
Anti-Kasha Behavior of 3-Hydroxyflavone and Its Derivatives


Abstract
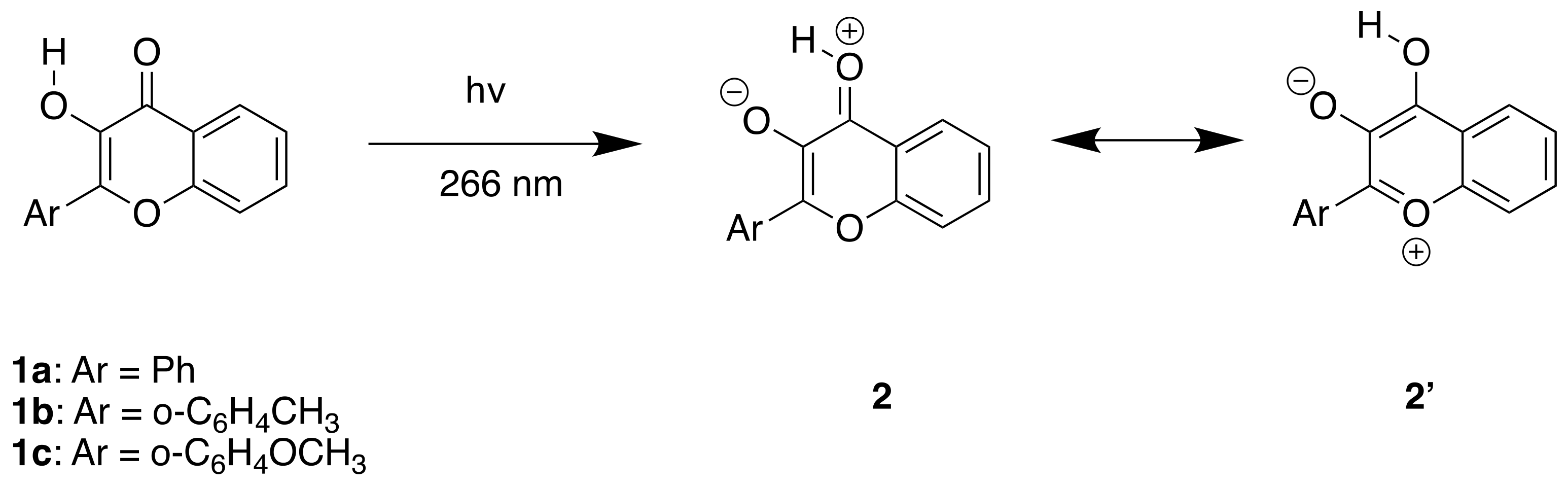
:1. Introduction
2. Results
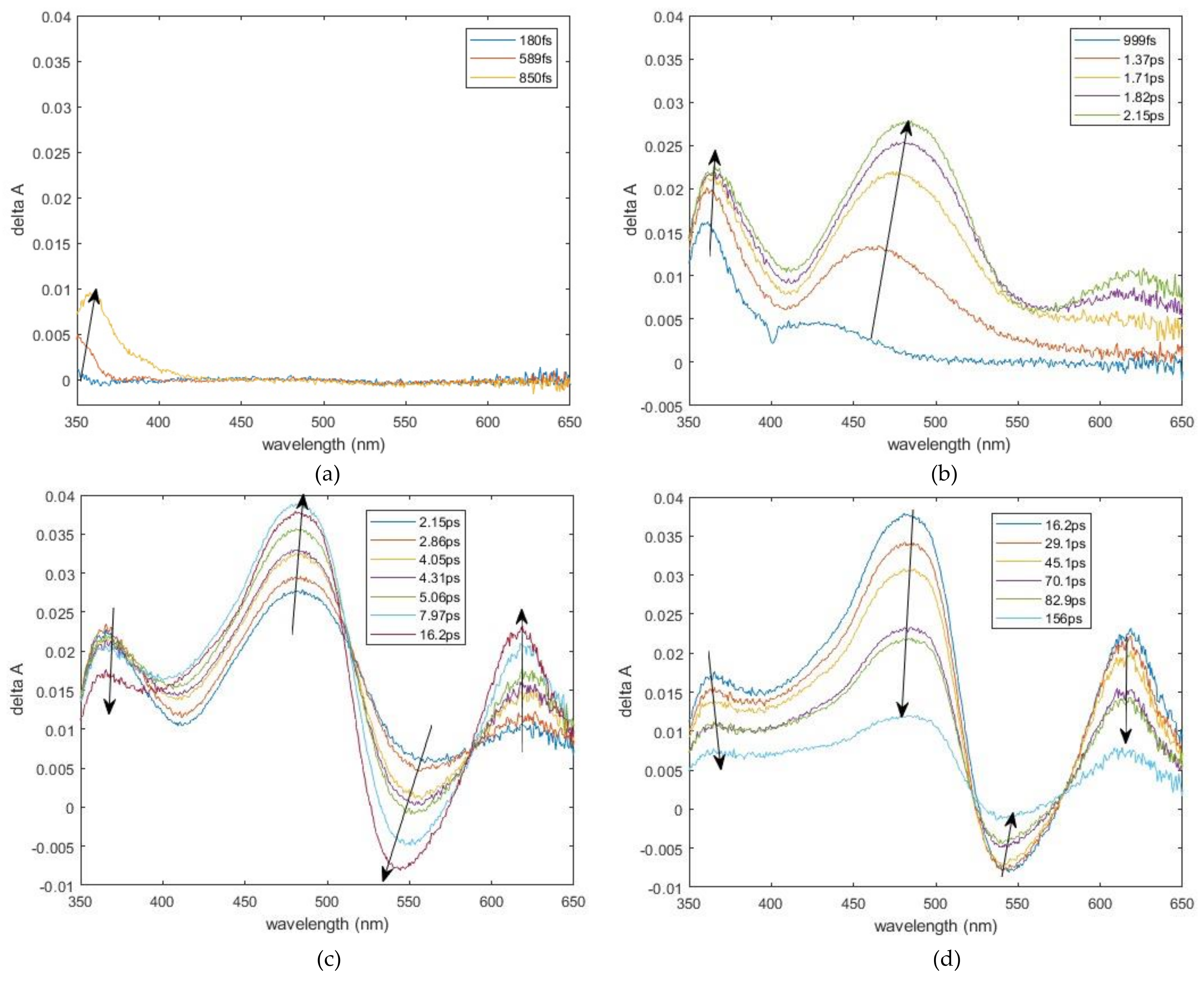
2.1. Experimental Results
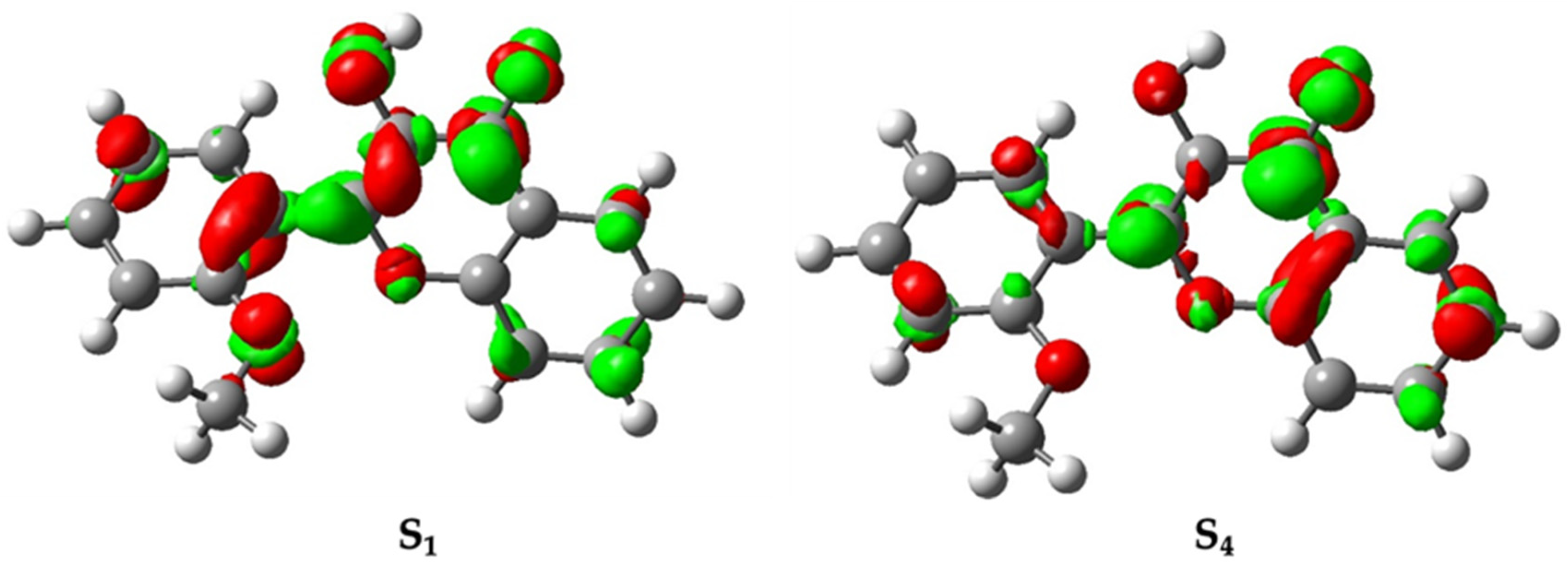
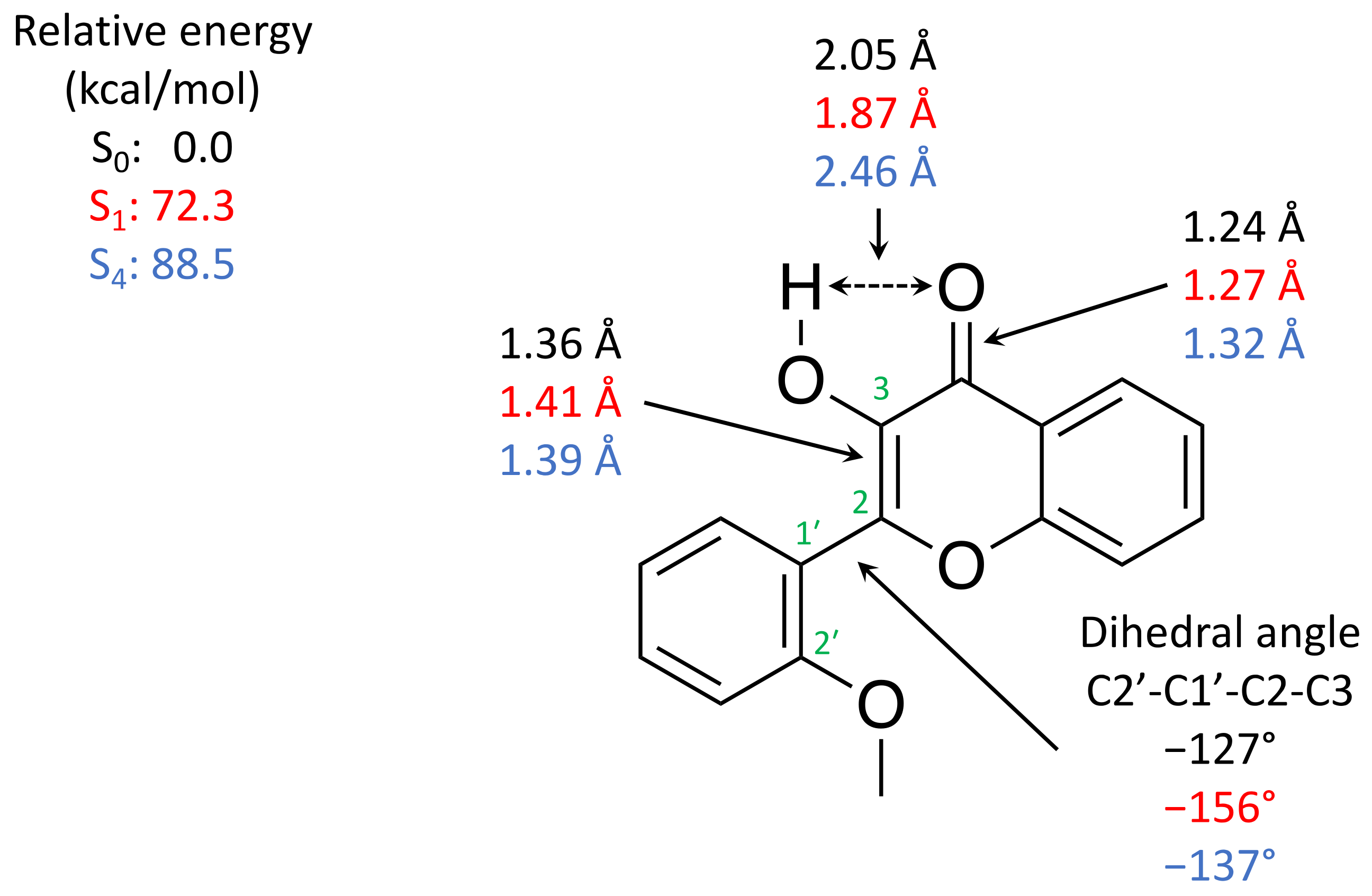
2.2. Computation Results
3. Discussion
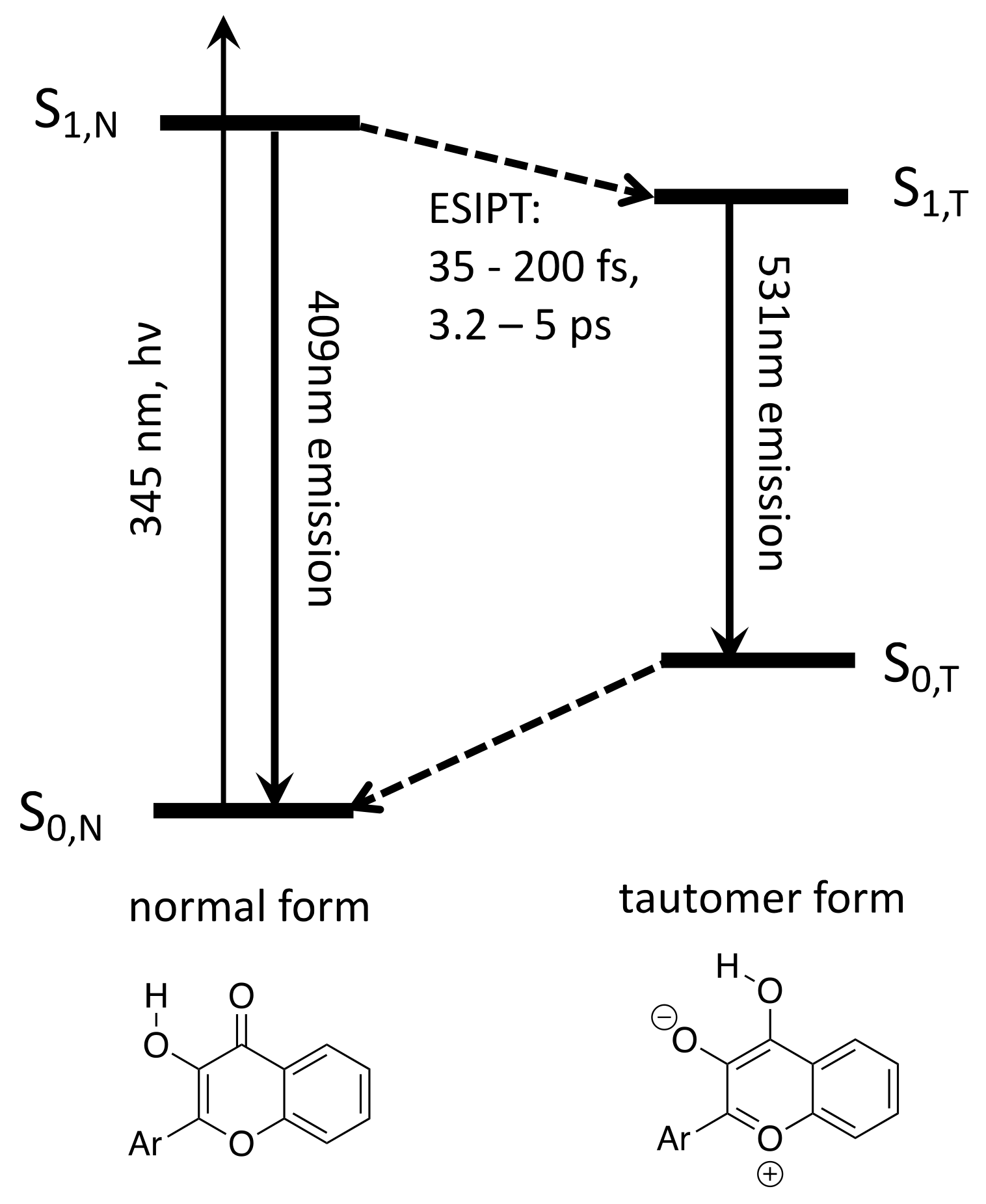
3.1. Band Assignments
3.2. Interpretation of Our Proposed Computational Model
3.3. The Long-Lived S4,N
4. Materials and Methods
4.1. Synthesis
4.2. Femtosecond Transient Absorption Experiment
4.3. Computational Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kasha, M. Characterization of Electronic Transitions in Complex Molecules. Discuss. Faraday Soc. 1950, 9, 14–19. [Google Scholar] [CrossRef]
- Kasha, M.; McGlynn, S.P. Molecular Electronic Spectroscopy. Annu. Rev. Phys. Chem. 1956, 7, 403–424. [Google Scholar] [CrossRef]
- Demchenko, A.P.; Tomin, V.I.; Chou, P.T. Breaking the Kasha Rule for More Efficient Photochemistry. Chem. Rev. 2017, 117, 13353–13381. [Google Scholar] [CrossRef]
- Sedgwick, A.C.; Wu, L.; Han, H.H.; Bull, S.D.; He, X.P.; James, T.D.; Sessler, J.L.; Tang, B.Z.; Tian, H.; Yoon, J. Excited-State Intramolecular Proton-Transfer (ESIPT) Based Fluorescence Sensors and Imaging Agents. Chem. Soc. Rev. 2018, 47, 8842–8880. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Ji, S.; Chen, Y.; Guo, H.; Yang, P. Excited State Intramolecular Proton Transfer (ESIPT): From Principal Photophysics to the Development of New Chromophores and Applications in Fluorescent Molecular Probes and Luminescent Materials. Phys. Chem. Chem. Phys. 2012, 14, 8803–8817. [Google Scholar] [CrossRef] [PubMed]
- Chou, P.T.; Martinez, M.L.; Clements, J.H. Reversal of Excitation Behavior of Proton-Transfer vs. Charge-Transfer by Dielectric Perturbation of Electronic Manifolds. J. Phys. Chem. 1993, 97, 2618–2622. [Google Scholar] [CrossRef]
- Swinney, T.C.; Kelley, D.F. Proton Transfer Dynamics in Substituted 3-hydroxyflavones: Solvent Polarization Effects. J. Chem. Phys. 1993, 99, 211–221. [Google Scholar] [CrossRef]
- Dharia, J.R.; Johnson, K.F.; Schlenoff, J.B. Synthesis and Characterization of Wavelength-Shifting Monomers and Polymers Based on 3-Hydroxyflavone. Macromolecules 2002, 27, 5167–5172. [Google Scholar] [CrossRef]
- Tormo, L.; Douhal, A. Caging Anionic Structure of a Proton Transfer Dye in a Hydrophobic Nanocavity with a Cooperative H-Bonding. J. Photochem. Photobiol. A Chem. 2005, 173, 358–364. [Google Scholar] [CrossRef]
- Banerjee, A.; Sengupta, P.K. Encapsulation of 3-Hydroxyflavone and Fisetin in β-Cyclodextrins: Excited State Proton Transfer Fluorescence and Molecular Mechanics Studies. Chem. Phys. Lett. 2006, 424, 379–386. [Google Scholar] [CrossRef]
