Biol. Chem. 2019, 294, 7615–7631. [Google Scholar] [CrossRef]
- Casey, E.; van Sinderen, D.; Mahony, J. In Vitro Characteristics of Phages to Guide ‘Real Life’ Phage Therapy Suitability. Viruses 2018, 10, 163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, R.; Matsumoto, T.; Sano, G.; Ishii, Y.; Tateda, K.; Sumiyama, Y.; Uchiyama, J.; Sakurai, S.; Matsuzaki, S.; Imai, S.; et al. Efficacy of bacteriophage therapy against gut-derived sepsis caused by Pseudomonas aeruginosa in mice. Antimicrob. Agents Chemother. 2007, 51, 446–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkins, C.; Harper, D.; Burch, D.; Anggard, E.; Soothill, J. Topical treatment of Pseudomonas aeruginosa otitis of dogs with a bacteriophage mixture: A before/after clinical trial. Vet. Microbiol. 2010, 146, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Saussereau, E.; Debarbieux, L. Bacteriophages in the experimental treatment of Pseudomonas aeruginosa infections in mice. Adv. Virus Res. 2012, 83, 123–141. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.K.; Turner, P.E.; Kim, S.; Mojibian, H.R.; Elefteriades, J.A.; Narayan, D. Phage treatment of an aortic graft infected with Pseudomonas aeruginosa. Evol. Med. Public Health 2018, 2018, 60–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jault, P.; Leclerc, T.; Jennes, S.; Pirnay, J.P.; Que, Y.A.; Resch, G.; Rousseau, A.F.; Ravat, F.; Carsin, H.; Le Floch, R.; et al. Efficacy and tolerability of a cocktail of bacteriophages to treat burn wounds infected by Pseudomonas aeruginosa (PhagoBurn): A randomised, controlled, double-blind phase 1/2 trial. Lancet Infect. Dis. 2019, 19, 35–45. [Google Scholar] [CrossRef]
- Wills, Q.F.; Kerrigan, C.; Soothill, J.S. Experimental bacteriophage protection against Staphylococcus aureus abscesses in a rabbit model. Antimicrob. Agents Chemother. 2005, 49, 1220–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chhibber, S.; Kaur, S.; Kumari, S. Therapeutic potential of bacteriophage in treating Klebsiella pneumoniae B5055-mediated lobar pneumonia in mice. J. Med. Microbiol. 2008, 57, 1508–1513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soothill, J.S. Treatment of experimental infections of mice with bacteriophages. J. Med. Microbiol. 1992, 37, 258–261. [Google Scholar] [CrossRef] [Green Version]
- Nale, J.Y.; Spencer, J.; Hargreaves, K.R.; Buckley, A.M.; Trzepinski, P.; Douce, G.R.; Clokie, M.R.J. Bacteriophage Combinations Significantly Reduce Clostridium difficile Growth In Vitro and Proliferation In Vivo. Antimicrob. Agents Chemother. 2016, 60, 968–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, Y.; Llorente, C.; Lang, S.; Brandl, K.; Chu, H.; Jiang, L.; White, R.C.; Clarke, T.H.; Nguyen, K.; Torralba, M.; et al. Bacteriophage targeting of gut bacterium attenuates alcoholic liver disease. Nature 2019, 575, 505–511. [Google Scholar] [CrossRef]
- Sheng, H.; Knecht, H.J.; Kudva, I.T.; Hovde, C.J. Application of bacteriophages to control intestinal Escherichia coli O157:H7 levels in ruminants. Appl. Environ. Microbiol. 2006, 72, 5359–5366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cislo, M.; Dabrowski, M.; Weber-Dabrowska, B.; Woyton, A. Bacteriophage treatment of suppurative skin infections. Arch. Immunol. Ther. Exp. 1987, 35, 175–183. [Google Scholar]
- Principi, N.; Silvestri, E.; Esposito, S. Advantages and Limitations of Bacteriophages for the Treatment of Bacterial Infections. Front. Pharmacol. 2019, 10, 513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, C.; Mills, S.; Ross, R.P. Phages & antibiotic resistance: Are the most abundant entities on earth ready for a comeback? Future Microbiol. 2018, 13, 711–726. [Google Scholar] [CrossRef] [PubMed]
- Fish, R.; Kutter, E.; Wheat, G.; Blasdel, B.; Kutateladze, M.; Kuhl, S. Bacteriophage treatment of intransigent diabetic toe ulcers: A case series. J. Wound Care 2016, 25, S27–S33. [Google Scholar] [CrossRef]
- Wright, A.; Hawkins, C.H.; Anggard, E.E.; Harper, D.R. A controlled clinical trial of a therapeutic bacteriophage preparation in chronic otitis due to antibiotic-resistant Pseudomonas aeruginosa; a preliminary report of efficacy. Clin. Otolaryngol. 2009, 34, 349–357. [Google Scholar] [CrossRef]
