P2Y11 Agonism Prevents Hypoxia/Reoxygenation- and Angiotensin II-Induced Vascular Dysfunction and Intimal Hyperplasia Development


 and
and
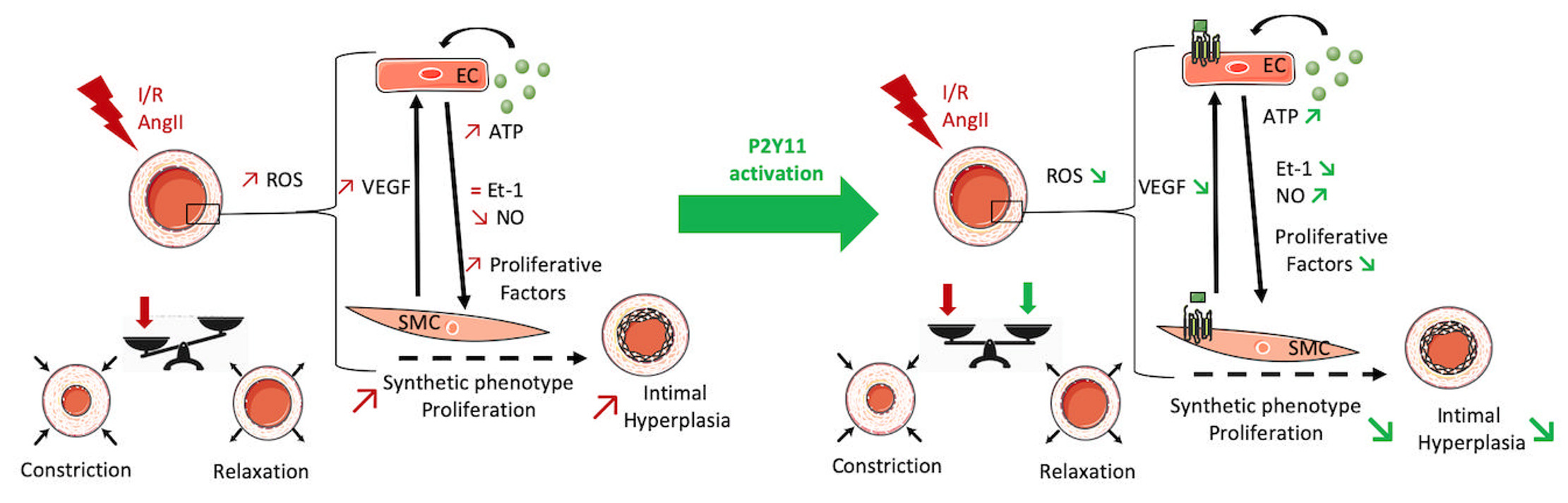
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
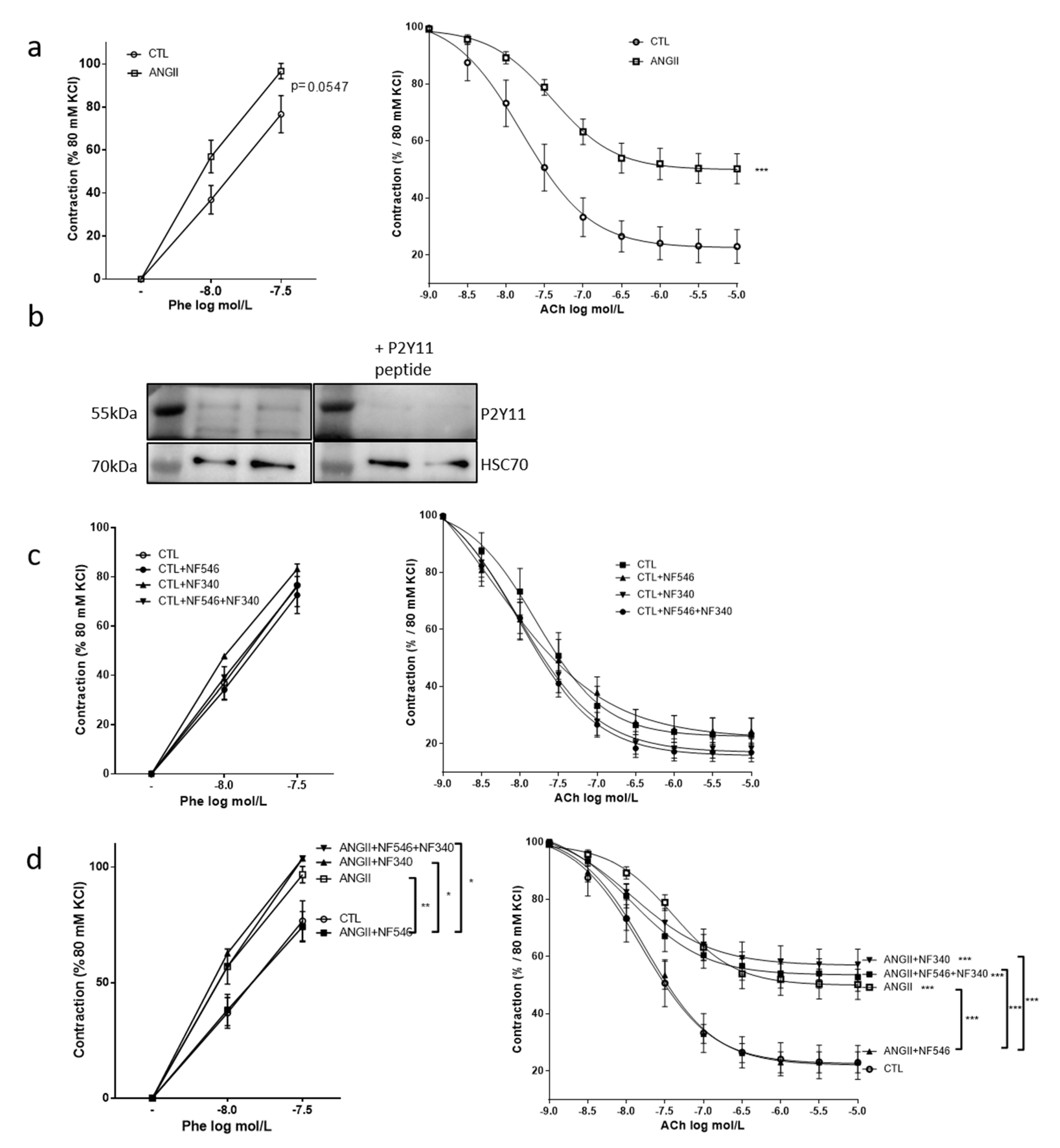
2.1. The P2Y11 Agonist NF546 Improves Angiotensin II—Induced Vascular Dysfunction
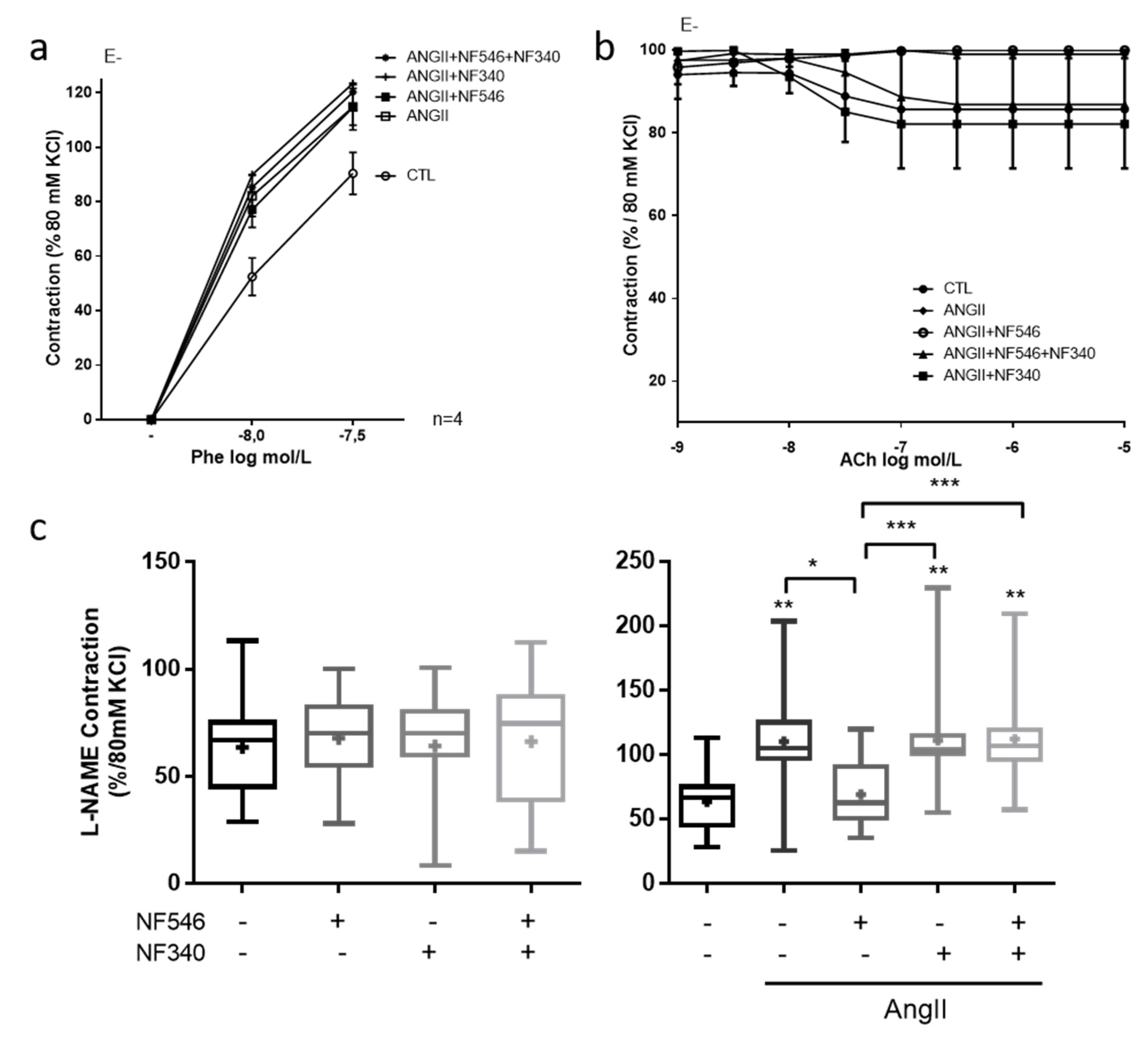
2.2. Beneficial Effect of P2Y11 Agonist Is Dependent on Endothelial Cells
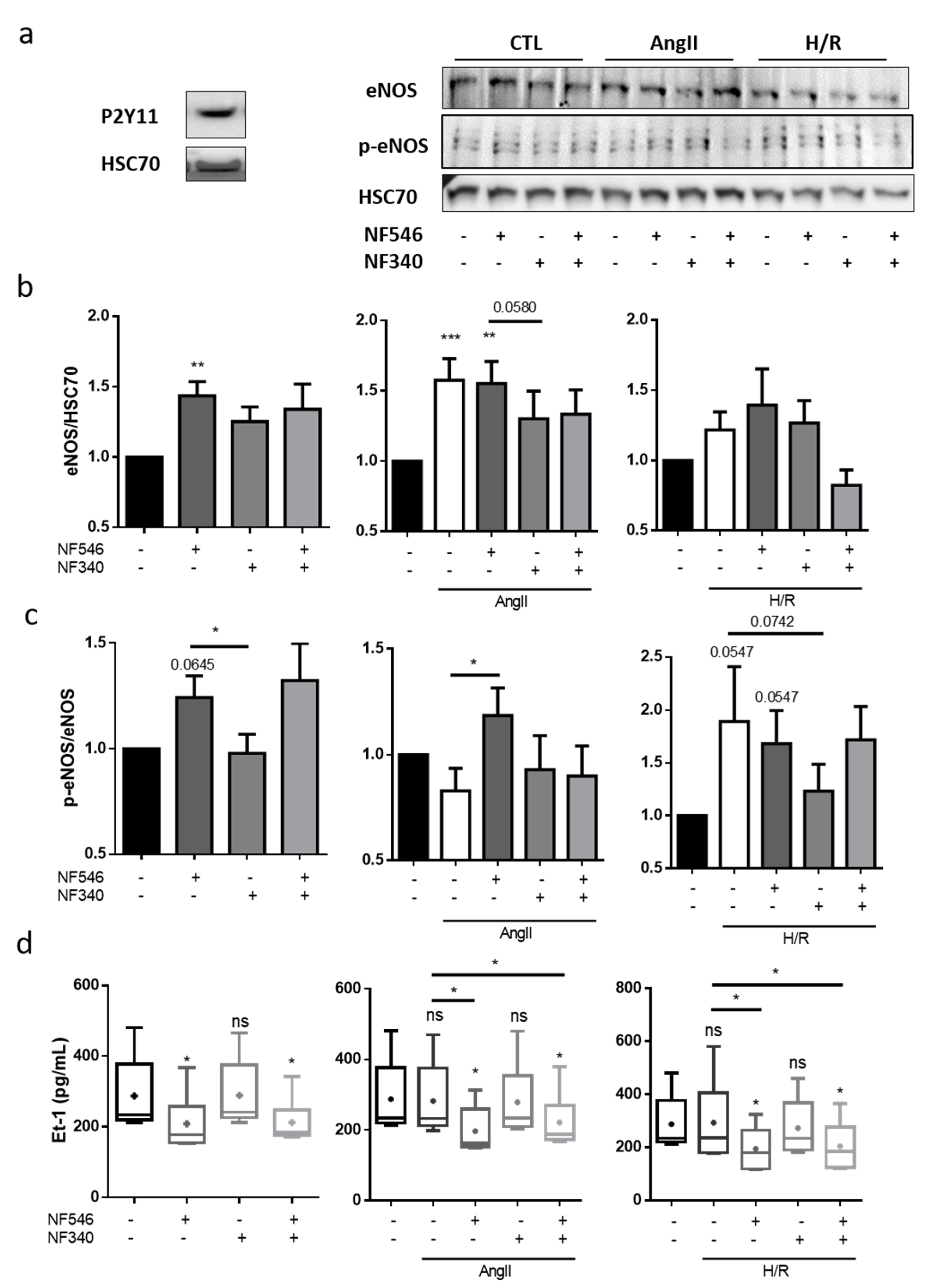
2.3. P2Y11 Agonist Improves Nitric Oxide Bioavailability
2.4. Vascular Tone Regulation by EC Is Modulated by P2Y11R in Favor of Relaxation
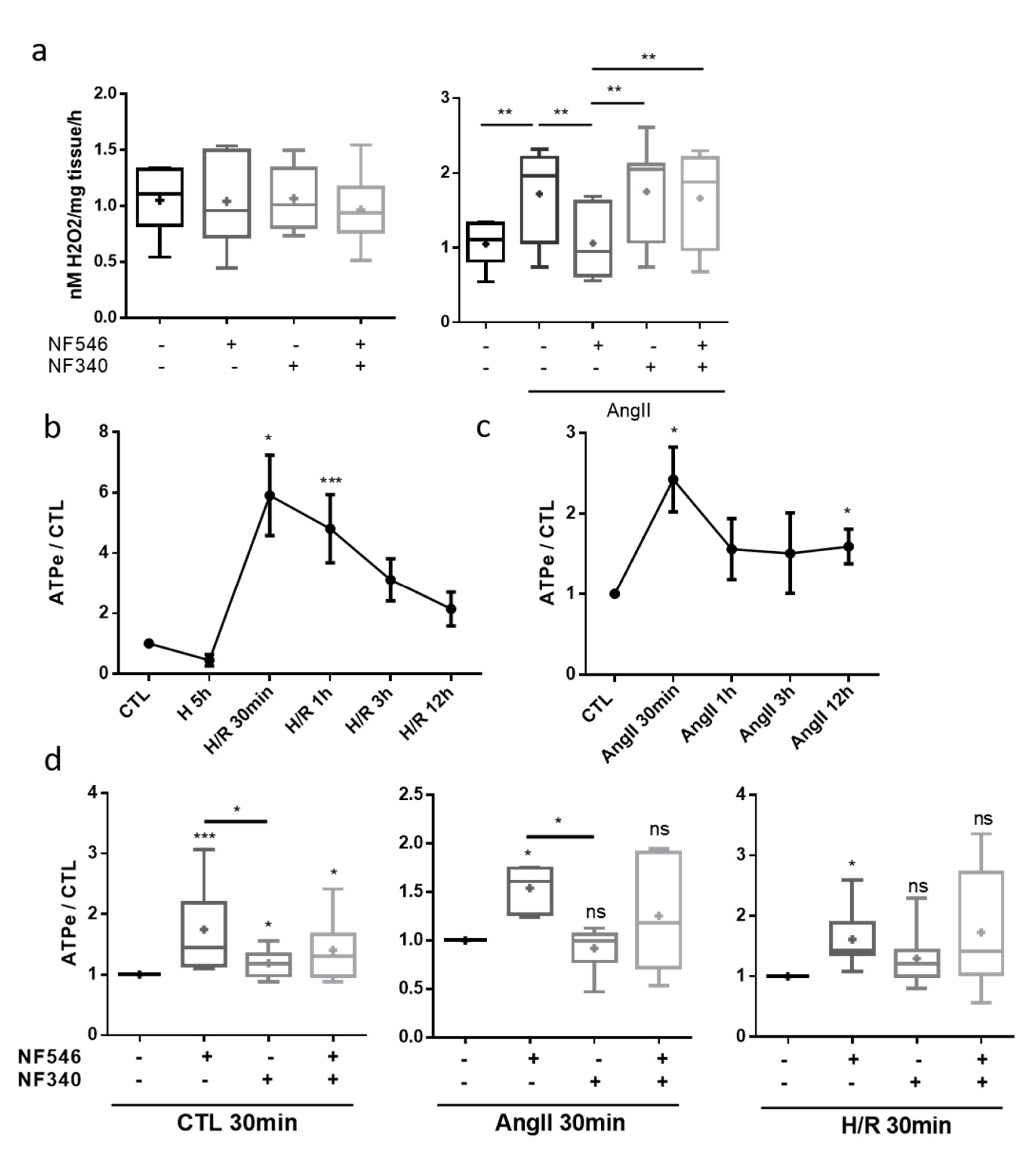
2.5. P2Y11 Agonist NF546 Decreases H2O2 Production in Response to Angiotensin II Exposure
2.6. Endothelial Cells Release eATP after Stress Exposure
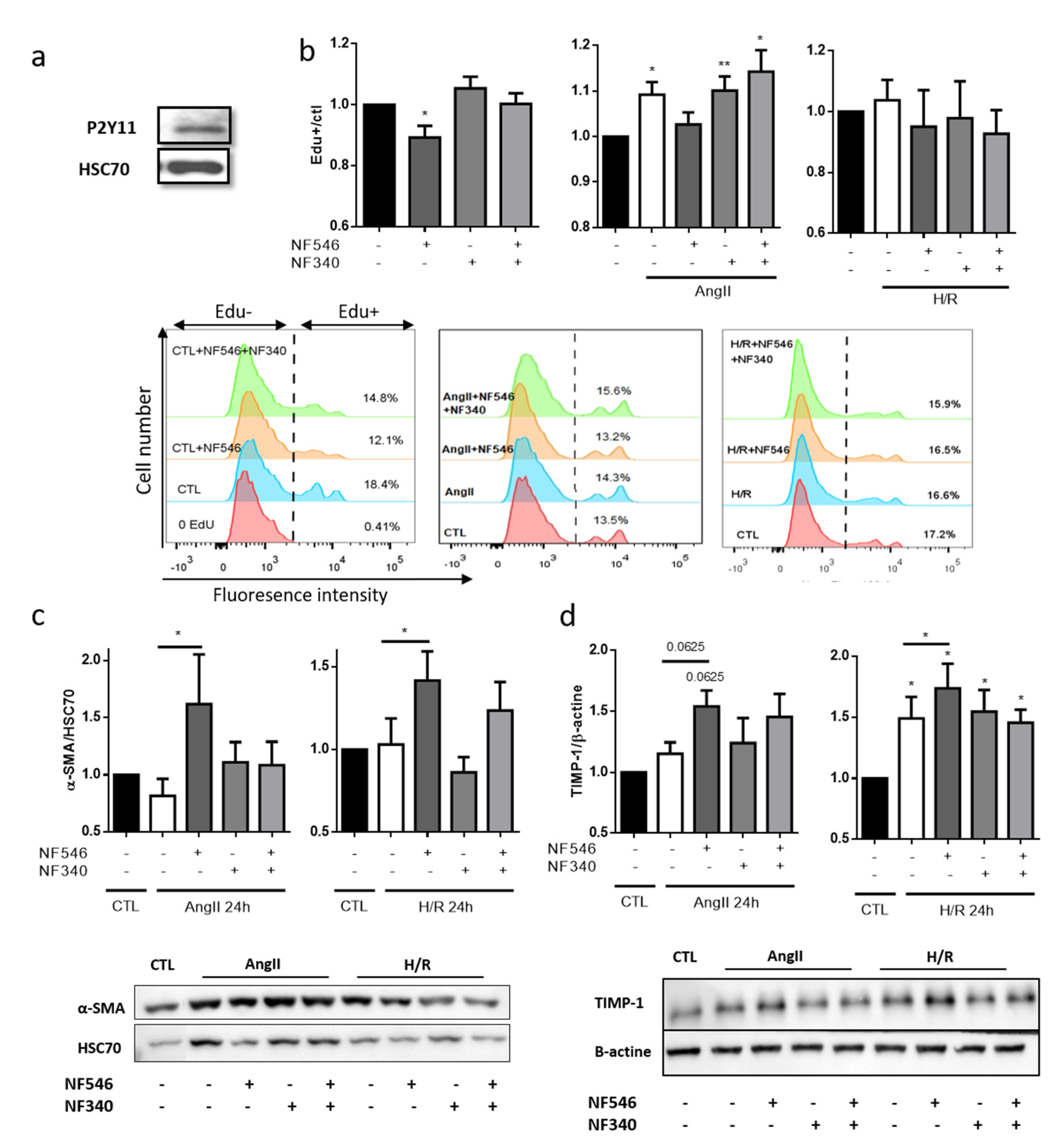
2.7. P2Y11R Activation Decreases SMC Proliferation in Basal and Stress Conditions but Not after Hypoxia
2.8. P2Y11 Activation Decreases SMC Phenotype Switch Toward Synthetic Phenotype
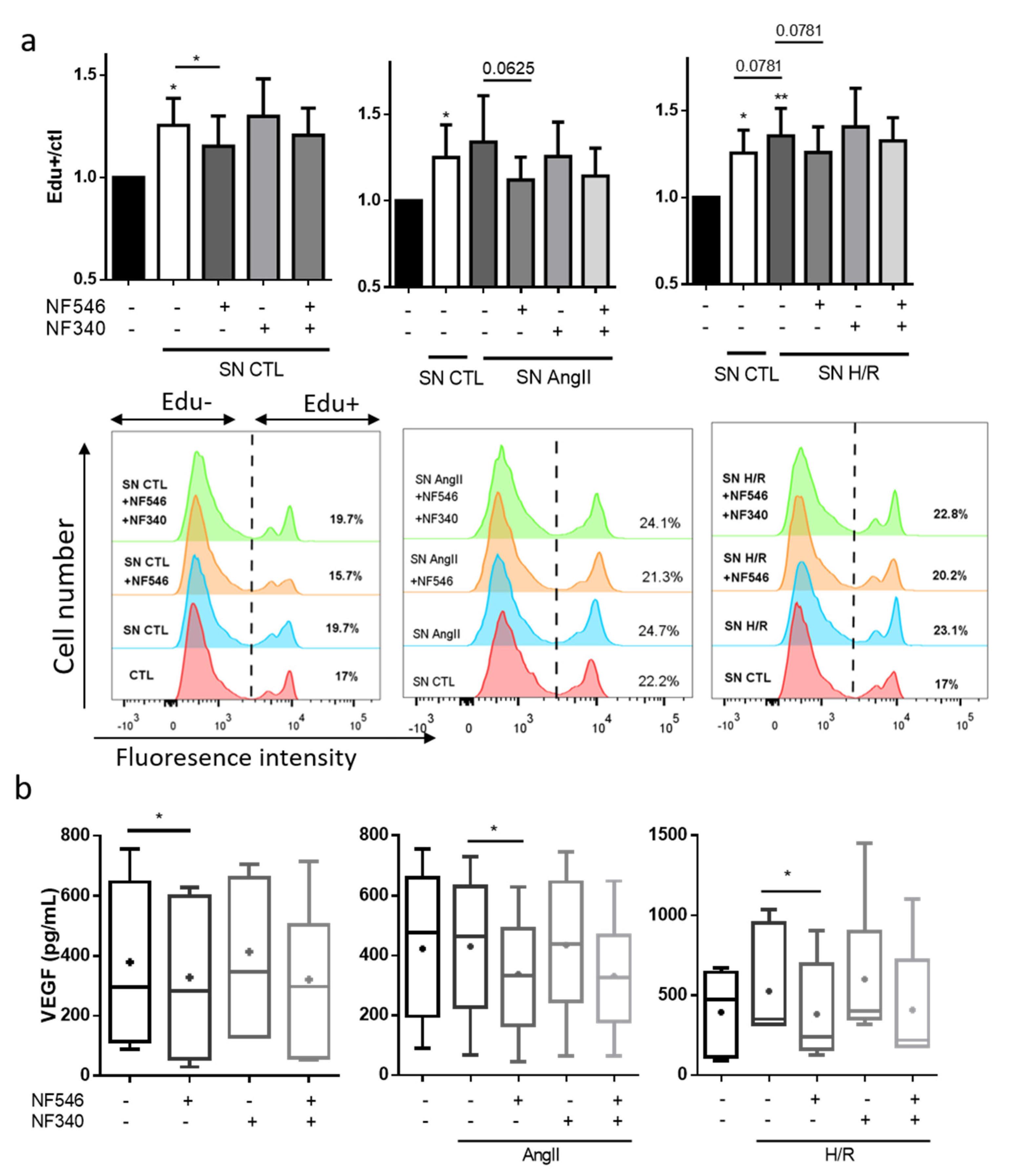
2.9. P2Y11 Activation Prevents SMC Proliferation Induced by Endothelial Cells Exposed to HR or AngII
2.10. SMC VEGF Secretion Is Modulated by P2Y11R
3. Discussion
4. Materials and Methods
4.1. Aortic Rings Preparation
4.2. Vascular Function Analysis
4.3. Cell Culture and Reagents
4.4. ROS Measurement
4.5. Secretome Analyses
4.6. Western Blot
4.7. Evaluation of HCASMC Proliferation
4.8. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AngII | angiotensin II |
