
Distinct Evolutionary Profiles and Functions of microRNA156 and microRNA529 in Land Plants

 , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
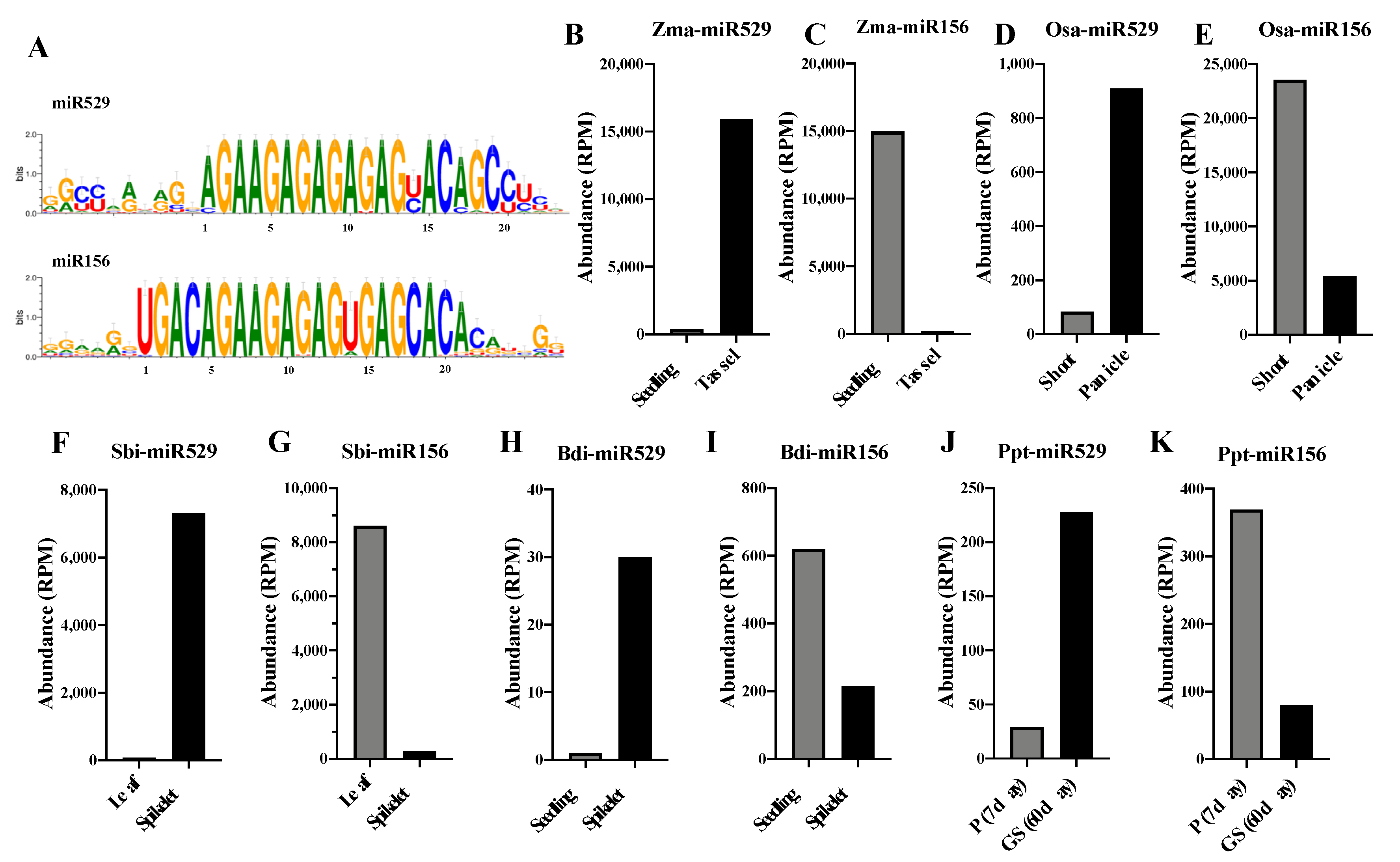
2.1. Sequence Comparison Reveals Differences in the Mismatch-Sensitive (MS) Regions of miR156 and miR529
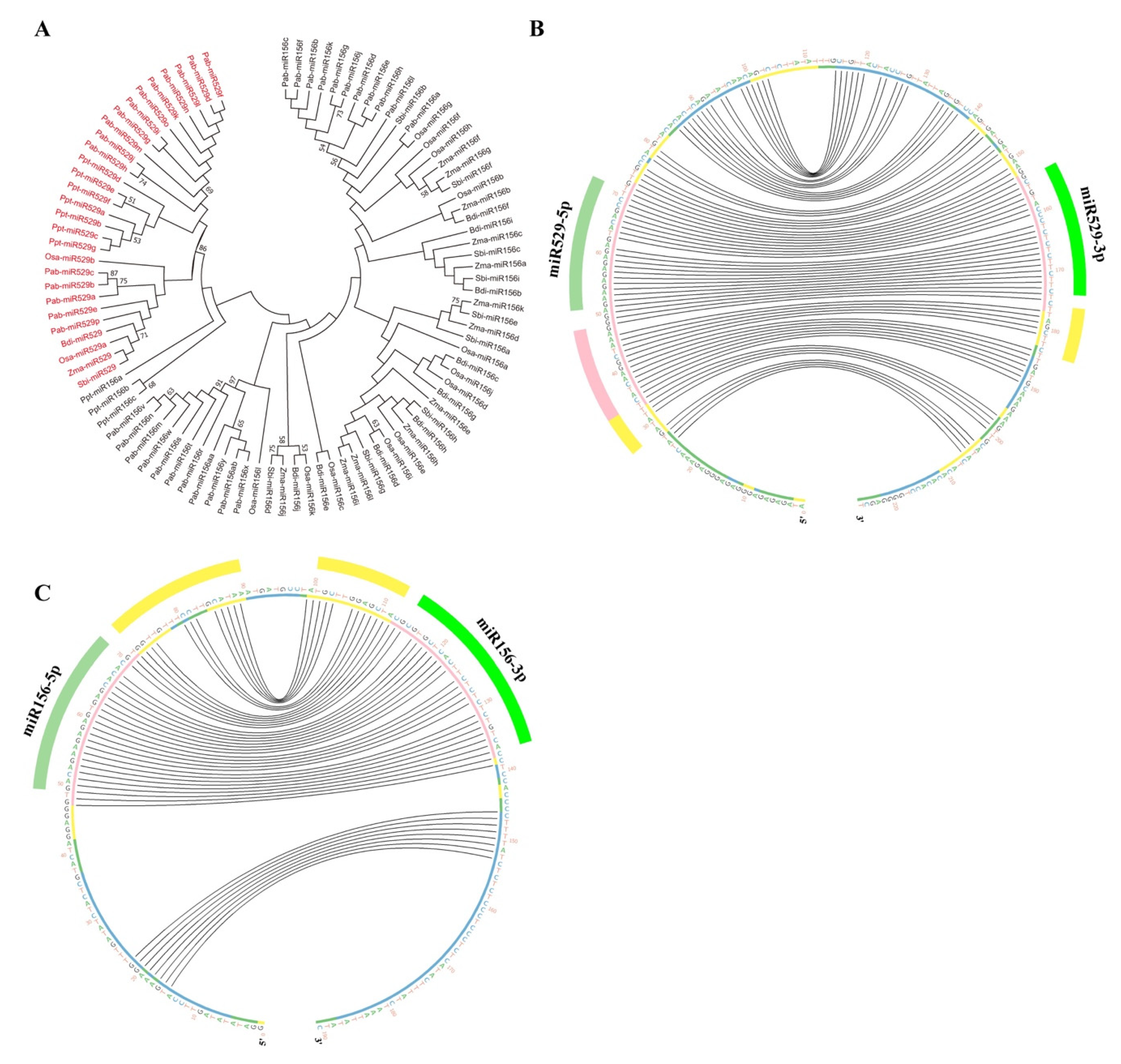
2.2. Phylogenetic Analysis Shows That MIR156 and MIR529 Evolved Independently and Exhibit Divergent Evolutionary Patterns
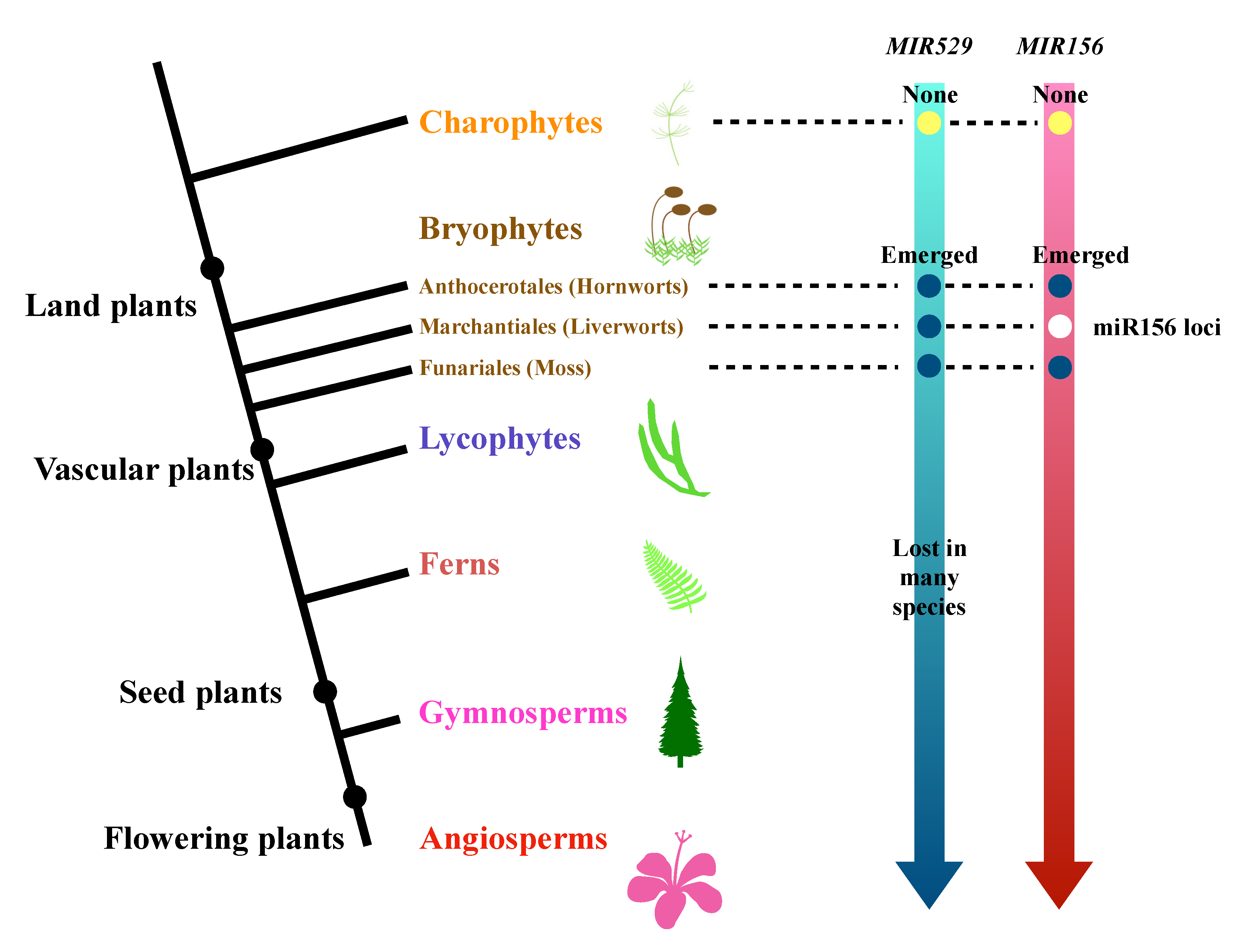
2.3. The Evolutionary History of miR529 and miR156 in Land Plants
2.4. A New Method to Distinguish miR529 from miR156 among Diverse Plant Species
2.5. Comparative Analysis of the Expression Patterns of miR156 and miR529 in Land Plants
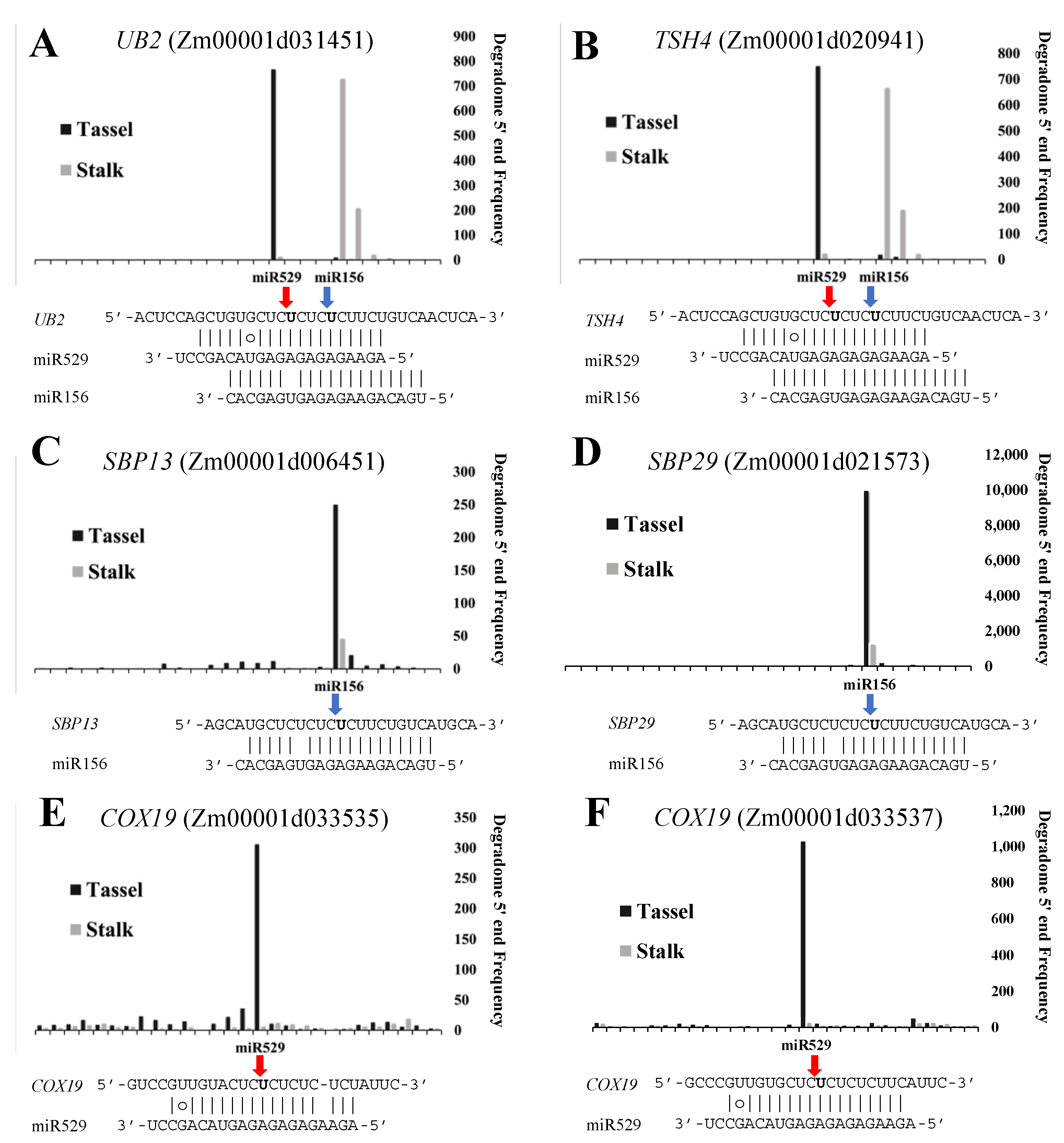
2.6. Common and Specific Target Genes of miR156 and miR529 in Land Plants
3. Discussion
3.1. The Distinction between miR529 and miR156
3.2. Comparative Analysis of the Origins and Continuity of miR156 and miR529 in Land Plants
3.3. miR156 and miR529 Exhibit Different Expression Patterns and Function Cooperatively or Independently by Regulating Common or Specific Targets
4. Materials and Methods
4.1. Prediction of miR156 and miR529 Family Members in Land Plants
4.2. Prediction of miRNA Targets
4.3. RNA Extraction and Library Preparation
4.4. sRNA-Seq Data Processing
4.5. PARE Analysis
4.6. Identification of Homologous Genes
4.7. Multiple Sequence Alignment and Visualization
4.8. Construction of Phylogenetic Trees
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Moran, Y.; Agron, M.; Praher, D.; Technau, U. The evolutionary origin of plant and animal microRNAs. Nat. Ecol. Evol. 2017, 1, 27. [Google Scholar] [CrossRef]
- Chen, X.M. Small RNAs and their roles in plant development. Annu. Rev. Cell Dev. Biol. 2009, 25, 21–44. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Li, Y.; Cao, X.; Qi, Y. MicroRNAs and their regulatory roles in plant–environment interactions. Ann. Rev. Plant Biol. 2019, 70, 489–525. [Google Scholar] [CrossRef]
- Rogers, K.; Chen, X. Biogenesis, turnover, and mode of action of plant microRNAs. Plant Cell 2013, 25, 2383–2399. [Google Scholar] [CrossRef] [Green Version]