- Jun, J.W.; Shin, T.H.; Kim, J.H.; Shin, S.P.; Han, J.E.; Heo, G.J.; De Zoysa, M.; Shin, G.W.; Chai, J.Y.; Park, S.C. Bacteriophage therapy of a Vibrio parahaemolyticus infection caused by a multiple-antibiotic-resistant O3:K6 pandemic clinical strain. J. Infect. Dis. 2014, 210, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.K.; Sistrom, M.; Wertz, J.E.; Kortright, K.E.; Narayan, D.; Turner, P.E. Phage selection restores antibiotic sensitivity in MDR Pseudomonas aeruginosa. Sci. Rep. 2016, 6, 26717. [Google Scholar] [CrossRef] [PubMed]
- Azimi, T.; Mosadegh, M.; Nasiri, M.J.; Sabour, S.; Karimaei, S.; Nasser, A. Phage therapy as a renewed therapeutic approach to mycobacterial infections: A comprehensive review. Infect. Drug Resist. 2019, 12, 2943–2959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sula, L.; Sulova, J.; Stolcpartova, M. Therapy of experimental tuberculosis in guinea pigs with mycobacterial phages DS-6A, GR-21 T, My-327. Czech. Med. 1981, 4, 209–214. [Google Scholar] [PubMed]
- Peng, L.; Luo, Y.; Chen, B.; Li, Y.; Shen, X.; Su, C.; Wang, G. Therapeutic effect of bacteriophage D29 in the treatment for guinea pigs infected with sensitive strain of Mycobacterium tuberculosis. Chin. J. Zoonoses 2009, 25, 733–736. [Google Scholar]
- Gordillo Altamirano, F.L.; Barr, J.J. Phage Therapy in the Postantibiotic Era. Clin. Microbiol. Rev. 2019, 32. [Google Scholar] [CrossRef] [Green Version]
- Ooi, M.L.; Drilling, A.J.; Morales, S.; Fong, S.; Moraitis, S.; Macias-Valle, L.; Vreugde, S.; Psaltis, A.J.; Wormald, P.J. Safety and Tolerability of Bacteriophage Therapy for Chronic Rhinosinusitis Due to Staphylococcus aureus. JAMA Otolaryngol. 2019, 145, 723–729. [Google Scholar] [CrossRef] [PubMed]
- Bogovazova, G.G.; Voroshilova, N.N.; Bondarenko, V.M. The efficacy of Klebsiella pneumoniae bacteriophage in the therapy of experimental Klebsiella infection. Zh. Mikrobiol. Epidemiol. Immunobiol. 1991, 5–8. [Google Scholar]
- Aziz, R.K.; Dwivedi, B.; Akhter, S.; Breitbart, M.; Edwards, R.A. Multidimensional metrics for estimating phage abundance, distribution, gene density, and sequence coverage in metagenomes. Front. Microbiol. 2015, 6, 381. [Google Scholar] [CrossRef] [Green Version]
- Hatfull, G.F. Bacteriophage genomics. Curr. Opin. Microbiol. 2008, 11, 447–453. [Google Scholar] [CrossRef] [Green Version]
- Endersen, L.; Coffey, A.; Neve, H.; McAuliffe, O.; Ross, R.P.; O’Mahony, J.M. Isolation and characterisation of six novel mycobacteriophages and investigation of their antimicrobial potential in milk. Int. Dairy J. 2013, 28, 8–14. [Google Scholar] [CrossRef]
- Caratenuto, R.A., 3rd; Ciabattoni, G.O.; DesGranges, N.J.; Drost, C.L.; Gao, L.; Gipson, B.; Kahler, N.C.; Kirven, N.A.; Melehani, J.C.; Patel, K.; et al. Genome Sequences of Six Cluster N Mycobacteriophages, Kevin1, Nenae, Parmesanjohn, ShrimpFriedEgg, Smurph, and SpongeBob, Isolated on Mycobacterium smegmatis mc(2)155. Microbiol. Resour. Announc. 2019, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levin, B.R.; Bull, J.J. Phage therapy revisited: The population biology of a bacterial infection and its treatment with bacteriophage and antibiotics. Am. Nat. 1996, 147, 881–898. [Google Scholar] [CrossRef]
- Young, R.; Gill, J.J. MICROBIOLOGY. Phage therapy redux--What is to be done? Science 2015, 350, 1163–1164. [Google Scholar] [CrossRef]
- Dedrick, R.M.; Guerrero Bustamante, C.A.; Garlena, R.A.; Pinches, R.S.; Cornely, K.; Hatfull, G.F. Mycobacteriophage ZoeJ: A broad host-range close relative of mycobacteriophage TM4. Tuberculosis 2019, 115, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Mayer, O.; Jain, P.; Weisbrod, T.R.; Biro, D.; Ho, L.; Jacobs-Sera, D.; Hatfull, G.F.; Jacobs, W.R., Jr. Fluorescent Reporter DS6A Mycobacteriophages Reveal Unique Variations in Infectibility and Phage Production in Mycobacteria. J. Bacteriol. 2016, 198, 3220–3232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rybniker, J.; Kramme, S.; Small, P.L. Host range of 14 mycobacteriophages in Mycobacterium ulcerans and seven other mycobacteria including Mycobacterium tuberculosis—Application for identification and susceptibility testing. J. Med. Microbiol. 2006, 55, 37–42. [Google Scholar] [CrossRef] [Green Version]
