
Oxygen Toxicity to the Immature Lung—Part I: Pathomechanistic Understanding and Preclinical Perspectives

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Link between Oxygen Exposure, ROS Production and BPD
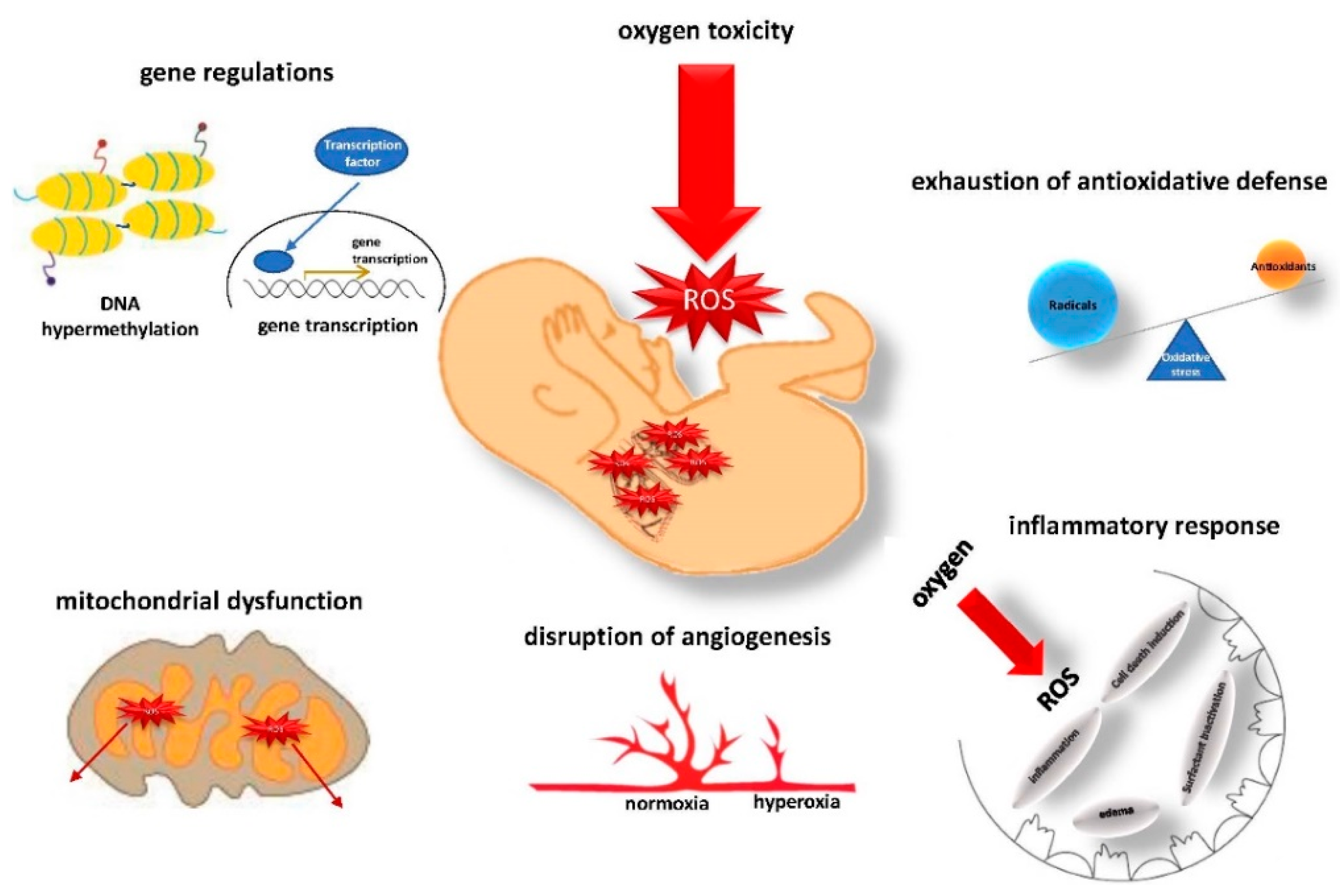
3. The Pathomechanisms of ROS Injury to the Immature Lung
3.1. Gene Regulation and Epigenetic Alterations
3.2. Antioxidative Defense and Mitochondrial Dysfunction
3.3. Disruption of Angiogenesis
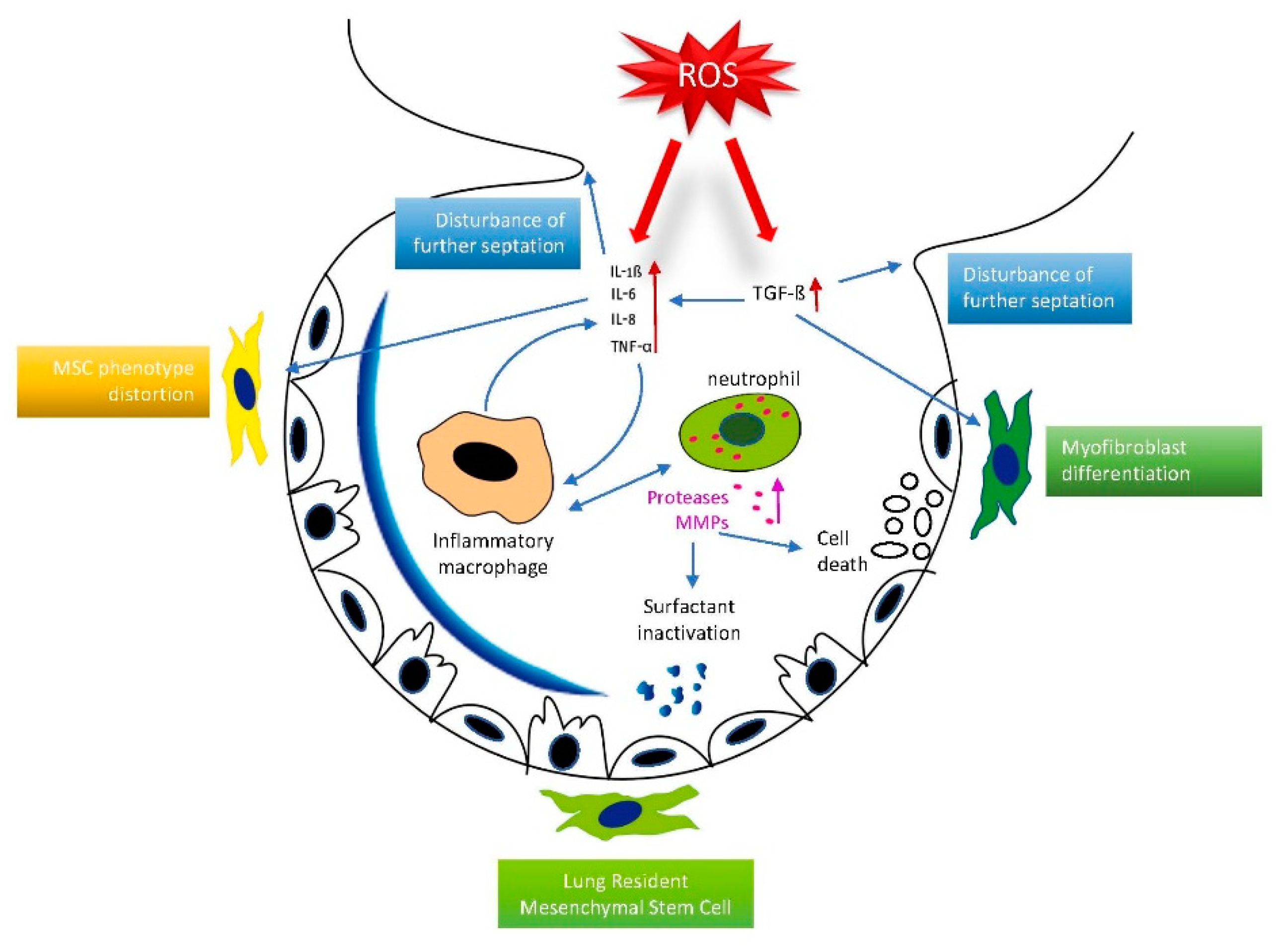
3.4. Inflammatory Response
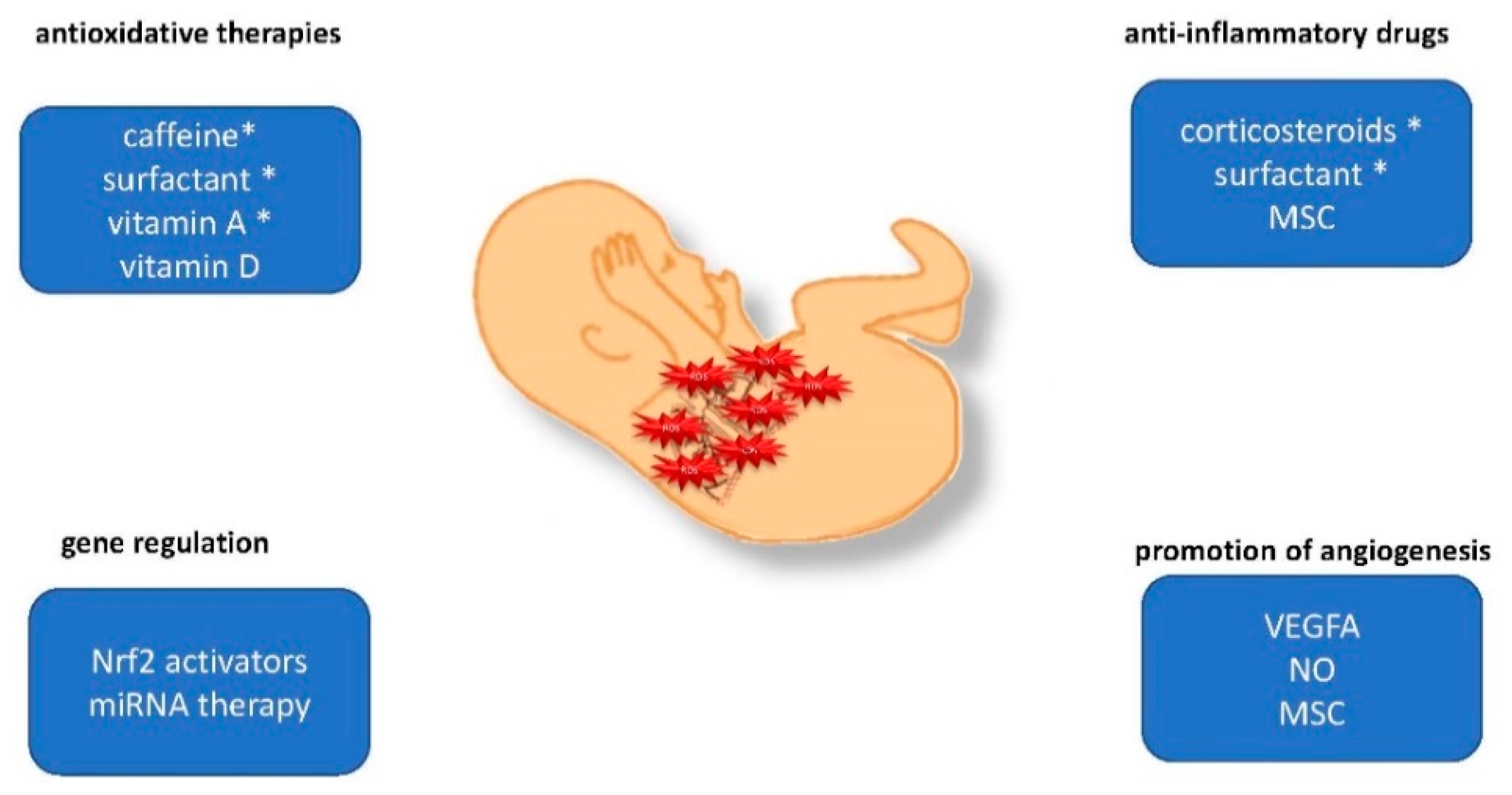
4. Preclinical Strategies to Prevent ROS-Induced Lung Injury
4.1. Gene Regulation and Epigenetic Alterations
4.2. Antioxidative Defense
4.3. Anti-Inflammatory Drugs
5. Outlook and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gitto, E.; Pellegrino, S.; D’Arrigo, S.; Barberi, I.; Reiter, R.J. Oxidative stress in resuscitation and in ventilation of newborns. Eur. Respir. J. 2009, 34, 1461–1469. [Google Scholar] [CrossRef]
- Saugstad, O.D.; Oei, J.-L.; Lakshminrusimha, S.; Vento, M. Oxygen therapy of the newborn from molecular understanding to clinical practice. Pediatr. Res. 2019, 85, 20–29. [Google Scholar] [CrossRef]
- Hilgendorff, A.; Reiss, I.; Ehrhardt, H.; Eickelberg, O.; Alvira, C.M. Chronic lung disease in the preterm infant. Lessons learned from animal models. Am. J. Respir. Cell Mol. Biol. 2014, 50, 233–245. [Google Scholar] [CrossRef]
- Kumar, V.H.S.; Wang, H.; Nielsen, L. Short-term perinatal oxygen exposure may impair lung development in adult mice. Biol. Res. 2020, 53, 51. [Google Scholar] [CrossRef]
- Bouch, S.; O’Reilly, M.; Harding, R.; Sozo, F. Neonatal exposure to mild hyperoxia causes persistent increases in oxidative stress and immune cells in the lungs of mice without altering lung structure. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L488–L496. [Google Scholar] [CrossRef] [Green Version]
- Rognlien, A.G.W.; Wollen, E.J.; Atneosen-Åsegg, M.; Saugstad, O.D. Temporal Patterns of Gene Expression Profiles in the Neonatal Mouse Lung after Hypoxia-Reoxygenation. Neonatology 2017, 111, 45–54. [Google Scholar] [CrossRef]
- Wollen, E.J.; Sejersted, Y.; Wright, M.S.; Bik-Multanowski, M.; Madetko-Talowska, A.; Günther, C.-C.; Nygård, S.; Kwinta, P.; Pietrzyk, J.J.; Saugstad, O.D. Transcriptome profiling of the newborn mouse lung after hypoxia and reoxygenation: Hyperoxic reoxygenation affects mTOR signaling pathway, DNA repair, and JNK-pathway regulation. Pediatr. Res. 2013, 74, 536–544. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhang, A.; Li, Y.; Xu, J.; Huang, F.; Zhao, M.; Wu, B.; He, S. Effect of intermittent hypoxia or hyperoxia on lung development in preterm rat neonates during constant oxygen therapy. J. Cell. Biochem. 2019, 120, 17545–17554. [Google Scholar] [CrossRef]
- Di Fiore, J.M.; Vento, M. Intermittent hypoxemia and oxidative stress in preterm infants. Respir. Physiol. Neurobiol. 2019, 266, 121–129. [Google Scholar] [CrossRef]
- Saugstad, O.D. Mechanisms of tissue injury by oxygen radicals: Implications for neonatal disease. Acta Paediatr. 1996, 85, 1–4. [Google Scholar] [CrossRef]