- Achkar, N.P.; Cambiagno, D.A.; Manavella, P.A. miRNA biogenesis: A dynamic pathway. Trends Plant Sci. 2016, 21, 1034–1044. [Google Scholar] [CrossRef] [PubMed]
- Manavella, P.A.; Yang, W.S.; Palatnik, J. Keep calm and carry on: miRNA biogenesis under stress. Plant J. 2019, 99, 832–843. [Google Scholar] [PubMed] [Green Version]
- Schwab, R.; Palatnik, J.F.; Riester, M.; Schommer, C.; Schmid, M.; Weigel, D. Specific effects of microRNAs on the plant transcriptome. Dev. Cell 2005, 8, 517–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, X.; Zhuang, Z.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server (2017 release). Nucleic Acids Res. 2018, 46, W49–W54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallory, A.C.; Reinhart, B.J.; Jones-Rhoades, M.W.; Tang, G.L.; Zamore, P.D.; Barton, M.K.; Bartel, D.P. MicroRNA control of PHABULOSA in leaf development: Importance of pairing to the microRNA 5′ region. EMBO J. 2004, 23, 3356–3364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Wang, F.; Axtell, M.J. Analysis of complementarity requirements for plant microRNA targeting using a Nicotiana benthamiana quantitative transient assay. Plant Cell 2014, 26, 741–753. [Google Scholar] [CrossRef] [Green Version]
- Meyers, B.C.; Axtell, M.J.; Bartel, B.; Bartel, D.P.; Baulcombe, D.; Bowman, J.L.; Cao, X.; Carrington, J.C.; Chen, X.; Green, P.J.; et al. Criteria for annotation of plant microRNAs. Plant Cell 2008, 20, 3186–3190. [Google Scholar] [CrossRef]
- Axtell, M.J.; Meyers, B.C. Revisiting criteria for plant microRNA annotation in the era of big data. Plant Cell 2018, 30, 272–284. [Google Scholar] [CrossRef] [Green Version]
- Klein, J.; Saedler, H.; Huijser, P. A new family of DNA binding proteins includes putative transcriptional regulators of the Antirrhinum majus floral meristem identity gene. Mol. Gen. Genet. 1996, 250, 7–16. [Google Scholar]