- Lapenkova, M.B.; Smirnova, N.S.; Rutkevich, P.N.; Vladimirsky, M.A. Evaluation of the Efficiency of Lytic Mycobacteriophage D29 on the Model of M. tuberculosis-Infected Macrophage RAW 264 Cell Line. Bull. Exp. Biol. Med. 2018, 164, 344–346. [Google Scholar] [CrossRef] [PubMed]
- Carrigy, N.B.; Larsen, S.E.; Reese, V.; Pecor, T.; Harrison, M.; Kuehl, P.J.; Hatfull, G.F.; Sauvageau, D.; Baldwin, S.L.; Finlay, W.H.; et al. Prophylaxis of Mycobacterium tuberculosis H37Rv Infection in a Preclinical Mouse Model via Inhalation of Nebulized Bacteriophage D29. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef]
- Liu, K.Y.; Yang, W.H.; Dong, X.K.; Cong, L.M.; Li, N.; Li, Y.; Wen, Z.B.; Yin, Z.; Lan, Z.J.; Li, W.P.; et al. Inhalation Study of Mycobacteriophage D29 Aerosol for Mice by Endotracheal Route and Nose-Only Exposure. J. Aerosol. Med. Pulm. Drug Deliv. 2016, 29, 393–405. [Google Scholar] [CrossRef]
- Jacobs-Sera, D.; Marinelli, L.J.; Bowman, C.; Broussard, G.W.; Guerrero Bustamante, C.; Boyle, M.M.; Petrova, Z.O.; Dedrick, R.M.; Pope, W.H.; (SEA-PHAGES) program; et al. On the nature of mycobacteriophage diversity and host preference. Virology 2012, 434, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.A. Genetic Diversity of Mycobacteriophages and the Unique Abilities of Cluster K. Corinthian 2017, 1, 5. [Google Scholar]
- de Jonge, P.A.; Nobrega, F.L.; Brouns, S.J.J.; Dutilh, B.E. Molecular and Evolutionary Determinants of Bacteriophage Host Range. Trends Microbiol. 2019, 27, 51–63. [Google Scholar] [CrossRef]
- Shao, Y.; Wang, I.N. Bacteriophage adsorption rate and optimal lysis time. Genetics 2008, 180, 471–482. [Google Scholar] [CrossRef] [Green Version]
- Cebria-Mendoza, M.; Sanjuan, R.; Domingo-Calap, P. Directed Evolution of a Mycobacteriophage. Antibiotics 2019, 8, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Kriakov, J.; Singh, A.; Jacobs, W.R., Jr.; Besra, G.S.; Bhatt, A. Defects in glycopeptidolipid biosynthesis confer phage I3 resistance in Mycobacterium smegmatis. Microbiology 2009, 155, 4050–4057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furuchi, A.; Tokunaga, T. Nature of the receptor substance of Mycobacterium smegmatis for D4 bacteriophage adsorption. J. Bacteriol. 1972, 111, 404–411. [Google Scholar] [CrossRef] [Green Version]
- Khoo, K.H.; Suzuki, R.; Dell, A.; Morris, H.R.; McNeil, M.R.; Brennan, P.J.; Besra, G.S. Chemistry of the lyxose-containing mycobacteriophage receptors of Mycobacterium phlei/Mycobacterium smegmatis. Biochemistry 1996, 35, 11812–11819. [Google Scholar] [CrossRef] [PubMed]
- Sassi, M.; Bebeacua, C.; Drancourt, M.; Cambillau, C. The first structure of a mycobacteriophage, the Mycobacterium abscessus subsp. bolletii phage Araucaria. J. Virol. 2013, 87, 8099–8109. [Google Scholar] [CrossRef] [Green Version]
- Arutyunov, D.; Singh, U.; El-Hawiet, A.; Seckler, H.D.; Nikjah, S.; Joe, M.; Bai, Y.; Lowary, T.L.; Klassen, J.S.; Evoy, S.; et al. Mycobacteriophage cell binding proteins for the capture of mycobacteria. Bacteriophage 2014, 4, e960346. [Google Scholar] [CrossRef] [PubMed]
- Kortright, K.E.; Chan, B.K.; Turner, P.E. High-throughput discovery of phage receptors using transposon insertion sequencing of bacteria. Proc. Natl. Acad. Sci. USA 2020, 117, 18670–18679. [Google Scholar] [CrossRef] [PubMed]
- Vilcheze, C.; Copeland, J.; Keiser, T.L.; Weisbrod, T.; Washington, J.; Jain, P.; Malek, A.; Weinrick, B.; Jacobs, W.R., Jr. Rational Design of Biosafety Level 2-Approved, Multidrug-Resistant Strains of Mycobacterium tuberculosis through Nutrient Auxotrophy. mBio 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunne, M.; Rupf, B.; Tala, M.; Qabrati, X.; Ernst, P.; Shen, Y.; Sumrall, E.; Heeb, L.; Pluckthun, A.; Loessner, M.J.; et al. Reprogramming Bacteriophage Host Range through Structure-Guided Design of Chimeric Receptor Binding Proteins. Cell Rep. 2019, 29, 1336–1350.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brabban, A.D.; Hite, E.; Callaway, T.R. Evolution of foodborne pathogens via temperate bacteriophage-mediated gene transfer. Foodborne Pathog. Dis. 2005, 2, 287–303. [Google Scholar] [CrossRef] [PubMed]
- Haaber, J.; Leisner, J.J.; Cohn, M.T.; Catalan-Moreno, A.; Nielsen, J.B.; Westh, H.; Penades, J.R.; Ingmer, H. Bacterial viruses enable their host to acquire antibiotic resistance genes from neighbouring cells. Nat. Commun. 2016, 7, 13333. [Google Scholar] [CrossRef]
