Functional Inactivation of Drosophila GCK Orthologs Causes Genomic Instability and Oxidative Stress in a Fly Model of MODY-2

 ,
,  , , , ,
, , , ,  ,
,  ,
,  and
and 
Abstract
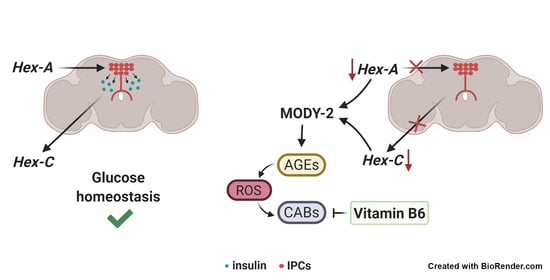
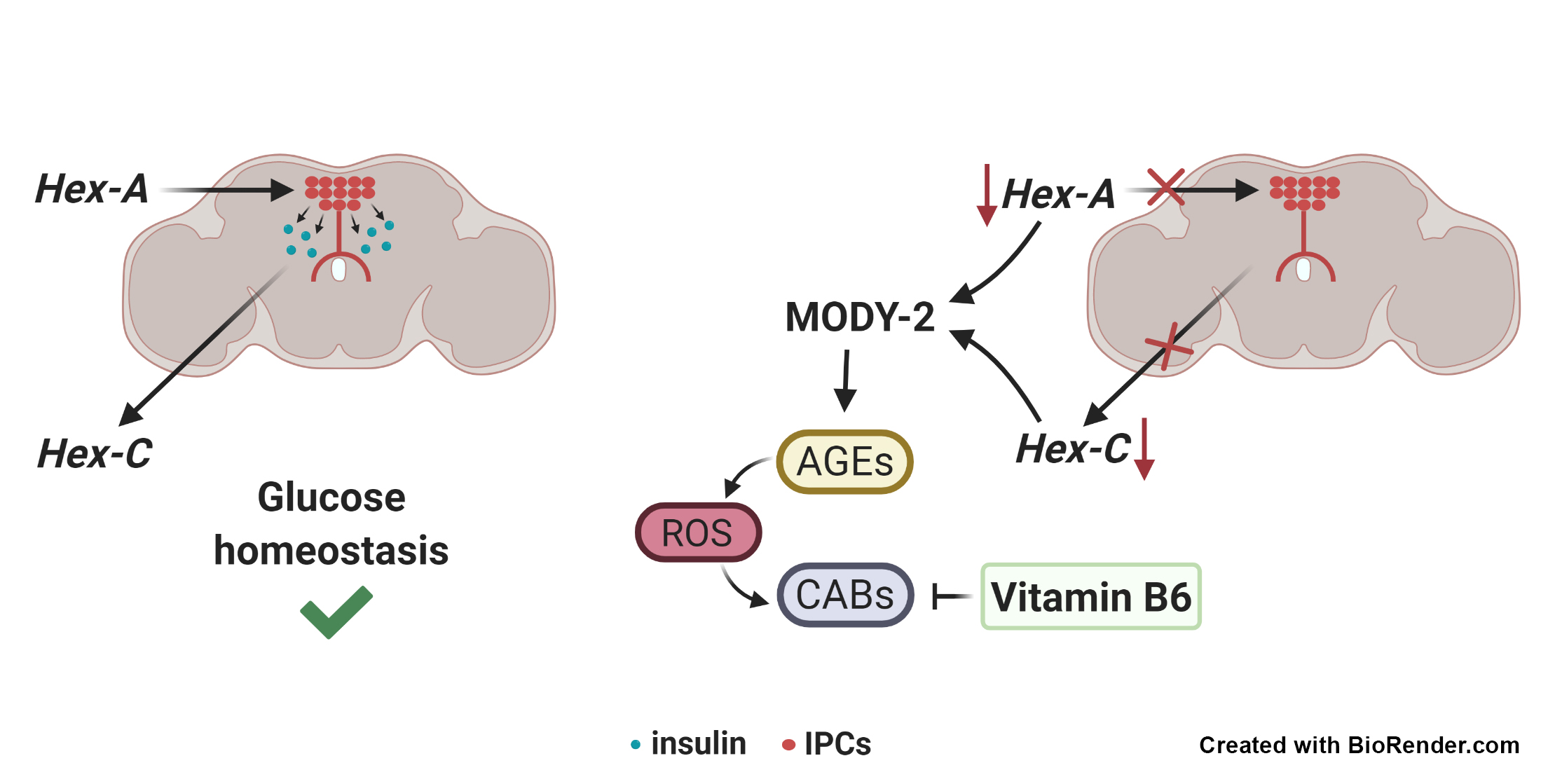
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Tissue Specific Silencing of Two Orthologs of Mammalian GCK Causes MODY-2 in Drosophila
2.2. Hex-A Is Involved in Insulin Secretion
2.3. Hex-A Regulates Hex-C Expression
2.4. The Depletion of Either Hex-A or Hex-C Causes Chromosome Aberrations
2.5. Hex-A- and Hex-C-Depleted Cells Accumulate Advanced Glycation End-Products (AGEs)
2.6. Vitamin B6 Prevents Chromosome Damage in Drosophila MODY-2
3. Discussion
4. Materials and Methods
4.1. Drosophila Stocks and Crosses
4.2. Fly Food Recipes
4.3. Chromosome Cytology
4.4. Immunostaining of Larval and Adult Drosophila Brains
4.5. Treatments of Larvae and Isolated Brains
4.6. Glucose Measurement and Weight Analysis
4.7. Lipid Droplet Measurement
4.8. Nitroblue Tetrazolium (NBT) Assay
4.9. RNA Extraction, Reverse Transcription, and RT-qPCR
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Conflicts of Interest
Abbreviations
| 4-DP | 4-deoxypiridoxine |
| AGEs | Advanced glycation end-products |
| CABs | Chromosome aberrations |
| DILP2 | Drosophila insulin-like protein 2 |
| GCK | Glucokinase |
| GKRP | Glucokinase regulatory protein |
| FB | Fat body |
| HSD | High sugar diet |
| IPCs | Insulin producing cells |
| LDs | Lipid droplets |
| MODY | Maturity-onset diabetes of the young |
| NBT | Nitroblue tetrazolium |
| PFK2/FBP2 | 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatase |
| PLP | Pyridoxal 5′-phosphate |
| ROS | Reactive oxygen species |
References
- Carmody, D.; Stoy, J.; Greeley, S.A.W.; Bell, G.I.; Philipson, L.H. Chapter 2—A clinical guide to monogenic diabetes. In Genetic Diagnosis of Endocrine Disorders (Second Edition); Weiss, R.E., Refetoff, S., Eds.; Academic Press: Cambridge, MA, USA, 2016; pp. 21–30. [Google Scholar]
- Association, A.D. 2. Classification and Diagnosis of Diabetes. Diabetes Care 2020, 43, S14–S31. [Google Scholar] [CrossRef] [Green Version]
- Delvecchio, M.; Pastore, C.; Giordano, P. Treatment Options for MODY Patients: A Systematic Review of Literature. Diabetes Ther. 2020, 11, 1667–1685. [Google Scholar] [CrossRef] [PubMed]
- Froguel, P.; Vaxillaire, M.; Sun, F.; Velho, G.; Zouali, H.; Butel, M.O.; Lesage, S.; Vionnet, N.; Clément, K.; Fougerousse, F. Close linkage of glucokinase locus on chromosome 7p to early-onset non-insulin-dependent diabetes mellitus. Nature 1992, 356, 162–164. [Google Scholar] [CrossRef] [PubMed]
- Vionnet, N.; Stoffel, M.; Takeda, J.; Yasuda, K.; Bell, G.I.; Zouali, H.; Lesage, S.; Velho, G.; Iris, F.; Passa, P. Nonsense mutation in the glucokinase gene causes early-onset non-insulin-dependent diabetes mellitus. Nature 1992, 356, 721–722. [Google Scholar] [CrossRef] [PubMed]