- Vogel, E.R.; Britt, R.D.; Trinidad, M.C.; Faksh, A.; Martin, R.J.; MacFarlane, P.M.; Pabelick, C.M.; Prakash, Y.S. Perinatal oxygen in the developing lung. Can. J. Physiol. Pharmacol. 2015, 93, 119–127. [Google Scholar] [CrossRef] [Green Version]
- Oak, P.; Pritzke, T.; Thiel, I.; Koschlig, M.; Mous, D.S.; Windhorst, A.; Jain, N.; Eickelberg, O.; Foerster, K.; Schulze, A.; et al. Attenuated PDGF signaling drives alveolar and microvascular defects in neonatal chronic lung disease. EMBO Mol. Med. 2017, 9, 1504–1520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millán, I.; Piñero-Ramos, J.D.; Lara, I.; Parra-Llorca, A.; Torres-Cuevas, I.; Vento, M. Oxidative Stress in the Newborn Period: Useful Biomarkers in the Clinical Setting. Antioxidants 2018, 7, 193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saugstad, O.D. Oxidative stress in the newborn—A 30-year perspective. Biol. Neonat. 2005, 88, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, K.; Zhang, F.; Zhang, Y.; Deng, C.; Guo, C. Ablation of glutaredoxin 1 promotes pulmonary angiogenesis and alveolar formation in hyperoxia-injured lungs by modifying HIF-1α stability and inhibiting the NF-κB pathway. Biochem. Biophys. Res. Commun. 2020, 525, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Lauer, T.; Behnke, J.; Oehmke, F.; Baecker, J.; Gentil, K.; Chakraborty, T.; Schloter, M.; Gertheiss, J.; Ehrhardt, H. Bacterial Colonization within the First Six Weeks of Life and Pulmonary Outcome in Preterm Infants <1000 g. JCM 2020, 9, 2240. [Google Scholar] [CrossRef]
- Casado, F.; Morty, R.E. The emergence of preclinical studies on the role of the microbiome in lung development and experimental animal models of bronchopulmonary dysplasia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 318, L402–L404. [Google Scholar] [CrossRef] [Green Version]
- Revhaug, C.; Bik-Multanowski, M.; Zasada, M.; Rognlien, A.G.W.; Günther, C.-C.; Ksiązek, T.; Madetko-Talowska, A.; Szewczyk, K.; Grabowska, A.; Kwinta, P.; et al. Immune System Regulation Affected by a Murine Experimental Model of Bronchopulmonary Dysplasia: Genomic and Epigenetic Findings. Neonatology 2019, 116, 269–277. [Google Scholar] [CrossRef]
- Chen, C.-M.; Liu, Y.-C.; Chen, Y.-J.; Chou, H.-C. Genome-Wide Analysis of DNA Methylation in Hyperoxia-Exposed Newborn Rat Lung. Lung 2017, 195, 661–669. [Google Scholar] [CrossRef]
- Bik-Multanowski, M.; Revhaug, C.; Grabowska, A.; Dobosz, A.; Madetko-Talowska, A.; Zasada, M.; Saugstad, O.D. Hyperoxia induces epigenetic changes in newborn mice lungs. Free Radic. Biol. Med. 2018, 121, 51–56. [Google Scholar] [CrossRef]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.-Y.; van Houten, B.; Wang, X.; Miller-DeGraff, L.; Fostel, J.; Gladwell, W.; Perrow, L.; Panduri, V.; Kobzik, L.; Yamamoto, M.; et al. Targeted deletion of nrf2 impairs lung development and oxidant injury in neonatal mice. Antioxid. Redox Signal. 2012, 17, 1066–1082. [Google Scholar] [CrossRef] [Green Version]
- Ratner, V.; Starkov, A.; Matsiukevich, D.; Polin, R.A.; Ten, V.S. Mitochondrial dysfunction contributes to alveolar developmental arrest in hyperoxia-exposed mice. Am. J. Respir. Cell Mol. Biol. 2009, 40, 511–518. [Google Scholar] [CrossRef] [Green Version]
