
Role of PI3K-AKT-mTOR Pathway as a Pro-Survival Signaling and Resistance-Mediating Mechanism to Therapy of Prostate Cancer



Abstract
:1. Introduction
2. PI3K-AKT-mTOR Signaling
2.1. PI3K
2.2. AKT
2.3. mTOR
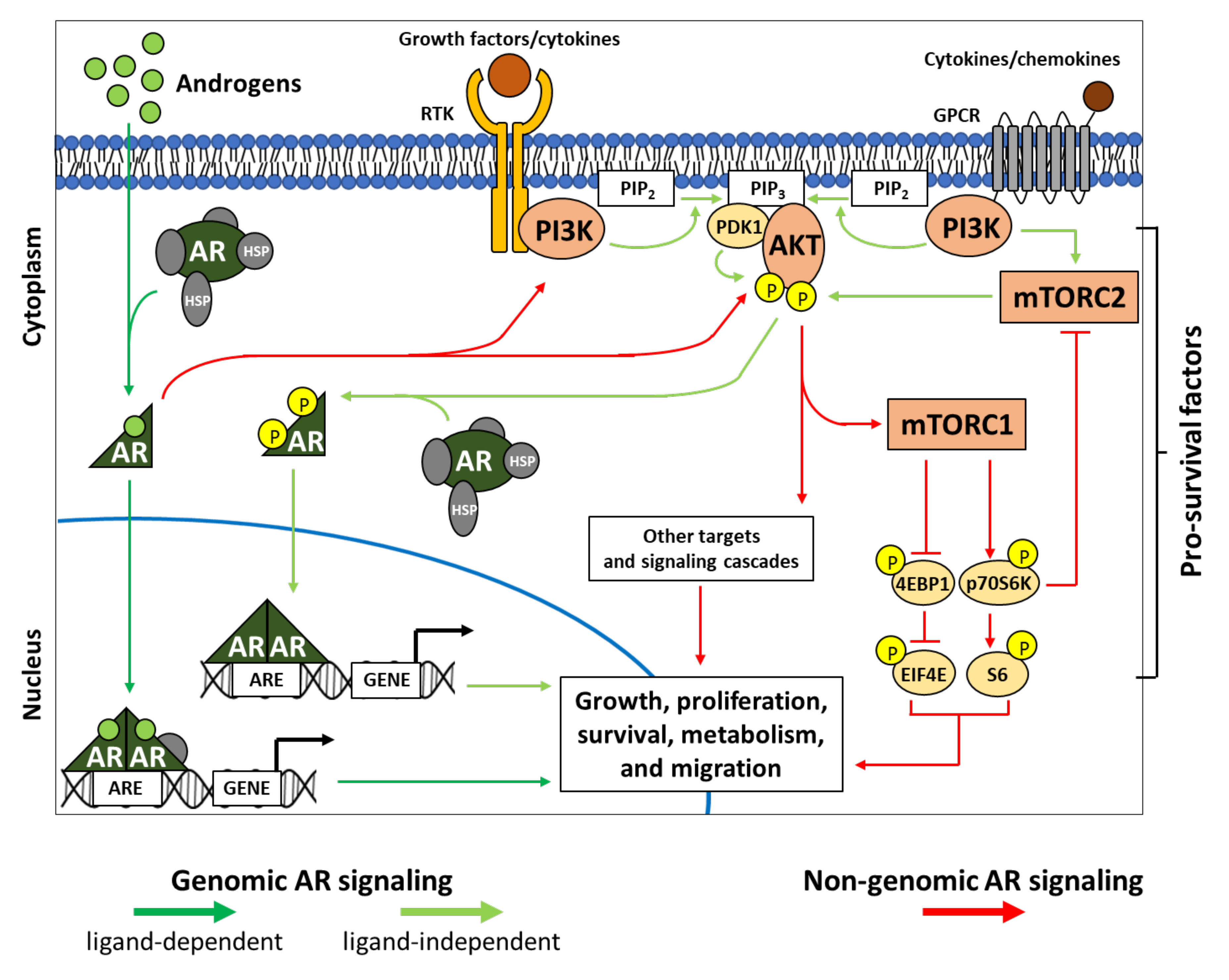
3. PI3K-AKT-mTOR Interplays in Genomic and Non-Genomic AR Signaling
3.1. AR and Genomic Signaling
3.2. AR and Non-Genomic Signaling
4. PI3K-AKT-mTOR in PCa Progression and AR-Targeted Therapy Resistance
5. PI3K-AKT-mTOR Signaling Pathway as Resistance Mechanism to Therapy of PCa
5.1. Deregulation of PI3K-AKT-mTOR Signaling in PCa
5.1.1. PTEN Loss of Function
5.1.2. PI3K Gain of Function
5.1.3. AKT Gain of Function
5.1.4. mTOR Gain of Function
5.2. PI3K-AKT-mTOR as a Pro-Survival/Anti-Apoptotic Signaling
6. Targeting PI3K-AKT-mTOR Signaling in PCa
6.1. PI3K Inhibitors
6.2. AKT Inhibitors
6.3. mTOR Inhibitors
6.4. Dual PI3K and mTORC1/2 Inhibitors
6.5. Combination Strategies with PI3K-AKT-mTOR Inhibitors
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| ADT | androgen deprivation therapy; |
| AML | acute myeloid leukemia; |
| AKT/PKB | protein kinase B; |
| AR | androgen receptor; |
| ARE | androgen response element; |
| ASK1 | apoptosis signal-regulating kinase; |
| BAT | bipolar androgen therapy; |
| CRPCa | castration-resistant prostate cancer; |
| CSPCa | castration-sensitive prostate cancer; |
| DAB2IP | disabled homolog 2-interacting protein; |
| HSP | heat shock protein; |
| LBD | ligand binding domain; |
| MEFs | mouse embryonic fibroblasts; |
| mTOR | mammalian target of Rapamycin; |
| mTORC1/2 | mTOR complex 1/2; |
| PCa | prostate cancer; |
| PDK1 | phosphoinositide-dependent kinase 1; |
| PH | pleckstrin homology; |
| PI3K | phosphatidylinositol-3-kinase; |
| PI(4,5)P2 | phosphatidylinositol-4,5-biphosphate |
| PI(3,4)P2 | phosphatidylinositol-3,4-biphosphate |
| PIP3 | phosphatidylinositol-3,4,5-triphosphate |
| PSA | prostate-specific antigen; |
| RTK | receptor tyrosine kinase; |
| p70S6K | p70S6 kinase; |
| SAL | supraphysiological androgen level; |
| SHIP1/2 | Src homology 2 domain containing inositol polyphosphate 5-phosphatase 1/2 |
References
- Jemal, A.; Siegel, R.; Ward, E.; Murray, T.; Xu, J.; Thun, M.J. Cancer Statistics, 2007. CA Cancer J. Clin. 2007, 57, 43–66. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Rebello, R.J.; Oing, C.; Knudsen, K.E.; Loeb, S.; Johnson, D.C.; Reiter, R.E.; Gillessen, S.; Van der Kwast, T.; Bristow, R.G. Prostate Cancer. Nat. Rev. Dis. Primers 2021, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Lonergan, P.E.; Tindall, D.J. Androgen Receptor Signaling in Prostate Cancer Development and Progression. J. Carcinog. 2011, 10, 20. [Google Scholar] [PubMed]
- La Vignera, S.; Condorelli, R.A.; Russo, G.I.; Morgia, G.; Calogero, A.E. Endocrine Control of Benign Prostatic Hyperplasia. Andrology 2016, 4, 404–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decker, K.F.; Zheng, D.; He, Y.; Bowman, T.; Edwards, J.R.; Jia, L. Persistent Androgen Receptor-Mediated Transcription in Castration-Resistant Prostate Cancer under Androgen-Deprived Conditions. Nucl. Acids Res. 2012, 40, 10765–10779. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.S. Molecular States Underlying Androgen Receptor Activation: A Framework for Therapeutics Targeting Androgen Signaling in Prostate Cancer. J. Clin. Oncol. 2012, 30, 644–646. [Google Scholar] [CrossRef] [PubMed]
- Perner, S.; Cronauer, M.V.; Schrader, A.J.; Klocker, H.; Culig, Z.; Baniahmad, A. Adaptive Responses of Androgen Receptor Signaling in Castration-Resistant Prostate Cancer. Oncotarget 2015, 6, 35542–35555. [Google Scholar] [CrossRef] [Green Version]
- Lakshmana, G.; Baniahmad, A. Interference with the Androgen Receptor Protein Stability in Therapy-Resistant Prostate Cancer. Int. J. Cancer 2018, 144, 1775–1779. [Google Scholar] [CrossRef]
- Ehsani, M.; David, F.; Baniahmad, A. Androgen Receptor-Dependent Mechanisms Mediating Drug Resistance in Prostate Cancer. Cancers 2021, 13, 1534. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, A.C.; Edlind, M.P. PI3K-AKT-mTOR Signaling in Prostate Cancer Progression and Androgen Deprivation Therapy Resistance. Asian J. Androl. 2014, 16, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.G.; Buel, G.R.; Blenis, J. Nutrient Regulation of the mTOR Complex 1 Signaling Pathway. Mol. Cells 2013, 35, 463–473. [Google Scholar] [CrossRef] [Green Version]
- Mori, S.; Nada, S.; Kimura, H.; Tajima, S.; Takahashi, Y.; Kitamura, A.; Oneyama, C.; Okada, M. The mTOR Pathway Controls Cell Proliferation by Regulating the FoxO3a Transcription Factor via SGK1 Kinase. PLoS ONE 2014, 9, e88891. [Google Scholar] [CrossRef] [Green Version]
- Castedo, M.; Ferri, K.F.; Kroemer, G. Mammalian Target of Rapamycin (mTOR): Pro- and Anti-apoptotic. Cell Death Differ. 2002, 9, 99–100. [Google Scholar] [CrossRef] [PubMed]
- Holroyd, A.K.; Michie, A.M. The Role of mTOR-Mediated Signaling in the Regulation of Cellular Migration. Immunol. Lett. 2018, 196, 74–79. [Google Scholar] [CrossRef] [Green Version]
- Jillson, L.; Yette, G.; Laajala, T.; Tilley, W.; Costello, J.; Cramer, S. Androgen Receptor Signaling in Prostate Cancer Genomic Subtypes. Cancers 2021, 13, 3272. [Google Scholar] [CrossRef] [PubMed]
- Toren, P.; Zoubeidi, A. Targeting the PI3K/Akt Pathway in Prostate Cancer: Challenges and Opportunities. Int. J. Oncol. 2014, 45, 1793–1801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shorning, B.Y.; Dass, M.S.; Smalley, M.J.; Pearson, H.B. The PI3K-AKT-mTOR Pathway and Prostate Cancer: At the Crossroads of AR, MAPK, and WNT Signaling. Int. J. Mol. Sci. 2020, 21, 4507. [Google Scholar] [CrossRef] [PubMed]
- Jia, S.; Liu, Z.; Zhang, S.; Liu, P.; Zhang, L.; Lee, S.H.; Zhang, J.; Signoretti, S.; Loda, M.; Roberts, T.M.; et al. Essential Roles of PI(3)K–p110beta in Cell Growth, Metabolism and Tumorigenesis. Nature 2008, 454, 776–779. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Chen, S.; Asara, J.M.; Balk, S.P. Phosphoinositide 3-Kinase Pathway Activation in Phosphate and Tensin Homolog (PTEN)-Deficient Prostate Cancer Cells Is In-Dependent of Receptor Tyrosine Kinases and Mediated by the p110beta and p110delta Catalytic Subunits. J. Biol. Chem. 2010, 285, 14980–14989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The Emerging Mechanisms of Isoform-Specific PI3K Signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. [Google Scholar] [CrossRef]
- Thorpe, L.; Yuzugullu, H.; Zhao, J.J. PI3K in Cancer: Divergent Roles of Isoforms, Modes of Activation and Therapeutic Targeting. Nat. Rev. Cancer 2014, 15, 7–24. [Google Scholar] [CrossRef]
- Liu, P.; Cheng, H.; Roberts, T.M.; Zhao, J.J. Targeting the Phosphoinositide 3-Kinase Pathway in Cancer. Nat. Rev. Drug Discov. 2009, 8, 627–644. [Google Scholar] [CrossRef] [Green Version]
- Guillermet-Guibert, J.; Bjorklof, K.; Salpekar, A.; Gonella, C.; Ramadani, F.; Bilancio, A.; Meek, S.; Smith, A.J.; Okkenhaug, K.; Vanhaesebroeck, B. The p110beta Isoform of Phosphoinositide 3-Kinase Signals Downstream of G Protein-Coupled Receptors and Is Functionally Redundant with p110gamma. Proc. Natl. Acad. Sci. USA 2008, 105, 8292–8297. [Google Scholar] [CrossRef] [Green Version]
- Gewinner, C.; Wang, Z.C.; Richardson, A.; Teruya-Feldstein, J.; Etemadmoghadam, D.; Bowtell, D.; Barretina, J.; Lin, W.M.; Rameh, L.; Salmena, L.; et al. Evidence that Inositol Polyphosphate 4-Phosphatase Type II Is a Tumor Suppressor that Inhibits PI3K Signaling. Cancer Cell 2009, 16, 115–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemmon, M.A. Pleckstrin Homology (PH) Domains and Phosphoinositides. Biochem. Soc. Symp. 2007, 74, 81–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papa, A.; Pandolfi, P.P. The PTEN–PI3K Axis in Cancer. Biomolecules 2019, 9, 153. [Google Scholar] [CrossRef] [Green Version]