- Sweere, J.M.; Van Belleghem, J.D.; Ishak, H.; Bach, M.S.; Popescu, M.; Sunkari, V.; Kaber, G.; Manasherob, R.; Suh, G.A.; Cao, X.; et al. Bacteriophage trigger antiviral immunity and prevent clearance of bacterial infection. Science 2019, 363. [Google Scholar] [CrossRef] [PubMed]
- Scordo, J.M.; Olmo-Fontanez, A.M.; Kelley, H.V.; Sidiki, S.; Arcos, J.; Akhter, A.; Wewers, M.D.; Torrelles, J.B. The human lung mucosa drives differential Mycobacterium tuberculosis infection outcome in the alveolar epithelium. Mucosal. Immunol. 2019, 12, 795–804. [Google Scholar] [CrossRef]
- Arcos, J.; Sasindran, S.J.; Moliva, J.I.; Scordo, J.M.; Sidiki, S.; Guo, H.; Venigalla, P.; Kelley, H.V.; Lin, G.; Diangelo, L.; et al. Mycobacterium tuberculosis cell wall released fragments by the action of the human lung mucosa modulate macrophages to control infection in an IL-10-dependent manner. Mucosal. Immunol. 2017, 10, 1248–1258. [Google Scholar] [CrossRef]
- Moliva, J.I.; Duncan, M.A.; Olmo-Fontanez, A.; Akhter, A.; Arnett, E.; Scordo, J.M.; Ault, R.; Sasindran, S.J.; Azad, A.K.; Montoya, M.J.; et al. The Lung Mucosa Environment in the Elderly Increases Host Susceptibility to Mycobacterium tuberculosis Infection. J. Infect. Dis. 2019, 220, 514–523. [Google Scholar] [CrossRef]
- Moliva, J.I.; Hossfeld, A.P.; Canan, C.H.; Dwivedi, V.; Wewers, M.D.; Beamer, G.; Turner, J.; Torrelles, J.B. Exposure to human alveolar lining fluid enhances Mycobacterium bovis BCG vaccine efficacy against Mycobacterium tuberculosis infection in a CD8(+) T-cell-dependent manner. Mucosal. Immunol. 2018, 11, 968–978. [Google Scholar] [CrossRef]
- Ehlers, S.; Schaible, U.E. The granuloma in tuberculosis: Dynamics of a host-pathogen collusion. Front. Immunol. 2012, 3, 411. [Google Scholar] [CrossRef] [Green Version]
- Serafini, A.; Tan, L.; Horswell, S.; Howell, S.; Greenwood, D.J.; Hunt, D.M.; Phan, M.D.; Schembri, M.; Monteleone, M.; Montague, C.R.; et al. Mycobacterium tuberculosis requires glyoxylate shunt and reverse methylcitrate cycle for lactate and pyruvate metabolism. Mol. Microbiol. 2019, 112, 1284–1307. [Google Scholar] [CrossRef] [Green Version]
- Bell, L.C.K.; Noursadeghi, M. Pathogenesis of HIV-1 and Mycobacterium tuberculosis co-infection. Nat. Rev. Microbiol. 2018, 16, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Segura-Cerda, C.A.; Lopez-Romero, W.; Flores-Valdez, M.A. Changes in Host Response to Mycobacterium tuberculosis Infection Associated With Type 2 Diabetes: Beyond Hyperglycemia. Front. Cell Infect. Microbiol. 2019, 9, 342. [Google Scholar] [CrossRef]
- Zykov, M.P.; Donets Iu, I.; Godovannyi, B.A. A study of the phage lysability of mycobacteria isolated in Africa. Zh Mikrobiol Epidemiol. Immunobiol. 1966, 43, 74–80. [Google Scholar]
- Rodriguez, D.C.; Ocampo, M.; Salazar, L.M.; Patarroyo, M.A. Quantifying intracellular Mycobacterium tuberculosis: An essential issue for in vitro assays. Microbiologyopen 2018, 7, e00588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, S.; Baker, K.; Padman, B.S.; Patwa, R.; Dunstan, R.A.; Weston, T.A.; Schlosser, K.; Bailey, B.; Lithgow, T.; Lazarou, M.; et al. Bacteriophage Transcytosis Provides a Mechanism To Cross Epithelial Cell Layers. mBio 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morello, E.; Saussereau, E.; Maura, D.; Huerre, M.; Touqui, L.; Debarbieux, L. Pulmonary bacteriophage therapy on Pseudomonas aeruginosa cystic fibrosis strains: First steps towards treatment and prevention. PLoS ONE 2011, 6, e16963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, S.; Harjai, K.; Chhibber, S. Bacteriophage-aided intracellular killing of engulfed methicillin-resistant Staphylococcus aureus (MRSA) by murine macrophages. Appl. Microbiol. Biotechnol. 2014, 98, 4653–4661. [Google Scholar] [CrossRef]
- Broxmeyer, L.; Sosnowska, D.; Miltner, E.; Chacon, O.; Wagner, D.; McGarvey, J.; Barletta, R.G.; Bermudez, L.E. Killing of Mycobacterium avium and Mycobacterium tuberculosis by a mycobacteriophage delivered by a nonvirulent mycobacterium: A model for phage therapy of intracellular bacterial pathogens. J. Infect. Dis. 2002, 186, 1155–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danelishvili, L.; Young, L.S.; Bermudez, L.E. In vivo efficacy of phage therapy for Mycobacterium avium infection as delivered by a nonvirulent mycobacterium. Microb. Drug Resist. 2006, 12, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, K.A.; Dao, D.N.; Goldberg, M.F.; Hsu, T.; Venkataswamy, M.M.; Henao-Tamayo, M.; Ordway, D.; Sellers, R.S.; Jain, P.; Chen, B.; et al. A recombinant Mycobacterium smegmatis induces potent bactericidal immunity against Mycobacterium tuberculosis. Nat. Med. 2011, 17, 1261–1268. [Google Scholar] [CrossRef] [Green Version]