- Budinger, G.R.S.; Mutlu, G.M.; Urich, D.; Soberanes, S.; Buccellato, L.J.; Hawkins, K.; Chiarella, S.E.; Radigan, K.A.; Eisenbart, J.; Agrawal, H.; et al. Epithelial cell death is an important contributor to oxidant-mediated acute lung injury. Am. J. Respir. Crit. Care Med. 2011, 183, 1043–1054. [Google Scholar] [CrossRef] [Green Version]
- Ilizarov, A.M.; Koo, H.C.; Kazzaz, J.A.; Mantell, L.L.; Li, Y.; Bhapat, R.; Pollack, S.; Horowitz, S.; Davis, J.M. Overexpression of manganese superoxide dismutase protects lung epithelial cells against oxidant injury. Am. J. Respir. Cell Mol. Biol. 2001, 24, 436–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, M.N.; Codipilly, C.; Hogg, N.; Auten, R.L. The protective effect of overexpression of extracellular superoxide dismutase on nitric oxide bioavailability in the lung after exposure to hyperoxia stress. Exp. Lung Res. 2011, 37, 10–17. [Google Scholar] [CrossRef] [PubMed]
- ter Horst, S.A.J.; Walther, F.J.; Poorthuis, B.J.H.M.; Hiemstra, P.S.; Wagenaar, G.T.M. Inhaled nitric oxide attenuates pulmonary inflammation and fibrin deposition and prolongs survival in neonatal hyperoxic lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L35–L44. [Google Scholar] [CrossRef] [Green Version]
- Datta, A.; Kim, G.A.; Taylor, J.M.; Gugino, S.F.; Farrow, K.N.; Schumacker, P.T.; Berkelhamer, S.K. Mouse lung development and NOX1 induction during hyperoxia are developmentally regulated and mitochondrial ROS dependent. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L369–L377. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Maturu, P.; Liang, Y.W.; Wang, L.; Lingappan, K.; Couroucli, X. Hyperoxia-mediated transcriptional activation of cytochrome P4501A1 (CYP1A1) and decreased susceptibility to oxygen-mediated lung injury in newborn mice. Biochem. Biophys. Res. Commun. 2018, 495, 408–413. [Google Scholar] [CrossRef] [PubMed]
- Couroucli, X.I.; Liang, Y.W.; Jiang, W.; Wang, L.; Barrios, R.; Yang, P.; Moorthy, B. Prenatal administration of the cytochrome P4501A inducer, Β-naphthoflavone (BNF), attenuates hyperoxic lung injury in newborn mice: Implications for bronchopulmonary dysplasia (BPD) in premature infants. Toxicol. Appl. Pharmacol. 2011, 256, 83–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maturu, P.; Wei-Liang, Y.; Jiang, W.; Wang, L.; Lingappan, K.; Barrios, R.; Liang, Y.; Moorthy, B.; Couroucli, X.I. Newborn Mice Lacking the Gene for Cyp1a1 Are More Susceptible to Oxygen-Mediated Lung Injury, and Are Rescued by Postnatal β-Naphthoflavone Administration: Implications for Bronchopulmonary Dysplasia in Premature Infants. Toxicol. Sci. 2017, 157, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Lingappan, K.; Maturu, P.; Liang, Y.W.; Jiang, W.; Wang, L.; Moorthy, B.; Couroucli, X.I. β-Naphthoflavone treatment attenuates neonatal hyperoxic lung injury in wild type and Cyp1a2-knockout mice. Toxicol. Appl. Pharmacol. 2018, 339, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Bouch, S.; O’Reilly, M.; de Haan, J.B.; Harding, R.; Sozo, F. Does lack of glutathione peroxidase 1 gene expression exacerbate lung injury induced by neonatal hyperoxia in mice? Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 313, L115–L125. [Google Scholar] [CrossRef] [Green Version]