| α-SMA | alpha-smooth muscle actin |
| AT1 | angiotensin II type 1 receptor |
| ATP | adenosine triphosphate |
| CTL | control |
| eATP | extracellular adenosine triphosphate |
| EC | endothelial cell |
| eNOS | endothelial nitric oxyde synthase |
| Et-1 | endotheline-1 |
| H/R | hypoxia/reoxygenation |
| HCASMC | human coronary artery smooth muscle cells |
| HSC70 | heat shock cognate 70 protein |
| HUVEC | human umbilical vein endothelial cells |
| I/R | ischemia/reperfusion |
| L-NAME | N(ω)-nitro-L-arginine methyl ester |
| LPS | lipopolysaccharide |
| MMP-2 | matrix metalloProteinase 2 |
| NO | nitric oxide |
| P2Y11R | P2Y11 receptor |
| TIMP-1 | tissue inhibitor of matrix metalloproteinases 1 |
References
- Wu, C.-H.; Mohammadmoradi, S.; Chen, J.Z.; Sawada, H.; Daugherty, A.; Lu, H.S. Renin-Angiotensin System and Cardiovascular Functions. Arter. Thromb. Vasc. Biol. 2018, 38, e108–e116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montezano, A.C.; Cat, A.N.D.; Rios, F.J.; Touyz, R.M. Angiotensin II and Vascular Injury. Curr. Hypertens. Rep. 2014, 16, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Li, D.; Phillips, M.I.; Mehta, P.; Mehta, J.L. Myocardial angiotensin II receptor expression and ischemia-reperfusion injury. Vasc. Med. 1998, 3, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, S.; Kawai, T.; Scalia, R.; Rizzo, V. Understanding Angiotensin II Type 1 Receptor Signaling in Vascular Pathophysiology. Hypertension 2018, 71, 804–810. [Google Scholar] [CrossRef]
