
New N-Substituted-1,2,4-triazole Derivatives of Pyrrolo[3,4-d]pyridazinone with Significant Anti-Inflammatory Activity—Design, Synthesis and Complementary In Vitro, Computational and Spectroscopic Studies

 ,
,  ,
,  , , and
, , and
Abstract
:1. Introduction
2. Results
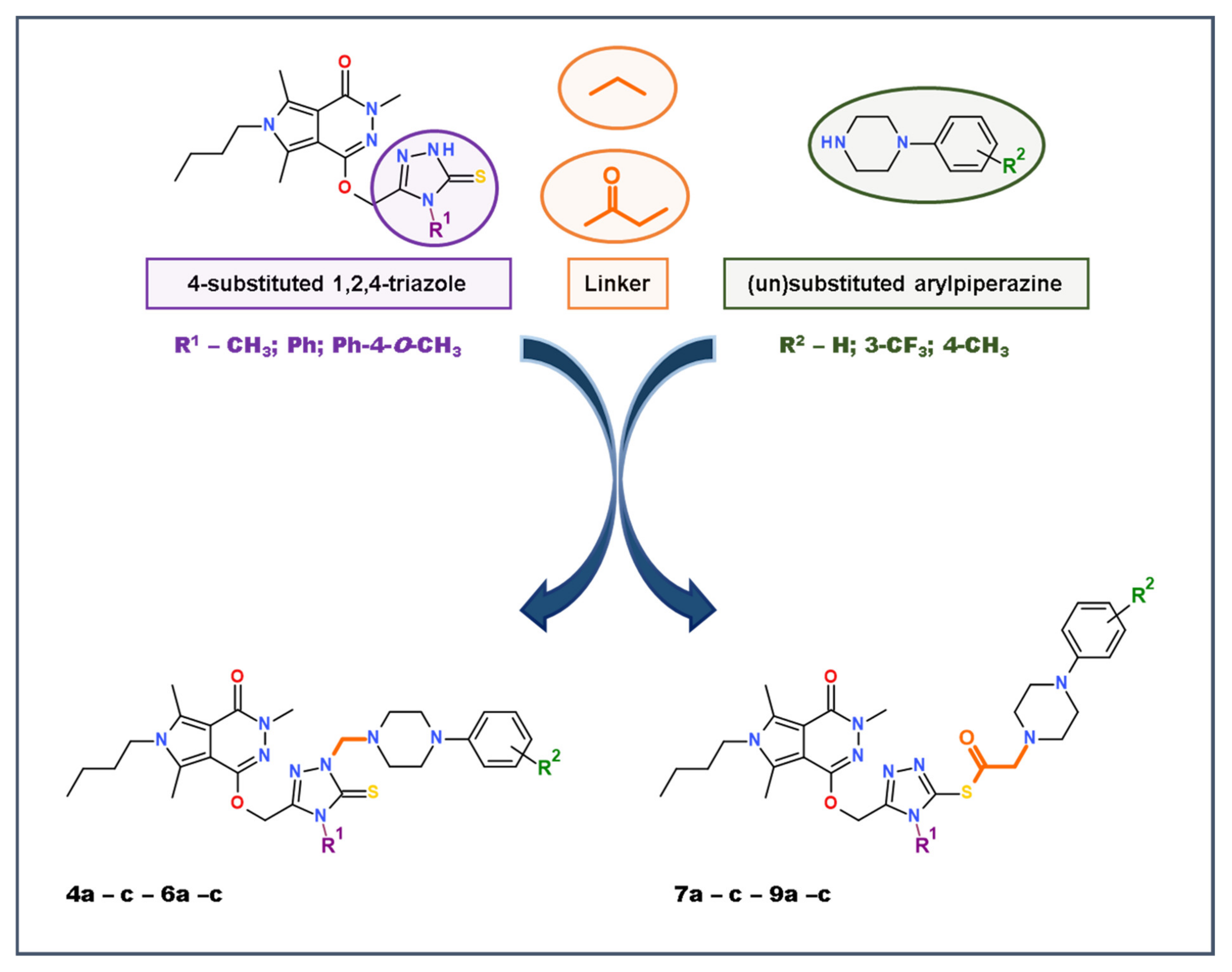
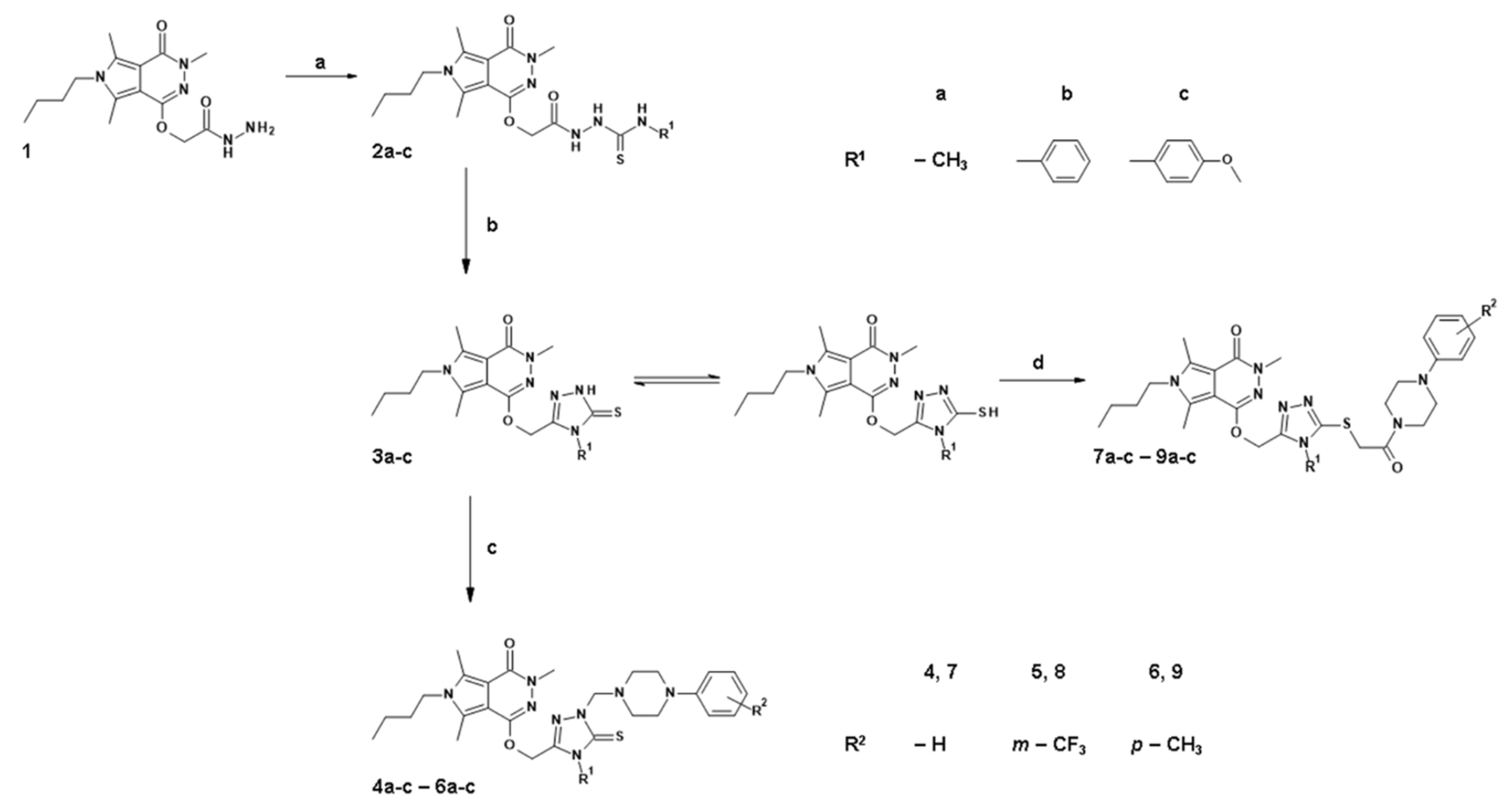
2.1. Chemistry
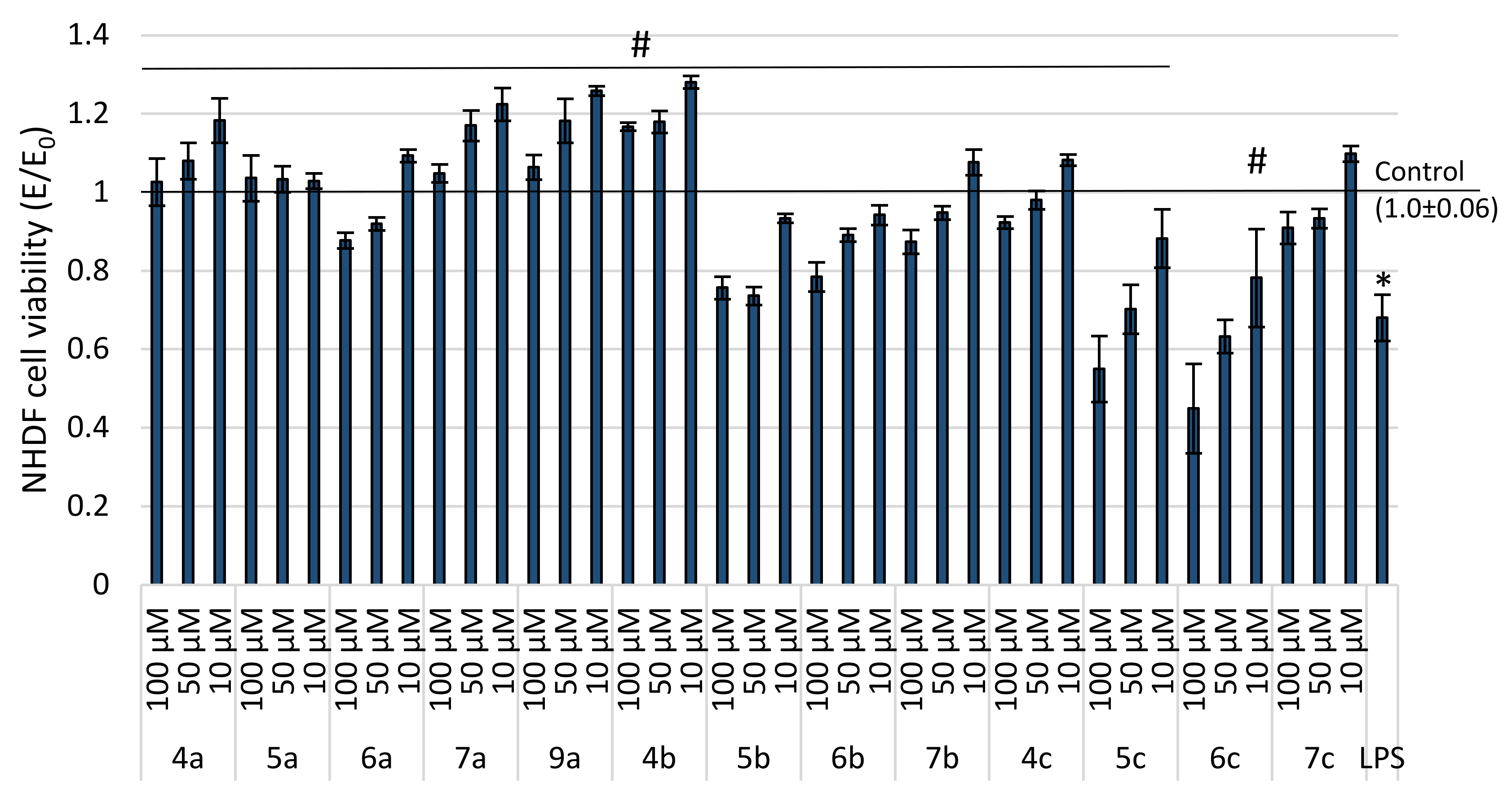
2.2. Evaluation of Viability
2.3. Cyclooxygenase (COX-1, COX-2) Inhibition Studies
2.3.1. In Vitro COX Inhibition Assay
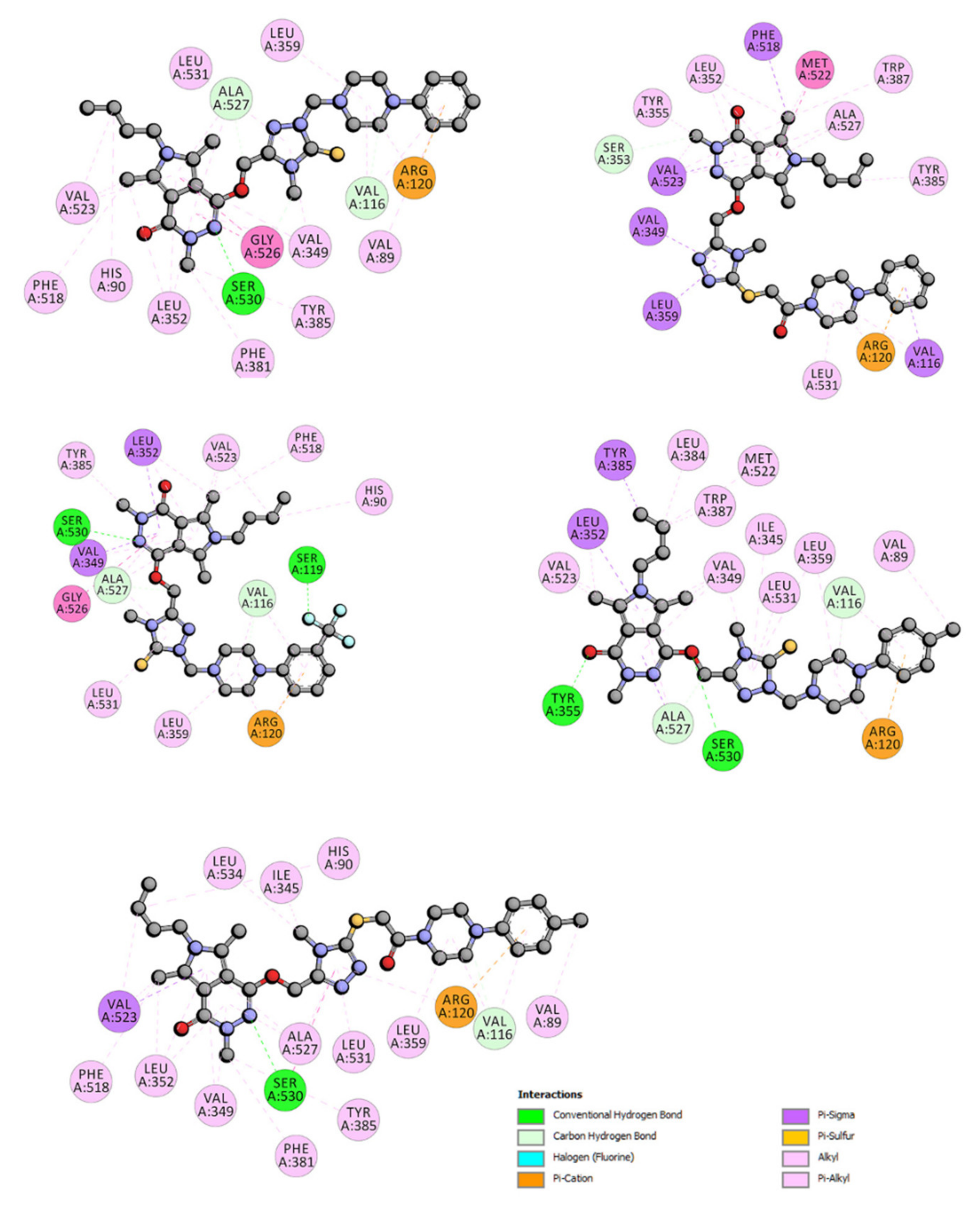
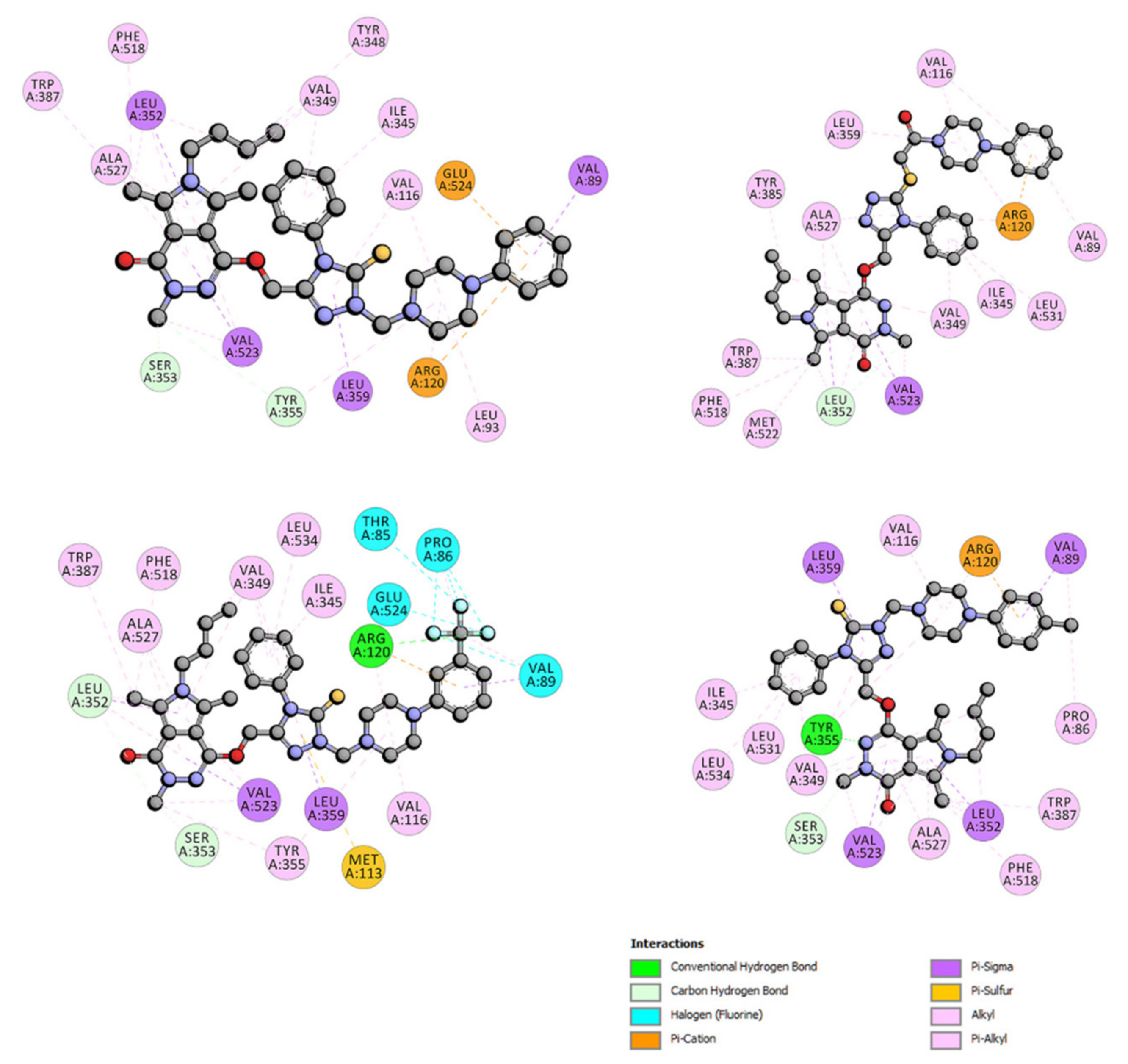
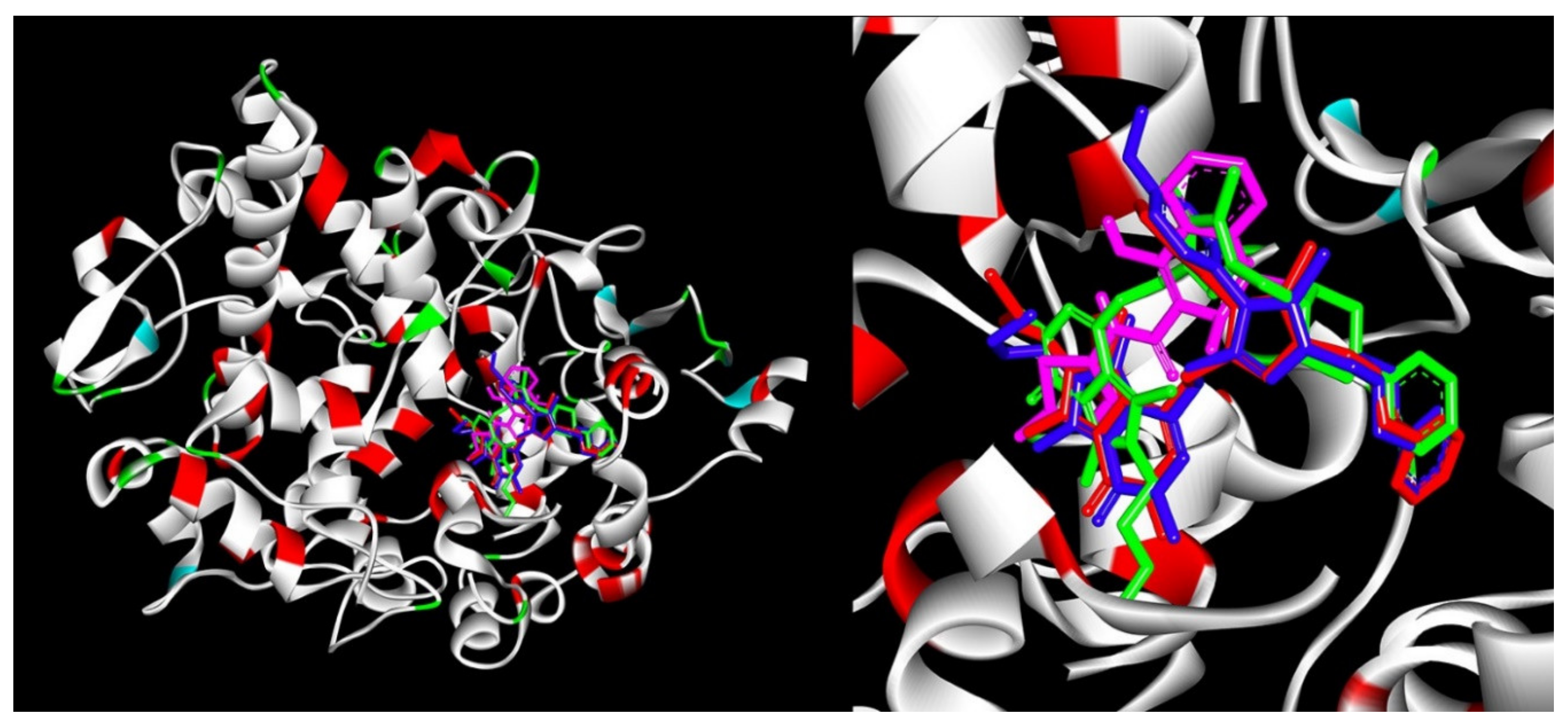
2.3.2. Molecular Docking Study
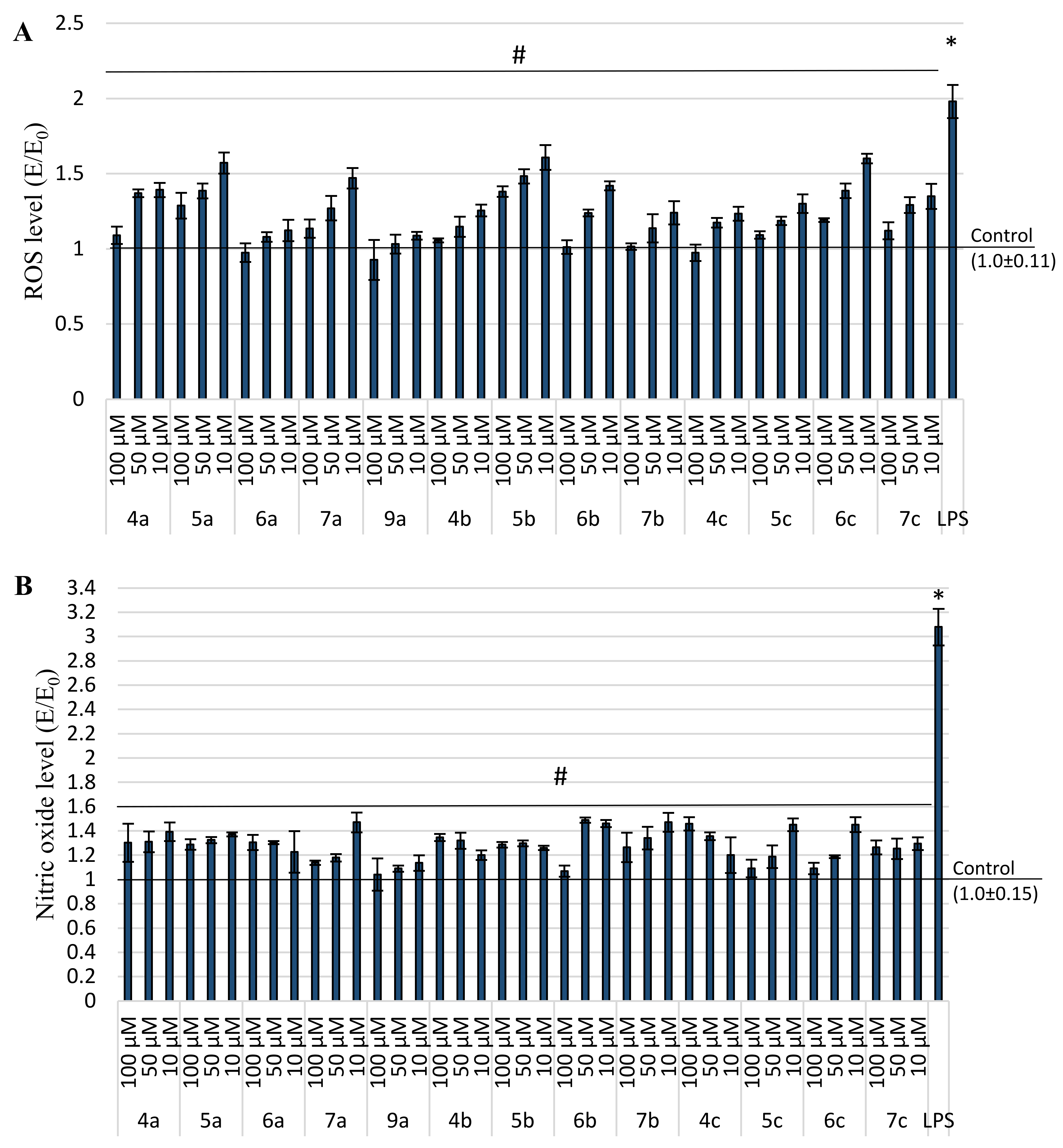
2.4. Anti-Inflammatory and Antioxidant Activity within Cells
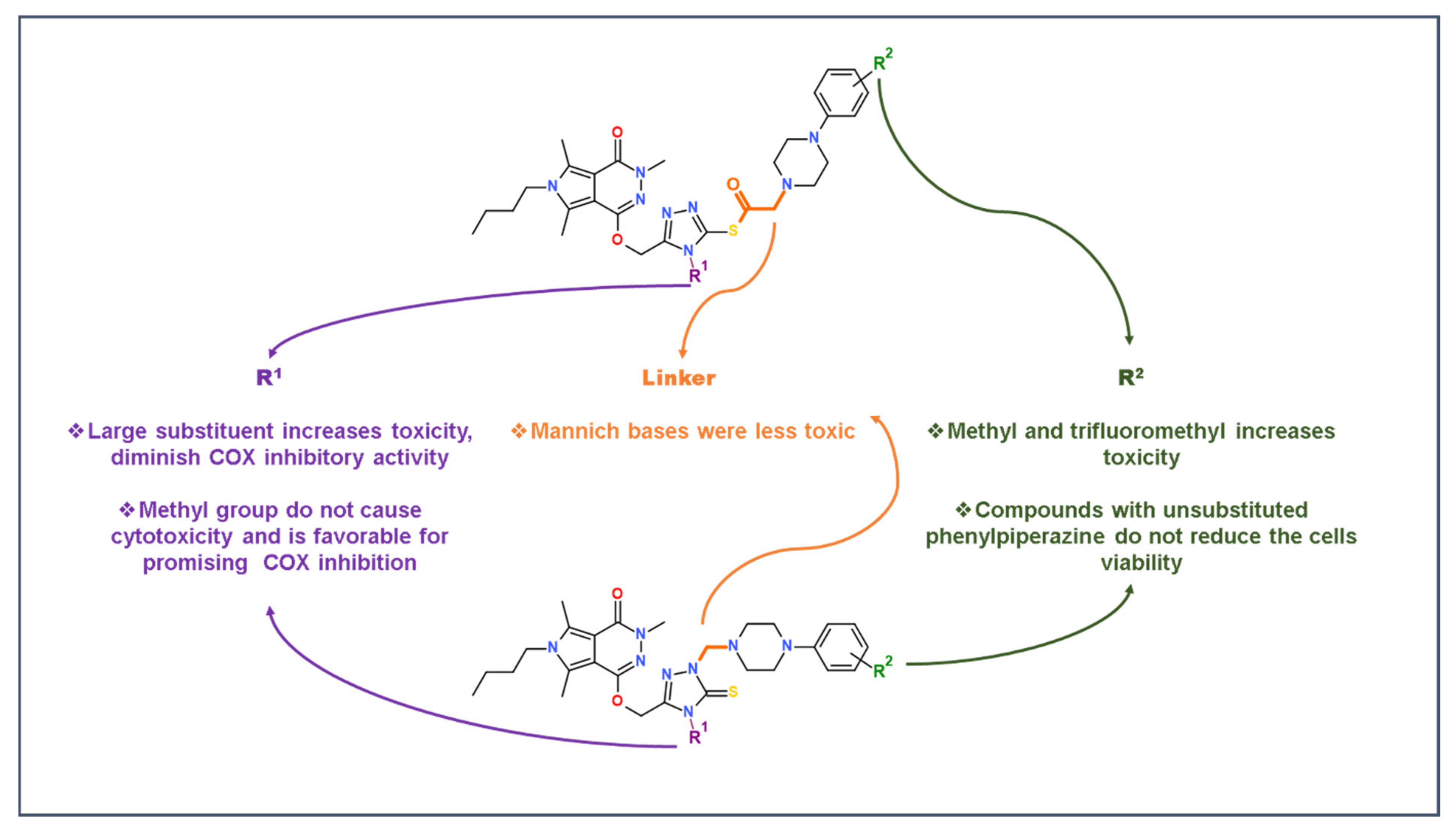
2.5. Structure-Activity Relationship Study
2.6. Bovine Serum Albumin (BSA) Ligand-Binding Assay
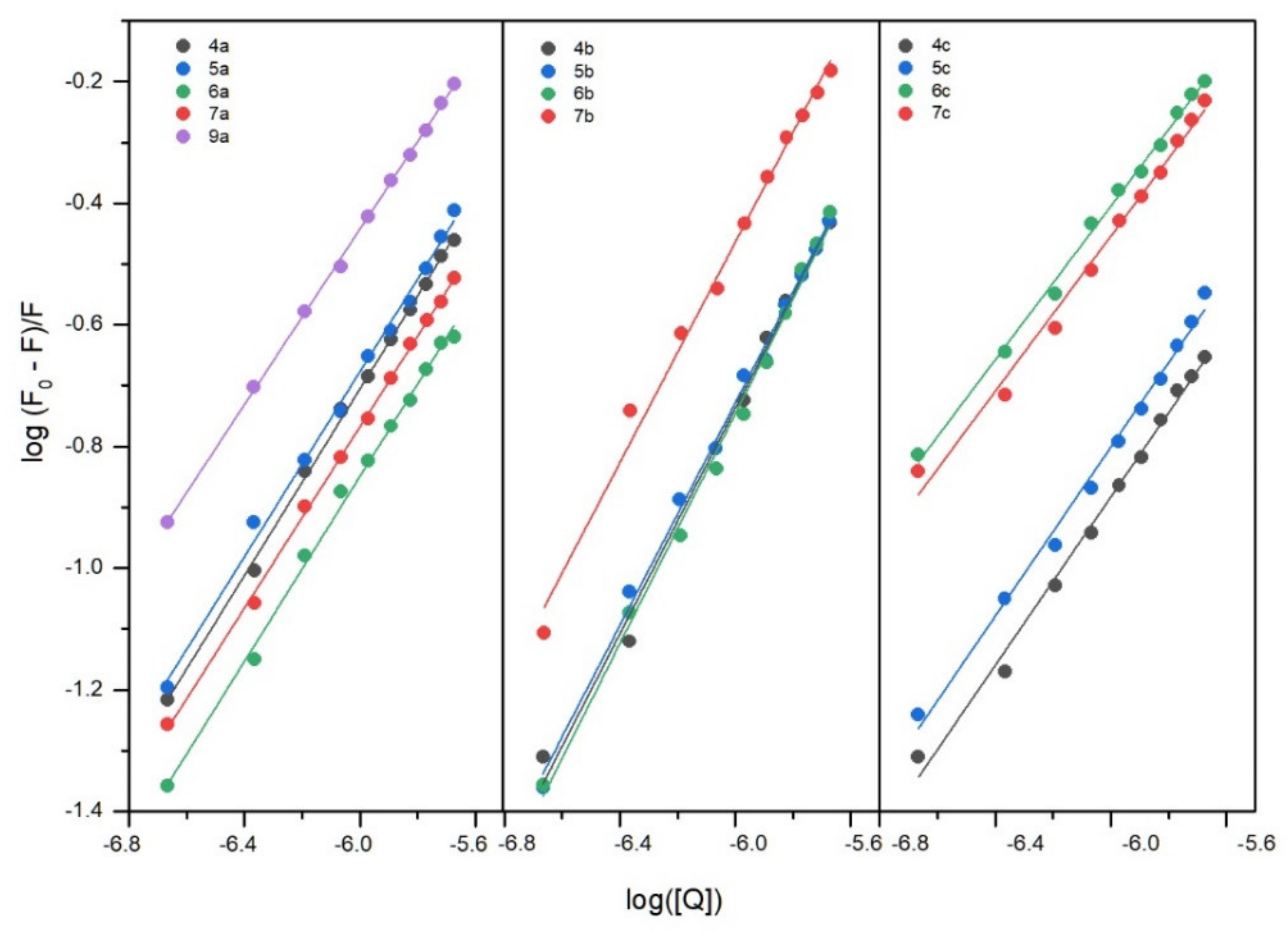
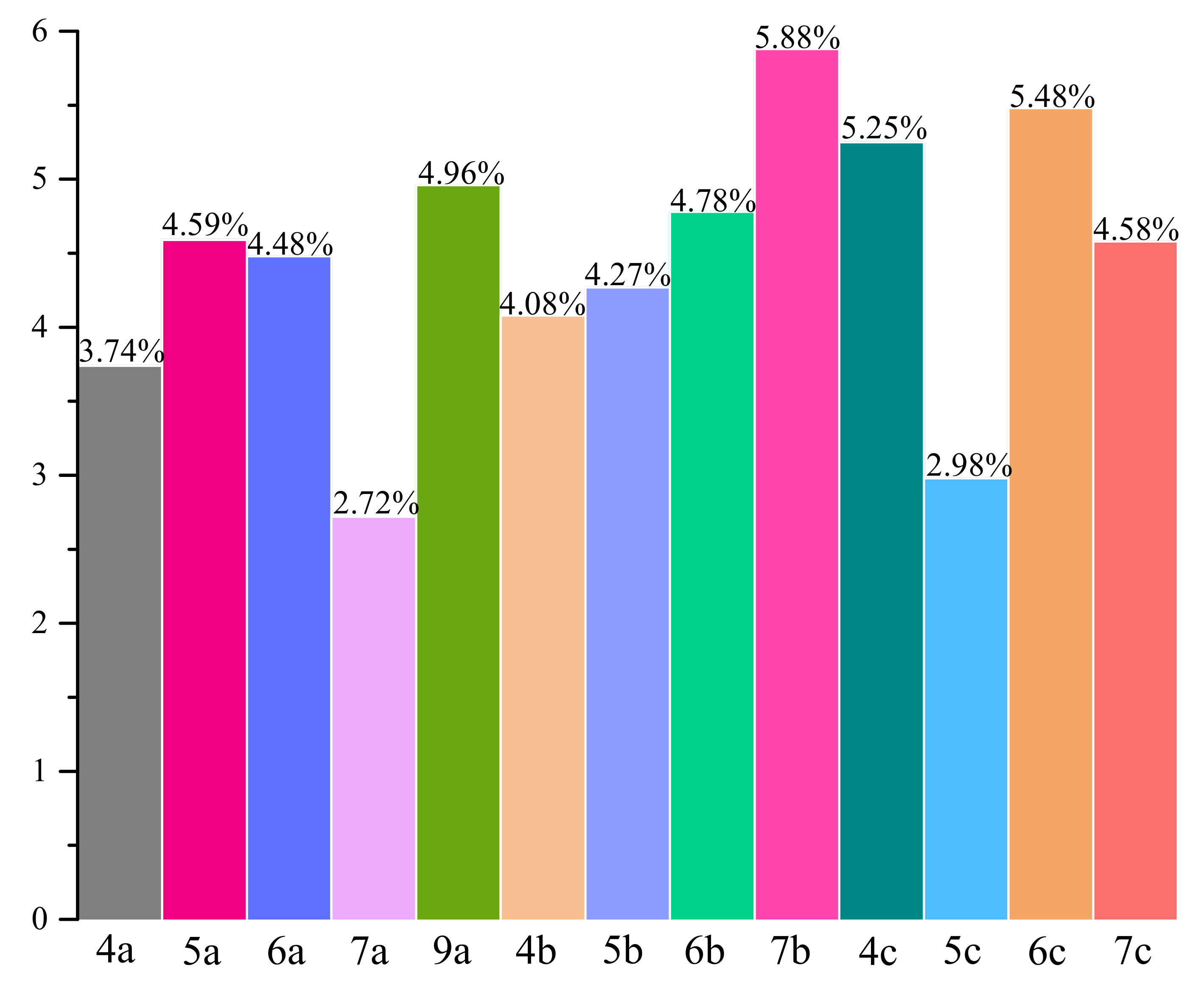
2.6.1. Fluorescence Quenching of BSA, Binding Constants, Thermodynamic Studies
2.6.2. Circular Dichroism Spectra
2.6.3. Fourier Transform Infrared Spectroscopic Measurements
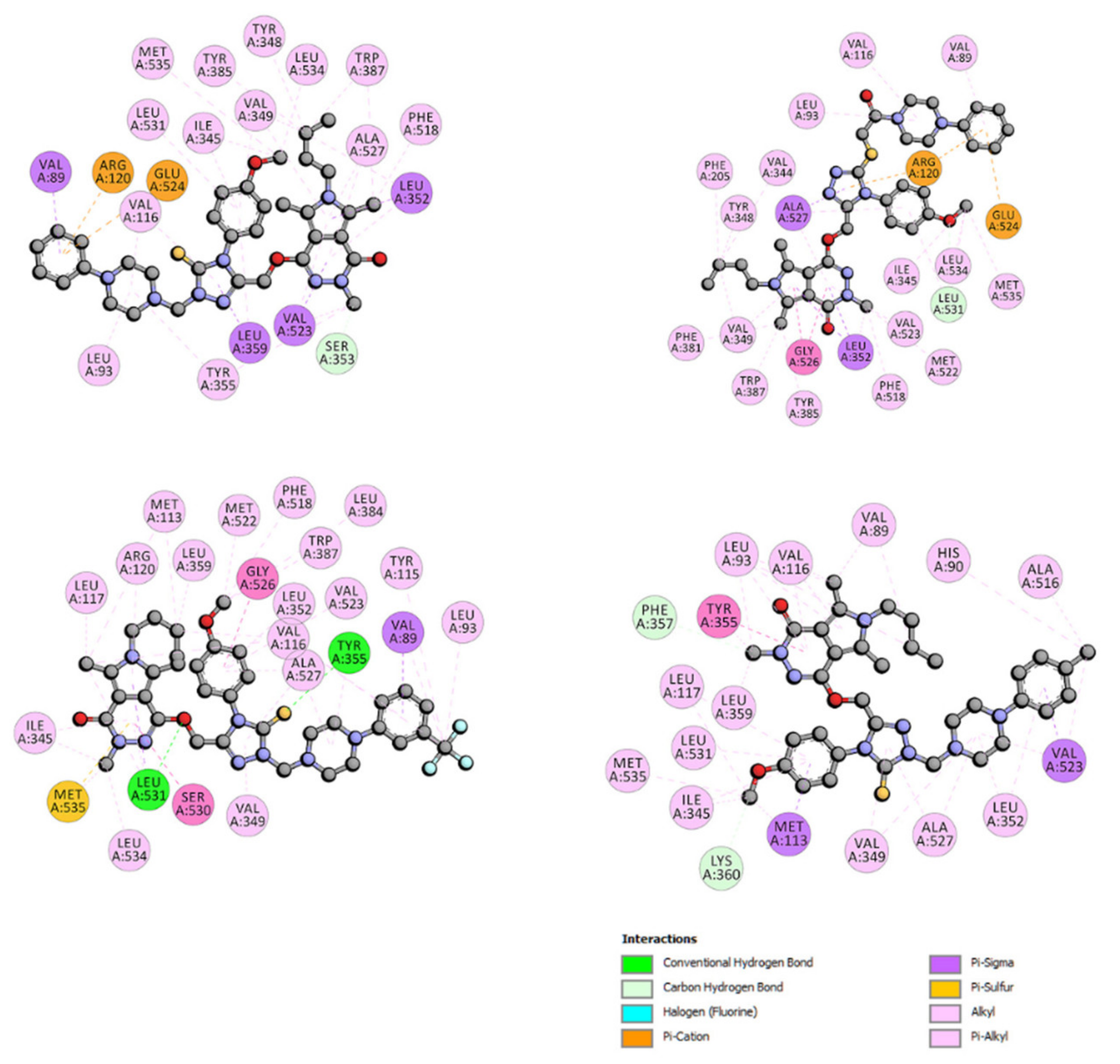
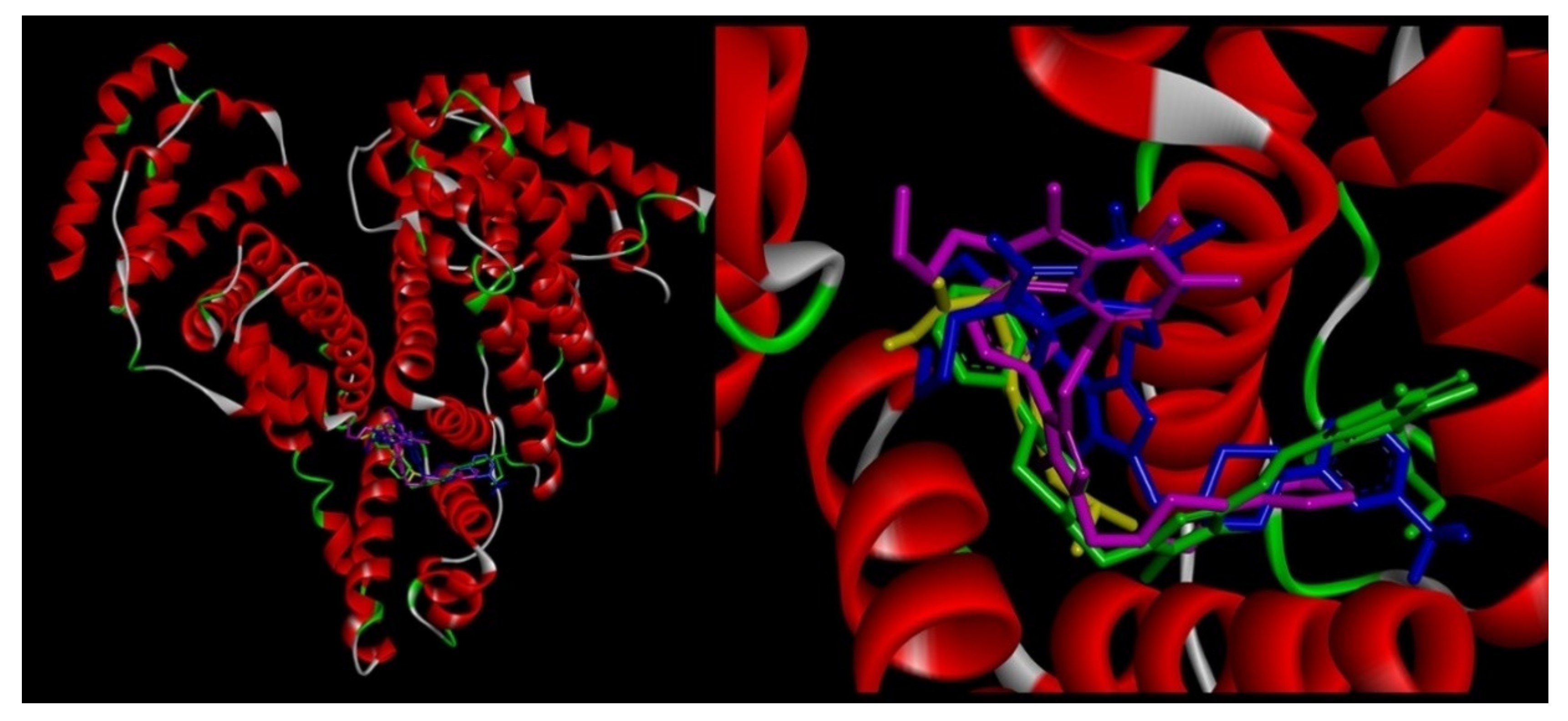
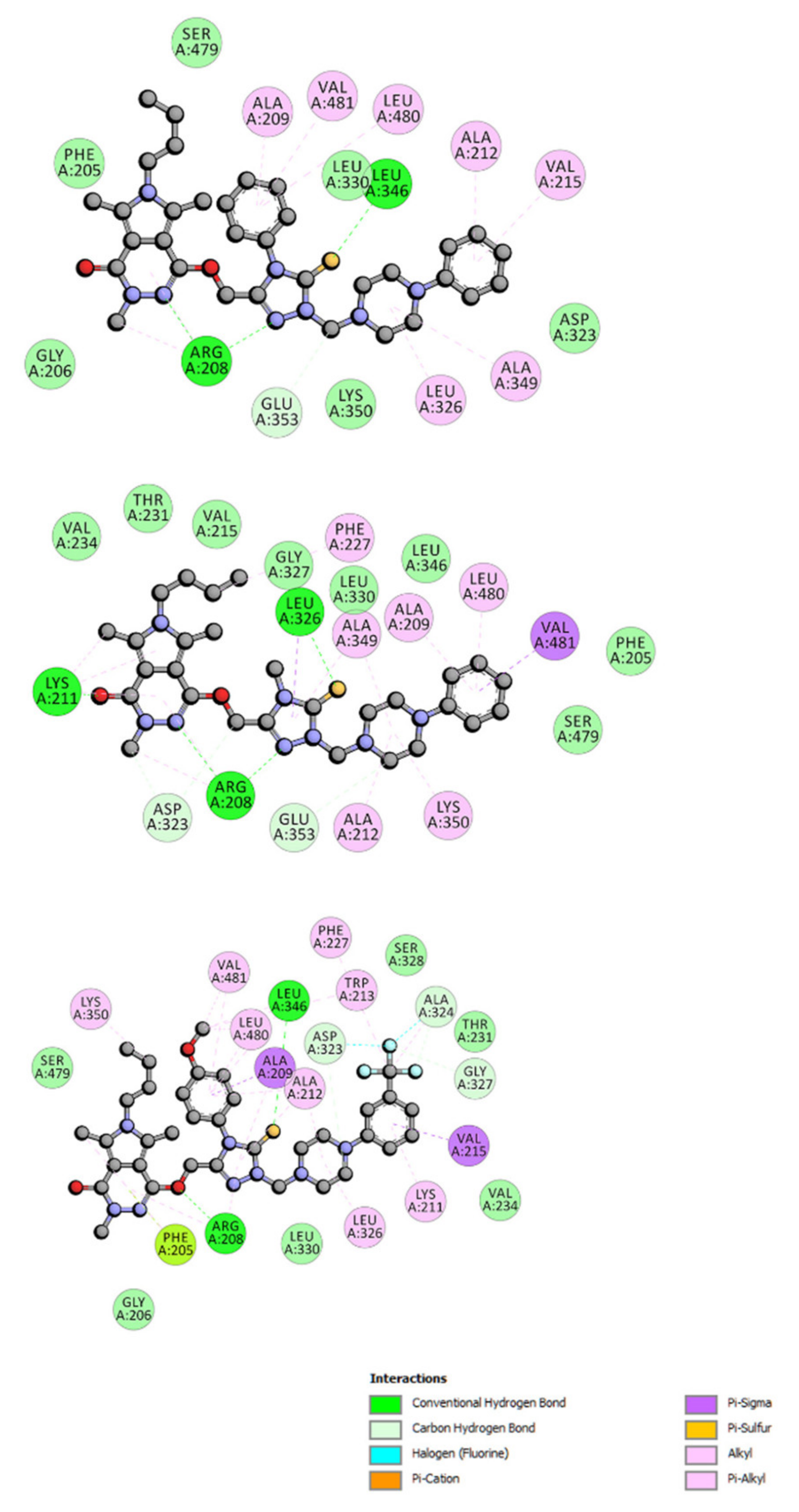
2.6.4. Site Markers Studies and Molecular Docking
2.7. In Silico Pharmacokinetic and Druglikeness Prediction
3. Materials and Methods
3.1. Chemistry
3.1.1. Instrumentation and Chemicals
3.1.2. Chemical Synthesis
General Procedure for Preparation of N-Substituted(aminothioxomethyl)hydrazide Derivatives of Pyrrolo[3,4-d]pyridazinone (2a-c)
General Procedure for Preparation of 4-Substituted-2H-1,2,4-triazole Derivatives of Pyrrolo[3,4-d]pyridazinone (3a-c)
General Procedure for Preparation of Mannich Base-Type Derivatives of Pyrrolo[3,4-d]pyridazinone (4a-c-6a-c)
General Procedure for Preparation of S-Substituted Derivatives of Pyrrolo[3,4-d]pyridazinone (7a-c-9a-c)
3.2. Biological Evaluation
3.2.1. Cell Line and Conditions
3.2.2. Tested Compounds
3.2.3. Cyclooxygenase Inhibition Assay
3.2.4. MTT Assay
3.2.5. Anti-Inflammatory and Antioxidant Activity