- Lingappan, K.; Jiang, W.; Wang, L.; Couroucli, X.I.; Moorthy, B. Sex-specific differences in hyperoxic lung injury in mice: Role of cytochrome P450 (CYP)1A. Toxicology 2015, 331, 14–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, C.W.; Lee, J.; Lee, H.J.; Park, H.-S.; Chun, Y.-S.; Kim, B.I. Deferoxamine Improves Alveolar and Pulmonary Vascular Development by Upregulating Hypoxia-inducible Factor-1α in a Rat Model of Bronchopulmonary Dysplasia. J. Korean Med. Sci. 2015, 30, 1295–1301. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Gao, S.; Yan, Y.; Qian, J.; Chen, H. Aerosolized deferoxamine administration in mouse model of bronchopulmonary dysplasia improve pulmonary development. Am. J. Transl. Res. 2018, 10, 325–332. [Google Scholar]
- Das, A.; Kole, L.; Wang, L.; Barrios, R.; Moorthy, B.; Jaiswal, A.K. BALT development and augmentation of hyperoxic lung injury in mice deficient in NQO1 and NQO2. Free Radic. Biol. Med. 2006, 40, 1843–1856. [Google Scholar] [CrossRef]
- Vadivel, A.; Aschner, J.L.; Rey-Parra, G.J.; Magarik, J.; Zeng, H.; Summar, M.; Eaton, F.; Thébaud, B. L-citrulline attenuates arrested alveolar growth and pulmonary hypertension in oxygen-induced lung injury in newborn rats. Pediatr. Res. 2010, 68, 519–525. [Google Scholar] [CrossRef] [Green Version]
- Lopez, E.; Boucherat, O.; Franco-Montoya, M.-L.; Bourbon, J.R.; Delacourt, C.; Jarreau, P.-H. Nitric oxide donor restores lung growth factor and receptor expression in hyperoxia-exposed rat pups. Am. J. Respir. Cell Mol. Biol. 2006, 34, 738–745. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.-J.; Markham, N.E.; Balasubramaniam, V.; Tang, J.-R.; Maxey, A.; Kinsella, J.P.; Abman, S.H. Inhaled nitric oxide enhances distal lung growth after exposure to hyperoxia in neonatal rats. Pediatr. Res. 2005, 58, 22–29. [Google Scholar] [CrossRef] [Green Version]
- Wedgwood, S.; Steinhorn, R.H.; Bunderson, M.; Wilham, J.; Lakshminrusimha, S.; Brennan, L.A.; Black, S.M. Increased hydrogen peroxide downregulates soluble guanylate cyclase in the lungs of lambs with persistent pulmonary hypertension of the newborn. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 289, L660–L666. [Google Scholar] [CrossRef] [Green Version]
- Afolayan, A.J.; Eis, A.; Alexander, M.; Michalkiewicz, T.; Teng, R.-J.; Lakshminrusimha, S.; Konduri, G.G. Decreased endothelial nitric oxide synthase expression and function contribute to impaired mitochondrial biogenesis and oxidative stress in fetal lambs with persistent pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 310, L40–L49. [Google Scholar] [CrossRef] [Green Version]
- Funk, A.J.; Mandrell, T.D.; Lokey, S.J.; Kosanke, S.D.; Li, C.-S.; Potters, C.F. Effects of leukotriene inhibition on pulmonary morphology in rat pup lungs exposed to hyperoxia. Comp. Med. 2007, 57, 186–192. [Google Scholar]
- Gronbach, J.; Shahzad, T.; Radajewski, S.; Chao, C.-M.; Bellusci, S.; Morty, R.E.; Reicherzer, T.; Ehrhardt, H. The Potentials and Caveats of Mesenchymal Stromal Cell-Based Therapies in the Preterm Infant. Stem Cells Int. 2018, 2018, 9652897. [Google Scholar] [CrossRef] [Green Version]