- Mu, D.N.; Dechend, R.; Fiebeler, A.; Park, J.; Schmidt, F.; Breu, V.; Mackman, N.; Luther, T.; Schneider, W.; Gulba, D.; et al. Angiotensin II (AT 1) Receptor Blockade Reduces Vascular Tissue Factor in Angiotensin II-Induced Cardiac Vasculopathy. Am. J. Pathol. 2000, 157, 111–122. [Google Scholar]
- Hausenloy, D.J.; Chilian, W.; Crea, F.; Davidson, S.M.; Bencsik, P.; Garcia-Dorado, D.; Van Royen, N.; Schulz, R.; Heusch, G. The coronary circulation in acute myocardial ischaemia/reperfusion injury: A target for cardioprotection. Cardiovasc. Res. 2019, 115, 1143–1155. [Google Scholar] [CrossRef]
- Zhao, Z.-Q.; Corvera, J.S.; Halkos, M.E.; Kerendi, F.; Wang, N.-P.; Guyton, R.A.; Vinten-Johansen, J. Inhibition of myocardial injury by ischemic postconditioning during reperfusion: Comparison with ischemic preconditioning. Am. J. Physiol. Circ. Physiol. 2003, 285, H579–H588. [Google Scholar] [CrossRef]
- Zhao, J.-L.; Yang, Y.; You, S.-J.; Cui, C.-J.; Gao, R. Different effects of postconditioning on myocardial no-reflow in the normal and hypercholesterolemic mini-swines. Microvasc. Res. 2007, 73, 137–142. [Google Scholar] [CrossRef]
- Galaup, A.; Gomez, E.; Souktani, R.; Durand, M.; Cazes, A.; Monnot, C.; Teillon, J.; Le Jan, S.; Bouleti, C.; Briois, G.; et al. Protection Against Myocardial Infarction and No-Reflow Through Preservation of Vascular Integrity by Angiopoietin-Like 4. Circulation 2012, 125, 140–149. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Mather, S.; Huang, Y.; Garland, C.J.; Yao, X. Extracellular ATP facilitates flow-induced vasodilatation in rat small mesenteric arteries. Am. J. Physiol. Circ. Physiol. 2004, 286, H1688–H1695. [Google Scholar] [CrossRef]