3.3. Molecular Docking
3.4. Spectroscopic Studies
3.4.1. Fluorescence
3.4.2. Circular Dichroism
3.4.3. FT-IR Measurement
3.5. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Nathan, C. Points of control in inflammation. Nature 2002, 420, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Savill, J. Resolution of inflammation: The beginning programs the end. Nat. Immunol. 2005, 6, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Negus, S.S.; Vanderah, T.W.; Brandt, M.R.; Bilsky, E.J.; Becerra, L.; Borsook, D. Preclinical Assessment of Candidate Analgesic Drugs: Recent Advances and Future Challenges. J. Pharmacol. Exp. Ther. 2006, 319, 507–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Leuti, A.; Fazio, D.; Fava, M.; Piccoli, A.; Oddi, S.; Maccarrone, M. Bioactive lipids, inflammation and chronic diseases. Adv. Drug Deliv. Rev. 2020, 159, 133–169. [Google Scholar] [CrossRef] [PubMed]
- Cashman, J.N. The Mechanisms of Action of NSAIDs in Analgesia. Drugs 1996, 52, 13–23. [Google Scholar] [CrossRef]
- Vane, J.R.; Botting, R.M. Mechanism of action of nonsteroidal anti-inflammatory drugs. Am. J. Med. 1998, 104, 25–85. [Google Scholar] [CrossRef]
- Marnett, L.J. Cyclooxygenase mechanisms. Curr. Opin. Chem. Biol. 2000, 4, 545–552. [Google Scholar] [CrossRef]
- Blobaum, A.L.; Marnett, L.J.; Hancock, A.B. PerspectiVe Structural and Functional Basis of Cyclooxygenase Inhibition. J. Med. Chem. 2006, 50, 1425–1441. [Google Scholar] [CrossRef] [Green Version]
- Soliva, R.; Almansa, C.; Kalko, S.G.; Luque, J.; Orozco, M. Theoretical Studies on the Inhibition Mechanism of Cyclooxygenase-2. Is There a Unique Recognition Site? J. Med. Chem. 2003, 46, 1372–1382. [Google Scholar] [CrossRef]
- Sostres, C.; Gargallo, C.J.; Arroyo, M.T.; Lanas, A. Adverse effects of non-steroidal anti-inflammatory drugs (NSAIDs, aspirin and coxibs) on upper gastrointestinal tract. Best Pract. Res. Clin. Gastroenterol. 2010, 24, 121–132. [Google Scholar] [CrossRef]
- Soll, A.H.; McCarthy, D. NSAID-related gastrointestinal complications. Clin. Cornerstone 1999, 1, 42–56. [Google Scholar] [CrossRef]