- Malek, M.; Kielkowska, A.; Chessa, T.; Anderson, K.E.; Barneda, D.; Pir, P.; Nakanishi, H.; Eguchi, S.; Koizumi, A.; Sasaki, J.; et al. PTEN Regulates PI(3,4)P 2 Signaling Downstream of Class I PI3K. Mol. Cell 2017, 68, 566–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phin, S.; Moore, M.W.; Cotter, P.D. Genomic Rearrangements of PTEN in Prostate Cancer. Front. Oncol. 2013, 3, 240. [Google Scholar] [CrossRef] [Green Version]
- Hodgson, M.C.; Shao, L.-J.; Frolov, A.; Li, R.; Peterson, L.E.; Ayala, G.; Ittmann, M.M.; Weigel, N.L.; Agoulnik, I.U. Decreased Expression and Androgen Regulation of the Tumor Suppressor Gene INPP4B in Prostate Cancer. Cancer Res. 2011, 71, 572–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faes, S.; Dormond, O. PI3K and AKT: Unfaithful Partners in Cancer. Int. J. Mol. Sci. 2015, 16, 21138–21152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majumder, P.K.; Yeh, J.J.; George, D.J.; Febbo, P.G.; Kum, J.; Xue, Q.; Bikoff, R.; Ma, H.; Kantoff, P.; Golub, T.R.; et al. Prostate Intraepithelial Neoplasia Induced by Prostate Restricted Akt Activation: The MPAKT Model. Proc. Natl. Acad. Sci. USA 2003, 100, 7841–7846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drake, J.M.; Graham, N.A.; Lee, J.K.; Stoyanova, T.; Faltermeier, C.M.; Sud, S.; Titz, B.; Huang, J.; Pienta, K.J.; Graeber, T.G.; et al. Metastatic Castration-Resistant Prostate Cancer Reveals Intrapatient Similarity and Interpatient Heterogeneity of Therapeutic Kinase Targets. Proc. Natl. Acad. Sci. USA 2013, 110, 4762–4769. [Google Scholar] [CrossRef] [Green Version]
- Balasuriya, N.; Kunkel, M.T.; Liu, X.; Biggar, K.K.; Li, S.S.; Newton, A.C.; O’Donoghue, P. Genetic Code Expansion and Live Cell Imaging Reveal That Thr-308 Phosphorylation Is Irreplaceable and Sufficient for Akt1 Activity. J. Biol. Chem. 2018, 293, 10744–10756. [Google Scholar] [CrossRef] [Green Version]
- Balasuriya, N.; Davey, N.E.; Johnson, J.L.; Liu, H.; Biggar, K.K.; Cantley, L.C.; Li, S.S.-C.; O’Donoghue, P. Phosphorylation-Dependent Substrate Selectivity of Protein Kinase B (AKT1). J. Biol. Chem. 2020, 295, 8120–8134. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [Green Version]
- Kremer, C.L.; Klein, R.R.; Mendelson, J.; Browne, W.; Samadzedeh, L.K.; Vanpatten, K.; Highstrom, L.; Pestano, G.A.; Nagle, R.B. Expression of mTOR Signaling Pathway Markers in Prostate Cancer Progression. Prostate 2006, 66, 1203–1212. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR Signaling in Growth Control and Disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [Green Version]
- Sancak, Y.; Thoreen, C.C.; Peterson, T.R.; Lindquist, R.A.; Kang, S.A.; Spooner, E.; Carr, S.A.; Sabatini, D.M. PRAS40 Is an Insulin-Regulated Inhibitor of the mTORC1 Protein Kinase. Mol. Cell 2007, 25, 903–915. [Google Scholar] [CrossRef]
- Haar, E.V.; Lee, S.-I.; Bandhakavi, S.; Griffin, T.; Kim, D.-H. Insulin Signalling to mTOR Mediated by the Akt/PKB Substrate PRAS40. Nature 2007, 9, 316–323. [Google Scholar] [CrossRef]
- Oshiro, N.; Takahashi, R.; Yoshino, K.-I.; Tanimura, K.; Nakashima, A.; Eguchi, S.; Miyamoto, T.; Hara, K.; Takehana, K.; Avruch, J.; et al. The Proline-rich Akt Substrate of 40 kDa (PRAS40) Is a Physiological Substrate of Mammalian Target of Rapamycin Complex 1. J. Biol. Chem. 2007, 282, 20329–20339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoki, K.; Zhu, T.; Guan, K.-L. TSC2 Mediates Cellular Energy Response to Control Cell Growth and Survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef] [Green Version]
- Inoki, K.; Ouyang, H.; Zhu, T.; Lindvall, C.; Wang, Y.; Zhang, X.; Yang, Q.; Bennett, C.; Harada, Y.; Stankunas, K.; et al. TSC2 Integrates Wnt and Energy Signals via a Coordinated Phosphorylation by AMPK and GSK3 to Regulate Cell Growth. Cell 2006, 126, 955–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, E.J.; Beal, P.A.; Keith, C.T.; Chen, J.; Shin, T.B.; Schreiber, S.L. Control of p70 S6 Kinase by Kinase Activity of FRAP In Vivo. Nature 1995, 377, 441–446. [Google Scholar] [CrossRef]
- Gingras, A.-C.; Kennedy, S.; O’Leary, M.A.; Sonenberg, N.; Hay, N. 4E-BP1, A Repressor of mRNA Translation, Is Phosphorylated and Inactivated by the Akt (PKB) Signaling Pathway. Genes Dev. 1998, 12, 502–513. [Google Scholar] [CrossRef]
- Jung, C.H.; Jun, C.B.; Ro, S.-H.; Kim, Y.-M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.-H. ULK-Atg13-FIP200 Complexes Mediate mTOR Signaling to the Autophagy Machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef] [Green Version]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.-I.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 Association with the ULK1–Atg13–FIP200 Complex Required for Autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef] [Green Version]
- Ganley, I.G.; Lam du, H.; Wang, J.; Ding, X.; Chen, S.; Jiang, X. ULK1.ATG13.FIP200 Complex Mediates mTOR Signaling and Is Essential for Autophagy. J. Biol. Chem. 2009, 284, 12297–12305. [Google Scholar] [CrossRef] [Green Version]
- Fu, W.; Hall, M.N. Regulation of mTORC2 Signaling. Genes 2020, 11, 1045. [Google Scholar] [CrossRef]
- Huang, J.; Dibble, C.C.; Matsuzaki, M.; Manning, B.D. The TSC1-TSC2 Complex Is Required for Proper Activation of mTOR Complex 2. Mol. Cell. Biol. 2008, 28, 4104–4115. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Wu, S.; Wu, C.-L.; Manning, B.D. Signaling Events Downstream of Mammalian Target of Rapamycin Complex 2 Are Attenuated in Cells and Tumors Deficient for the Tuberous Sclerosis Complex Tumor Suppressors. Cancer Res. 2009, 69, 6107–6114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dibble, C.C.; Asara, J.M.; Manning, B.D. Characterization of Rictor Phosphorylation Sites Reveals Direct Regulation of mTOR Complex 2 by S6K1. Mol. Cell. Biol. 2009, 29, 5657–5670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Julien, L.-A.; Carrière, A.; Moreau, J.; Roux, P.P. mTORC1-Activated S6K1 Phosphorylates Rictor on Threonine 1135 and Regulates mTORC2 Signaling. Mol. Cell. Biol. 2010, 30, 908–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and Regulation of Akt/PKB by the Rictor-mTOR Complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guertin, D.A.; Stevens, D.M.; Saitoh, M.; Kinkel, S.; Crosby, K.; Sheen, J.-H.; Mullholland, D.J.; Magnuson, M.; Wu, H.; Sabatini, D.M. mTOR Complex 2 Is Required for the Development of Prostate Cancer Induced by Pten Loss in Mice. Cancer Cell 2009, 15, 148–159. [Google Scholar] [CrossRef] [Green Version]
- Ellwood-Yen, K.; Keilhack, H.; Kunii, K.; Dolinski, B.; Connor, Y.; Hu, K.; Nagashima, K.; O’Hare, E.; Erkul, Y.; Di Bacco, A.; et al. PDK1 Attenuation Fails to Prevent Tumor Formation in PTEN-Deficient Transgenic Mouse Models. Cancer Res. 2011, 71, 3052–3065. [Google Scholar] [CrossRef] [Green Version]
- Malik, S.N.; Brattain, M.; Ghosh, P.M.; Troyer, D.A.; Prihoda, T.; Bedolla, R.; Kreisberg, J.I. Immunohisto-Chemical Demonstration of Phospho-Akt in High Gleason Grade Prostate Cancer. Clin. Cancer Res. 2002, 8, 1168–1171. [Google Scholar]
- Liao, Y.; Grobholz, R.; Abel, U.; Trojan, L.; Michel, M.S.; Angel, P.; Mayer, D. Increase of AKT/PKB Expression Correlates with Gleason Pattern in Human Prostate Cancer. Int. J. Cancer 2003, 107, 676–680. [Google Scholar] [CrossRef]
- Evren, S.; Dermen, A.; Lockwood, G.; Fleshner, N.; Sweet, J. mTOR-RAPTOR and 14-3-3σ Immunohistochemical Expression in High Grade Prostatic Intraepithelial Neoplasia and Prostatic Adenocarcinomas: A Tissue Microarray Study. J. Clin. Pathol. 2011, 64, 683–688. [Google Scholar] [CrossRef]
- Sutherland, S.I.; Pe Benito, R.; Henshall, S.M.; Horvath, L.G.; Kench, J.G. Expression of Phosphorylated-mTOR during the Development of Prostate Cancer. Prostate 2014, 74, 1231–1239. [Google Scholar] [CrossRef]