- Wang, L.; Sun, S.; Jin, J.; Fu, D.; Yang, X.; Weng, X.; Xu, C.; Li, X.; Xiao, J.; Zhang, Q. Coordinated regulation of vegetative and reproductive branching in rice. Proc. Natl. Acad. Sci. USA 2015, 112, 15504–15509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, G.; Poethig, R.S. Temporal regulation of shoot development in Arabidopsis thaliana by miR156 and its target SPL3. Development 2006, 133, 3539–3547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Wang, H. The miR156-SPL module, a regulatory hub and versatile toolbox, gears up crops for enhanced agronomic traits. Mol. Plant 2015, 8, 677–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, E.K.; Li, C.; Li, Y.; Liu, Z.; Xu, J.H. MiR529a modulates panicle architecture through regulating SQUAMOSA PROMOTER BINDING-LIKE genes in rice (Oryza sativa). Plant Mol. Biol. 2017, 94, 469–480. [Google Scholar] [CrossRef]
- Xie, Y.; Zhou, Q.; Zhao, Y.; Li, Q.; Liu, Y.; Ma, M.; Wang, B.; Shen, R.; Zheng, Z.; Wang, H. FHY3 and FAR1 integrate light signals with the miR156-SPL module-mediated aging pathway to regulate Arabidopsis flowering. Mol. Plant 2020, 13, 483–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stief, A.; Altmann, S.; Hoffmann, K.; Pant, B.D.; Scheible, W.R.; Baurle, I. Arabidopsis miR156 regulates tolerance to recurring environmental stress through SPL transcription factors. Plant Cell 2014, 26, 1792–1807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Studer, A.J.; Zhao, Q.; Meeley, R.; Doebley, J.F. Evidence that the origin of naked kernels during maize domestication was caused by a single amino acid substitution in tga1. Genetics 2015, 200, 965–974. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Yu, H.; Xiong, G.; Lu, Z.; Jiao, Y.; Meng, X.; Liu, G.; Chen, X.; Wang, Y.; Li, J. Tissue-specific ubiquitination by IPA1 INTERACTING PROTEIN1 modulates IPA1 protein levels to regulate plant architecture in rice. Plant Cell 2017, 29, 697–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, L.Z.; Zhang, S.D. Exploring the evolutionary differences of SBP-box genes targeted by miR156 and miR529 in plants. Genetica 2012, 140, 317–324. [Google Scholar] [CrossRef]
- Zhang, S.D.; Ling, L.Z.; Zhang, Q.F.; Xu, J.D.; Cheng, L. Evolutionary comparison of two combinatorial regulators of SBP-Box genes, miR156 and miR529, in plants. PLoS ONE 2015, 10, e0124621. [Google Scholar] [CrossRef] [Green Version]