- Wallace, J.L. NSAID gastropathy and enteropathy: Distinct pathogenesis likely necessitates distinct prevention strategies. Br. J. Pharmacol. 2012, 165, 67–74. [Google Scholar] [CrossRef] [Green Version]
- Laine, L. Gastrointestinal effects of NSAIDs and coxibs. J. Pain Symptom Manag. 2003, 25, 32–40. [Google Scholar] [CrossRef]
- Wallace, J.L.; Devchand, P.R. Emerging roles for cyclooxygenase-2 in gastrointestinal mucosal defense. Br. J. Pharmacol. 2005, 145, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Cannon, C.P.; Cannon, P.J. Physiology. COX-2 inhibitors and cardiovascular risk. Science 2012, 336, 1386–1387. [Google Scholar] [CrossRef]
- Dogné, J.-M.; Supuran, C.T.; Pratico, D. Adverse Cardiovascular Effects of the Coxibs. J. Med. Chem. 2005, 48, 2251–2257. [Google Scholar] [CrossRef] [PubMed]
- Palkar, M.B.; Singhai, A.S.; Ronad, P.M.; Vishwanathswamy, A.H.M.; Boreddy, T.S.; Veerapur, V.P.; Shaikh, M.S.; Rane, R.A.; Karpoormath, R. Synthesis, pharmacological screening and in silico studies of new class of Diclofenac analogues as a promising anti-inflammatory agents. Bioorganic Med. Chem. 2014, 22, 2855–2866. [Google Scholar] [CrossRef] [PubMed]
- Avci, A.; Taşci, H.; Kandemir, Ü.; Can, Ö.D.; Gökhan-Kelekçi, N.; Tozkoparan, B. Synthesis, characterization, and in vivo pharmacological evaluation of novel mannich bases derived from 1,2,4-triazole containing a naproxen moiety. Bioorg. Chem. 2020, 100, 103892. [Google Scholar] [CrossRef]
- Alsayed, S.S.R.; Elshemy, H.A.H.; Abdelgawad, M.A.; Abdel-Latif, M.S.; Abdellatif, K.R.A. Design, synthesis and biological screening of some novel celecoxib and etoricoxib analogs with promising COX-2 selectivity, anti-inflammatory activity and gastric safety profile. Bioorg. Chem. 2017, 70, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Manjunatha, K.; Poojary, B.; Lobo, P.L.; Fernandes, J.; Kumari, N.S. Synthesis and biological evaluation of some 1,3,4-oxadiazole derivatives. Eur. J. Med. Chem. 2010, 45, 5225–5233. [Google Scholar] [CrossRef] [PubMed]
- El-Sayed, N.A.; Nour, M.S.; Salem, M.A.; Arafa, R.K. New oxadiazoles with selective-COX-2 and EGFR dual inhibitory activity: Design, synthesis, cytotoxicity evaluation and in silico studies. Eur. J. Med. Chem. 2019, 183, 111693. [Google Scholar] [CrossRef]