- Reicherzer, T.; Häffner, S.; Shahzad, T.; Gronbach, J.; Mysliwietz, J.; Hübener, C.; Hasbargen, U.; Gertheiss, J.; Schulze, A.; Bellusci, S.; et al. Activation of the NF-κB pathway alters the phenotype of MSCs in the tracheal aspirates of preterm infants with severe BPD. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 315, L87–L101. [Google Scholar] [CrossRef]
- Chao, C.-M.; Chong, L.; Chu, X.; Shrestha, A.; Behnke, J.; Ehrhardt, H.; Zhang, J.; Chen, C.; Bellusci, S. Targeting Bronchopulmonary Dysplasia-Associated Pulmonary Hypertension (BPD-PH): Potential Role of the FGF Signaling Pathway in the Development of the Pulmonary Vascular System. Cells 2020, 9, 1875. [Google Scholar] [CrossRef] [PubMed]
- Saugstad, O.D. Bronchopulmonary dysplasia and oxidative stress: Are we closer to an understanding of the pathogenesis of BPD? Acta Paediatr. 1997, 86, 1277–1282. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, C.; Sakurai, R.; Wang, Y.; Guo, P.; Ambalavanan, N.; Torday, J.S.; Rehan, V.K. Hyperoxia-induced neonatal rat lung injury involves activation of TGF-{beta} and Wnt signaling and is protected by rosiglitazone. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 296, L1031–L1041. [Google Scholar] [CrossRef] [PubMed]
- Iosef, C.; Alastalo, T.-P.; Hou, Y.; Chen, C.; Adams, E.S.; Lyu, S.-C.; Cornfield, D.N.; Alvira, C.M. Inhibiting NF-κB in the developing lung disrupts angiogenesis and alveolarization. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L1023–L1036. [Google Scholar] [CrossRef] [Green Version]
- Ehrhardt, H.; Pritzke, T.; Oak, P.; Kossert, M.; Biebach, L.; Förster, K.; Koschlig, M.; Alvira, C.M.; Hilgendorff, A. Absence of TNF-α enhances inflammatory response in the newborn lung undergoing mechanical ventilation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 310, L909–L918. [Google Scholar] [CrossRef] [Green Version]
- Zheng, D.; Liwinski, T.; Elinav, E. Inflammasome activation and regulation: Toward a better understanding of complex mechanisms. Cell Discov. 2020, 6, 1–22. [Google Scholar] [CrossRef]
- Dapaah-Siakwan, F.; Zambrano, R.; Luo, S.; Duncan, M.R.; Kerr, N.; Donda, K.; de Rivero Vaccari, J.P.; Keane, R.W.; Dietrich, W.D.; Benny, M.; et al. Caspase-1 Inhibition Attenuates Hyperoxia-induced Lung and Brain Injury in Neonatal Mice. Am. J. Respir. Cell Mol. Biol. 2019, 61, 341–354. [Google Scholar] [CrossRef]
- Lignelli, E.; Palumbo, F.; Myti, D.; Morty, R.E. Recent advances in our understanding of the mechanisms of lung alveolarization and bronchopulmonary dysplasia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 317, L832–L887. [Google Scholar] [CrossRef]
- Behnke, J.; Dippel, C.M.; Choi, Y.; Rekers, L.; Schmidt, A.; Lauer, T.; Dong, Y.; Behnke, J.; Zimmer, K.-P.; Bellusci, S.; et al. Oxygen Toxicity to the Immature Lung—Part II: The Unmet Clinical Need for Causal Therapy. Int. J. Mol. Sci. 2021, 22, 10694. [Google Scholar] [CrossRef]
- Tamatam, C.M.; Reddy, N.M.; Potteti, H.R.; Ankireddy, A.; Noone, P.M.; Yamamoto, M.; Kensler, T.W.; Reddy, S.P. Preconditioning the immature lung with enhanced Nrf2 activity protects against oxidant-induced hypoalveolarization in mice. Sci. Rep. 2020, 10, 19034. [Google Scholar] [CrossRef] [PubMed]
