
Primary Human Hepatocyte Spheroids as Tools to Study the Hepatotoxic Potential of Non-Pharmaceutical Chemicals

 , , ,
, , , 
Abstract
:1. Introduction
2. Results
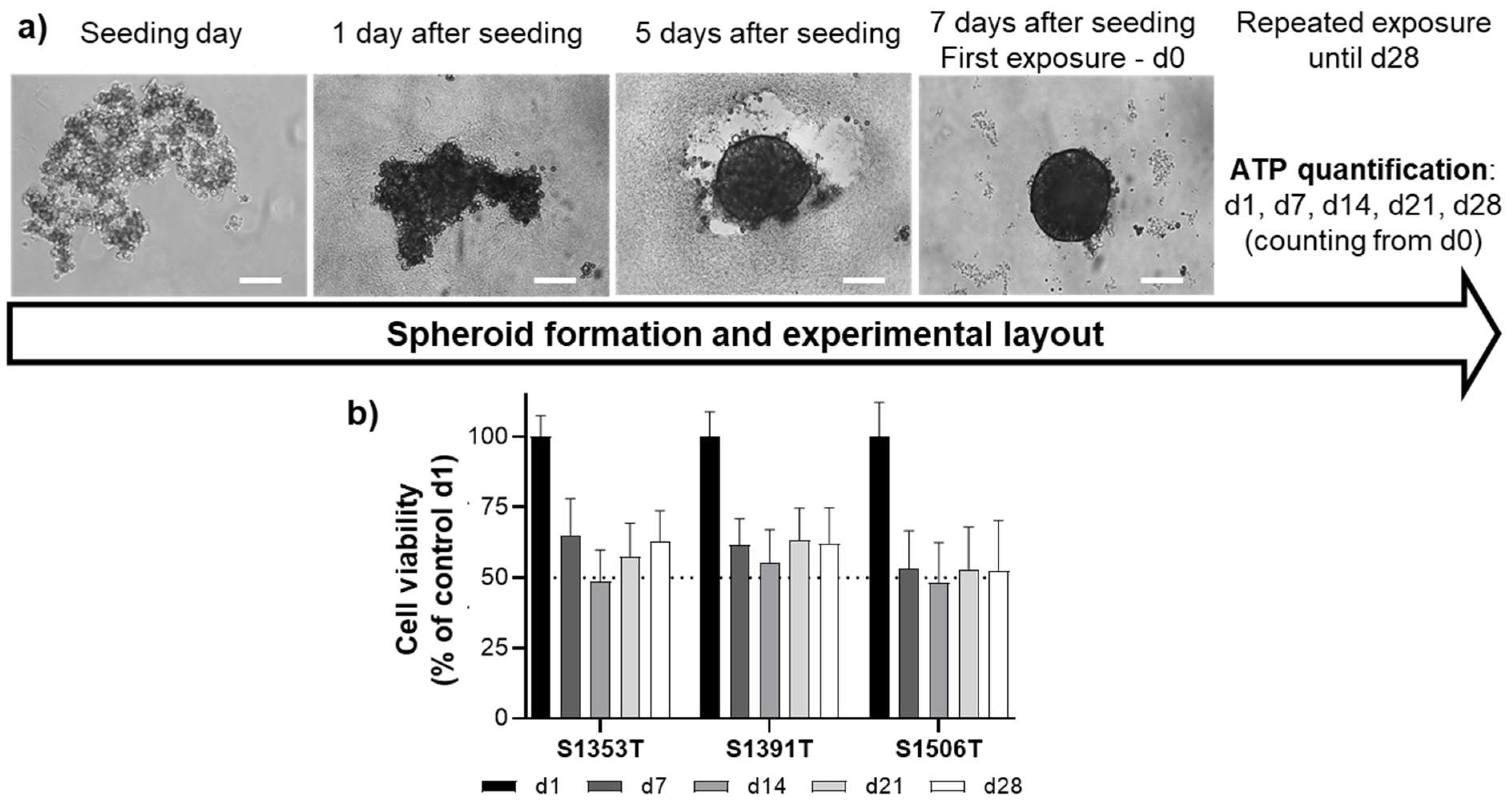
2.1. Spheroid Generation
2.2. Concentration Selection and Concentration–Response Curves
2.3. Synergistic Effects between Test Compounds and Bile Acids
2.4. Transcriptomic Analysis
3. Discussion
4. Materials and Methods
4.1. Reagents and Chemicals
4.2. Primary Human Hepatocytes Seeding and Spheroid Generation
4.3. Adenosine Triphosphate Quantification
4.4. Concentration–Response Curves
4.5. Evaluation of Synergistic Effects and Cholestatic Index Calculation
4.6. RNA Extraction
4.6.1. Reverse Transcription Quantitative Polymerase Chain Reaction
4.6.2. Microarray Analysis and Transcriptomic Data Processing
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Padda, M.S.; Sanchez, M.; Akhtar, A.J.; Boyer, J.L. Drug-induced cholestasis. Hepatology 2011, 53, 1377–1387. [Google Scholar] [CrossRef] [Green Version]
- Pauli-Magnus, C.; Meier, P.J. Hepatobiliary transporters and drug-induced cholestasis. Hepatology 2006, 44, 778–787. [Google Scholar] [CrossRef]
- MacDonald, J.S.; Robertson, R.T. Toxicity testing in the 21st century: A view from the pharmaceutical industry. Toxicol. Sci. 2009, 110, 40–46. [Google Scholar] [CrossRef] [Green Version]
- Olson, H.; Betton, G.; Robinson, D.; Thomas, K.; Monro, A.; Kolaja, G.; Lilly, P.; Sanders, J.; Sipes, G.; Bracken, W.; et al. Concordance of the toxicity of pharmaceuticals in humans and in animals. Regul. Toxicol. Pharm. 2000, 32, 56–67. [Google Scholar] [CrossRef]
- Lauschke, V.M.; Hendriks, D.F.; Bell, C.C.; Andersson, T.B.; Ingelman-Sundberg, M. Novel 3D culture systems for studies of human liver function and assessments of the hepatotoxicity of drugs and drug candidates. Chem. Res. Toxicol. 2016, 29, 1936–1955. [Google Scholar] [CrossRef]
- Richert, L.; Liguori, M.J.; Abadie, C.; Heyd, B.; Mantion, G.; Halkic, N.; Waring, J.F. Gene expression in human hepatocytes in suspension after isolation is similar to the liver of origin, is not affected by hepatocyte cold storage and cryopreservation, but is strongly changed after hepatocyte plating. Drug Metab. Dispos. 2006, 34, 870–879. [Google Scholar] [CrossRef] [Green Version]
- Parmentier, C.; Couttet, P.; Wolf, A.; Zaccharias, T.; Heyd, B.; Bachellier, P.; Uteng, M.; Richert, L. Evaluation of transcriptomic signature as a valuable tool to study drug-induced cholestasis in primary human hepatocytes. Arch. Toxicol. 2017, 91, 2879–2893. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.C.; Hendriks, D.F.; Moro, S.M.; Ellis, E.; Walsh, J.; Renblom, A.; Fredriksson Puigvert, L.; Dankers, A.C.; Jacobs, F.; Snoeys, J.; et al. Characterization of primary human hepatocyte spheroids as a model system for drug-induced liver injury, liver function and disease. Sci. Rep. 2016, 6, 25187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baze, A.; Parmentier, C.; Hendriks, D.F.G.; Hurrell, T.; Heyd, B.; Bachellier, P.; Schuster, C.; Ingelman-Sundberg, M.; Richert, L. Three-dimensional spheroid primary human hepatocytes in monoculture and coculture with nonparenchymal cells. Tissue Eng. Part C Methods 2018, 24, 534–545. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, D.F.G.; Fredriksson Puigvert, L.; Messner, S.; Mortiz, W.; Ingelman-Sundberg, M. Hepatic 3D spheroid models for the detection and study of compounds with cholestatic liability. Sci. Rep. 2016, 6, 35434. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.C.; Dankers, A.C.A.; Lauschke, V.M.; Sison-Young, R.; Jenkins, R.; Rowe, C.; Goldring, C.E.; Park, K.; Regan, S.L.; Walker, T.; et al. Comparison of hepatic 2D sandwich cultures and 3D spheroids for long-term toxicity applications: A multicenter study. Toxicol. Sci. 2018, 162, 655–666. [Google Scholar] [CrossRef] [Green Version]
- Vorrink, S.U.; Ullah, S.; Schmidt, S.; Nandania, J.; Velagapudi, V.; Beck, O.; Ingelman-Sundberg, M.; Lauschke, V.M. Endogenous and xenobiotic metabolic stability of primary human hepatocytes in long-term 3D spheroid cultures revealed by a combination of targeted and untargeted metabolomics. FASEB J. 2017, 31, 2696–2708. [Google Scholar] [CrossRef] [Green Version]
- Oorts, M.; Baze, A.; Bachellier, P.; Heyd, B.; Zacharias, T.; Annaert, P.; Richert, L. Drug-induced cholestasis risk assessment in sandwich-cultured human hepatocytes. Toxicol. Vitr. 2016, 34, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Parmentier, C.; Hendriks, D.F.G.; Heyd, B.; Bachellier, P.; Ingelman-Sundberg, M.; Richert, L. Inter-individual differences in the susceptibility of primary human hepatocytes towards drug-induced cholestasis are compound and time dependent. Toxicol. Lett. 2018, 295, 187–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, S.; Richert, L.; Augustijns, P.; Annaert, P. Hepatocyte-based in vitro model for assessment of drug-induced cholestasis. Toxicol. Appl. Pharm. 2014, 274, 124–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richert, L.; Baze, A.; Parmentier, C.; Gerets, H.H.J.; Sison-Young, R.; Dorau, M.; Lovatt, C.; Czich, A.; Goldring, C.; Park, B.K.; et al. Cytotoxicity evaluation using cryopreserved primary human hepatocytes in various culture formats. Toxicol. Lett. 2016, 258, 207–215. [Google Scholar] [CrossRef]