- Iynedjian, P.B. Molecular physiology of mammalian glucokinase. Cell. Mol. Life Sci. 2009, 66, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Iynedjian, P.B. Mammalian glucokinase and its gene. Biochem. J. 1993, 293, 1–13. [Google Scholar] [CrossRef]
- Magnuson, M.A. Glucokinase gene structure. Functional implications of molecular genetic studies. Diabetes 1990, 39, 523–527. [Google Scholar] [CrossRef]
- Postic, C.; Shiota, M.; Magnuson, M.A. Cell-specific roles of glucokinase in glucose homeostasis. Recent Prog. Horm. Res. 2001, 56, 195–217. [Google Scholar] [CrossRef] [Green Version]
- Agius, L. Glucokinase and molecular aspects of liver glycogen metabolism. Biochem. J. 2008, 414, 1–18. [Google Scholar] [CrossRef] [Green Version]
- De Backer, I.; Hussain, S.S.; Bloom, S.R.; Gardiner, J.V. Insights into the role of neuronal glucokinase. Am. J. Physiol. Endocrinol. Metab. 2016, 311, E42–E55. [Google Scholar] [CrossRef] [Green Version]
- Girard, J.; Ferré, P.; Foufelle, F. Mechanisms by which carbohydrates regulate expression of genes for glycolytic and lipogenic enzymes. Annu. Rev. Nutr. 1997, 17, 325–352. [Google Scholar] [CrossRef] [PubMed]
- Irwin, D.M.; Tan, H. Evolution of glucose utilization: Glucokinase and glucokinase regulator protein. Mol. Phylogenet. Evol. 2014, 70, 195–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agius, L. Hormonal and Metabolite Regulation of Hepatic Glucokinase. Annu. Rev. Nutr. 2016, 36, 389–415. [Google Scholar] [CrossRef] [PubMed]
- Sternisha, S.M.; Miller, B.G. Molecular and cellular regulation of human glucokinase. Arch. Biochem. Biophys. 2019, 663, 199–213. [Google Scholar] [CrossRef]
- Rizzo, M.A.; Magnuson, M.A.; Drain, P.F.; Piston, D.W. A functional link between glucokinase binding to insulin granules and conformational alterations in response to glucose and insulin. J. Biol. Chem. 2002, 277, 34168–34175. [Google Scholar] [CrossRef] [Green Version]
- Arden, C.; Harbottle, A.; Baltrusch, S.; Tiedge, M.; Agius, L. Glucokinase is an integral component of the insulin granules in glucose-responsive insulin secretory cells and does not translocate during glucose stimulation. Diabetes 2004, 53, 2346–2352. [Google Scholar] [CrossRef] [Green Version]
- Velho, G.; Blanché, H.; Vaxillaire, M.; Bellanné-Chantelot, C.; Pardini, V.C.; Timsit, J.; Passa, P.; Deschamps, I.; Robert, J.J.; Weber, I.T.; et al. Identification of 14 new glucokinase mutations and description of the clinical profile of 42 MODY-2 families. Diabetologia 1997, 40, 217–224. [Google Scholar] [CrossRef] [Green Version]
- Velho, G.; Froguel, P.; Clement, K.; Pueyo, M.E.; Rakotoambinina, B.; Zouali, H.; Passa, P.; Cohen, D.; Robert, J.J. Primary pancreatic beta-cell secretory defect caused by mutations in glucokinase gene in kindreds of maturity onset diabetes of the young. Lancet 1992, 340, 444–448. [Google Scholar] [CrossRef]