- Nieth, A.; Verseux, C.; Barnert, S.; Suss, R.; Romer, W. A first step toward liposome-mediated intracellular bacteriophage therapy. Expert Opin. Drug Deliv. 2015, 12, 1411–1424. [Google Scholar] [CrossRef] [PubMed]
- Di Giovine, M.; Salone, B.; Martina, Y.; Amati, V.; Zambruno, G.; Cundari, E.; Failla, C.M.; Saggio, I. Binding properties, cell delivery, and gene transfer of adenoviral penton base displaying bacteriophage. Virology 2001, 282, 102–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pontow, S.E.; Kery, V.; Stahl, P.D. Mannose receptor. Int. Rev. Cytol. 1992, 137B, 221–244. [Google Scholar] [CrossRef]
- Astarie-Dequeker, C.; N’Diaye, E.N.; Le Cabec, V.; Rittig, M.G.; Prandi, J.; Maridonneau-Parini, I. The mannose receptor mediates uptake of pathogenic and nonpathogenic mycobacteria and bypasses bactericidal responses in human macrophages. Infect. Immun. 1999, 67, 469–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piuri, M.; Rondon, L.; Urdaniz, E.; Hatfull, G.F. Generation of affinity-tagged fluoromycobacteriophages by mixed assembly of phage capsids. Appl. Environ. Microbiol. 2013, 79, 5608–5615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohde, C.; Resch, G.; Pirnay, J.P.; Blasdel, B.G.; Debarbieux, L.; Gelman, D.; Gorski, A.; Hazan, R.; Huys, I.; Kakabadze, E.; et al. Expert Opinion on Three Phage Therapy Related Topics: Bacterial Phage Resistance, Phage Training and Prophages in Bacterial Production Strains. Viruses 2018, 10, 178. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.S. Microbial warfare against viruses. Science 2018, 359, 993. [Google Scholar] [CrossRef]
- Seed, K.D. Battling Phages: How Bacteria Defend against Viral Attack. PLoS Pathog 2015, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labrie, S.J.; Samson, J.E.; Moineau, S. Bacteriophage resistance mechanisms. Nat. Rev. Microbiol. 2010, 8, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Hampton, H.G.; Watson, B.N.J.; Fineran, P.C. The arms race between bacteria and their phage foes. Nature 2020, 577, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, K.L. Phages Fight Back: Inactivation of the CRISPR-Cas Bacterial Immune System by Anti-CRISPR Proteins. PLoS Pathog 2016, 12, e1005282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldfarb, T.; Sberro, H.; Weinstock, E.; Cohen, O.; Doron, S.; Charpak-Amikam, Y.; Afik, S.; Ofir, G.; Sorek, R. BREX is a novel phage resistance system widespread in microbial genomes. Embo J. 2015, 34, 169–183. [Google Scholar] [CrossRef] [PubMed]
- Ofir, G.; Melamed, S.; Sberro, H.; Mukamel, Z.; Silverman, S.; Yaakov, G.; Doron, S.; Sorek, R. DISARM is a widespread bacterial defence system with broad anti-phage activities. Nat. Microbiol. 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres-Barcelo, C. Phage Therapy Faces Evolutionary Challenges. Viruses 2018, 10, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, Q.H.; Contamin, L.; Nguyen, T.V.A.; Banuls, A.L. Insights into the processes that drive the evolution of drug resistance in Mycobacterium tuberculosis. Evol. Appl. 2018, 11, 1498–1511. [Google Scholar] [CrossRef] [PubMed]
- Capparelli, R.; Nocerino, N.; Lanzetta, R.; Silipo, A.; Amoresano, A.; Giangrande, C.; Becker, K.; Blaiotta, G.; Evidente, A.; Cimmino, A.; et al. Bacteriophage-resistant Staphylococcus aureus mutant confers broad immunity against staphylococcal infection in mice. PLoS ONE 2010, 5, e11720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, T.J.; Trauner, A.; Komitopoulou, E.; Salmond, G.P. Exploitation of a new flagellatropic phage of Erwinia for positive selection of bacterial mutants attenuated in plant virulence: Towards phage therapy. J. Appl. Microbiol. 2010, 108, 676–685. [Google Scholar] [CrossRef]