- Vinken, M.; Landesmann, B.; Goumenou, M.; Vinken, S.; Shah, I.; Jaeschke, H.; Willett, C.; Whelan, M.; Rogiers, V. Development of an adverse outcome pathway from drug-mediated bile salt export pump inhibition to cholestatic liver injury. Toxicol. Sci. 2013, 136, 97–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gijbels, E.; Vilas-Boas, V.; Annaert, P.; Vanhaecke, T.; Devisscher, L.; Vinken, M. Robustness testing and optimization of an adverse outcome pathway on cholestatic liver injury. Arch. Toxicol. 2020, 94, 1151–1172. [Google Scholar] [CrossRef]
- Gijbels, E.; Vilas-Boas, V.; Deferm, N.; Devisscher, L.; Jaeschke, H.; Annaert, P.; Vinken, M. Mechanisms and in vitro models of drug-induced cholestasis. Arch. Toxicol. 2019, 93, 1169–1186. [Google Scholar] [CrossRef]
- Gijbels, E.; Devisscher, L.; Vinken, M. Testing in vitro tools for the prediction of cholestatic liver injury induced by non-pharmaceutical chemicals. Food Chem. Toxicol. 2021, 152, 112165. [Google Scholar] [CrossRef]
- Boedeker, W.; Watts, M.; Clausing, P.; Marquez, E. The global distribution of acute unintentional pesticide poisoning: Estimations based on a systematic review. BMC Public Health 2020, 20, 1875. [Google Scholar] [CrossRef]
- Bataller, R.; Bragulat, E.; Nogué, S.; Görbig, M.N.; Bruguera, M.; Rodés, J. Prolonged cholestasis after acute paraquat poisoning through skin absorption. Am. J. Gastroenterol. 2000, 95, 1340–1343. [Google Scholar] [CrossRef]
- Vilas-Boas, V.; Gijbels, E.; Cooreman, A.; Van Campenhout, R.; Gustafson, E.; Leroy, K.; Vinken, M. Industrial, biocide, and cosmetic chemical inducers of cholestasis. Chem. Res. Toxicol. 2019, 32, 1327–1334. [Google Scholar] [CrossRef] [PubMed]
- Vilas-Boas, V.; Gijbels, E.; Jonckheer, J.; De Waele, E.; Vinken, M. Cholestatic liver injury induced by food additives, dietary supplements and parenteral nutrition. Environ. Int. 2020, 136, 105422. [Google Scholar] [CrossRef] [PubMed]
- EFSA Panel on Food Additives Nutrient Sources Added to Food. Scientific Opinion on the re-evaluation Tartrazine (E 102). EFSA J. 2009, 7, 1331. [Google Scholar] [CrossRef]
- El-Wahab, H.M.; Moram, G.S. Toxic effects of some synthetic food colorants and/or flavor additives on male rats. Toxicol. Ind. Health 2013, 29, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Meyer, S.K.; Probert, P.M.E.; Lakey, A.F.; Axon, A.R.; Leitch, A.C.; Williams, F.M.; Jowsey, P.A.; Blain, P.G.; Kass, G.E.N.; Wright, M.C. Hepatic effects of tartrazine (E 102) after systemic exposure are independent of oestrogen receptor interactions in the mouse. Toxicol. Lett. 2017, 273, 55–68. [Google Scholar] [CrossRef]