- Sağlık, B.N.; Osmaniye, D.; Levent, S.; Çevik, U.A.; Çavuşoğlu, B.K.; Özkay, Y.; Kaplancıklı, Z.A. Design, synthesis and biological assessment of new selective COX-2 inhibitors including methyl sulfonyl moiety. Eur. J. Med. Chem. 2021, 209, 112918. [Google Scholar] [CrossRef]
- Jacob, P.J.; Manju, S.L. Identification and development of thiazole leads as COX-2/5-LOX inhibitors through in-vitro and in-vivo biological evaluation for anti-inflammatory activity. Bioorg. Chem. 2020, 100, 103882. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Aziz, S.A.; Taher, E.S.; Lan, P.; Asaad, G.F.; Gomaa, H.A.M.; El-Koussi, N.A.; Youssif, B.G.M. Design, synthesis, and biological evaluation of new pyrimidine-5-carbonitrile derivatives bearing 1,3-thiazole moiety as novel anti-inflammatory EGFR inhibitors with cardiac safety profile. Bioorg. Chem. 2021, 111, 104890. [Google Scholar] [CrossRef] [PubMed]
- Abdellatif, K.R.A.; Abdelall, E.K.A.; Lamie, P.F.; Labib, M.B.; El-Nahaas, E.S.; Abdelhakeem, M.M. New pyrazole derivatives possessing amino/methanesulphonyl pharmacophore with good gastric safety profile: Design, synthesis, cyclooxygenase inhibition, anti-inflammatory activity and histopathological studies. Bioorg. Chem. 2020, 95, 103540. [Google Scholar] [CrossRef] [PubMed]
- Abdellatif, K.R.; Abdelall, E.K.; Elshemy, H.A.; Lamie, P.F.; Elnahaas, E.; Amin, D.M. Design, synthesis of new anti-inflammatory agents with a pyrazole core: COX-1/COX-2 inhibition assays, anti-inflammatory, ulcerogenic, histopathological, molecular Modeling, and ADME studies. J. Mol. Struct. 2021, 1240, 130554. [Google Scholar] [CrossRef]
- Abuo-Rahma, G.E.D.A.A.; Abdel-Aziz, M.; Farag, N.A.; Kaoud, T.S. Novel 1-[4-(Aminosulfonyl)phenyl]-1H-1,2,4-triazole derivatives with remarkable selective COX-2 inhibition: Design, synthesis, molecular docking, anti-inflammatory and ulcerogenicity studies. Eur. J. Med. Chem. 2014, 83, 398–408. [Google Scholar] [CrossRef]