- Stride, A.; Vaxillaire, M.; Tuomi, T.; Barbetti, F.; Njølstad, P.R.; Hansen, T.; Costa, A.; Conget, I.; Pedersen, O.; Søvik, O.; et al. The genetic abnormality in the beta cell determines the response to an oral glucose load. Diabetologia 2002, 45, 427–435. [Google Scholar] [CrossRef]
- Velho, G.; Petersen, K.F.; Perseghin, G.; Hwang, J.H.; Rothman, D.L.; Pueyo, M.E.; Cline, G.W.; Froguel, P.; Shulman, G.I. Impaired hepatic glycogen synthesis in glucokinase-deficient (MODY-2) subjects. J. Clin. Investig. 1996, 98, 1755–1761. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.; Bellanné-Chantelot, C.; Deschamps, I.; Froguel, P.; Robert, J.J.; Velho, G. Long-term follow-up of oral glucose tolerance test-derived glucose tolerance and insulin secretion and insulin sensitivity indexes in subjects with glucokinase mutations (MODY2). Diabetes Care 2008, 31, 1321–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellard, S.; Bellanné-Chantelot, C.; Hattersley, A.T.; European Molecular Genetics Quality Network (EMQN) MODY group. Best practice guidelines for the molecular genetic diagnosis of maturity-onset diabetes of the young. Diabetologia 2008, 51, 546–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gill-Carey, O.; Hattersley, A.T. Genetics and type 2 diabetes in youth. Pediatr. Diabetes 2007, 8, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Sagen, J.V.; Bjørkhaug, L.; Molnes, J.; Raeder, H.; Grevle, L.; Søvik, O.; Molven, A.; Njølstad, P.R. Diagnostic screening of MODY2/GCK mutations in the Norwegian MODY Registry. Pediatr. Diabetes 2008, 9, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Steele, A.M.; Shields, B.M.; Wensley, K.J.; Colclough, K.; Ellard, S.; Hattersley, A.T. Prevalence of vascular complications among patients with glucokinase mutations and prolonged, mild hyperglycemia. JAMA 2014, 311, 279–286. [Google Scholar] [CrossRef] [Green Version]
- Efrat, S.; Leiser, M.; Wu, Y.J.; Fusco-DeMane, D.; Emran, O.A.; Surana, M.; Jetton, T.L.; Magnuson, M.A.; Weir, G.; Fleischer, N. Ribozyme-mediated attenuation of pancreatic beta-cell glucokinase expression in transgenic mice results in impaired glucose-induced insulin secretion. Proc. Natl. Acad. Sci. USA 1994, 91, 2051–2055. [Google Scholar] [CrossRef] [Green Version]
- Grupe, A.; Hultgren, B.; Ryan, A.; Ma, Y.H.; Bauer, M.; Stewart, T.A. Transgenic knockouts reveal a critical requirement for pancreatic beta cell glucokinase in maintaining glucose homeostasis. Cell 1995, 83, 69–78. [Google Scholar] [CrossRef] [Green Version]
- Bali, D.; Svetlanov, A.; Lee, H.W.; Fusco-DeMane, D.; Leiser, M.; Li, B.; Barzilai, N.; Surana, M.; Hou, H.; Fleischer, N. Animal model for maturity-onset diabetes of the young generated by disruption of the mouse glucokinase gene. J. Biol. Chem. 1995, 270, 21464–21467. [Google Scholar] [CrossRef] [Green Version]