- Li, F.W.; Nishiyama, T.; Waller, M.; Frangedakis, E.; Keller, J.; Li, Z.; Fernandez-Pozo, N.; Barker, M.S.; Bennett, T.; Blázquez, M.A.; et al. Anthoceros genomes illuminate the origin of land plants and the unique biology of hornworts. Nat. Plants 2020, 6, 259–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Fu, X.X.; Li, R.Q.; Zhao, X.; Liu, Y.; Li, M.H.; Zwaenepoel, A.; Ma, H.; Goffinet, B.; Guan, Y.L.; et al. The hornwort genome and early land plant evolution. Nat. Plants 2020, 6, 107–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuperus, J.T.; Fahlgren, N.; Carrington, J.C. Evolution and functional diversification of MIRNA genes. Plant Cell 2011, 23, 432–442. [Google Scholar] [CrossRef] [Green Version]
- Chorostecki, U.; Moro, B.; Rojas, A.M.L.; Debernardi, J.M.; Schapire, A.L.; Notredame, C.; Palatnik, J.F. Evolutionary footprints reveal insights into plant microRNA biogenesis. Plant Cell 2017, 29, 1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morea, E.G.O.; da Silva, E.M.; Silva, G.F.F.E.; Valente, G.T.; Rojas, C.H.B.; Vincentz, M.; Nogueira, F.T.S. Functional and evolutionary analyses of the miR156 and miR529 families in land plants. BMC Plant Biol. 2016, 16, 40. [Google Scholar]
- Sunkar, R.; Girke, T.; Jain, P.K.; Zhu, J.K. Cloning and characterization of microRNAs from rice. Plant Cell 2005, 17, 1397–1411. [Google Scholar] [CrossRef] [Green Version]
- Allen, E.; Xie, Z.; Gustafson, A.M.; Sung, G.H.; Spatafora, J.W.; Carrington, J.C. Evolution of microRNA genes by inverted duplication of target gene sequences in Arabidopsis thaliana. Nat. Genet. 2004, 36, 1282–1290. [Google Scholar] [CrossRef]
- Bustamante, A.; Marques, M.C.; Sanz-Carbonell, A.S.; Mulet, J.M.; Gomez, G. Alternative processing of its precursor is related to miR319 decreasing in melon plants exposed to cold. Sci. Rep. 2018, 8, 15538. [Google Scholar] [CrossRef]