- Abdel-Aziz, M.; Beshr, E.A.; Abdel-Rahman, I.M.; Ozadali, K.; Tan, O.U.; Aly, O.M. 1-(4-Methoxyphenyl)-5-(3,4,5-trimethoxyphenyl)-1H-1,2,4-triazole-3-carboxamides: Synthesis, molecular modeling, evaluation of their anti-inflammatory activity and ulcerogenicity. Eur. J. Med. Chem. 2014, 77, 155–165. [Google Scholar] [CrossRef]
- Abdelazeem, A.H.; El-Din, A.G.S.; Arab, H.H.; El-Saadi, M.T.; El-Moghazy, S.M.; Amin, N.H. Design, synthesis and anti-inflammatory/analgesic evaluation of novel di-substituted urea derivatives bearing diaryl-1,2,4-triazole with dual COX-2/sEH inhibitory activities. J. Mol. Struct. 2021, 1240, 130565. [Google Scholar] [CrossRef]
- Abdellatif, K.R.A.; Abdelall, E.K.A.; Elshemy, H.A.H.; Philoppes, J.N.; Hassanein, E.H.M.; Kahk, N.M. Optimization of pyrazole-based compounds with 1,2,4-triazole-3-thiol moiety as selective COX-2 inhibitors cardioprotective drug candidates: Design, synthesis, cyclooxygenase inhibition, anti-inflammatory, ulcerogenicity, cardiovascular evaluation, and molecular modeling studies. Bioorg. Chem. 2021, 114, 105122. [Google Scholar]
- Mohassab, A.M.; Hassan, H.A.; Abdelhamid, D.; Gouda, A.M.; Gomaa, H.A.M.; Youssif, B.G.M.; Radwan, M.O.; Fujita, M.; Otsuka, M.; Abdel-Aziz, M. New quinoline/1,2,4-triazole hybrids as dual inhibitors of COX-2/5-LOX and inflammatory cytokines: Design, synthesis, and docking study. J. Mol. Struct. 2021, 1244, 130948. [Google Scholar] [CrossRef]
- Cai, H.; Huang, X.; Xu, S.; Shen, H.; Zhang, P.; Huang, Y.; Jiang, J.; Sun, Y.; Jiang, B.; Wu, X.; et al. Discovery of novel hybrids of diaryl-1,2,4-triazoles and caffeic acid as dual inhibitors of cyclooxygenase-2 and 5-lipoxygenase for cancer therapy. Eur. J. Med. Chem. 2016, 108, 89–103. [Google Scholar] [CrossRef]
- Szczukowski, Ł.; Redzicka, A.; Wiatrak, B.; Krzyżak, E.; Marciniak, A.; Gębczak, K.; Gębarowski, T.; Świątek, P. Design, synthesis, biological evaluation and in silico studies of novel pyrrolo[3,4-d]pyridazinone derivatives with promising anti-inflammatory and antioxidant activity. Bioorg. Chem. 2020, 102, 104035. [Google Scholar] [CrossRef]