- Klymchenko, A.S.; Demchenko, A.P. Probing AOT Reverse Micelles with Two-Color Fluorescence Dyes Based on 3-Hydroxychromone. Langmuir 2002, 18, 5637–5639. [Google Scholar] [CrossRef]
- Sarkar, M.; Guha Ray, J.; Sengupta, P.K. Effect of Reverse Micelles on the Intramolecular Excited State Proton Transfer (ESPT) and Dual Luminescence Behaviour of 3-Hydroxyflavone. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 1996, 52, 275–278. [Google Scholar] [CrossRef]
- Bondar, O.P.; Pivovarenko, V.G.; Rowe, E.S. Flavonols--New Fluorescent Membrane Probes for Studying the Interdigitation of Lipid Bilayers. Biochim. Biophys. Acta 1998, 1369, 119–130. [Google Scholar] [CrossRef] [Green Version]
- Shynkar, V.V.; Klymchenko, A.S.; Duportail, G.; Demchenko, A.P.; Mély, Y. Two-Color Fluorescent Probes for Imaging the Dipole Potential of Cell Plasma Membranes. Biochim. Biophys. Acta—Biomembr. 2005, 1712, 128–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klymchenko, A.S.; Avilov, S.V.; Demchenko, A.P. Resolution of Cys and Lys Labeling of α-Crystallin with Site-Sensitive Fluorescent 3-Hydroxyflavone Dye. Anal. Biochem. 2004, 329, 43–57. [Google Scholar] [CrossRef]