- Saranyutanon, S.; Srivastava, S.K.; Pai, S.; Singh, S.; Singh, A.P. Therapies Targeted to Androgen Receptor Signaling Axis in Prostate Cancer: Progress, Challenges, and Hope. Cancers 2019, 12, 51. [Google Scholar] [CrossRef] [Green Version]
- Liao, R.S.; Ma, S.; Miao, L.; Li, R.; Yin, Y.; Raj, G.V. Androgen Receptor-Mediated Non-Genomic Regulation of Prostate Cancer Cell Proliferation. Transl. Androl. Urol. 2013, 2, 187–196. [Google Scholar]
- Wilson, S.; Qi, J.; Filipp, F.V. Refinement of the Androgen Response Element Based on ChIP-Seq in Andro-Gen-Insensitive and Androgen-Responsive Prostate Cancer Cell Lines. Sci. Rep. 2016, 6, 32611. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Wei, T.; Ye, Z.; Orme, J.J.; Lin, D.; Sheng, H.; Fazli, L.; Karnes, R.J.; Jimenez, R.; Wang, L.; et al. A Noncanonical AR Addiction Drives Enzalutamide Resistance in Prostate Cancer. Nat. Commun. 2021, 12, 1–14. [Google Scholar]
- Ueda, T.; Bruchovsky, N.; Sadar, M. Activation of the Androgen Receptor N-terminal Domain by Interleukin-6 via MAPK and STAT3 Signal Transduction Pathways. J. Biol. Chem. 2002, 277, 7076–7085. [Google Scholar] [CrossRef] [Green Version]
- Traish, A.M.; Morgentaler, A. Epidermal Growth Factor Receptor Expression Escapes Androgen Regulation in Prostate Cancer: A Potential Molecular Switch for Tumour Growth. Br. J. Cancer 2009, 101, 1949–1956. [Google Scholar] [CrossRef] [Green Version]
- Culig, Z.; Hobisch, A.; Cronauer, M.V.; Radmayr, C.; Trapman, J.; Hittmair, A.; Bartsch, G.; Klocker, H. Androgen Receptor Activation in Prostatic Tumor Cell Lines by Insulin-Like Growth Factor-I, Keratinocyte Growth Factor, and EP-Idermal Growth Factor. Cancer Res. 1994, 54, 5474–5478. [Google Scholar] [PubMed]
- Wen, S.; Niu, Y.; Huang, H. Posttranslational Regulation of Androgen Dependent and Independent Androgen Receptor Activities in Prostate Cancer. Asian J. Urol. 2019, 7, 203–218. [Google Scholar] [CrossRef]
- Peterziel, H.; Mink, S.; Schonert, A.; Becker, M.; Klocker, H.; Cato, A. Rapid Signalling by Androgen Receptor in Prostate Cancer Cells. Oncogene 1999, 18, 6322–6329. [Google Scholar] [CrossRef] [Green Version]
- Leung, J.; Sadar, M.D. Non-Genomic Actions of the Androgen Receptor in Prostate Cancer. Front. Endocrinol. 2017, 8, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Migliaccio, A.; Castoria, G.; Di Domenico, M.; De Falco, A.; Bilancio, A.; Lombardi, M.; Barone, M.V.; Ametrano, D.; Zannini, M.S.; Abbondanza, C.; et al. Steroid-Induced Androgen Receptor-Oestradiol Receptor Beta-Src Complex Triggers Prostate Cancer Cell Proliferation. EMBO J. 2000, 19, 5406–5417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinlein, C.A.; Chang, C. The Roles of Androgen Receptors and Androgen-Binding Proteins in Nongenomic Androgen Actions. Mol. Endocrinol. 2002, 16, 2181–2187. [Google Scholar] [CrossRef]
- Baron, S.; Manin, M.; Beaudoin, C.; Leotoing, L.; Communal, Y.; Veyssiere, G.; Morel, L. Androgen Receptor Mediates Non-genomic Activation of Phosphatidylinositol 3-OH Kinase in Androgen-Sensitive Epithelial Cells. J. Biol. Chem. 2004, 279, 14579–14586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatson, J.W.; Kaur, P.; Singh, M. Dihydrotestosterone Differentially Modulates the Mitogen-Activated Protein Kinase and the Phosphoinositide 3-Kinase/Akt Pathways through the Nuclear and Novel Membrane Androgen Receptor in C6 Cells. Endocrinology 2006, 147, 2028–2034. [Google Scholar] [CrossRef] [Green Version]
- Cinar, B.; Mukhopadhyay, N.K.; Meng, G.; Freeman, M.R. Phosphoinositide 3-Kinase-independent Non-genomic Signals Transit from the Androgen Receptor to Akt1 in Membrane Raft Microdomains. J. Biol. Chem. 2007, 282, 29584–29593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kokal, M.; Mirzakhani, K.; Pungsrinont, T.; Baniahmad, A. Mechanisms of Androgen Receptor Agonist- and Antagonist-Mediated Cellular Senescence in Prostate Cancer. Cancers 2020, 12, 1833. [Google Scholar] [CrossRef] [PubMed]
- Roediger, J.; Hessenkemper, W.; Bartsch, S.; Manvelyan, M.; Huettner, S.S.; Liehr, T.; Esmaeili, M.; Foller, S.; Petersen, I.; Grimm, M.-O.; et al. Supraphysiological Androgen Levels Induce Cellular Senescence in Human Prostate Cancer Cells through the Src-Akt Pathway. Mol. Cancer 2014, 13, 214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pungsrinont, T.; Sutter, M.F.; Ertingshausen, M.C.C.M.; Lakshmana, G.; Kokal, M.; Khan, A.S.; Baniahmad, A. Senolytic Compounds Control a Distinct Fate of Androgen Receptor Agonist- and Antagonist-Induced Cellular Senescent LNCaP Prostate Cancer Cells. Cell Biosci. 2020, 10, 59. [Google Scholar] [CrossRef] [Green Version]
- Wen, Y.; Hu, M.C.; Makino, K.; Spohn, B.; Bartholomeusz, G.; Yan, D.H.; Hung, M.C. HER-2/Neu Promotes Androgen-Independent Survival and Growth of Prostate Cancer Cells through the Akt Pathway. Cancer Res. 2000, 60, 6841–6845. [Google Scholar] [PubMed]
- Koryakina, Y.; Ta, H.Q.; Gioeli, D. Androgen Receptor Phosphorylation: Biological Context and Functional Consequences. Endocr. Relat. Cancer 2014, 21, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Brawley, S.; Mohan, R.; Nein, C.D. Localized Prostate Cancer: Treatment Options. Am. Fam. Phys. 2018, 97, 798–805. [Google Scholar]
- Huggins, C.; Hodges, C.V. Studies on Prostatic Cancer: I. the Effect of Castration, of Estrogen and of Androgen Injection on Serum Phosphatases in Meta-Static Carcinoma of the Prostate. Cancer Res. 1941, 1, 293–297. [Google Scholar]
- Sharifi, N.; Gulley, J.L.; Dahut, W.L. Androgen Deprivation Therapy for Prostate Cancer. JAMA 2005, 294, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Harris, W.P.; Mostaghel, E.A.; Nelson, P.S.; Montgomery, B. Androgen Deprivation Therapy: Progress in Understanding Mechanisms of Resistance and Optimizing Androgen Depletion. Nat. Clin. Pract. Urol. 2009, 6, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Mills, I. Maintaining and Reprogramming Genomic Androgen Receptor Activity in Prostate Cancer. Nat. Rev. Cancer 2014, 14, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Del Re, M.; Crucitta, S.; Restante, G.; Rofi, E.; Arrigoni, E.; Biasco, E.; Sbrana, A.; Coppi, E.; Galli, L.; Bracarda, S.; et al. Pharmacogenetics of Androgen Signaling in Prostate Cancer: Focus on Castration Resistance and Predictive Biomarkers of Response to Treatment. Crit. Rev. Oncol. Hematol. 2018, 125, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Xin, L.; Teitell, M.A.; Lawson, D.A.; Kwon, A.; Mellinghoff, I.K.; Witte, O.N. Progression of Prostate Cancer by Synergy of AKT with Genotropic and Nongenotropic Actions of the Androgen Receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 7789–7794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helsen, C.; Broeck, T.V.D.; Voet, A.; Prekovic, S.; Van Poppel, H.; Joniau, S.; Claessens, F. Androgen Receptor Antagonists for Prostate Cancer Therapy. Endocr. Relat. Cancer 2014, 21, 105–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maylin, Z.R.; Nicolescu, R.C.; Pandha, H.; Asim, M. Breaking Androgen Receptor Addiction of Prostate Cancer by Targeting Different Functional Domains in the Treatment of Advanced Disease. Transl. Oncol. 2021, 14, 101115. [Google Scholar] [CrossRef]
- Denmeade, S.R.; Isaacs, J.T. Bipolar Androgen Therapy: The Rationale for Rapid Cycling of Supraphysiologic Androgen/Ablation in Men with Castration Resistant Prostate Cancer. Prostate 2010, 70, 1600–1607. [Google Scholar] [CrossRef] [Green Version]
- Szmulewitz, R.; Mohile, S.; Posadas, E.; Kunnavakkam, R.; Karrison, T.; Manchen, E.; Stadler, W.M. A Randomized Phase 1 Study of Testosterone Replacement for Patients with Low-Risk Castration-Resistant Prostate Cancer. Eur. Urol. 2009, 56, 97–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, M.J.; Huang, D.; Kelly, W.K.; Slovin, S.F.; Stephenson, R.D.; Eicher, C.; De La Cruz, A.; Curley, T.; Schwartz, L.H.; Scher, H.I. Phase 1 Trial of High-Dose Exogenous Testosterone in Patients with Castration-Resistant Metastatic Prostate Cancer. Eur. Urol. 2009, 56, 237–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leone, G.; Buttigliero, C.; Pisano, C.; Di Stefano, R.F.; Tabbò, F.; Turco, F.; Vignani, F.; Scagliotti, G.V.; Di Maio, M.; Tucci, M. Bipolar Androgen Therapy in Prostate Cancer: Current Evidences and Future Perspectives. Crit. Rev. Oncol. 2020, 152, 102994. [Google Scholar] [CrossRef]