- Szczukowski, Ł.; Krzyżak, E.; Zborowska, A.; Zając, P.; Potyrak, K.; Peregrym, K.; Wiatrak, B.; Marciniak, A.; Świątek, P. Design, Synthesis and Comprehensive Investigations of Pyrrolo[3,4-d]pyridazinone-Based 1,3,4-Oxadiazole as New Class of Selective COX-2 Inhibitors. Int. J. Mol. Sci. 2020, 21, 9623. [Google Scholar] [CrossRef]
- Szandruk-Bender, M.; Wiatrak, B.; Szczukowski, Ł.; Świątek, P.; Rutkowska, M.; Dzimira, S.; Merwid-Ląd, A.; Danielewski, M.; Szeląg, A. Oxadiazole Derivatives of Pyrrolo[3,4-d]pyridazinone Exert Antinociceptive Activity in the Tail-Flick and Formalin Test in Rodents and Reveal Reduced Gastrotoxicity. Int. J. Mol. Sci. 2020, 21, 9685. [Google Scholar] [CrossRef] [PubMed]
- Dogruer, D.S.; Kupeli, E.; Yesilada, E.; Sahin, M.F. Synthesis of New 2-[1(2H)-Phthalazinon-2-yl]acetamide and 3-[1(2H)-Phthalazinon-2-yl]propanamide Derivatives as Antinociceptive and Anti-inflammatory Agents. Arch. Pharm. (Weinheim) 2004, 337, 303–310. [Google Scholar] [CrossRef]
- Gupta, S.; Pandey, D.; Mandalapu, D.; Bala, V.; Sharma, V.; Shukla, M.; Yadav, S.K.; Singh, N.; Jaiswal, S.; Maikhuri, J.P.; et al. Design, synthesis and biological profiling of aryl piperazine based scaffolds for the management of androgen sensitive prostatic disorders. Medchemcomm 2016, 7, 2111–2121. [Google Scholar] [CrossRef]
- McGarry, T.; Biniecka, M.; Veale, D.J.; Fearon, U. Hypoxia, oxidative stress and inflammation. Free Radic. Biol. Med. 2018, 125, 15–24. [Google Scholar] [CrossRef]
- Burdon, C.; Mann, C.; Cindrova-Davies, T.; Ferguson-Smith, A.C.; Burton, G.J. Oxidative Stress and the Induction of Cyclooxygenase Enzymes and Apoptosis in the Murine Placenta. Placenta 2007, 28, 724–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Zhu, M.; Xu, C.; Ji, B. Characterization of the baicalein-bovine serum albumin complex without or with Cu 2+or Fe 3+ by spectroscopic approaches. Eur. J. Med. Chem. 2011, 46, 588–599. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.H.; Pan, D.Q.; Wang, X.X.; Liu, T.T.; Jiang, M.; Wang, Q. Characterizing the binding interaction between antimalarial artemether (AMT) and bovine serum albumin (BSA): Spectroscopic and molecular docking methods. J. Photochem. Photobiol. B Biol. 2016, 162, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; He, W.; Liu, J.; Sheng, F.; Hu, Z.; Chen, X. Binding of the bioactive component Jatrorrhizine to human serum albumin. Biochim. Biophys. Acta—Gen. Subj. 2005, 1722, 15–21. [Google Scholar] [CrossRef]