- Krom, N.; Ramakrishna, W. Comparative analysis of divergent and convergent gene pairs and their expression patterns in rice, Arabidopsis, and Populus. Plant Physiol. 2008, 147, 1763–1773. [Google Scholar] [CrossRef] [Green Version]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Li, P.C.; Li, X.; Liu, C.Y.; Cao, S.Y.; Chu, C.C.; Cao, X.F. Loss of function of OsDCL1 affects microRNA accumulation and causes developmental defects in rice. Plant Physiol. 2005, 139, 296–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Axtell, M.J.; Snyder, J.A.; Bartell, D.P. Common functions for diverse small RNAs of land plants. Plant Cell 2007, 19, 1750–1769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.Y.; Xu, Y.Y.; Huan, Q.; Chong, K. Deep sequencing of Brachypodium small RNAs at the global genome level identifies microRNAs involved in cold stress response. BMC Genom. 2009, 10, 449. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.F.; Chia, J.M.; Kumari, S.; Stein, J.C.; Liu, Z.J.; Narechania, A.; Maher, C.A.; Guill, K.; McMullen, M.D.; Ware, D. A Genome-wide characterization of microRNA genes in maize. PloS Genet 2009, 5, e1000716. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Zheng, Y.; Jagadeeswaran, G.; Li, Y.F.; Gowdu, K.; Sunkar, R. Identification and temporal expression analysis of conserved and novel microRNAs in Sorghum. Genom. 2011, 98, 460–468. [Google Scholar] [CrossRef] [Green Version]
- Bertolini, E.; Verelst, W.; Horner, D.S.; Gianfranceschi, L.; Piccolo, V.; Inze, D.; Pe, M.E.; Mica, E. Addressing the role of microRNAs in reprogramming leaf growth during drought stress in Brachypodium distachyon. Mol. Plant 2013, 6, 423–443. [Google Scholar] [CrossRef] [Green Version]
- Xia, R.; Xu, J.; Arikit, S.; Meyers, B.C. Extensive families of miRNAs and PHAS loci in Norway Spruce demonstrate the origins of complex phasiRNA networks in seed plants. Mol. Biol. Evol. 2015, 32, 2905–2918. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.J.; Qin, C.; Chen, Z.; Zuo, T.; Yang, X.R.; Zhou, H.K.; Xu, M.; Cao, S.L.; Shen, Y.O.; Lin, H.J.; et al. Identification of miRNAs and their target genes in developing maize ears by combined small RNA and degradome sequencing. BMC Genom. 2014, 15, 25. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Jiang, Z.; Gao, L.; You, C.; Ma, X.; Wang, X.; Xu, X.; Mo, B.; Chen, X.; Liu, L. Genome-wide transcript and small RNA profiling reveals transcriptomic responses to heat stress. Plant Physiol. 2019, 181, 609–629. [Google Scholar] [CrossRef] [Green Version]