- Ameer-Beg, S.; Ormson, S.M.; Brown, R.G.; Matousek, P.; Towrie, M.; Nibbering, E.T.J.; Foggi, P.; Neuwahl, F.V.R. Ultrafast Measurements of Excited State Intramolecular Proton Transfer (ESIPT) in Room Temperature Solutions of 3-Hydroxyflavone and Derivatives. J. Phys. Chem. A 2001, 105, 3709–3718. [Google Scholar] [CrossRef]
- Tomin, V.I.; Jaworski, R. ESIPT Reaction Starting from S2 and S3 Singlet States in 3-Hydroxyflavone. Eur. Phys. J. Spec. Top. 2007, 144, 123–128. [Google Scholar] [CrossRef]
- Tomin, V.I.; Javorski, R. Investigation of Reactions from the Highest Excited States of Molecules by Fluorescent Spectroscopy Methods. Opt. Spectrosc. (Engl. Transl. Opt. Spektrosk.) 2008, 104, 40–49. [Google Scholar] [CrossRef]
- Woolfe, G.J.; Thistlethwaite, P.J. Direct Observation of Excited State Intramolecular Proton Transfer Kinetics in 3-Hydroxyflavone. J. Am. Chem. Soc. 1981, 103, 6916–6923. [Google Scholar] [CrossRef]
- Brucker, G.A.; Swinney, T.C.; Kelley, D.F. Proton-Transfer and Solvent Polarization Dynamics in 3-Hydroxyflavone. J. Phys. Chem. 1991, 95, 3190–3195. [Google Scholar] [CrossRef]
- Brucker, G.A.; Kelley, D.F. Role of Phenyl Torsion in the Excited-State Dynamics of 3-Hydroxyflavone. J. Phys. Chem. 1988, 92, 3805–3809. [Google Scholar] [CrossRef]