- Wani, T.A.; Bakheit, A.H.; Zargar, S.; Bhat, M.A.; Al-Majed, A.A. Molecular docking and experimental investigation of new indole derivative cyclooxygenase inhibitor to probe its binding mechanism with bovine serum albumin. Bioorg. Chem. 2019, 89, 103010. [Google Scholar] [CrossRef]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 3rd ed.; Springer: Boston, MA, USA, 2006; ISBN 978-0-387-31278-1. [Google Scholar]
- Ware, W.R. Oxygen quenching of fluorescence in solution: An experimental study of the diffusion process. J. Phys. Chem. 1962, 66, 455–458. [Google Scholar] [CrossRef]
- Dufour, C.; Dangles, O. Flavonoid-serum albumin complexation: Determination of binding constants and binding sites by fluorescence spectroscopy. Biochim. Biophys. Acta—Gen. Subj. 2005, 1721, 164–173. [Google Scholar] [CrossRef]
- Abdelhameed, A.S.; Bakheit, A.H.; Mohamed, M.S.; Eldehna, W.M.; Abdel-Aziz, H.A.; Attia, M.I. Synthesis and biophysical insights into the binding of a potent anti-proliferative non-symmetric bis-isatin derivative with bovine serum albumin: Spectroscopic and molecular docking approaches. Appl. Sci. 2017, 7, 617. [Google Scholar] [CrossRef]
- Suryawanshi, V.D.; Walekar, L.S.; Gore, A.H.; Anbhule, P.V.; Kolekar, G.B. Spectroscopic analysis on the binding interaction of biologically active pyrimidine derivative with bovine serum albumin. J. Pharm. Anal. 2016, 6, 56–63. [Google Scholar] [CrossRef] [Green Version]
- Wani, T.A.; Bakheit, A.H.; Al-Majed, A.R.A.; Bhat, M.A.; Zargar, S. Study of the interactions of bovine serum albumin with the new anti-inflammatory agent 4-(1,3-dsioxo-1,3-dihydro-2H-isoindol-2-yl)-N-[(4-ethoxy-phenyl) methylidene]benzohydrazide using a multi-spectroscopic approach and molecular docking. Molecules 2017, 22, 1258. [Google Scholar] [CrossRef] [Green Version]