- Axon, A.; May, F.E.; Gaughan, L.E.; Williams, F.M.; Blain, P.G.; Wright, M.C. Tartrazine and sunset yellow are xenoestrogens in a new screening assay to identify modulators of human oestrogen receptor transcriptional activity. Toxicology 2012, 298, 40–51. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, Y.; Moore, R.; Hess, H.A.; Guo, G.L.; Gonzalez, F.J.; Korach, K.S.; Maronpot, R.R.; Negishi, M. Estrogen receptor alpha mediates 17alpha-ethynylestradiol causing hepatotoxicity. J. Biol. Chem. 2006, 281, 16625–16631. [Google Scholar] [CrossRef] [Green Version]
- Vinken, M.; Pauwels, M.; Ates, G.; Vivier, M.; Vanhaecke, T.; Rogiers, V. Screening of repeated dose toxicity data present in SCC(NF)P/SCCS safety evaluations of cosmetic ingredients. Arch. Toxicol. 2012, 86, 405–412. [Google Scholar] [CrossRef]
- Weatherly, L.M.; Gosse, J.A. Triclosan exposure, transformation, and human health effects. J. Toxicol. Environ. Health B Crit. Rev. 2017, 20, 447–469. [Google Scholar] [CrossRef]
- Calafat, A.M.; Ye, X.; Wong, L.Y.; Reidy, J.A.; Needham, L.L. Urinary concentrations of triclosan in the U.S. population: 2003–2004. Environ. Health Perspect. 2008, 116, 303–307. [Google Scholar] [CrossRef]
- Sandborgh-Englund, G.; Adolfsson-Erici, M.; Odham, G.; Ekstrand, J. Pharmacokinetics of triclosan following oral ingestion in humans. J. Toxicol. Environ. Health A 2006, 69, 1861–1873. [Google Scholar] [CrossRef]
- Rotroff, D.M.; Wetmore, B.A.; Dix, D.J.; Ferguson, S.S.; Clewell, H.J.; Houck, K.A.; Lecluyse, E.L.; Andersen, M.E.; Judson, R.S.; Smith, C.M.; et al. Incorporating human dosimetry and exposure into high-throughput in vitro toxicity screening. Toxicol. Sci. 2010, 117, 348–358. [Google Scholar] [CrossRef] [Green Version]
- Vorrink, S.U.; Zhou, Y.; Ingelman-Sundberg, M.; Lauschke, V.M. Prediction of drug-induced hepatotoxicity using long-term stable primary hepatic 3D spheroid cultures in chemically defined conditions. Toxicol. Sci. 2018, 163, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Ahn, L.Y.; Kim, S.E.; Yi, S.; Dingemanse, J.; Lim, K.S.; Jang, I.J.; Yu, K.S. Pharmacokinetic-pharmacodynamic relationships of macitentan, a new endothelin receptor antagonist, after multiple dosing in healthy Korean subjects. Am. J. Cardiovasc. Drugs 2014, 14, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Gulati, R.; Sharma, S.; Gupta, U. Pharmacokinetics of cyclosporine from conventional and new microemulsion formulations in healthy volunteers. J. Assoc. Phys. India 1998, 46, 860–863. [Google Scholar]
- Datta, P.; Lundin-Schiller, S. Estrogenicity of the synthetic food colorants tartrazine, erythrosin B, and sudan I in an estrogen-responsive human breast cancer cell line. J. Tenn. Acad. Sci. 2008, 83, 45–51. [Google Scholar]
- Luckert, C.; Braeuning, A.; de Sousa, G.; Durinck, S.; Katsanou, E.S.; Konstantinidou, P.; Machera, K.; Milani, E.S.; Peijnenburg, A.; Rahmani, R.; et al. Adverse outcome pathway-driven analysis of liver steatosis in vitro: A case study with cyproconazole. Chem. Res. Toxicol. 2018, 31, 784–798. [Google Scholar] [CrossRef]