- Postic, C.; Shiota, M.; Niswender, K.D.; Jetton, T.L.; Chen, Y.; Moates, J.M.; Shelton, K.D.; Lindner, J.; Cherrington, A.D.; Magnuson, M.A. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J. Biol. Chem. 1999, 274, 305–315. [Google Scholar] [CrossRef] [Green Version]
- Toye, A.A.; Moir, L.; Hugill, A.; Bentley, L.; Quarterman, J.; Mijat, V.; Hough, T.; Goldsworthy, M.; Haynes, A.; Hunter, A.J.; et al. A new mouse model of type 2 diabetes, produced by N-ethyl-nitrosourea mutagenesis, is the result of a missense mutation in the glucokinase gene. Diabetes 2004, 53, 1577–1583. [Google Scholar] [CrossRef] [Green Version]
- Inoue, M.; Sakuraba, Y.; Motegi, H.; Kubota, N.; Toki, H.; Matsui, J.; Toyoda, Y.; Miwa, I.; Terauchi, Y.; Kadowaki, T.; et al. A series of maturity onset diabetes of the young, type 2 (MODY2) mouse models generated by a large-scale ENU mutagenesis program. Hum. Mol. Genet. 2004, 13, 1147–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenner, D.; Odili, S.; Hong, H.K.; Kobayashi, Y.; Kohsaka, A.; Siepka, S.M.; Vitaterna, M.H.; Chen, P.; Zelent, B.; Grimsby, J.; et al. Generation of N-ethyl-N-nitrosourea (ENU) diabetes models in mice demonstrates genotype-specific action of glucokinase activators. J. Biol. Chem. 2011, 286, 39560–39572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aigner, B.; Rathkolb, B.; Herbach, N.; Hrabé de Angelis, M.; Wanke, R.; Wolf, E. Diabetes models by screen for hyperglycemia in phenotype-driven ENU mouse mutagenesis projects. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E232–E240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owusu-Ansah, E.; Perrimon, N. Modeling metabolic homeostasis and nutrient sensing in Drosophila: Implications for aging and metabolic diseases. Dis. Model. Mech. 2014, 7, 343–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padmanabha, D.; Baker, K.D. Drosophila gains traction as a repurposed tool to investigate metabolism. Trends Endocrinol. Metab. 2014, 25, 518–527. [Google Scholar] [CrossRef]
- Teleman, A.A.; Ratzenböck, I.; Oldham, S. Drosophila: A model for understanding obesity and diabetic complications. Exp. Clin. Endocrinol. Diabetes 2012, 120, 184–185. [Google Scholar] [CrossRef]
- Pendse, J.; Ramachandran, P.V.; Na, J.; Narisu, N.; Fink, J.L.; Cagan, R.L.; Collins, F.S.; Baranski, T.J. A Drosophila functional evaluation of candidates from human genome-wide association studies of type 2 diabetes and related metabolic traits identifies tissue-specific roles for dHHEX. BMC Genom. 2013, 14, 136. [Google Scholar] [CrossRef] [Green Version]
- Barry, W.E.; Thummel, C.S. The Drosophila HNF4 nuclear receptor promotes glucose-stimulated insulin secretion and mitochondrial function in adults. Elife 2016, 5. [Google Scholar] [CrossRef]