- Schweizer, M.T.; Wang, H.; Luber, B.; Nadal, R.; Spitz, A.; Rosen, D.M.; Cao, H.; Antonarakis, E.S.; Eisenberger, M.A.; Carducci, M.A.; et al. Bipolar Androgen Therapy for Men with Androgen Ablation Naïve Prostate Cancer: Results from the Phase II BATMAN Study. Prostate 2016, 76, 1218–1226. [Google Scholar] [CrossRef] [PubMed]
- Teply, B.A.; Wang, H.; Luber, B.; Sullivan, R.; Rifkind, I.; Bruns, A.; Spitz, A.; DeCarli, M.; Sinibaldi, V.; Pratz, C.F.; et al. Bipolar Androgen Therapy in Men with Metastatic Castration-Resistant Prostate Cancer after Progression on Enzalutamide: An Open-Label, Phase 2, Multicohort Study. Lancet Oncol. 2017, 19, 76–86. [Google Scholar] [CrossRef] [Green Version]
- Denmeade, S.R. Bipolar Androgen Therapy in the Treatment of Prostate Cancer. Clin. Adv. Hematol. Oncol. 2018, 16, 408–411. [Google Scholar] [PubMed]
- Wach, S.; Taubert, H.; Cronauer, M. Role of Androgen Receptor Splice Variants, Their Clinical Relevance and Treatment Options. World J. Urol. 2019, 38, 647–656. [Google Scholar] [CrossRef]
- Fine, S.W. Neuroendocrine Tumors of the Prostate. Mod. Pathol. 2018, 31, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-H.; Zhang, Y.-Q.; Huang, J.-T. Neuroendocrine Cells of Prostate Cancer: Biologic Functions and Molecular Mechanisms. Asian J. Androl. 2019, 21, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Puca, L.; Vlachostergios, P.; Beltran, H. Neuroendocrine Differentiation in Prostate Cancer: Emerging Biology, Models, and Therapies. Cold Spring Harb. Perspect. Med. 2018, 9, a030593. [Google Scholar] [CrossRef] [PubMed]
- Shiota, M.; Fujimoto, N.; Kashiwagi, E.; Eto, M. The Role of Nuclear Receptors in Prostate Cancer. Cells 2019, 8, 602. [Google Scholar] [CrossRef] [Green Version]
- Puhr, M.; Hoefer, J.; Eigentler, A.; Ploner, C.; Handle, F.; Schaefer, G.; Kroon, J.; Leo, A.; Heidegger, I.M.; Eder, E.I.; et al. The Glucocorticoid Receptor Is a Key Player for Prostate Cancer Cell Survival and a Target for Improved Antiandrogen Therapy. Clin. Cancer Res. 2017, 24, 927–938. [Google Scholar] [CrossRef] [Green Version]
- Grindstad, T.; Andersen, S.; Al-Saad, S.; Donnem, T.; Kiselev, Y.; Nordahl Melbø-Jørgensen, C.; Skjefstad, K.; Busund, L.T.; Bremnes, R.M.; Richardsen, E. High Progesterone Receptor Expression in Prostate Cancer Is Associated with Clinical Failure. PLoS ONE 2015, 10, e0116691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, E.C.; Cambio, A.J.; Yang, J.C.; Ok, J.H.; Lara, P.N., Jr.; Evans, C.P. Clinical Implications of Neuroendocrine Differentiation in Prostate Cancer. Prostate Cancer Prost. Dis. 2007, 10, 6–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bland, T.; Wang, J.; Yin, L.; Pu, T.; Li, J.; Gao, J.; Lin, T.-P.; Gao, A.C.; Wu, B.J. WLS-Wnt Signaling Promotes Neuroendocrine Prostate Cancer. iScience 2020, 24, 101970. [Google Scholar] [CrossRef] [PubMed]
- Nevedomskaya, E.; Baumgart, S.J.; Haendler, B. Recent Advances in Prostate Cancer Treatment and Drug Discovery. Int. J. Mol. Sci. 2018, 19, 1359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumanasuriya, S.; De Bono, J. Treatment of Advanced Prostate Cancer—A Review of Current Therapies and Future Promise. Cold Spring Harb. Perspect. Med. 2017, 8, a030635. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative Genomic Profiling of Human Prostate Cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, D.; Van Allen, E.M.; Wu, Y.-M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.-M.; Montgomery, B.; Taplin, M.-E.; Pritchard, C.C.; Attard, G.; et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [Green Version]
- Abida, W.; Cyrta, J.; Heller, G.; Prandi, D.; Armenia, J.; Coleman, I.; Cieslik, M.; Benelli, M.; Robinson, D.; Van Allen, E.M.; et al. Genomic Correlates of Clinical Outcome in Advanced Prostate Cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 11428–11436. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Kreisberg, J.I.; Ghosh, P.M. Cross-Talk between the Androgen Receptor and the Phosphatidylinositol 3-Kinase/Akt Pathway in Prostate Cancer. Curr. Cancer Drug Targets 2007, 7, 591–604. [Google Scholar] [CrossRef] [PubMed]
- McMenamin, M.E.; Soung, P.; Perera, S.; Kaplan, I.; Loda, M.; Sellers, W.R. Loss of PTEN Expression in Paraffin-Embedded Primary Prostate Cancer Correlates with High Gleason Score and Advanced Stage. Cancer Res. 1999, 59, 4291–4296. [Google Scholar] [PubMed]
- Geybels, M.S.; Fang, M.; Wright, J.L.; Qu, X.; Bibikova, M.; Klotzle, B.; Fan, J.-B.; Feng, Z.; Ostrander, E.A.; Nelson, P.S.; et al. PTEN Loss Is Associated with Prostate Cancer Recurrence and Alterations in Tumor DNA Methylation Profiles. Oncotarget 2017, 8, 84338–84348. [Google Scholar] [CrossRef] [Green Version]
- Pearson, H.B.; Li, J.; Meniel, V.S.; Fennell, C.M.; Waring, P.; Montgomery, K.G.; Rebello, R.J.; Macpherson, A.A.; Koushyar, S.; Furic, L.; et al. Identification of Pik3ca Mutation as a Genetic Driver of Prostate Cancer That Cooperates with Pten Loss to Accelerate Progression and Castration-Resistant Growth. Cancer Discov. 2018, 8, 764–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamaspishvili, T.; Berman, D.; Ross, A.E.; Scher, H.I.; De Marzo, A.M.; Squire, J.; Lotan, T.L. Clinical Implications of PTEN Loss in Prostate Cancer. Nat. Rev. Urol. 2018, 15, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Gao, J.; Lei, Q.-Y.; Rozengurt, N.; Pritchard, C.; Jiao, J.; Thomas, G.; Li, G.; Roy-Burman, P.; Nelson, P.S.; et al. Prostate-Specific Deletion of the Murine Pten Tumor Suppressor Gene Leads to Metastatic Prostate Cancer. Cancer Cell 2003, 4, 209–221. [Google Scholar] [CrossRef] [Green Version]
- Sarker, D.; Reid, A.H.; Yap, T.A.; de Bono, J.S. Targeting the PI3K/AKT Pathway for the Treatment of Prostate Cancer. Clin. Cancer Res. 2009, 15, 4799–4805. [Google Scholar] [CrossRef] [Green Version]
- Mulholland, D.J.; Tran, L.M.; Li, Y.; Cai, H.; Morim, A.; Wang, S.; Plaisier, S.; Garraway, I.P.; Huang, J.; Graeber, T.; et al. Cell Autonomous Role of PTEN in Regulating Castration-Resistant Prostate Cancer Growth. Cancer Cell 2011, 19, 792–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulholland, D.J.; Kobayashi, N.; Ruscetti, M.; Zhi, A.; Tran, L.M.; Huang, J.; Gleave, M.; Wu, H. Pten Loss and RAS/MAPK Activation Cooperate to Promote EMT and Metastasis Initiated from Prostate Cancer Stem/Progenitor Cells. Cancer Res. 2012, 72, 1878–1889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwak, M.K.; Johnson, D.T.; Zhu, C.; Lee, S.H.; Ye, D.-W.; Luong, R.; Sun, Z. Conditional Deletion of the Pten Gene in the Mouse Prostate Induces Prostatic Intraepithelial Neoplasms at Early Ages but a Slow Progression to Prostate Tumors. PLoS ONE 2013, 8, e53476. [Google Scholar] [CrossRef] [PubMed]
- Carver, B.S.; Chapinski, C.; Wongvipat, J.; Hieronymus, H.; Chen, Y.; Chandarlapaty, S.; Arora, V.K.; Le, C.; Koutcher, J.; Scher, H.; et al. Reciprocal Feedback Regulation of PI3K and Androgen Receptor Signaling in PTEN-Deficient Prostate Cancer. Cancer Cell 2011, 19, 575–586. [Google Scholar] [CrossRef] [Green Version]
- Jessen, K.; Kessler, L.; Kucharski, J.; Guo, X.; Staunton, J.; Janes, M.; Elia, M.; Banerjee, U.; Lan, L.; Wang, S.; et al. Abstract A171: A Potent and Selective PI3K Inhibitor, INK1117, Targets Human Cancers Harboring Oncogenic PIK3CA Mutations. Mol. Cancer Ther. 2011, 10, 171. [Google Scholar]