- Douhal, A.; Sanz, M.; Tormo, L.; Organero, J.A. Femtochemistry of Inter- and Intramolecular Hydrogen Bonds. ChemPhysChem 2005, 6, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Douhal, A.; Sanz, M.; Carranza, M.A.; Organero, J.A.; Santos, L. Femtosecond Observation of Intramolecular Charge- and Proton-Transfer Reactions in a Hydroxyflavone Derivative. Chem. Phys. Lett. 2004, 394, 54–60. [Google Scholar] [CrossRef]
- Tomin, V.I.; Jaworski, R. ESIPT from S2 Singlet State in 3-Hydroxyflavone. J. Mol. Struct. 2009, 924–926, 461–465. [Google Scholar] [CrossRef]
- Doroshenko, A.O. Comments on the Paper “ESIPT from S2 Singlet State in 3-Hydroxyflavone” by V.I. Tomin and R. Jaworski [J. Mol. Struct. 924–926 (2009) 461–465]. J. Mol. Struct. 2009, 933, 169–171. [Google Scholar] [CrossRef]
- Tomin, V.I.; Jaworski, R. ESIPT May Proceed in 3HF at Excitation in S2 State. J. Mol. Struct. 2012, 1030, 104–106. [Google Scholar] [CrossRef]
- Chevalier, K.; Wolf, M.M.N.; Funk, A.; Andres, M.; Gerhards, M.; Diller, R. Transient IR Spectroscopy and Ab Initio Calculations on ESIPT in 3-Hydroxyflavone Solvated in Acetonitrile. Phys. Chem. Chem. Phys. 2012, 14, 15007–15020. [Google Scholar] [CrossRef]
- Klymchenko, A.S.; Yushchenko, D.A.; Mély, Y. Tuning Excited State Intramolecular Proton Transfer in 3-Hydroxyflavone Derivative by Reaction of Its Isothiocyanate Group with an Amine. J. Photochem. Photobiol. A Chem. 2007, 192, 93–97. [Google Scholar] [CrossRef]