- Zhan, Z.; Chen, Y.; Duan, Y.; Li, L.; Mew, K.; Hu, P.; Ren, H.; Peng, M. Identification of key genes, pathways and potential therapeutic agents for liver fibrosis using an integrated bioinformatics analysis. PeerJ 2019, 7, e6645. [Google Scholar] [CrossRef]
- Yanguas, S.C.; Cogliati, B.; Willebrords, J.; Maes, M.; Colle, I.; van den Bossche, B.; de Oliveira, C.P.M.S.; Andraus, W.; Alves, V.A.F.; Leclercq, I.; et al. Experimental models of liver fibrosis. Arch. Toxicol. 2016, 90, 1025–1048. [Google Scholar] [CrossRef] [Green Version]
- Willebrords, J.; Pereira, I.V.; Maes, M.; Crespo Yanguas, S.; Colle, I.; Van Den Bossche, B.; Da Silva, T.C.; de Oliveira, C.P.; Andraus, W.; Alves, V.A.; et al. Strategies, models and biomarkers in experimental non-alcoholic fatty liver disease research. Prog. Lipid Res. 2015, 59, 106–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozyra, M.; Johansson, I.; Nordling, Å.; Ullah, S.; Lauschke, V.M.; Ingelman-Sundberg, M. Human hepatic 3D spheroids as a model for steatosis and insulin resistance. Sci. Rep. 2018, 8, 14297. [Google Scholar] [CrossRef] [Green Version]
- Hurrell, T.; Kastrinou-Lampou, V.; Fardellas, A.; Hendriks, D.F.G.; Nordling, Å.; Johansson, I.; Baze, A.; Parmentier, C.; Richert, L.; Ingelman-Sundberg, M. Human liver spheroids as a model to study aetiology and treatment of hepatic fibrosis. Cells 2020, 9, 964. [Google Scholar] [CrossRef] [Green Version]
- Noreikaitė, A.; Saint-Marcoux, F.; Kaduševičius, E.; Stankevičius, E. Cyclosporine therapeutic window evaluation by Chebyshev’s inequality method in kidney recipients. Medicina 2014, 50, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Burbank, M.G.; Sharanek, A.; Burban, A.; Mialanne, H.; Aerts, H.; Guguen-Guillouzo, C.; Weaver, R.J.; Guillouzo, A. Mechanistic insights in the cytotoxic and cholestatic potential of the endothelial receptor antagonists using HepaRG cells. Toxicol. Sci. 2017, 157, 451–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pulido, T.; Adzerikho, I.; Channick, R.N.; Delcroix, M.; Galiè, N.; Ghofrani, H.A.; Jansa, P.; Jing, Z.C.; Le Brun, F.O.; Mehta, S.; et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N. Engl. J. Med. 2013, 369, 809–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soares, B.M.; Araújo, T.M.; Ramos, J.A.; Pinto, L.C.; Khayat, B.M.; De Oliveira Bahia, M.; Montenegro, R.C.; Burbano, R.M.; Khayat, A.S. Effects on DNA repair in human lymphocytes exposed to the food dye tartrazine yellow. Anticancer Res. 2015, 35, 1465–1474. [Google Scholar]