- Fritsch, C.M.; Schnell, C.; Chatenay-Rivauday, C.; Guthy, D.A.; De Pover, A.; Wartmann, M.; Brachmann, S.; Maira, S.M.; Huang, A.; Quadt, C.; et al. NVP-BYL719, a Novel PI3Kalpha Selective In-Hibitor with All the Characteristics Required for Clinical Development as an Anti-Cancer Agent. Cancer Res. 2012, 72. [Google Scholar] [CrossRef]
- Juric, D.; Rodon, J.; Tabernero, J.; Janku, F.; Burris, H.A.; Schellens, J.H.; Middleton, M.R.; Berlin, J.; Schuler, M.; Gil-Martin, M.; et al. Phosphatidylinositol 3-Kinase α–Selective Inhibition with Alpelisib (BYL719) in PIK3CA-Altered Solid Tumors: Results from the First-in-Human Study. J. Clin. Oncol. 2018, 36, 1291–1299. [Google Scholar] [CrossRef] [PubMed]
- Tzenaki, N.; Andreou, M.; Stratigi, K.; Vergetaki, A.; Makrigiannakis, A.; Vanhaesebroeck, B.; Papakonstanti, E.A. High Levels of p110δ PI3K Expression in Solid Tumor Cells Suppress PTEN Activity, Generating Cellular Sensitivity to p110δ Inhibitors through PTEN Activation. FASEB J. 2012, 26, 2498–2508. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Youn, H.; Tang, J.; Tawfik, O.; Dennis, K.; Terranova, P.F.; Du, J.; Raynal, P.; Thrasher, J.B.; Li, B. Phosphoinositide 3-OH Kinase p85alpha and p110beta Are Essential for Androgen Receptor Transactivation and Tumor Progression in Prostate Cancers. Oncogene 2008, 27, 4569–4579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Sun, A.; Youn, H.; Hong, Y.; Terranova, P.F.; Thrasher, J.; Xu, P.; Spencer, D. Conditional Akt Activation Promotes Androgen-Independent Progression of Prostate Cancer. Carcinogenesis 2006, 28, 572–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Pratt, C.; Zeeman, M.E.; Schultz, N.; Taylor, B.S.; O’Neill, A.; Castillo-Martin, M.; Nowak, D.G.; Naguib, A.; Grace, D.M.; et al. Identification of PHLPP1 as a Tumor Suppressor Reveals the Role of Feedback Activation in PTEN-Mutant Prostate Cancer Progression. Cancer Cell 2011, 20, 173–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, P.; Seshacharyulu, P.; Das, S.; Rachagani, S.; Ponnusamy, M.P.; Yan, Y.; Johansson, S.L.; Datta, K.; Lin, M.F.; Batra, S.K. Impaired Expression of Protein Phosphatase 2A Subunits Enhances Metastatic Potential of Human Prostate Cancer Cells through Activation of AKT Pathway. Br. J. Cancer 2013, 108, 2590–2600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, H.; Li, L.; Fridley, B.L.; Jenkins, G.D.; Kalari, K.; Lingle, W.; Petersen, G.; Lou, Z.; Wang, L. FKBP51 Affects Cancer Cell Response to Chemotherapy by Negatively Regulating Akt. Cancer Cell 2009, 16, 259–266. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.; Lee, H.-J.; Wu, S.-P.; Lin, S.-C.; Lanz, R.B.; Creighton, C.J.; DeMayo, F.; Tsai, S.Y.; Tsai, M.-J. Androgen Deprivation–Induced NCoA2 Promotes Metastatic and Castration-Resistant Prostate Cancer. J. Clin. Investig. 2014, 124, 5013–5026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammarsten, P.; Cipriano, M.; Josefsson, A.; Stattin, P.; Egevad, L.; Granfors, T.; Fowler, C.J. Phospho-Akt Immunoreactivity in Prostate Cancer: Relationship to Disease Severity and Outcome, Ki67 and Phosphorylated EGFR Expression. PLoS ONE 2012, 7, e47994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCall, P.; Gemmell, L.K.; Mukherjee, R.; Bartlett, J.M.S.; Edwards, J. Phosphorylation of the Androgen Receptor Is Associated with Reduced Survival in Hormone-Refractory Prostate Cancer Patients. Br. J. Cancer 2008, 98, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Bedolla, R.; Prihoda, T.J.; Kreisberg, J.I.; Malik, S.N.; Krishnegowda, N.K.; Troyer, D.A.; Ghosh, P.M. Determining Risk of Biochemical Recurrence in Prostate Cancer by Immunohistochemical Detection of PTEN Expression and Akt Activation. Clin. Cancer Res. 2007, 13, 3860–3867. [Google Scholar] [CrossRef] [Green Version]
- Kladney, R.D.; Cardiff, R.D.; Kwiatkowski, D.J.; Chiang, G.; Weber, J.; Arbeit, J.M.; Lu, Z.H. Tuberous Sclerosis Complex 1: An Epithelial Tumor Suppressor Essential to Prevent Spontaneous Prostate Cancer in Aged Mice. Cancer Res. 2010, 70, 8937–8947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.; Teruya-Feldstein, J.; Behrendt, N.; Chen, Z.; Noda, T.; Hino, O.; Cordon-Cardo, C.; Pandolfi, P.P. Genetic Analysis of Pten and Tsc2 Functional Interactions in the Mouse Reveals Asymmetrical Haploinsufficiency in Tumor Sup-Pression. Genes Dev. 2005, 19, 1779–1786. [Google Scholar] [CrossRef] [Green Version]
- Peterson, T.R.; Laplante, M.; Thoreen, C.C.; Sancak, Y.; Kang, S.A.; Kuehl, W.M.; Gray, N.S.; Sabatini, D.M. DEPTOR Is an mTOR Inhibitor Frequently Overexpressed in Multiple Myeloma Cells and Required for Their Survival. Cell 2009, 137, 873–886. [Google Scholar] [CrossRef] [Green Version]
- Catena, V.; Bruno, T.; De Nicola, F.; Goeman, F.; Pallocca, M.; Iezzi, S.; Sorino, C.; Cigliana, G.; Floridi, A.; Blandino, G.; et al. Deptor Transcriptionally Regulates Endoplasmic Reticulum Homeostasis in Multiple Myeloma Cells. Oncotarget 2016, 7, 70546–70558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Zhou, Y.; Rychahou, P.; Harris, J.W.; Zaytseva, Y.Y.; Liu, J.; Wang, C.; Weiss, H.L.; Liu, C.; Lee, E.Y.; et al. Deptor Is a Novel Target of Wnt/β-Catenin/c-Myc and Contributes to Colorectal Cancer Cell Growth. Cancer Res. 2018, 78, 3163–3175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, G.; Ouyang, G.; Bao, S. The Activation of Akt/PKB Signaling Pathway and Cell Survival. J. Cell. Mol. Med. 2005, 9, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; Greenberg, M.E. Akt Phosphorylation of BAD Couples Survival Signals to the Cell-Intrinsic Death Machinery. Cell 1997, 91, 231–241. [Google Scholar] [CrossRef] [Green Version]
- Pilling, A.B.; Hwang, C. Targeting Prosurvival BCL2 Signaling through Akt Blockade Sensitizes Castration-Resistant Prostate Cancer Cells to Enzalutamide. Prostate 2019, 79, 1347–1359. [Google Scholar] [CrossRef] [PubMed]
- Cardone, M.H.; Roy, N.; Stennicke, H.R.; Salvesen, G.S.; Franke, T.F.; Stanbridge, E.; Frisch, S.; Reed, J.C. Regulation of Cell Death Protease Caspase-9 by Phosphorylation. Science 1998, 282, 1318–1321. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.H.; Khursigara, G.; Sun, X.; Franke, T.F.; Chao, M.V. Akt Phosphorylates and Negatively Regulates Apoptosis Signal-Regulating Kinase 1. Mol. Cell. Biol. 2001, 21, 893–901. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Luo, D.; Miao, R.; Bai, L.; Ge, Q.; Sessa, W.C.; Min, W. Hsp90–Akt Phosphorylates ASK1 and Inhibits ASK1-Mediated Apoptosis. Oncogene 2005, 24, 3954–3963. [Google Scholar] [CrossRef] [Green Version]
- Barthwal, M.; Sathyanarayana, P.; Kundu, C.N.; Rana, B.; Pradeep, A.; Sharma, C.; Woodgett, J.; Rana, A. Negative Regulation of Mixed Lineage Kinase 3 by Protein Kinase B/AKT Leads to Cell Survival. J. Biol. Chem. 2003, 278, 3897–3902. [Google Scholar] [CrossRef] [Green Version]
- Park, H.-S.; Kim, M.-S.; Huh, S.-H.; Park, J.; Chung, J.; Kang, S.S.; Choi, E.-J. Akt (Protein Kinase B) Negatively Regulates SEK1 by Means of Protein Phosphorylation. J. Biol. Chem. 2002, 277, 2573–2578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, D.; Gore, C.; Zhou, J.; Pong, R.-C.; Zhang, H.; Yu, L.; Vessella, R.L.; Min, W.; Hsieh, J.-T. DAB2IP Coordinates Both PI3K-Akt and ASK1 Pathways for Cell Survival and Apoptosis. Proc. Natl. Acad. Sci. USA 2009, 106, 19878–19883. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Toyooka, S.; Gazdar, A.F.; Hsieh, J.-T. Epigenetic Regulation of a Novel Tumor Suppressor Gene (hDAB2IP) in Prostate Cancer Cell Lines. J. Biol. Chem. 2003, 278, 3121–3130. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Tu, S.-W.; Hsieh, J.-T. Down-regulation of Human DAB2IP Gene Expression Mediated by Polycomb Ezh2 Complex and Histone Deacetylase in Prostate Cancer. J. Biol. Chem. 2005, 280, 22437–22444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basu, S.; Totty, N.F.; Irwin, M.S.; Sudol, M.; Downward, J. Akt Phosphorylates the Yes-Associated Protein, YAP, to Induce Interaction with 14-3-3 and Attenuation of p73-Mediated Apoptosis. Mol. Cell 2003, 11, 11–23. [Google Scholar] [CrossRef]