- Gunduz, S.; Goren, A.C.; Ozturk, T. Facile Syntheses of 3-Hydroxyflavones. Org. Lett. 2012, 14, 1576–1579. [Google Scholar] [CrossRef]
- Ma, J.; Su, T.; Li, M.-D.; Du, W.; Huang, J.; Guan, X.; Phillips, D.L. How and When Does an Unusual and Efficient Photoredox Reaction of 2-(1-Hydroxyethyl) 9,10-Anthraquinone Occur? A Combined Time-Resolved Spectroscopic and DFT Study. J. Am. Chem. Soc. 2012, 134, 14858–14868. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision B.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Sichula, V.; Kucheryavy, P.; Khatmullin, R.; Hu, Y.; Mirzakulova, E.; Vyas, S.; Manzer, S.F.; Hadad, C.M.; Glusac, K.D. Electronic Properties of N(5)-Ethyl Flavinium Ion. J. Phys. Chem. A 2010, 114, 12138–12147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vyas, S.; Onchoke, K.K.; Rajesh, C.S.; Hadad, C.M.; Dutta, P.K. Optical Spectroscopic Studies of Mononitrated Benzo [a]Pyrenes. J. Phys. Chem. A 2009, 113, 12558–12565. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transient Absorptions (nm) | Growth Lifetime | Decay Lifetime |
|---|---|---|
| τr, ps (Ar) | τ1, ps (A1) τ2, ps (A2) | |
| 361 | <0.1 | 6.5 ± 0.6 (4.3 × 10−3) 100 ± 2.5 (1.4 × 10−2) |
| 482 | 2.6 ± 0.4 (4.8 × 10−2) | 113 ± 1.3 (3.8 × 10−2) |
| 540 | 4.4 ± 0.1 (−1.8 × 10−2) | 113 ± 2.4 (−1.0 × 10−2) |