- Nardiello, C.; Morty, R.E. MicroRNA in late lung development and bronchopulmonary dysplasia: The need to demonstrate causality. Mol. Cell. Pediatr. 2016, 3, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugar, S.S.; Heyob, K.M.; Cheng, X.; Lee, R.J.; Rogers, L.K. Perinatal inflammation alters histone 3 and histone 4 methylation patterns: Effects of MiR-29b supplementation. Redox Biol. 2021, 38, 101783. [Google Scholar] [CrossRef] [PubMed]
- Coarfa, C.; Grimm, S.L.; Katz, T.; Zhang, Y.; Jangid, R.K.; Walker, C.L.; Moorthy, B.; Lingappan, K. Epigenetic response to hyperoxia in the neonatal lung is sexually dimorphic. Redox Biol. 2020, 37, 101718. [Google Scholar] [CrossRef]
- Jiao, B.; Tang, Y.; Liu, S.; Guo, C. Tetrandrine attenuates hyperoxia-induced lung injury in newborn rats via NF-κB p65 and ERK1/2 pathway inhibition. Ann. Transl. Med. 2020, 8, 1018. [Google Scholar] [CrossRef]
- Li, Q.; Wall, S.B.; Ren, C.; Velten, M.; Hill, C.L.; Locy, M.L.; Rogers, L.K.; Tipple, T.E. Thioredoxin Reductase Inhibition Attenuates Neonatal Hyperoxic Lung Injury and Enhances Nuclear Factor E2-Related Factor 2 Activation. Am. J. Respir. Cell Mol. Biol. 2016, 55, 419–428. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Chong, L.; Shao, Y.; Chen, S.; Li, C. Lipoxin A4 reduces hyperoxia-induced lung injury in neonatal rats through PINK1 signaling pathway. Int. Immunopharmacol. 2019, 73, 414–423. [Google Scholar] [CrossRef]
- Muramatsu, Y.; Ito, M.; Oshima, T.; Kojima, S.; Ohno, K. Hydrogen-rich water ameliorates bronchopulmonary dysplasia (BPD) in newborn rats. Pediatr. Pulmonol. 2016, 51, 928–935. [Google Scholar] [CrossRef]
- Maturu, P.; Wei-Liang, Y.; Androutsopoulos, V.P.; Jiang, W.; Wang, L.; Tsatsakis, A.M.; Couroucli, X.I. Quercetin attenuates the hyperoxic lung injury in neonatal mice: Implications for Bronchopulmonary dysplasia (BPD). Food Chem. Toxicol. 2018, 114, 23–33. [Google Scholar] [CrossRef]
- Chen, Y.; Li, Q.; Liu, Y.; Shu, L.; Wang, N.; Wu, Y.; Sun, X.; Wang, L. Attenuation of hyperoxia-induced lung injury in neonatal rats by 1α,25-Dihydroxyvitamin D3. Exp. Lung Res. 2015, 41, 344–352. [Google Scholar] [CrossRef]
- Kose, M.; Bastug, O.; Sonmez, M.F.; Per, S.; Ozdemir, A.; Kaymak, E.; Yahşi, H.; Ozturk, M.A. Protective effect of vitamin D against hyperoxia-induced lung injury in newborn rats. Pediatr. Pulmonol. 2017, 52, 69–76. [Google Scholar] [CrossRef]
- James, M.L.; Ross, A.C.; Nicola, T.; Steele, C.; Ambalavanan, N. VARA attenuates hyperoxia-induced impaired alveolar development and lung function in newborn mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 304, L803–L812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, T.M.; Frei, B.; Rifai, N.; Avery, M.E.; Suh, J.; Yoder, B.A.; Coalson, J.J. Early high dose antioxidant vitamins do not prevent bronchopulmonary dysplasia in premature baboons exposed to prolonged hyperoxia: A pilot study. Pediatr. Res. 1998, 43, 719–726. [Google Scholar] [CrossRef] [Green Version]