- Mayo, L.D.; Donner, D.B. A Phosphatidylinositol 3-Kinase/Akt Pathway Promotes Translocation of Mdm2 from the Cytoplasm to the Nucleus. Proc. Natl. Acad. Sci. USA 2001, 98, 11598–11603. [Google Scholar] [CrossRef] [Green Version]
- Gottlieb, T.M.; Leal, J.F.; Seger, R.; Taya, Y.; Oren, M. Cross-Talk between Akt, p53 and Mdm2: Possible Implications for the Regulation of Apoptosis. Oncogene 2002, 21, 1299–1303. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.P.; Hung, M.C. Novel Targets of Akt, p21CipI/WAF1, and MDM2. Semin Oncol. 2002, 29, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Slingerland, J.M. Multiple Roles of the PI3K/PKB (Akt) Pathway in Cell Cycle Progression. Cell Cycle 2003, 2, 336–342. [Google Scholar] [CrossRef] [Green Version]
- Testa, J.R.; Bellacosa, A. AKT Plays a Central Role in Tumorigenesis. Proc. Natl. Acad. Sci. USA 2001, 98, 10983–10985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellacosa, A.; Kumar, C.C.; Di Cristofano, A.; Testa, J.R. Activation of AKT Kinases in Cancer: Implications for Therapeutic Targeting. Adv Cancer Res 2005, 94, 29–86. [Google Scholar]
- Khan, M.; Biswas, D.; Ghosh, M.; Mandloi, S.; Chakrabarti, S.; Chakrabarti, P. mTORC2 Controls Cancer Cell Survival by Modulating Gluconeogenesis. Cell Death Discov. 2015, 1, 15016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goncharova, E.A.; Li, H.; Pimtong, W.; Lu, S.; Khavin, I.; Krymskaya, V.P. mTORC2 Is Required for Proliferation and Survival of TSC2-Null Cells. Mol. Cell. Biol. 2011, 31, 2484–2498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazyken, D.; Magnuson, B.; Bodur, C.; Acosta-Jaquez, H.A.; Zhang, D.; Tong, X.; Barnes, T.M.; Steinl, G.K.; Patterson, N.E.; Altheim, C.H.; et al. AMPK Directly Activates mTORC2 to Promote Cell Survival during Acute Energetic Stress. Sci. Signal. 2019, 12, eaav3249. [Google Scholar] [CrossRef]
- Ruggero, D.; Sonenberg, N. The Akt of translational control. Oncogene 2005, 24, 7426–7434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Q.; Thompson, J.E.; Carroll, M. mTOR Regulates Cell Survival after Etoposide Treatment in Primary AML Cells. Blood 2005, 106, 4261–4268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Xu, H.; An, M. mTORC1 Regulates Apoptosis and Cell Proliferation in Pterygium via Targeting Autophagy and FGFR3. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morita, M.; Prudent, J.; Basu, K.; Goyon, V.; Katsumura, S.; Hulea, L.; Pearl, D.; Siddiqui, N.; Strack, S.; McGuirk, S.; et al. mTOR Controls Mitochondrial Dynamics and Cell Survival via MTFP1. Mol. Cell 2017, 67, 922–935. [Google Scholar] [CrossRef] [Green Version]
- Oki, T.; Mercier, F.; Kato, H.; Jung, Y.; McDonald, T.O.; Spencer, J.A.; Mazzola, M.C.; van Gastel, N.; Lin, C.P.; Michor, F.; et al. Imaging Dynamic mTORC1 Pathway Activity In Vivo Reveals Marked Shifts That Support Time-Specific Inhibitor Therapy in AML. Nat. Commun. 2021, 12, 1–13. [Google Scholar] [CrossRef]
- Meyuhas, O. Ribosomal Protein S6 Phosphorylation: Four Decades of Research. Int. Rev. Cell Mol. Biol. 2015, 320, 41–73. [Google Scholar]
- Jeon, Y.-J.; Kim, I.K.; Hong, S.-H.; Nan, H.; Kim, H.-J.; Lee, H.-J.; Masuda, E.S.; Meyuhas, O.; Oh, B.-H.; Jung, Y.-K. Ribosomal Protein S6 Is a Selective Mediator of TRAIL-Apoptotic Signaling. Oncogene 2008, 27, 4344–4352. [Google Scholar] [CrossRef] [Green Version]
- Wittenberg, A.D.; Azar, S.; Klochendler, A.; Stolovich-Rain, M.; Avraham, S.; Birnbaum, L.; Gallimidi, A.B.; Katz, M.; Dor, Y.; Meyuhas, O. Phosphorylated Ribosomal Protein S6 Is Required for Akt-Driven Hyperplasia and Malignant Transformation, but Not for Hypertrophy, Aneuploidy and Hyperfunction of Pancreatic β-Cells. PLoS ONE 2016, 11, e0149995. [Google Scholar] [CrossRef] [Green Version]
- Bitting, R.; Armstrong, A.J. Targeting the PI3K/Akt/mTOR Pathway in Castration-Resistant Prostate Cancer. Endocr. Relat. Cancer 2013, 20, 83–99. [Google Scholar] [CrossRef] [Green Version]
- Maira, S.M.; Pecchi, S.; Huang, A.; Burger, M.; Knapp, M.; Sterker, D.; Schnell, C.; Guthy, D.; Nagel, T.; Wiesmann, M.; et al. Identification and Characterization of NVP-BKM120, an Orally Available Pan-Class I PI3-Kinase Inhibitor. Mol. Cancer Ther. 2012, 11, 317–328. [Google Scholar] [CrossRef] [Green Version]
- Yadav, S.S.; Li, J.; Stockert, J.A.; O’Connor, J.; Herzog, B.; Elaiho, C.; Galsky, M.D.; Tewari, A.K.; Yadav, K.K. Combination Effect of Therapies Targeting the PI3K- and AR-Signaling Pathways in Prostate Cancer. Oncotarget 2016, 7, 76181–76196. [Google Scholar] [CrossRef] [PubMed]
- Bendell, J.C.; Rodon, J.; Burris, H.A.; de Jonge, M.; Verweij, J.; Birle, D.; Demanse, D.; De Buck, S.S.; Ru, Q.C.; Peters, M.; et al. Phase I, Dose-Escalation Study of BKM120, an Oral Pan-Class I PI3K Inhibitor, in Patients with Advanced Solid Tumors. J. Clin. Oncol. 2012, 30, 282–290. [Google Scholar] [CrossRef]
- Wipf, P.; Minion, D.J.; Halter, R.J.; Berggren, M.I.; Ho, C.B.; Chiang, G.G.; Kirkpatrick, L.; Abraham, R.; Powis, G. Synthesis and Biological Evaluation of Synthetic Viridins Derived From C(20)-Heteroalkylation of the Steroidal PI-3-Kinase Inhibitor Wortmannin. Org. Biomol. Chem. 2004, 2, 1911–1920. [Google Scholar] [CrossRef] [PubMed]
- Hotte, S.J.; Chi, K.N.; Joshua, A.; Tu, D.; Macfarlane, R.J.; Gregg, R.W.; Ruether, J.D.; Basappa, N.S.; Finch, D.; Salim, M.; et al. A Phase II Study of PX-866 in Patients with Recurrent or Metastatic Castration-resistant Prostate Cancer: Canadian Cancer Trials Group Study IND205. Clin. Genitourin. Cancer 2019, 17, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Bhutani, J.; Sheikh, A.; Niazi, A.K. Akt Inhibitors: Mechanism of Action and Implications for Anticancer Therapeutics. Infect. Agent Cancer 2013, 8, 49. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.S.; Banerji, U. Maximising the Potential of AKT Inhibitors as Anti-Cancer Treatments. Pharmacol. Ther. 2016, 172, 101–115. [Google Scholar] [CrossRef]
- Hu, Y.; Qiao, L.; Wang, S.; Rong, S.-B.; Meuillet, E.J.; Berggren, M.; Gallegos, A.; Powis, G.; Kozikowski, A.P. 3-(Hydroxymethyl)-Bearing Phosphatidylinositol Ether Lipid Analogues and Carbonate Surrogates Block PI3-K, Akt, and Cancer Cell Growth. J. Med. Chem. 2000, 43, 3045–3051. [Google Scholar] [CrossRef]
- Luo, Y.; Smith, R.A.; Guan, R.; Liu, X.; Klinghofer, V.; Shen, J.; Hutchins, C.; Richardson, P.; Holzman, T.; Rosenberg, S.H.; et al. Pseudosubstrate Peptides Inhibit Akt and Induce Cell Growth Inhibition. Biochemistry 2004, 43, 1254–1263. [Google Scholar] [CrossRef]
- Barnett, S.F.; Bilodeau, M.T.; Lindsley, C.W. The Akt/PKB Family of Protein Kinases: A Review of Small Molecule Inhibitors and Progress towards Target Validation. Curr. Top. Med. Chem. 2005, 5, 109–125. [Google Scholar] [CrossRef]
- Floryk, D.; Thompson, T.C. Perifosine Induces Differentiation and Cell Death in Prostate Cancer Cells. Cancer Lett. 2008, 266, 216–226. [Google Scholar] [CrossRef] [Green Version]
- Festuccia, C.; Gravina, G.L.; Muzi, P.; Millimaggi, D.; Dolo, V.; Vicentini, C.; Bologna, M. Akt Down-Modulation Induces Apoptosis of Human Prostate Cancer Cells and Synergizes with EGFR Tyrosine Kinase Inhibitors. Prostate 2008, 68, 965–974. [Google Scholar] [CrossRef]
- Posadas, E.M.; Gulley, J.L.; Arlen, P.M.; Trout, A.; Parnes, H.L.; Wright, J.; Lee, M.-J.; Chung, E.J.; Trepel, J.B.; Sparreboom, A.; et al. A Phase II Study of Perifosine in Androgen Independent Prostate Cancer. Cancer Biol. Ther. 2005, 4, 1133–1137. [Google Scholar] [CrossRef] [Green Version]