| 617 | 3.1 ± 0.2 (2.5 × 10−2) | 111 ± 1.9 (2.2 × 10−2) |
| Excited State | Energy (eV) (Wavelength (nm)) | Major Character (% Contributions) | Oscillator Strength |
|---|---|---|---|
| S1 | 3.60 | HOMO → LUMO (90%) | 0.2858 |
| (345) | |||
| S2 | 3.92 | HOMO-1 → LUMO (89%) | 0.0453 |
| (317) | |||
| S3 | 4.18 | HOMO-3 → LUMO (44%) | 0.0017 |
| (296) | HOMO-4 → LUMO (39%) | ||
| S4 | 4.44 | HOMO-2 → LUMO (76%) | 0.1108 |
| (279) | |||
| S5 | 4.63 | HOMO → LUMO + 1 (74%) | 0.0323 |
| (268) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, K.W.; Luk, H.L.; Phillips, D.L. Anti-Kasha Behavior of 3-Hydroxyflavone and Its Derivatives. Int. J. Mol. Sci. 2021, 22, 11103. https://doi.org/10.3390/ijms222011103
Fan KW, Luk HL, Phillips DL. Anti-Kasha Behavior of 3-Hydroxyflavone and Its Derivatives. International Journal of Molecular Sciences. 2021; 22(20):11103. https://doi.org/10.3390/ijms222011103
Chicago/Turabian StyleFan, Ka Wa, Hoi Ling Luk, and David Lee Phillips. 2021. "Anti-Kasha Behavior of 3-Hydroxyflavone and Its Derivatives" International Journal of Molecular Sciences 22, no. 20: 11103. https://doi.org/10.3390/ijms222011103
APA StyleFan, K. W., Luk, H. L., & Phillips, D. L. (2021). Anti-Kasha Behavior of 3-Hydroxyflavone and Its Derivatives. International Journal of Molecular Sciences, 22(20), 11103. https://doi.org/10.3390/ijms222011103