- Dănilă, M.D.; Privistirescu, A.; Duicu, O.M.; Rațiu, C.D.; Angoulvant, D.; Muntean, D.M.; Sturza, A. The effect of purinergic signaling via the P2Y11 receptor on vascular function in a rat model of acute inflammation. Mol. Cell. Biochem. 2017, 431, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Benoist, L.; Chadet, S.; Genet, T.; Lefort, C.; Heraud, A.; Danila, M.D.; Muntean, D.M.; Baron, C.; Angoulvant, D.; Babuty, D.; et al. Stimulation of P2Y11 receptor protects human cardiomyocytes against Hypoxia/Reoxygenation injury and involves PKCε signaling pathway. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefort, C.; Benoist, L.; Chadet, S.; Piollet, M.; Heraud, A.; Babuty, M.; Baron, C.; Ivanes, F.; Angoulvant, D. Stimulation of P2Y11 receptor modulates cardiac fibroblasts secretome toward immunomodulatory and protective roles after Hypoxia/Reoxygenation injury. J. Mol. Cell. Cardiol. 2018, 121, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Bourguignon, T.; Benoist, L.; Chadet, S.; Miquelestorena-Standley, E.; Fromont, G.; Ivanes, F.; Angoulvant, D. Stimulation of murine P2Y11-like purinoreceptor protects against hypoxia/reoxygenation injury and decreases heart graft rejection lesions. J. Thorac. Cardiovasc. Surg. 2019, 158, 780–790. [Google Scholar] [CrossRef]
- Chadet, S.; Ivanes, F.; Benoist, L.; Gandonnière, C.S.; Guibon, R.; Velge-Roussel, F.; Babuty, M.; Baron, C.; Roger, S.; Angoulvant, D. Hypoxia/Reoxygenation Inhibits P2Y11 Receptor Expression and Its Immunosuppressive Activity in Human Dendritic Cells. J. Immunol. 2015, 195, 651–660. [Google Scholar] [CrossRef] [Green Version]