- Van Summeren, A.; Renes, J.; Bouwman, F.G.; Noben, J.P.; van Delft, J.H.; Kleinjans, J.C.; Mariman, E.C. Proteomics investigations of drug-induced hepatotoxicity in HepG2 cells. Toxicol. Sci. 2011, 120, 109–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharanek, A.; Azzi, P.B.; Al-Attrache, H.; Savary, C.C.; Humbert, L.; Rainteau, D.; Guguen-Guillouzo, C.; Guillouzo, A. Different dose-dependent mechanisms are involved in early cyclosporine A-induced cholestatic effects in HepaRG cells. Toxicol. Sci. 2014, 141, 244–253. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Chitranshi, P.; Loukotková, L.; Gamboa da Costa, G.; Beland, F.A.; Zhang, J.; Fang, J.L. Cytochrome P450-mediated metabolism of triclosan attenuates its cytotoxicity in hepatic cells. Arch. Toxicol. 2017, 91, 2405–2423. [Google Scholar] [CrossRef] [PubMed]
- Tolosa, L.; Gómez-Lechón, M.J.; Jiménez, N.; Hervás, D.; Jover, R.; Donato, M.T. Advantageous use of HepaRG cells for the screening and mechanistic study of drug-induced steatosis. Toxicol. Appl Pharm. 2016, 302, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Yueh, M.-F.; He, F.; Chen, C.; Vu, C.; Tripathi, A.; Knight, R.; Karin, M.; Chen, S.; Tukey, R.H. Triclosan leads to dysregulation of the metabolic regulator FGF21 exacerbating high fat diet-induced nonalcoholic fatty liver disease. Proc. Natl. Acad. Sci. USA 2020, 117, 31259. [Google Scholar] [CrossRef] [PubMed]
- Yueh, M.F.; Taniguchi, K.; Chen, S.; Evans, R.M.; Hammock, B.D.; Karin, M.; Tukey, R.H. The commonly used antimicrobial additive triclosan is a liver tumor promoter. Proc. Natl. Acad. Sci. USA 2014, 111, 17200–17205. [Google Scholar] [CrossRef] [Green Version]
- Atashpour, S.; Kargar Jahromi, H.; Kargar Jahromi, Z.; Zarei, S. Antioxidant effects of aqueous extract of Salep on Paraquat-induced rat liver injury. World J. Hepatol. 2017, 9, 209–216. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Gijbels, E.; Devisscher, L.; Vinken, M. Dataset on transcriptomic profiling of cholestatic liver injury in an in vitro and in vivo animal model. Data Brief. 2020, 32, 106156. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Donor | Gender | Age | Pathology |
|---|---|---|---|
| S1353T | Male | 62 | Sigmoid adenocarcinoma |
| S1391T | Female | 54 | Non-transplantable liver |
| S1506T | Female | 47 | Adenocarcinoma |
| Test Compound | Tested Concentration (µM) | Human-Relevant Concentration (µM) | |
|---|---|---|---|
| Concentration-Response | CIx | ||
| BOS | 0; 0.015; 0.075; 0.15; 0.75; 1.5; 7.5; 15; 75 | 0; 0.15; 1.5; 7.5; 75 | 7.5 * |
| CyA | 0; 0.01; 0.05; 0.1; 0.5; 1; 2.5; 5; 10; 25 | 0; 0.01; 0.05; 0.5; 5 | 0.8 * |
| MAC | 0; 0.005; 0.05; 0.25; 0.5; 1; 2.5; 5; 10 | 0; 0.005; 0.5; 5; 10 | 0.5 * |
| PQ | 0; 0.02; 0.2; 1; 2; 10; 20; 80; 200; 500 | 0; 0.2; 1; 2; 10 | 2.0 # |