- Hirai, H.; Sootome, H.; Nakatsuru, Y.; Miyama, K.; Taguchi, S.; Tsujioka, K.; Ueno, Y.; Hatch, H.; Majumder, P.K.; Pan, B.-S.; et al. MK-2206, an Allosteric Akt Inhibitor, Enhances Antitumor Efficacy by Standard Chemotherapeutic Agents or Molecular Targeted Drugs In vitro and In vivo. Mol. Cancer Ther. 2010, 9, 1956–1967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, C.; Lamoureux, F.; Crafter, C.; Davies, B.R.; Beraldi, E.; Fazli, L.; Kim, S.; Thaper, D.; Gleave, M.E.; Zoubeidi, A. Synergistic Targeting of PI3K/AKT Pathway and Androgen Receptor Axis Significantly Delays Castration-Resistant Prostate Cancer Progression In Vivo. Mol. Cancer Ther. 2013, 12, 2342–2355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, B.R.; Greenwood, H.; Dudley, P.; Crafter, C.; Yu, D.-H.; Zhang, J.; Li, J.; Gao, B.; Ji, Q.; Maynard, J.; et al. Preclinical Pharmacology of AZD5363, an Inhibitor of AKT: Pharmacodynamics, Antitumor Activity, and Correlation of Monotherapy Activity with Genetic Background. Mol. Cancer Ther. 2012, 11, 873–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolinsky, M.; Rescigno, P.; Bianchini, D.; Zafeiriou, Z.; Mehra, N.; Mateo, J.; Michalarea, V.; Riisnaes, R.; Crespo, M.; Figueiredo, I.; et al. A Phase I Dose-Escalation Study of Enzalutamide in Combination with the AKT Inhibitor AZD5363 (Capivasertib) in Patients with Metastatic Castration-Resistant Prostate Cancer. Ann. Oncol. 2020, 31, 619–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crabb, S.J.; Birtle, A.J.; Martin, K.; Downs, N.; Ratcliffe, I.; Maishman, T.; Ellis, M.; Griffiths, G.; Thompson, S.; Ksiazek, L.; et al. ProCAID: A Phase I Clinical Trial to Combine the AKT Inhibitor AZD5363 with Docetaxel and Prednisolone Chemotherapy for Metastatic Castration Resistant Prostate Cancer. Investig. New Drugs 2017, 35, 599–607. [Google Scholar] [CrossRef] [Green Version]
- Burris, H.A.; Siu, L.L.; Infante, J.R.; Wheler, J.J.; Kurkjian, C.; Opalinska, J.; Smith, D.A.; Antal, J.M.; Gauvin, J.L.; Gonzalez, T.; et al. Safety, Pharmacokinetics (PK), Pharmacodynamics (PD), and Clinical Activity of the Oral AKT Inhibitor GSK2141795 (GSK795) in a Phase I First-in-Human Study. J. Clin. Oncol. 2011, 29, 3003. [Google Scholar] [CrossRef]
- Aghajanian, C.; Bell-McGuinn, K.M.; Burris, H.A., 3rd; Siu, L.L.; Stayner, L.A.; Wheler, J.J.; Hong, D.S.; Kurkjian, C.; Pant, S.; Santiago-Walker, A.; et al. A Phase I, Open-Label, Two-Stage Study to Investigate the Safety, Tolerability, Pharmacokinetics, and Pharmaco-Dynamics of the Oral AKT Inhibitor GSK2141795 in Patients with Solid Tumors. Investig. New Drugs 2018, 36, 1016–1025. [Google Scholar] [CrossRef]
- Chandarlapaty, S.; Sawai, A.; Scaltriti, M.; Rodrik-Outmezguine, V.; Grbovic-Huezo, O.; Serra, V.; Majumder, P.K.; Baselga, J.; Rosen, N. AKT Inhibition Relieves Feedback Suppression of Receptor Tyrosine Kinase Expression and Activity. Cancer Cell 2011, 19, 58–71. [Google Scholar] [CrossRef] [Green Version]
- Majumder, P.K.; Febbo, P.G.; Bikoff, R.; Berger, R.; Xue, Q.; McMahon, L.M.; Manola, J.; Brugarolas, J.; McDonnell, T.J.; Golub, T.R.; et al. mTOR Inhibition Reverses Akt-Dependent Prostate Intraepithelial Neoplasia through Regulation of Apoptotic and HIF-1-Dependent Pathways. Nat. Med. 2004, 10, 594–601. [Google Scholar] [CrossRef] [PubMed]
- Amato, R.J.; Jac, J.; Mohammad, T.; Saxena, S. Pilot Study of Rapamycin in Patients with Hormone-Refractory Prostate Cancer. Clin. Genitourin. Cancer 2008, 6, 97–102. [Google Scholar] [CrossRef]
- George, D.J.; Halabi, S.; Healy, P.; Jonasch, D.; Anand, M.; Rasmussen, J.; Wood, S.Y.; Spritzer, C.; Madden, J.F.; Armstrong, A.J. Phase 2 Clinical Trial of TORC1 Inhibition with Everolimus in Men with Metastatic Castration-Resistant Prostate Cancer. Urol. Oncol. 2020, 38, 79.e15–79.e22. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.J.; Netto, G.J.; Rudek, M.A.; Halabi, S.; Wood, D.P.; Creel, P.A.; Mundy, K.; Davis, S.L.; Wang, T.; Albadine, R.; et al. A Pharmacodynamic Study of Rapamycin in Men with Intermediate- to High-Risk Localized Prostate Cancer. Clin. Cancer Res. 2010, 16, 3057–3066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.-H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged Rapamycin Treatment Inhibits mTORC2 Assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Yip, C.K.; Murata, K.; Walz, T.; Sabatini, D.M.; Kang, S.A. Structure of the Human mTOR Complex I and Its Implications for Rapamycin Inhibition. Mol. Cell 2010, 38, 768–774. [Google Scholar] [CrossRef] [Green Version]
- Sparks, C.A.; Guertin, D.A. Targeting mTOR: Prospects for mTOR Complex 2 Inhibitors in Cancer Therapy. Oncogene 2010, 29, 3733–3744. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, A.C.; Liu, Y.; Edlind, M.P.; Ingolia, N.T.; Janes, M.R.; Sher, A.; Shi, E.Y.; Stumpf, C.; Christensen, C.; Bonham, M.J.; et al. The Translational Landscape of mTOR Signalling Steers Cancer Initiation and Metastasis. Nature 2012, 485, 55–61. [Google Scholar] [CrossRef] [Green Version]
- Graham, L.; Banda, K.; Torres, A.; Carver, B.S.; Chen, Y.; Pisano, K.; Shelkey, G.; Curley, T.; Scher, H.I.; Lotan, T.; et al. A Phase II Study of the Dual mTOR Inhibitor MLN0128 in Patients with Metastatic Castration Resistant Prostate Cancer. Investig. New Drugs 2018, 36, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Sheng, J.; Liu, Z.; Fan, Y.; Zhang, C.; Lv, T.; Hu, S.; Jin, J.; Yu, W.; Song, Y. Potent Antitumour of the mTORC1/2 Dual Inhibitor AZD2014 in Docetaxel-Sensitive and Docetaxel-Resistant Castration-Resistant Prostate Cancer Cells. J. Cell Mol. Med. 2021, 25, 2436–2449. [Google Scholar] [CrossRef]
- Maira, S.-M.; Stauffer, F.; Brueggen, J.; Furet, P.; Schnell, C.; Fritsch, C.; Brachmann, S.; Chène, P.; De Pover, A.; Schoemaker, K.; et al. Identification and Characterization of NVP-BEZ235, a New Orally Available Dual Phosphatidylinositol 3-Kinase/Mammalian Target of Rapamycin Inhibitor with Potent In Vivo Antitumor Activity. Mol. Cancer Ther. 2008, 7, 1851–1863. [Google Scholar] [CrossRef] [Green Version]
- Wallin, J.J.; Edgar, K.A.; Guan, J.; Berry, M.; Prior, W.W.; Lee, L.; Lesnick, J.D.; Lewis, C.; Nonomiya, J.; Pang, J.; et al. GDC-0980 Is a Novel Class I PI3K/mTOR Kinase Inhibitor with Robust Activity in Cancer Models Driven by the PI3K Pathway. Mol. Cancer Ther. 2011, 10, 2426–2436. [Google Scholar] [CrossRef] [Green Version]
- Luszczak, S.; Simpson, B.S.; Stopka-Farooqui, U.; Sathyadevan, V.K.; Echeverria, L.M.C.; Kumar, C.; Costa, H.; Haider, A.; Freeman, A.; Jameson, C.; et al. Co-Targeting PIM and PI3K/mTOR Using Multikinase Inhibitor AUM302 and a Combination of AZD-1208 and BEZ235 in Prostate Cancer. Sci. Rep. 2020, 10, 14380. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.; Rodon, J.; Sharma, S.; Herbst, R.S.; Tabernero, J.; Infante, J.R.; Silva, A.; Demanse, D.; Hackl, W.; Baselga, J. First-in-Human Phase I Study of the Oral PI3K Inhibitor BEZ235 in Patients (Pts) with Advanced Solid Tumors. J. Clin. Oncol. 2010, 28, 3005. [Google Scholar] [CrossRef]
- Massard, C.; Chi, K.N.; Castellano, D.; de Bono, J.; Gravis, G.; Dirix, L.; Machiels, J.-P.; Mita, A.; Mellado, B.; Turri, S.; et al. Phase Ib Dose-Finding Study of Abiraterone Acetate Plus Buparlisib (BKM120) or Dactolisib (BEZ235) in Patients with Castration-Resistant Prostate Cancer. Eur. J. Cancer 2017, 76, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.J.; Bendell, J.C.; Dolly, S.; Morgan, J.A.; Ware, J.A.; Fredrickson, J.; Mazina, K.E.; Lauchle, J.O.; Burris, H.A.; De Bono, J.S. A First-in-Human Phase I Study to Evaluate GDC-0980, an Oral PI3K/mTOR Inhibitor, Administered QD in Patients with Advanced Solid Tumors. J. Clin. Oncol. 2011, 29, 3020. [Google Scholar] [CrossRef]