- Balogh, J.; Wihlborg, A.-K.; Isackson, H.; Joshi, B.V.; Jacobson, K.A.; Arner, A.; Erlinge, D. Phospholipase C and cAMP-dependent positive inotropic effects of ATP in mouse cardiomyocytes via P2Y-like receptors. J. Mol. Cell. Cardiol. 2005, 39, 223–230. [Google Scholar] [CrossRef] [Green Version]
- Böhm, F.; Pernow, J. The importance of endothelin-1 for vascular dysfunction in cardiovascular disease. Cardiovasc. Res. 2007, 76, 8–18. [Google Scholar] [CrossRef] [Green Version]
- Geraghty, J.G.; Stoltenberg, R.L.; Sollinger, H.W.; Hullett, D.A. Vascular Smooth Muscle Cells And Neointimal Hyperplasia In Chronic Transplant Rejection. Transplantation 1996, 62, 502–509. [Google Scholar] [CrossRef]
- Prada, M.P.; Syed, A.U.; Buonarati, O.R.; Reddy, G.R.; Nystoriak, M.A.; Ghosh, D.; Simó, S.; Sato, D.; Sasse, K.C.; Ward, S.M.; et al. A Gs-coupled purinergic receptor boosts Ca2+ influx and vascular contractility during diabetic hyperglycemia. eLife 2019, 8, 1–37. [Google Scholar] [CrossRef]
- Tousoulis, D.; Kampoli, A.-M.; Papageorgiou, C.T.N.; Stefanadis, C. The Role of Nitric Oxide on Endothelial Function. Curr. Vasc. Pharmacol. 2012, 10, 4–18. [Google Scholar] [CrossRef]
- Suzuki, H.; Eguchi, K.; Ohtsu, H.; Higuchi, S.; Dhobale, S.; Frank, G.D.; Motley, E.D.; Eguchi, S. Activation of Endothelial Nitric Oxide Synthase by the Angiotensin II Type 1 Receptor. Endocrinology 2006, 147, 5914–5920. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; He, G.-W.; Xue, H.