- Goetz, M.J.; Kremer, S.; Behnke, J.; Staude, B.; Shahzad, T.; Holzfurtner, L.; Chao, C.-M.; Morty, R.E.; Bellusci, S.; Ehrhardt, H. MSC Based Therapies to Prevent or Treat BPD-A Narrative Review on Advances and Ongoing Challenges. IJMS 2021, 22, 1138. [Google Scholar] [CrossRef] [PubMed]
- Endesfelder, S.; Strauß, E.; Scheuer, T.; Schmitz, T.; Bührer, C. Antioxidative effects of caffeine in a hyperoxia-based rat model of bronchopulmonary dysplasia. Respir. Res. 2019, 20, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Wu, Q.; Zhong, D.; Li, C.; Du, L. Caffeine prevents hyperoxia-induced lung injury in neonatal mice through NLRP3 inflammasome and NF-κB pathway. Respir. Res. 2020, 21, 140. [Google Scholar] [CrossRef] [PubMed]
- Teng, R.-J.; Jing, X.; Michalkiewicz, T.; Afolayan, A.J.; Wu, T.-J.; Konduri, G.G. Attenuation of endoplasmic reticulum stress by caffeine ameliorates hyperoxia-induced lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 312, L586–L598. [Google Scholar] [CrossRef]
- Masood, A.; Yi, M.; Lau, M.; Belcastro, R.; Li, J.; Kantores, C.; Pace-Asciak, C.R.; Jankov, R.P.; Tanswell, A.K. Cyclooxygenase-2 inhibition partially protects against 60% O2 -mediated lung injury in neonatal rats. Pediatr. Pulmonol. 2014, 49, 991–1002. [Google Scholar] [CrossRef]
- Britt, R.D.; Velten, M.; Tipple, T.E.; Nelin, L.D.; Rogers, L.K. Cyclooxygenase-2 in newborn hyperoxic lung injury. Free Radic. Biol. Med. 2013, 61, 502–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mankouski, A.; Kantores, C.; Wong, M.J.; Ivanovska, J.; Jain, A.; Benner, E.J.; Mason, S.N.; Tanswell, A.K.; Auten, R.L.; Jankov, R.P. Intermittent hypoxia during recovery from neonatal hyperoxic lung injury causes long-term impairment of alveolar development: A new rat model of BPD. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 312, L208–L216. [Google Scholar] [CrossRef] [PubMed]
- Kothe, T.B.; Kemp, M.W.; Schmidt, A.; Royse, E.; Salomone, F.; Clarke, M.W.; Musk, G.C.; Jobe, A.H.; Hillman, N.H. Surfactant plus budesonide decreases lung and systemic inflammation in mechanically ventilated preterm sheep. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 316, L888–L893. [Google Scholar] [CrossRef] [PubMed]
- Dani, C.; Buonocore, G.; Longini, M.; Felici, C.; Rodriguez, A.; Corsini, I.; Rubaltelli, F.F. Superoxide dismutase and catalase activity in naturally derived commercial surfactants. Pediatr. Pulmonol. 2009, 44, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Backhaus, S.; Zakrzewicz, A.; Richter, K.; Damm, J.; Wilker, S.; Fuchs-Moll, G.; Küllmar, M.; Hecker, A.; Manzini, I.; Ruppert, C.; et al. Surfactant inhibits ATP-induced release of interleukin-1β via nicotinic acetylcholine receptors. J. Lipid Res. 2017, 58, 1055–1066. [Google Scholar] [CrossRef] [Green Version]
- Collins, J.J.P.; Tibboel, D.; de Kleer, I.M.; Reiss, I.K.M.; Rottier, R.J. The Future of Bronchopulmonary Dysplasia: Emerging Pathophysiological Concepts and Potential New Avenues of Treatment. Front. Med. 2017, 4, 61. [Google Scholar] [CrossRef] [Green Version]