- Goodstein, D.M.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 2012, 40, D1178–D1186. [Google Scholar] [CrossRef]
- Chang, J.M.; Di, T.P.; Lefort, V.; Gascuel, O.; Notredame, C. TCS: A web server for multiple sequence alignment evaluation and phylogenetic reconstruction. Nucleic Acids Res. 2015, 43, W3–W6. [Google Scholar] [CrossRef] [PubMed]
- Kenrick, P.; Crane, P.R. The origin and early evolution of plants on land. Nature 1997, 389, 33–39. [Google Scholar] [CrossRef]
- Christenhusz, M.J.M.; Reveal, J.L.; Farjon, A.; Gardner, M.F.; Mill, R.R.; Chase, M.W. A new classification and linear sequence of extant gymnosperms. Phytotaxa 2011, 19, 55–70. [Google Scholar] [CrossRef]
- Angiosperm Phylogeny Group. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 2016, 181, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Pteridophyte Phylogeny Group I. A community-derived classification for extant lycophytes and ferns. J. Syst. Evol. 2016, 54, 563–603. [Google Scholar]
- Ligrone, R.; Duckett, J.G.; Renzaglia, K.S. Major transitions in the evolution of early land plants: A bryological perspective. Ann. Bot. 2012, 109, 851–871. [Google Scholar] [CrossRef] [PubMed]
- You, C.J.; Cui, J.; Wang, H.; Qi, X.P.; Kuo, L.Y.; Ma, H.; Gao, L.; Mo, B.X.; Chen, X.M. Conservation and divergence of small RNA pathways and microRNAs in land plants. Genome Biol. 2017, 18, 158. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Althoff, F.; Su, G.; Lee, G.; Takenaka, M.; Hong, S.; Lu, C.; Lin, P.; Lin, S.; Zachgo, S.; et al. Identification of miRNAs and their targets in the liverwort Marchantia polymorpha by integrating RNA-Seq and degradome analyses. Plant Cell Physiol. 2016, 57, 339–358. [Google Scholar]