-M.; Yao, X.; Liu, X.-C.; Underwood, M.J.; Yang, Q. TRPC3 channel contributes to nitric oxide release: Significance during normoxia and hypoxia–reoxygenation. Cardiovasc. Res. 2011, 91, 472–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, M.; Ding, J.; Jiang, H.; Kong, L.; Sun, Z.; Chen, J.-W.; Miao, C. Propofol ameliorates endothelial inflammation induced by hypoxia/reoxygenation in human umbilical vein endothelial cells: Role of phosphatase A2. Vasc. Pharmacol. 2015, 73, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Kalinowski, L.; Dobrucki, L.W.; Szczepanska-Konkel, M.; Jankowski, M.; Martyniec, L.; Angielski, S.; Malinski, T. Third-Generation β-Blockers Stimulate Nitric Oxide Release From Endothelial Cells Through ATP Efflux. Circulation 2003, 107, 2747–2752. [Google Scholar] [CrossRef] [Green Version]
- Da Silva, C.G.; Specht, A.; Wegiel, B.; Ferran, C.; Kaczmarek, E. Mechanism of Purinergic Activation of Endothelial Nitric Oxide Synthase in Endothelial Cells. Circulation 2009, 119, 871–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, H.; Harrison, D.G. Endothelial Dysfunction in Cardiovascular Diseases: The Role of Oxidant Stress. Circ. Res. 2000, 87, 840–844. [Google Scholar] [CrossRef] [Green Version]
- Görlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, R.N.; Libby, P. Vascular Remodeling in Transplant Vasculopathy. Circ. Res. 2007, 100, 967–978. [Google Scholar] [CrossRef]
- Waybill, P.N.; Chinchilli, V.M.; Ballermann, B.J. Smooth Muscle Cell Proliferation in Response to Co-culture with Venous and Arterial Endothelial Cells. J. Vasc. Interv. Radiol. 1997, 8, 375–381. [Google Scholar] [CrossRef]