- Liao, J.; Kapadia, V.S.; Brown, L.S.; Cheong, N.; Longoria, C.; Mija, D.; Ramgopal, M.; Mirpuri, J.; McCurnin, D.C.; Savani, R.C. The NLRP3 inflammasome is critically involved in the development of bronchopulmonary dysplasia. Nat. Commun. 2015, 6, 8977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hummler, J.K.; Dapaah-Siakwan, F.; Vaidya, R.; Zambrano, R.; Luo, S.; Chen, S.; Kerr, N.; de Rivero Vaccari, J.P.; Keane, R.W.; Dietrich, W.D.; et al. Inhibition of Rac1 Signaling Downregulates Inflammasome Activation and Attenuates Lung Injury in Neonatal Rats Exposed to Hyperoxia. Neonatology 2017, 111, 280–288. [Google Scholar] [CrossRef]
- Tokuriki, S.; Igarashi, A.; Okuno, T.; Ohta, G.; Naiki, H.; Ohshima, Y. Treatment with Geranylgeranylacetone Induces Heat Shock Protein 70 and Attenuates Neonatal Hyperoxic Lung Injury in a Model of Bronchopulmonary Dysplasia. Lung 2017, 195, 469–476. [Google Scholar] [CrossRef] [Green Version]
- Meyer, S.; Gortner, L. Early postnatal additional high-dose oral vitamin A supplementation versus placebo for 28 days for preventing bronchopulmonary dysplasia or death in extremely low birth weight infants. Neonatology 2014, 105, 182–188. [Google Scholar] [CrossRef]
- Brattström, P.; Russo, C.; Ley, D.; Bruschettini, M. High-versus low-dose caffeine in preterm infants: A systematic review and meta-analysis. Acta Paediatr. 2019, 108, 401–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morty, R.E. Using Experimental Models to Identify Pathogenic Pathways and Putative Disease Management Targets in Bronchopulmonary Dysplasia. Neonatology 2020, 117, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Thébaud, B.; Goss, K.N.; Laughon, M.; Whitsett, J.A.; Abman, S.H.; Steinhorn, R.H.; Aschner, J.L.; Davis, P.G.; McGrath-Morrow, S.A.; Soll, R.F.; et al. Bronchopulmonary dysplasia. Nat. Rev. Dis. Primers 2019, 5, 78. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, Y.; Rekers, L.; Dong, Y.; Holzfurtner, L.; Goetz, M.J.; Shahzad, T.; Zimmer, K.-P.; Behnke, J.; Behnke, J.; Bellusci, S.; et al. Oxygen Toxicity to the Immature Lung—Part I: Pathomechanistic Understanding and Preclinical Perspectives. Int. J. Mol. Sci. 2021, 22, 11006. https://doi.org/10.3390/ijms222011006
Choi Y, Rekers L, Dong Y, Holzfurtner L, Goetz MJ, Shahzad T, Zimmer K-P, Behnke J, Behnke J, Bellusci S, et al. Oxygen Toxicity to the Immature Lung—Part I: Pathomechanistic Understanding and Preclinical Perspectives. International Journal of Molecular Sciences. 2021; 22(20):11006. https://doi.org/10.3390/ijms222011006
Chicago/Turabian StyleChoi, Yesi, Lisa Rekers, Ying Dong, Lena Holzfurtner, Maurizio J. Goetz, Tayyab Shahzad, Klaus-Peter Zimmer, Judith Behnke, Jonas Behnke, Saverio Bellusci, and et al. 2021. "Oxygen Toxicity to the Immature Lung—Part I: Pathomechanistic Understanding and Preclinical Perspectives" International Journal of Molecular Sciences 22, no. 20: 11006. https://doi.org/10.3390/ijms222011006
APA StyleChoi, Y., Rekers, L., Dong, Y., Holzfurtner, L., Goetz, M. J., Shahzad, T., Zimmer, K. -P., Behnke, J., Behnke, J., Bellusci, S., & Ehrhardt, H. (2021). Oxygen Toxicity to the Immature Lung—Part I: Pathomechanistic Understanding and Preclinical Perspectives. International Journal of Molecular Sciences, 22(20), 11006. https://doi.org/10.3390/ijms222011006