- Qian, D.Z.; Rademacher, B.L.; Pittsenbarger, J.; Huang, C.-Y.; Myrthue, A.; Higano, C.S.; Garzotto, M.; Nelson, P.S.; Beer, T.M. CCL2 Is Induced by Chemotherapy and Protects Prostate Cancer Cells from Docetaxel-Induced Cytotoxicity. Prostate 2009, 70, 433–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bozulic, L.; Surucu, B.; Hynx, D.; Hemmings, B.A. PKBalpha/Akt1 Acts Downstream of DNA-PK in the DNA Double-Strand Break Response and Promotes Survival. Mol. Cell 2008, 30, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.K.; Montaser-Kouhsari, L.; Beck, A.H.; Toker, A. MERIT40 Is an Akt Substrate that Promotes Resolution of DNA Damage Induced by Chemotherapy. Cell Rep. 2015, 11, 1358–1366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wendel, H.-G.; De Stanchina, E.; Fridman, J.S.; Malina, A.; Ray, S.; Kogan, S.; Cordon-Cardo, C.; Pelletier, J.; Lowe, S.W. Survival Signalling by Akt and eIF4E in Oncogenesis and Cancer Therapy. Nature 2004, 428, 332–337. [Google Scholar] [CrossRef] [PubMed]
- VanderWeele, D.; Zhou, R.; Rudin, C.M. Akt Up-Regulation Increases Resistance to Microtubule-Directed Chemotherapeutic Agents through Mammalian Target of Rapamycin. Mol. Cancer Ther. 2004, 3, 1605–1613. [Google Scholar] [PubMed]
- Schayowitz, A.; Sabnis, G.; Goloubeva, O.; Njar, V.C.; Brodie, A.M. Prolonging Hormone Sensitivity in Prostate Cancer Xenografts through Dual Inhibition of AR and mTOR. Br. J. Cancer 2010, 103, 1001–1007. [Google Scholar] [CrossRef]
- Nakabayashi, M.; Werner, L.; Courtney, K.D.; Buckle, G.; Oh, W.K.; Bubley, G.J.; Hayes, J.H.; Weckstein, D.; Elfiky, A.; Sims, D.M.; et al. Phase II Trial of RAD001 and Bicalutamide for Castration-Resistant Prostate Cancer. BJU Int. 2012, 110, 1729–1735. [Google Scholar] [CrossRef]
- Chow, H.; Ghosh, P.M.; deVere White, R.; Evans, C.P.; Dall’Era, M.A.; Yap, S.A.; Li, Y.; Beckett, L.A.; Lara, P.N., Jr.; Pan, C.X. A Phase 2 Clinical Trial of Everolimus Plus Bicalutamide for Castration-Resistant Prostate Cancer. Cancer 2016, 122, 1897–1904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bono, J.S.; De Giorgi, U.; Rodrigues, D.N.; Massard, C.; Bracarda, S.; Font, A.; Arija, J.A.A.; Shih, K.C.; Radavoi, G.D.; Xu, N.; et al. Randomized Phase II Study Evaluating Akt Blockade with Ipatasertib, in Combination with Abiraterone, in Patients with Metastatic Prostate Cancer with and without PTEN Loss. Clin. Cancer Res. 2018, 25, 928–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braglia, L.; Zavatti, M.; Vinceti, M.; Martelli, A.M.; Marmiroli, S. Deregulated PTEN/PI3K/AKT/mTOR Signaling in Prostate Cancer: Still a Potential Druggable Target? Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118731. [Google Scholar] [CrossRef] [PubMed]
- Bertacchini, J.; Mediani, L.; Beretti, F.; Guida, M.; Ghalali, A.; Brugnoli, F.; Bertagnolo, V.; Petricoin, E.; Poti, F.; Arioli, J.; et al. Clusterin Enhances AKT2-Mediated Motility of Normal and Cancer Prostate Cells through a PTEN and PHLPP1 Circuit. J. Cell. Physiol. 2018, 234, 11188–11199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crumbaker, M.; Khoja, L.; Joshua, A.M. AR Signaling and the PI3K Pathway in Prostate Cancer. Cancers 2017, 9, 34. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Target | Agent | Phase | Regimen | Population | Status | Registry |
|---|---|---|---|---|---|---|
| Pan-PI3K inhibitors | BKM120 (Buparlisib) | I | +Abiraterone acetate (CYP17A1 inhibitor) | CRPCa progressed on Abiraterone acetate | Completed | NCT01634061 |
| I | +Abiraterone acetate | Docetaxel -pretreated metastatic CRPCa | Terminated | NCT01741753 | ||
| II | Monotherapy | Metastatic CRPCa progressed following ADT and chemotherapy | Terminated | NCT01385293 | ||
| II | Monotherapy | High-risk, localized prostate cancer prior to radical prostatectomy | Terminated | NCT01695473 | ||
| PX866 (Sonolisib) | II | Monotherapy | Metastatic CRPCa progressed following ADT | Completed | NCT01331083 | |
| Dual PI3K/ mTOR inhibitors | BEZ235 | I | +Abiraterone acetate | CRPCa progressed on Abiraterone acetate | Completed | NCT01634061 |
| GDC-0980 | II | +Abiraterone acetate | Docetaxel pre-treated CRPCa | Active, not recruiting | NCT01485861 | |
| LY3023414 | II | +Enzalutamide | Metastatic CRPCa | Completed | NCT02407054 | |
| AKT inhibitors | AZD5363 (capivasertib) | I | Monotherapy | Metastatic CRPCa | Completed | NCT01692262 |
| I | +Enzalutamide or Abiraterone | Metastatic CRPCa | Completed | NCT04087174 | ||
| I/II | +Docetaxel and Prednisolone (glucocorticoid) | Metastatic CRPCa | Active, not recruiting | NCT02121639 | ||
| GSK2141795 (Uprosertib) | I | Monotherapy | Castration-resistant, locally advanced or metastatic with/without PTEN loss | Completed | NCT00920257 | |
| MK2206 | II | +Bicalutamide (anti-androgen) | PCa patients with biochemical relapse and rising PSA after primary therapy | Active, not recruiting | NCT01251861 | |
| I | +Hydroxychloroquine | Stage III PCa | Active, not recruiting | NCT01480154 | ||
| GDC-0068 (Ipatasertib) | II | +Abiraterone acetate and Prednisone | Metastatic or advanced prostate carcinoma | Active, not recruiting | NCT01485861 | |
| Ib | +Atezolizumab and Docetaxel | Metastatic CRPCa | Recruiting | NCT04404140 | ||
| III | +Abiraterone acetate + Prednisone/Prednisolone | Metastatic CRPCa | Active, not recruiting | NCT03072238 | ||
| Perifosine | II | Monotherapy | Metastatic androgen-independent PCa | Completed | NCT00060437 | |
| mTORC1 inhibitors | Everolimus | II | Monotherapy | Metastatic CRPCa | Completed | NCT00629525 |
| I | +Radiation therapy | Biochemical recurrence after radical prostatectomy | Completed | NCT01548807 | ||
| II | +Pasireotide (somatostatin) | Chemotherapy-naive CRPCa | Terminated | NCT01313559 | ||
| I/II | +Docetaxel, Bevacizumab (VEGF inhibitor) | Metastatic CRPCa | Completed | NCT00574769 | ||
| I/II | +Docetaxel | Metastatic CRPCa | Completed | NCT00459186 | ||
| II | +Carboplatin and Predisone | Metastatic CRPCa progressed after Docetaxel | Completed | NCT01051570 | ||
| II | +Bicalutamide | Recurrent or metastatic CRPCa after first-line ADT | Completed | NCT00814788 | ||
| Temsirolimus | I/II | +Bevacizumab | Chemotherapy-treated metastatic CRPCa | Completed | NCT01083368 | |
| II | Monotherapy | Chemotherapy-treated metastatic CRPCa | Terminated | NCT00887640 | ||
| II | Monotherapy | Chemotherapy-naive metastatic CRPCa | Completed | NCT00919035 | ||
| I | +Vorinostat (HDAC inhibitor) | Metastatic CRPCa | Terminated | NCT01174199 | ||
| I/II | +Docetaxel | CRPC receiving first-line docetaxel | Completed | NCT01206036 | ||
| I/II | +Cixutumumab | Metastatic CRPCa | Completed | NCT01026623 | ||
| Dual mTORC1/2 inhibitors | MLN0128 | II | Monotherapy | Metastatic CRPCa | Completed | NCT02091531 |
| AZD2014 | I | Monotherapy | High-risk PCa before radical prostatectomy | Completed | NCT02064608 | |
| I | Monotherapy/+ Abiraterone acetate | CRPCa | Completed | NCT01884285 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pungsrinont, T.; Kallenbach, J.; Baniahmad, A. Role of PI3K-AKT-mTOR Pathway as a Pro-Survival Signaling and Resistance-Mediating Mechanism to Therapy of Prostate Cancer. Int. J. Mol. Sci. 2021, 22, 11088. https://doi.org/10.3390/ijms222011088
Pungsrinont T, Kallenbach J, Baniahmad A. Role of PI3K-AKT-mTOR Pathway as a Pro-Survival Signaling and Resistance-Mediating Mechanism to Therapy of Prostate Cancer. International Journal of Molecular Sciences. 2021; 22(20):11088. https://doi.org/10.3390/ijms222011088
Chicago/Turabian StylePungsrinont, Thanakorn, Julia Kallenbach, and Aria Baniahmad. 2021. "Role of PI3K-AKT-mTOR Pathway as a Pro-Survival Signaling and Resistance-Mediating Mechanism to Therapy of Prostate Cancer" International Journal of Molecular Sciences 22, no. 20: 11088. https://doi.org/10.3390/ijms222011088
APA StylePungsrinont, T., Kallenbach, J., & Baniahmad, A. (2021). Role of PI3K-AKT-mTOR Pathway as a Pro-Survival Signaling and Resistance-Mediating Mechanism to Therapy of Prostate Cancer. International Journal of Molecular Sciences, 22(20), 11088. https://doi.org/10.3390/ijms222011088