- Wright, R.C.T.; Friman, V.P.; Smith, M.C.M.; Brockhurst, M.A. Cross-resistance is modular in bacteria-phage interactions. PLoS Biol. 2018, 16. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.R.; De Vos, D.; Friman, V.P.; Pirnay, J.P.; Buckling, A. Effects of sequential and simultaneous applications of bacteriophages on populations of Pseudomonas aeruginosa in vitro and in wax moth larvae. Appl. Environ. Microbiol. 2012, 78, 5646–5652. [Google Scholar] [CrossRef] [Green Version]
- Chaudhry, W.N.; Concepcion-Acevedo, J.; Park, T.; Andleeb, S.; Bull, J.J.; Levin, B.R. Synergy and Order Effects of Antibiotics and Phages in Killing Pseudomonas aeruginosa Biofilms. PLoS ONE 2017, 12, e0168615. [Google Scholar] [CrossRef] [PubMed]
- Torres-Barcelo, C.; Hochberg, M.E. Evolutionary Rationale for Phases as Complements of Antibiotics. Trends Microbiol. 2016, 24, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Oechslin, F.; Piccardi, P.; Mancini, S.; Gabard, J.; Moreillon, P.; Entenza, J.M.; Resch, G.; Que, Y.A. Synergistic Interaction Between Phage Therapy and Antibiotics Clears Pseudomonas Aeruginosa Infection in Endocarditis and Reduces Virulence. J. Infect. Dis. 2017, 215, 703–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.G.; Buckling, A. Phages limit the evolution of bacterial antibiotic resistance in experimental microcosms. Evol. Appl. 2012, 5, 575–582. [Google Scholar] [CrossRef]
- Ryan, E.M.; Alkawareek, M.Y.; Donnelly, R.F.; Gilmore, B.F. Synergistic phage-antibiotic combinations for the control of Escherichia coli biofilms in vitro. FEMS Immunol. Med. Microbiol. 2012, 65, 395–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cairns, J.; Becks, L.; Jalasvuori, M.; Hiltunen, T. Sublethal streptomycin concentrations and lytic bacteriophage together promote resistance evolution. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamal, F.; Dennis, J.J. Burkholderia cepacia complex Phage-Antibiotic Synergy (PAS): Antibiotics stimulate lytic phage activity. Appl. Environ. Microbiol. 2015, 81, 1132–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trasta, A. Personalized medicine and proper dosage: Over- and undertreatment of chronic diseases endanger patients’ health and strain public health systems. EMBO Rep. 2018, 19. [Google Scholar] [CrossRef]
- Graham, F.; Hatfull, R.V. Respirable Bacteriophage Aerosols for the Prevention and Treatment of Tuberculosis. In Drug Delivery Systems for Tuberculosis Prevention and Treatment; Hickey, A.J., Ed.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2016. [Google Scholar]
- Pride, D.T.; Salzman, J.; Haynes, M.; Rohwer, F.; Davis-Long, C.; White, R.A.; Loomer, P.; Armitage, G.C.; Relman, D.A. Evidence of a robust resident bacteriophage population revealed through analysis of the human salivary virome. ISME J. 2012, 6, 915–926. [Google Scholar] [CrossRef] [PubMed]
- Santiago-Rodriguez, T.M.; Ly, M.; Bonilla, N.; Pride, D.T. The human urine virome in association with urinary tract infections. Front. Microbiol. 2015, 6, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro, F.; Muniesa, M. Phages in the Human Body. Front. Microbiol. 2017, 8, 566. [Google Scholar] [CrossRef] [Green Version]
- Lim, E.S.; Zhou, Y.; Zhao, G.; Bauer, I.K.; Droit, L.; Ndao, I.M.; Warner, B.B.; Tarr, P.I.; Wang, D.; Holtz, L.R. Early life dynamics of the human gut virome and bacterial microbiome in infants. Nat. Med. 2015, 21, 1228–1234. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.W.; Schmieder, R.; Haynes, M.; Willner, D.; Furlan, M.; Youle, M.; Abbott, K.; Edwards, R.; Evangelista, J.; Conrad, D.; et al. Metagenomics and metatranscriptomics: Windows on CF-associated viral and microbial communities. J. Cyst. Fibros. 2013, 12, 154–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krut, O.; Bekeredjian-Ding, I. Contribution of the Immune Response to Phage Therapy. J. Immunol. 2018, 200, 3037–3044. [Google Scholar] [CrossRef] [PubMed]
- Barr, J.J.; Auro, R.; Furlan, M.; Whiteson, K.L.; Erb, M.L.; Pogliano, J.; Stotland, A.; Wolkowicz, R.; Cutting, A.S.; Doran, K.S.; et al. Bacteriophage adhering to mucus provide a non-host-derived immunity. Proc. Natl. Acad. Sci. USA 2013, 110, 10771–10776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraser, J.S.; Yu, Z.; Maxwell, K.L.; Davidson, A.R. Ig-like domains on bacteriophages: A tale of promiscuity and deceit. J. Mol. Biol. 2006, 359, 496–507. [Google Scholar] [CrossRef] [PubMed]