- Rulifson, E.J.; Kim, S.K.; Nusse, R. Ablation of insulin-producing neurons in flies: Growth and diabetic phenotypes. Science 2002, 296, 1118–1120. [Google Scholar] [CrossRef]
- Böhni, R.; Riesgo-Escovar, J.; Oldham, S.; Brogiolo, W.; Stocker, H.; Andruss, B.F.; Beckingham, K.; Hafen, E. Autonomous control of cell and organ size by CHICO, a Drosophila homolog of vertebrate IRS1-4. Cell 1999, 97, 865–875. [Google Scholar] [CrossRef] [Green Version]
- Musselman, L.P.; Fink, J.L.; Narzinski, K.; Ramachandran, P.V.; Hathiramani, S.S.; Cagan, R.L.; Baranski, T.J. A high-sugar diet produces obesity and insulin resistance in wild-type Drosophila. Dis. Model. Mech. 2011, 4, 842–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merigliano, C.; Mascolo, E.; La Torre, M.; Saggio, I.; Vernì, F. Protective role of vitamin B6 (PLP) against DNA damage in Drosophila models of type 2 diabetes. Sci. Rep. 2018, 8, 11432. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, F.M.; Harrison, D.E.; Ashcroft, S.J. Glucose induces closure of single potassium channels in isolated rat pancreatic beta-cells. Nature 1984, 312, 446–448. [Google Scholar] [CrossRef] [PubMed]
- Giacco, F.; Du, X.; Carratú, A.; Gerfen, G.J.; D’Apolito, M.; Giardino, I.; Rasola, A.; Marin, O.; Divakaruni, A.S.; Murphy, A.N.; et al. GLP-1 Cleavage Product Reverses Persistent ROS Generation After Transient Hyperglycemia by Disrupting an ROS-Generating Feedback Loop. Diabetes 2015, 64, 3273–3284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volpe, C.M.O.; Villar-Delfino, P.H.; Dos Anjos, P.M.F.; Nogueira-Machado, J.A. Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell Death Dis. 2018, 9, 119. [Google Scholar] [CrossRef] [PubMed]
- Mascolo, E.; Vernì, F. Vitamin B6 and Diabetes: Relationship and Molecular Mechanisms. Int. J. Mol. Sci. 2020, 21, 3669. [Google Scholar] [CrossRef]
- Mishra, P.K.; Ekielski, A.; Mukherjee, S.; Sahu, S.; Chowdhury, S.; Mishra, M.; Talegaonkar, S.; Siddiqui, L.; Mishra, H. Wood-Based Cellulose Nanofibrils: Haemocompatibility and Impact on the Development and Behaviour of. Biomolecules 2019, 9, 363. [Google Scholar] [CrossRef] [Green Version]
- Chaudhuri, J.; Bains, Y.; Guha, S.; Kahn, A.; Hall, D.; Bose, N.; Gugliucci, A.; Kapahi, P. The Role of Advanced Glycation End Products in Aging and Metabolic Diseases: Bridging Association and Causality. Cell Metab. 2018, 28, 337–352. [Google Scholar] [CrossRef] [Green Version]
- Nowotny, B.; Zahiragic, L.; Bierwagen, A.; Kabisch, S.; Groener, J.B.; Nowotny, P.J.; Fleitmann, A.K.; Herder, C.; Pacini, G.; Erlund, I.; et al. Low-energy diets differing in fibre, red meat and coffee intake equally improve insulin sensitivity in type 2 diabetes: A randomised feasibility trial. Diabetologia 2015, 58, 255–264. [Google Scholar] [CrossRef] [Green Version]
- Marzio, A.; Merigliano, C.; Gatti, M.; Vernì, F. Sugar and chromosome stability: Clastogenic effects of sugars in vitamin B6-deficient cells. PLoS Genet. 2014, 10, e1004199. [Google Scholar] [CrossRef] [Green Version]