- Tsuzuki, M.; Nishihama, R.; Ishizaki, K.; Kurihara, Y.; Matsui, M.; Bowman, J.L.; Kohchi, T.; Hamada, T.; Watanabe, Y. Profiling and characterization of small RNAs in the liverwort, Marchantia polymorpha, belonging to the first diverged land plants. Plant Cell Physiol. 2016, 57, 359–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, G.; Park, M.Y.; Conway, S.R.; Wang, J.W.; Weigel, D.; Poethig, R.S. The sequential action of miR156 and miR172 regulates developmental timing in Arabidopsis. Cell 2009, 138, 750–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Modjtahedi, N.; Tokatlidis, K.; Dessen, P.; Kroemer, G. Mitochondrial proteins containing Coiled-Coil-Helix-Coiled-Coil-Helix (CHCH) domains in health and disease. Trends Biochem Sci. 2016, 41, 245–260. [Google Scholar] [CrossRef] [Green Version]
- Tsuzuki, M.; Futagami, K.; Shimamura, M.; Inoue, C.; Kunimoto, K.; Oogami, T.; Tomita, Y.; Inoue, K.; Kohchi, T.; Yamaoka, S.; et al. An early arising role of the microRNA156/529-SPL module in reproductive development revealed by the liverwort Marchantia polymorpha. Curr. Biol. 2019, 29, 3307–3314.e5. [Google Scholar] [CrossRef]
- Sun, M.; Shen, Y.; Li, H.; Yang, J.; Cai, X.; Zheng, G.; Zhu, Y.; Jia, B.; Sun, X. The multiple roles of OsmiR535 in modulating plant height, panicle branching and grain shape. Plant Sci. 2019, 283, 60–69. [Google Scholar] [CrossRef]
- Jeong, D.H.; Park, S.; Zhai, J.; Gurazada, S.; Paoli, E.; Meyers, B.C.; Green, P.J. Massive analysis of rice small RNAs: Mechanistic implications of regulated microRNAs and variants for differential target RNA cleavage. Plant Cell 2011, 23, 4185–4207. [Google Scholar] [CrossRef] [Green Version]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.W.; Brouwer, P.; Carretero-Paulet, L.; Cheng, S.; de Vries, J.; Delaux, P.M.; Eily, A.; Koppers, N.; Kuo, L.Y.; Li, Z.; et al. Fern genomes elucidate land plant evolution and cyanobacterial symbioses. Nat. Plants 2018, 4, 460–472. [Google Scholar] [CrossRef] [Green Version]
- Nystedt, B.; Street, N.R.; Wetterbom, A.; Zuccolo, A.; Lin, Y.C.; Scofield, D.G.; Vezzi, F.; Delhomme, N.; Giacomello, S.; Alexeyenko, A.; et al. The Norway spruce genome sequence and conifer genome evolution. Nature 2013, 497, 579–584. [Google Scholar] [CrossRef] [Green Version]