| TAR | 0; 0.0001; 0.001; 0.01; 0.1; 1; 10; 100; 1000 | 0; 0.0001; 0.01; 10; 1000 | 0.01 $ |
| TRI | 0; 0.01; 0.1; 1; 10; 100; 1000 | 0; 0.1; 1; 10; 100 | 10 & |
| CyA | PQ | TAR | TRI | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| µM | 0.01 | 0.05 | 0.5 | 5 | 0.2 | 1 | 2 | 10 | 0.0001 | 0.01 | 10 | 1000 | 0.1 | 1 | 10 | 100 | |
| S1353T | d1 | 0.92 ± 0.00 | 0.93 ± 0.04 | 0.96 ± 0.16 | 0.97 ± 0.08 | 0.98 ± 0.06 | 0.95 ± 0.08 | 0.96 ± 0.11 | 0.88 ± 0.00 | 0.97 ± 0.07 | 1.00 ± 0.02 | 1.01 ± 0.16 | 1.06 ± 0.09 | 0.98 ± 0.02 | 0.91 ± 0.02 | 1.02 ± 0.09 | 0.99 ± 0.05 |
| d7 | 0.92 ± 0.06 | 0.94 ± 0.07 | 0.94 ± 0.18 | 0.33 ± 0.18 | 0.90 ± 0.10 | 0.98 ± 0.09 | 1.06 ± 0.18 | 0.94 ± 0.08 | 0.98 ± 0.00 | 0.98 ± 0.13 | 1.04 ± 0.20 | 0.90 ± 0.05 | 1.02 ± 0.05 | 0.91 ± 0.13 | 0.97 ± 0.00 | 1.01 ± 0.08 | |
| d14 | 0.94 ± 0.07 | 0.96 ± 0.16 | 1.03 ± 0.11 | 0.11 ± 0.04 | 0.94 ± 0.01 | 0.94 ± 0.01 | 0.90 ± 0.01 | 1.40 ± 0.08 | 0.93 ± 0.11 | 1.01 ± 0.00 | 1.08 ± 0.19 | 0.90 ± 0.00 | 1.05 ± 0.11 | 0.94 ± 0.14 | 0.93 ± 0.00 | 0.82 ± 0.07 | |
| d21 | 1.00 ± 0.04 | 0.99 ± 0.00 | 1.06 ± 0.07 | 0.06 ± 0.07 | 1.00 ± 0.20 | 0.94 ± 0.09 | 0.68 ± 0.07 | 0.54 ± 0.16 | 1.06 ± 0.00 | 0.94 ± 0.12 | 0.98 ± 0.04 | 0.87 ± 0.01 | 0.98 ± 0.07 | 0.91 ± 0.00 | 0.87 ± 0.12 | 0.01 ± 0.01 | |
| d28 | 1.00 ± 0.23 | 0.91 ± 0.13 | 0.82 ± 0.01 | 0.04 ± 0.02 | 0.84 ± 0.07 | 0.52 ± 0.30 | 0.32 ± 0.08 | 0.28 ± 0.04 | 0.89 ± 0.07 | 0.95 ± 0.15 | 0.96 ± 0.05 | 0.74 ± 0.10 | 0.89 ± 0.05 | 0.84 ± 0.05 | 0.89 ± 0.11 | 0.01 ± 0.00 | |
| S1391T | d1 | 1.03 ± 0.14 | 0.98 ± 0.09 | 1.01 ± 0.09 | 0.98 ± 0.07 | 1.00 ± 0.12 | 0.93 ± 0.14 | 0.99 ± 0.01 | 0.92 ± 0.09 | 1.03 ± 0.08 | 0.92 ± 0.05 | 0.92 ± 0.01 | 0.88 ± 0.18 | 1.05 ± 0.12 | 1.02 ± 0.05 | 0.97 ± 0.09 | 1.04 ± 0.09 |
| d7 | 0.90 ± 0.02 | 0.88 ± 0.02 | 0.95 ± 0.09 | 0.93 ± 0.08 | 0.93 ± 0.13 | 0.90 ± 0.12 | 0.84 ±0.04 | 0.93 ± 0.07 | 0.98 ± 0.12 | 0.94 ± 0.08 | 0.90 ± 0.05 | 0.73 ± 0.07 | 0.85 ± 0.02 | 0.89 ± 0.11 | 1.01 ± 0.14 | 0.85 ± 0.04 | |
| d14 | 0.81 ± 0.09 | 0.83 ± 0.12 | 1.01 ± 0.06 | 0.21 ± 0.13 | 0.82 ± 0.02 | 0.80 ± 0.02 | 0.94 ± 0.16 | 1.05 ± 0.15 | 0.87 ± 0.00 | 0.82 ± 0.01 | 0.95 ± 0.08 | 0.77 ± 0.02 | 0.87 ± 0.03 | 1.08 ± 0.27 | 0.96 ± 0.14 | 1.11 ± 0.32 | |
| d21 | 0.80 ± 0.02 | 0.83 ± 0.21 | 0.77 ± 0.18 | 0.09 ± 0.07 | 0.83 ± 0.04 | 0.85 ± 0.00 | 0.91 ± 0.08 | 1.08 ± 0.05 | 0.87 ± 0.10 | 0.83 ± 0.02 | 0.97 ± 0.05 | 0.73 ± 0.16 | 0.79 ± 0.02 | 0.90 ± 0.13 | 0.80 ± 0.01 | 1.10 ± 0.16 | |
| d28 | 0.83 ± 0.02 | 0.79 ± 0.03 | 0.73 ± 0.16 | 0.08 ± 0.04 | 1.06 ± 0.16 | 0.93 ± 0.17 | 0.94 ± 0.13 | 1.07 ± 0.16 | 1.01 ± 0.07 | 0.80 ± 0.14 | 0.75 ± 0.04 | 0.67 ± 0.13 | 0.84 ± 0.00 | 0.90 ± 0.04 | 0.48 ± 0.05 | 0.54 ± 0.13 | |