- Mascolo, E.; Barile, A.; Mecarelli, L.S.; Amoroso, N.; Merigliano, C.; Massimi, A.; Saggio, I.; Hansen, T.; Tramonti, A.; Di Salvo, M.L.; et al. The expression of four pyridoxal kinase (PDXK) human variants in Drosophila impacts on genome integrity. Sci. Rep. 2019, 9, 14188. [Google Scholar] [CrossRef] [PubMed]
- Contestabile, R.; di Salvo, M.L.; Bunik, V.; Tramonti, A.; Vernì, F. The multifaceted role of vitamin B. Open Biol. 2020, 10, 200034. [Google Scholar] [CrossRef] [PubMed]
- Graham, P.; Pick, L. Drosophila as a Model for Diabetes and Diseases of Insulin Resistance. Curr. Top. Dev. Biol. 2017, 121, 397–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Liu, J.; Li, C.R.; Momen, B.; Kohanski, R.A.; Pick, L. Deletion of Drosophila insulin-like peptides causes growth defects and metabolic abnormalities. Proc. Natl. Acad. Sci. USA 2009, 106, 19617–19622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brownlee, M. The pathobiology of diabetic complications: A unifying mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodarzi, M.T.; Navidi, A.A.; Rezaei, M.; Babahmadi-Rezaei, H. Oxidative damage to DNA and lipids: Correlation with protein glycation in patients with type 1 diabetes. J. Clin. Lab. Anal. 2010, 24, 72–76. [Google Scholar] [CrossRef]
- Tatsch, E.; Bochi, G.V.; Piva, S.J.; De Carvalho, J.A.; Kober, H.; Torbitz, V.D.; Duarte, T.; Signor, C.; Coelho, A.C.; Duarte, M.M.; et al. Association between DNA strand breakage and oxidative, inflammatory and endothelial biomarkers in type 2 diabetes. Mutat. Res. 2012, 732, 16–20. [Google Scholar] [CrossRef]
- Martínez-Pérez, L.M.; Cerda-Flores, R.M.; Gallegos-Cabriales, E.C.; Dávila-Rodríguez, M.I.; Ibarra-Costilla, E.; Cortés-Gutiérrez, E.I. Frequency of micronuclei in Mexicans with type 2 diabetes mellitus. Prague Med. Rep. 2007, 108, 248–255. [Google Scholar]
- Nakamura, S.; Niwa, T. Pyridoxal phosphate and hepatocyte growth factor prevent dialysate-induced peritoneal damage. J. Am. Soc. Nephrol. 2005, 16, 144–150. [Google Scholar] [CrossRef]
- Millan, A.L.; Trobo, S.I.; de Dios, A.; Cerrato García, M.; Pérez, M.S.; Cerrone, G.E.; Frechtel, G.D.; López, A.P. MODY patients exhibit shorter telomere length than non-diabetic subjects. Diabetes Metab. Res. Rev. 2020, e3374. [Google Scholar] [CrossRef]
- Salpea, K.D.; Talmud, P.J.; Cooper, J.A.; Maubaret, C.G.; Stephens, J.W.; Abelak, K.; Humphries, S.E. Association of telomere length with type 2 diabetes, oxidative stress and UCP2 gene variation. Atherosclerosis 2010, 209, 42–50. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Dong, X.; Cao, L.; Sun, Y.; Qiu, Y.; Zhang, Y.; Cao, R.; Covasa, M.; Zhong, L. Association between telomere length and diabetes mellitus: A meta-analysis. J. Int. Med. Res. 2016, 44, 1156–1173. [Google Scholar] [CrossRef] [PubMed]