- Gruber, A.R.; Bernhart, S.H.; Lorenz, R. The ViennaRNA web services. Methods Mol. Biol. 2015, 1269, 307–326. [Google Scholar] [PubMed]
- Zhai, J.X.; Arikit, S.; Simon, S.A.; Kingham, B.F.; Meyers, B.C. Rapid construction of parallel analysis of RNA end (PARE) libraries for Illumina sequencing. Methods 2014, 67, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Addo-Quaye, C.; Miller, W.; Axtell, M.J. CleaveLand: A pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics 2009, 25, 130–131. [Google Scholar] [CrossRef] [PubMed]
- Portwood, J.L.; Woodhouse, M.R.; Cannon, E.K.; Gardiner, J.M.; Harper, L.C.; Schaeffer, M.L.; Walsh, J.R.; Sen, T.Z.; Cho, K.T.; Schott, D.A.; et al. MaizeGDB 2018: The maize multi-genome genetics and genomics database. Nucleic Acids Res. 2019, 47, D1146–D1154. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, Q.; Wang, X.; He, J.; Lan, T.; Zheng, J.; Li, Y.; Pan, J.; Lin, L.; Zhao, J.; Li, J.; et al. Distinct Evolutionary Profiles and Functions of microRNA156 and microRNA529 in Land Plants. Int. J. Mol. Sci. 2021, 22, 11100. https://doi.org/10.3390/ijms222011100
Xie Q, Wang X, He J, Lan T, Zheng J, Li Y, Pan J, Lin L, Zhao J, Li J, et al. Distinct Evolutionary Profiles and Functions of microRNA156 and microRNA529 in Land Plants. International Journal of Molecular Sciences. 2021; 22(20):11100. https://doi.org/10.3390/ijms222011100
Chicago/Turabian StyleXie, Qi, Xufeng Wang, Juan He, Ting Lan, Jiayu Zheng, Yupeng Li, Jinkang Pan, Ling Lin, Junyi Zhao, Jing Li, and et al. 2021. "Distinct Evolutionary Profiles and Functions of microRNA156 and microRNA529 in Land Plants" International Journal of Molecular Sciences 22, no. 20: 11100. https://doi.org/10.3390/ijms222011100
APA StyleXie, Q., Wang, X., He, J., Lan, T., Zheng, J., Li, Y., Pan, J., Lin, L., Zhao, J., Li, J., Yu, Y., Mo, B., Chen, X., Gao, L., & Liu, L. (2021). Distinct Evolutionary Profiles and Functions of microRNA156 and microRNA529 in Land Plants. International Journal of Molecular Sciences, 22(20), 11100. https://doi.org/10.3390/ijms222011100