- Pope, W.H.; Jacobs-Sera, D.; Russell, D.A.; Peebles, C.L.; Al-Atrache, Z.; Alcoser, T.A.; Alexander, L.M.; Alfano, M.B.; Alford, S.T.; Amy, N.E.; et al. Expanding the diversity of mycobacteriophages: Insights into genome architecture and evolution. PLoS ONE 2011, 6, e16329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abedon, S.T. Phage therapy of pulmonary infections. Bacteriophage 2015, 5, e1020260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levin, B.R.; Bull, J.J. Population and evolutionary dynamics of phage therapy. Nat. Rev. Microbiol. 2004, 2, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Malik, D.J.; Sokolov, I.J.; Vinner, G.K.; Mancuso, F.; Cinquerrui, S.; Vladisavljevic, G.T.; Clokie, M.R.J.; Garton, N.J.; Stapley, A.G.F.; Kirpichnikova, A. Formulation, stabilisation and encapsulation of bacteriophage for phage therapy. Adv. Colloid Interface Sci. 2017, 249, 100–133. [Google Scholar] [CrossRef] [Green Version]
- Huh, H.; Wong, S.; St Jean, J.; Slavcev, R. Bacteriophage interactions with mammalian tissue: Therapeutic applications. Adv. Drug Deliv. Rev. 2019, 145, 4–17. [Google Scholar] [CrossRef] [PubMed]
- Dabrowska, K. Phage therapy: What factors shape phage pharmacokinetics and bioavailability? Systematic and critical review. Med. Res. Rev. 2019, 39, 2000–2025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrigy, N.B.; Chang, R.Y.; Leung, S.S.Y.; Harrison, M.; Petrova, Z.; Pope, W.H.; Hatfull, G.F.; Britton, W.J.; Chan, H.K.; Sauvageau, D.; et al. Anti-Tuberculosis Bacteriophage D29 Delivery with a Vibrating Mesh Nebulizer, Jet Nebulizer, and Soft Mist Inhaler. Pharm. Res. 2017, 34, 2084–2096. [Google Scholar] [CrossRef] [PubMed]
- Jonczyk-Matysiak, E.; Weber-Dabrowska, B.; Owczarek, B.; Miedzybrodzki, R.; Lusiak-Szelachowska, M.; Lodej, N.; Gorski, A. Phage-Phagocyte Interactions and Their Implications for Phage Application as Therapeutics. Viruses 2017, 9, 150. [Google Scholar] [CrossRef] [Green Version]
- Dabrowska, K.; Miernikiewicz, P.; Piotrowicz, A.; Hodyra, K.; Owczarek, B.; Lecion, D.; Kazmierczak, Z.; Letarov, A.; Gorski, A. Immunogenicity studies of proteins forming the T4 phage head surface. J. Virol. 2014, 88, 12551–12557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miedzybrodzki, R.; Borysowski, J.; Klak, M.; Jonczyk-Matysiak, E.; Obminska-Mrukowicz, B.; Suszko-Pawlowska, A.; Bubak, B.; Weber-Dabrowska, B.; Gorski, A. In Vivo Studies on the Influence of Bacteriophage Preparations on the Autoimmune Inflammatory Process. Biomed Res. Int. 2017, 2017, 3612015. [Google Scholar] [CrossRef] [Green Version]
- Hodyra-Stefaniak, K.; Miernikiewicz, P.; Drapala, J.; Drab, M.; Jonczyk-Matysiak, E.; Lecion, D.; Kazmierczak, Z.; Beta, W.; Majewska, J.; Harhala, M.; et al. Mammalian Host-Versus-Phage immune response determines phage fate in vivo. Sci. Rep. 2015, 5, 14802. [Google Scholar] [CrossRef] [Green Version]
- Singla, S.; Harjai, K.; Katare, O.P.; Chhibber, S. Encapsulation of Bacteriophage in Liposome Accentuates Its Entry in to Macrophage and Shields It from Neutralizing Antibodies. PLoS ONE 2016, 11, e0153777. [Google Scholar] [CrossRef] [Green Version]
- Vandenheuvel, D.; Lavigne, R.; Brussow, H. Bacteriophage Therapy: Advances in Formulation Strategies and Human Clinical Trials. Annu. Rev. Virol. 2015, 2, 599–618. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.Y.K.; Wallin, M.; Lin, Y.; Leung, S.S.Y.; Wang, H.; Morales, S.; Chan, H.K. Phage therapy for respiratory infections. Adv. Drug Deliv. Rev. 2018, 133, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Hanif, S.N.; Garcia-Contreras, L. Pharmaceutical aerosols for the treatment and prevention of tuberculosis. Front. Cell Infect. Microbiol. 2012, 2, 118. [Google Scholar] [CrossRef] [Green Version]
- Roach, D.R.; Leung, C.Y.; Henry, M.; Morello, E.; Singh, D.; Di Santo, J.P.; Weitz, J.S.; Debarbieux, L. Synergy between the Host Immune System and Bacteriophage Is Essential for Successful Phage Therapy against an Acute Respiratory Pathogen. Cell Host Microbe 2017, 22, 38–47.e4. [Google Scholar] [CrossRef] [PubMed]