- Page, R.C.; Hattersley, A.T.; Levy, J.C.; Barrow, B.; Patel, P.; Lo, D.; Wainscoat, J.S.; Permutt, M.A.; Bell, G.I.; Turner, R.C. Clinical characteristics of subjects with a missense mutation in glucokinase. Diabet. Med. 1995, 12, 209–217. [Google Scholar] [CrossRef]
- Sturis, J.; Kurland, I.J.; Byrne, M.M.; Mosekilde, E.; Froguel, P.; Pilkis, S.J.; Bell, G.I.; Polonsky, K.S. Compensation in pancreatic beta-cell function in subjects with glucokinase mutations. Diabetes 1994, 43, 718–723. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Sui, T.; Zhang, Y.; Wang, Y.; Chen, M.; Deng, J.; Chai, Z.; Lai, L.; Li, Z. Genetic deletion of a short fragment of glucokinase in rabbit by CRISPR/Cas9 leading to hyperglycemia and other typical features seen in MODY-2. Cell. Mol. Life Sci. 2020, 77, 3265–3277. [Google Scholar] [CrossRef] [PubMed]
- Gatti, M.; Goldberg, M.L. Mutations affecting cell division in Drosophila. Methods Cell Biol. 1991, 35, 543–586. [Google Scholar]
- Bonaccorsi, S.; Giansanti, M.G.; Gatti, M. Spindle assembly in Drosophila neuroblasts and ganglion mother cells. Nat. Cell Biol. 2000, 2, 54–56. [Google Scholar] [CrossRef]
- Wu, J.S.; Luo, L. A protocol for dissecting Drosophila melanogaster brains for live imaging or immunostaining. Nat. Protoc. 2006, 1, 2110–2115. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mascolo, E.; Liguori, F.; Stufera Mecarelli, L.; Amoroso, N.; Merigliano, C.; Amadio, S.; Volonté, C.; Contestabile, R.; Tramonti, A.; Vernì, F. Functional Inactivation of Drosophila GCK Orthologs Causes Genomic Instability and Oxidative Stress in a Fly Model of MODY-2. Int. J. Mol. Sci. 2021, 22, 918. https://doi.org/10.3390/ijms22020918
Mascolo E, Liguori F, Stufera Mecarelli L, Amoroso N, Merigliano C, Amadio S, Volonté C, Contestabile R, Tramonti A, Vernì F. Functional Inactivation of Drosophila GCK Orthologs Causes Genomic Instability and Oxidative Stress in a Fly Model of MODY-2. International Journal of Molecular Sciences. 2021; 22(2):918. https://doi.org/10.3390/ijms22020918
Chicago/Turabian StyleMascolo, Elisa, Francesco Liguori, Lorenzo Stufera Mecarelli, Noemi Amoroso, Chiara Merigliano, Susanna Amadio, Cinzia Volonté, Roberto Contestabile, Angela Tramonti, and Fiammetta Vernì. 2021. "Functional Inactivation of Drosophila GCK Orthologs Causes Genomic Instability and Oxidative Stress in a Fly Model of MODY-2" International Journal of Molecular Sciences 22, no. 2: 918. https://doi.org/10.3390/ijms22020918
APA StyleMascolo, E., Liguori, F., Stufera Mecarelli, L., Amoroso, N., Merigliano, C., Amadio, S., Volonté, C., Contestabile, R., Tramonti, A., & Vernì, F. (2021). Functional Inactivation of Drosophila GCK Orthologs Causes Genomic Instability and Oxidative Stress in a Fly Model of MODY-2. International Journal of Molecular Sciences, 22(2), 918. https://doi.org/10.3390/ijms22020918