- Zhou, Y.; Dirksen, W.P.; Babu, G.J.; Periasamy, M. Differential vasoconstrictions induced by angiotensin II: Role of AT1 and AT2 receptors in isolated C57BL/6J mouse blood vessels. Am. J. Physiol. Circ. Physiol. 2003, 285, H2797–H2803. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, C. P2Y11 Receptors: Properties, Distribution and Functions. Cannabinoids Neuropsychiatr. Disord. 2017, 1051, 107–122. [Google Scholar] [CrossRef] [Green Version]
- Dănilă, M.-D.; Piollet, M.; Aburel, O.-M.; Angoulvant, D.; Lefort, C.; Chadet, S.; Roger, S.; Muntean, D.M.; Ivanes, F. Modulation of P2Y11-related purinergic signaling in inflammation and cardio-metabolic diseases. Eur. J. Pharmacol. 2020, 876, 173060. [Google Scholar] [CrossRef] [PubMed]
- Xie, N.; Wang, C.; Lian, Y.; Wu, C.; Zhang, H.; Zhang, Q. Inhibition of mitochondrial fission attenuates Aβ-induced microglia apoptosis. Neuroscience 2014, 256, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Brandes, R.P. Endothelial Dysfunction and Hypertension. Hypertension 2014, 64, 924–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meis, S.; Hamacher, A.; Hongwiset, D.; Marzian, C.; Wiese, M.; Eckstein, N.; Royer, H.-D.; Communi, D.; Boeynaems, J.-M.; Hausmann, R.; et al. NF546 [4,4′-(Carbonylbis(imino-3,1-phenylene-carbonylimino-3,1-(4-methyl-phenylene)-carbonylimino))-bis(1,3-xylene-α,α′-diphosphonic Acid) Tetrasodium Salt] Is a Non-Nucleotide P2Y11 Agonist and Stimulates Release of Interleukin-8 from Human Monocyte-Derived Dendritic Cells. J. Pharmacol. Exp. Ther. 2009, 332, 238–247. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piollet, M.; Sturza, A.; Chadet, S.; Gabillard-Lefort, C.; Benoist, L.; Muntean, D.-M.; Aburel, O.-M.; Angoulvant, D.; Ivanes, F. P2Y11 Agonism Prevents Hypoxia/Reoxygenation- and Angiotensin II-Induced Vascular Dysfunction and Intimal Hyperplasia Development. Int. J. Mol. Sci. 2021, 22, 855. https://doi.org/10.3390/ijms22020855
Piollet M, Sturza A, Chadet S, Gabillard-Lefort C, Benoist L, Muntean D-M, Aburel O-M, Angoulvant D, Ivanes F. P2Y11 Agonism Prevents Hypoxia/Reoxygenation- and Angiotensin II-Induced Vascular Dysfunction and Intimal Hyperplasia Development. International Journal of Molecular Sciences. 2021; 22(2):855. https://doi.org/10.3390/ijms22020855
Chicago/Turabian StylePiollet, Marie, Adrian Sturza, Stéphanie Chadet, Claudie Gabillard-Lefort, Lauriane Benoist, Danina-Mirela Muntean, Oana-Maria Aburel, Denis Angoulvant, and Fabrice Ivanes. 2021. "P2Y11 Agonism Prevents Hypoxia/Reoxygenation- and Angiotensin II-Induced Vascular Dysfunction and Intimal Hyperplasia Development" International Journal of Molecular Sciences 22, no. 2: 855. https://doi.org/10.3390/ijms22020855
APA StylePiollet, M., Sturza, A., Chadet, S., Gabillard-Lefort, C., Benoist, L., Muntean, D. -M., Aburel, O. -M., Angoulvant, D., & Ivanes, F. (2021). P2Y11 Agonism Prevents Hypoxia/Reoxygenation- and Angiotensin II-Induced Vascular Dysfunction and Intimal Hyperplasia Development. International Journal of Molecular Sciences, 22(2), 855. https://doi.org/10.3390/ijms22020855