| S1506T | d1 | 0.98 ± 0.22 | 1.10 ± 0.03 | 1.01 ± 0.02 | 1.03 ± 0.13 | 1.01 ± 0.12 | 0.95 ± 0.04 | 1.01 ± 0.22 | 0.92 ± 0.04 | 1.01 ± 0.14 | 1.06 ± 0.02 | 1.18 ± 0.11 | 0.95 ± 0.06 | 1.05 ± 0.11 | 0.93 ± 0.10 | 1.11 ± 0.12 | 0.87 ± 0.07 |
| d7 | 1.07 ± 0.03 | 1.06 ± 0.13 | 1.09 ± 0.15 | 0.77 ± 0.08 | 0.91 ± 0.07 | 1.11 ± 0.01 | 1.06 ± 0.02 | 1.05 ± 0.15 | 1.01 ± 0.12 | 1.00 ± 0.04 | 0.98 ± 0.05 | 0.98 ± 0.25 | 1.08 ± 0.15 | 1.09 ± 0.16 | 1.12 ± 0.09 | 1.04 ± 0.25 | |
| d14 | 0.88 ± 0.01 | 1.13 ± 0.00 | 1.01 ± 0.20 | 0.12 ± 0.05 | 0.99 ± 0.13 | 0.99 ± 0.08 | 1.18 ± 0.01 | 1.03 ± 0.00 | 1.18 ± 0.04 | 1.05 ± 0.26 | 1.03 ± 0.19 | 0.83 ± 0.06 | 1.00 ± 0.26 | 0.96 ± 0.14 | 1.12 ± 0.07 | 0.90 ± 0.11 | |
| d21 | 0.94 ± 0.15 | 1.02 ± 0.13 | 1.01 ± 0.20 | 0.02 ± 0.01 | 0.91 ± 0.13 | 0.96 ± 0.18 | 0.93 ± 0.07 | 0.20 ± 0.04 | 1.05 ± 0.07 | 1.02 ± 0.08 | 1.04 ± 0.15 | 1.03 ± 0.15 | 1.08 ± 0.22 | 1.06 ± 0.08 | 1.05 ± 0.02 | 0.27 ± 0.47 | |
| d28 | 0.88 ± 0.06 | 1.06 ± 0.28 | 1.04 ± 0.02 | 0.02 ± 0.02 | 0.87 ± 0.07 | 0.45 ± 0.10 | 0.45 ± 0.04 | 0.22 ± 0.13 | 0.87 ± 0.06 | 1.00 ± 0.21 | 1.25 ± 0.12 | 0.87 ± 0.15 | 0.86 ± 0.06 | 0.96 ± 0.23 | 0.84 ± 0.35 | 0.35 ± 0.60 | |
| d7 | d14 | d21 | d28 | |||||
|---|---|---|---|---|---|---|---|---|
| CyA Conc. µM * | SMCHOL | CyA Conc. µM * | SMCHOL | CyA Conc. µM * | SMCHOL | CyA Conc. µM * | SMCHOL | |
| S1353T | 5 | 6.50 | 5 | 6.50 | 5 | 6.50 | 5 | 6.50 |
| S1391T | - | - | 5 | 6.50 | 0.5 | 0.65 | 0.05 | 0.06 |
| S1506T | 5 | 6.50 | 5 | 6.50 | 5 | 6.50 | 5 | 6.50 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vilas-Boas, V.; Gijbels, E.; Leroy, K.; Pieters, A.; Baze, A.; Parmentier, C.; Vinken, M. Primary Human Hepatocyte Spheroids as Tools to Study the Hepatotoxic Potential of Non-Pharmaceutical Chemicals. Int. J. Mol. Sci. 2021, 22, 11005. https://doi.org/10.3390/ijms222011005
Vilas-Boas V, Gijbels E, Leroy K, Pieters A, Baze A, Parmentier C, Vinken M. Primary Human Hepatocyte Spheroids as Tools to Study the Hepatotoxic Potential of Non-Pharmaceutical Chemicals. International Journal of Molecular Sciences. 2021; 22(20):11005. https://doi.org/10.3390/ijms222011005
Chicago/Turabian StyleVilas-Boas, Vânia, Eva Gijbels, Kaat Leroy, Alanah Pieters, Audrey Baze, Céline Parmentier, and Mathieu Vinken. 2021. "Primary Human Hepatocyte Spheroids as Tools to Study the Hepatotoxic Potential of Non-Pharmaceutical Chemicals" International Journal of Molecular Sciences 22, no. 20: 11005. https://doi.org/10.3390/ijms222011005
APA StyleVilas-Boas, V., Gijbels, E., Leroy, K., Pieters, A., Baze, A., Parmentier, C., & Vinken, M. (2021). Primary Human Hepatocyte Spheroids as Tools to Study the Hepatotoxic Potential of Non-Pharmaceutical Chemicals. International Journal of Molecular Sciences, 22(20), 11005. https://doi.org/10.3390/ijms222011005