- Van Belleghem, J.D.; Clement, F.; Merabishvili, M.; Lavigne, R.; Vaneechoutte, M. Pro- and anti-inflammatory responses of peripheral blood mononuclear cells induced by Staphylococcus aureus and Pseudomonas aeruginosa phages. Sci. Rep. 2017, 7, 8004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gogokhia, L.; Buhrke, K.; Bell, R.; Hoffman, B.; Brown, D.G.; Hanke-Gogokhia, C.; Ajami, N.J.; Wong, M.C.; Ghazaryan, A.; Valentine, J.F.; et al. Expansion of Bacteriophages Is Linked to Aggravated Intestinal Inflammation and Colitis. Cell Host Microbe 2019, 25, 285–299.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, X.; Zhang, H.M.; Wu, T.T.; Xu, L.; Gan, Y.L.; Jiang, L.S.; Zhang, L.; Guo, S.L. Titer dynamic analysis of D29 within MTB-infected macrophages and effect on immune function of macrophages. Exp. Lung Res. 2014, 40, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Sarker, S.A.; McCallin, S.; Barretto, C.; Berger, B.; Pittet, A.C.; Sultana, S.; Krause, L.; Huq, S.; Bibiloni, R.; Bruttin, A.; et al. Oral T4-like phage cocktail application to healthy adult volunteers from Bangladesh. Virology 2012, 434, 222–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrovic Fabijan, A.; Lin, R.C.Y.; Ho, J.; Maddocks, S.; Ben Zakour, N.L.; Iredell, J.R.; Westmead Bacteriophage Therapy Team. Safety of bacteriophage therapy in severe Staphylococcus aureus infection. Nat. Microbiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Pelfrene, E.; Willebrand, E.; Cavaleiro Sanches, A.; Sebris, Z.; Cavaleri, M. Bacteriophage therapy: A regulatory perspective. J. Antimicrob. Chemother. 2016, 71, 2071–2074. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Borg, R.E.; Dow, L.P.; Pruitt, B.L.; Chen, I.A. Controlled phage therapy by photothermal ablation of specific bacterial species using gold nanorods targeted by chimeric phages. Proc. Natl. Acad. Sci. USA 2020, 117, 1951–1961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luong, T.; Salabarria, A.C.; Edwards, R.A.; Roach, D.R. Standardized bacteriophage purification for personalized phage therapy. Nat. Protoc. 2020, 15, 2867–2890. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Applications | Notes |
|---|---|
| Typing of clinical Mycobacterium isolates | • Use of pre-defined host-specific mycobacteriophage sets to identify new M. tuberculosis isolates. |
| TB diagnostics | • Use of reporter phages (e.g., LRP, Φ2DRMs, etc) to determine the presence of viable bacilli (active disease), M. tuberculosis drug susceptibility, and TB treatment efficacy (e.g., identifying presence of persister bacilli). |
| Mycobacterial genomic tools |
|
| Prophylaxis and therapeutics |
|
| Challenges and Limitations | Potential Solutions |
|---|---|
| Host specificity |
|
| Unknown impact of human ALF on the M.tb cell envelope | • Identify how the M.tb cell envelope adapts (changes) to the different environments that encounters at different stages of infection [e.g., contact with ALF, within the phagosome, extracellular, within granulomas or cavities, or when being transmitted (in sputum)]. |
| Phages access to intracellular M.tb |
|
| M.tb resistance to phages |
|
| Overactivation of the mammalian host immune system and risk of anaphylaxis |
|
| Lack of phage therapy regulations | • Standardize global regulations for phage production (under GMP conditions). |
| Phage cytotoxicity to the human host |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Allué-Guardia, A.; Saranathan, R.; Chan, J.; Torrelles, J.B. Mycobacteriophages as Potential Therapeutic Agents against Drug-Resistant Tuberculosis. Int. J. Mol. Sci. 2021, 22, 735. https://doi.org/10.3390/ijms22020735
Allué-Guardia A, Saranathan R, Chan J, Torrelles JB. Mycobacteriophages as Potential Therapeutic Agents against Drug-Resistant Tuberculosis. International Journal of Molecular Sciences. 2021; 22(2):735. https://doi.org/10.3390/ijms22020735
Chicago/Turabian StyleAllué-Guardia, Anna, Rajagopalan Saranathan, John Chan, and Jordi B. Torrelles. 2021. "Mycobacteriophages as Potential Therapeutic Agents against Drug-Resistant Tuberculosis" International Journal of Molecular Sciences 22, no. 2: 735. https://doi.org/10.3390/ijms22020735
APA StyleAllué-Guardia, A., Saranathan, R., Chan, J., & Torrelles, J. B. (2021). Mycobacteriophages as Potential Therapeutic Agents against Drug-Resistant Tuberculosis. International Journal of Molecular Sciences, 22(2), 735. https://doi.org/10.3390/ijms22020735