- Mohammadnia, F.; Fatemi, M.H.; Taghizadeh, S.M. Study on the interaction of anti-inflammatory drugs with human serum albumin using molecular docking, quantitative structure–activity relationship, and fluorescence spectroscopy. Luminescence 2020, 35, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Klotz, I.M.; Urquhart, J.M. The Binding of Organic Ions by Proteins. Effect of Temperature. J. Am. Chem. Soc. 1949, 71, 847–851. [Google Scholar] [CrossRef]
- Kelly, S.M.; Jess, T.J.; Price, N.C. How to study proteins by circular dichroism. Biochim. Biophys. Acta—Proteins Proteom. 2005, 1751, 119–139. [Google Scholar] [CrossRef]
- Kelly, S.; Price, N. The Use of Circular Dichroism in the Investigation of Protein Structure and Function. Curr. Protein Pept. Sci. 2005, 1, 349–384. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.X.; Cui, T.; Shi, Q.L. Applications of Circular Dichroism (CD) and Optical Rotatory Dispersion (ORD) in Molecular Biology, 1st ed.; Science Press: Beijing, China, 1987. [Google Scholar]
- Byler, D.M.; Susi, H. Examination of the secondary structure of proteins by deconvolved FTIR spectra. Biopolymers 1986, 25, 469–487. [Google Scholar] [CrossRef]
- Sudlow, G.; Birkett, D.J.; Wade, D.N. The Characterization of Two Specific Drug Binding Sites on Human Serum Albumin. Mol. Pharmacol. 1975, 11, 824–832. [Google Scholar]
- Ghuman, J.; Zunszain, P.A.; Petitpas, I.; Bhattacharya, A.A.; Otagiri, M.; Curry, S. Structural basis of the drug-binding specificity of human serum albumin. J. Mol. Biol. 2005, 353, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominny, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- FVeber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar]
- Ertl, P.; Rohde, B.; Selzer, P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef]
- Ahmed, A.; Tajmir-Riahi, H.A.; Carpentier, R. A quantitative secondary structure analysis of the 33 kDa extrinsic polypeptide of photosystem II by FTIR spectroscopy. FEBS Lett. 1995, 363, 65–68. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Xie, M.X.; Kang, J.; Zheng, D. Studies on the interaction of total saponins of panax notoginseng and human serum albumin by Fourier transform infrared spectroscopy. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2003, 59, 2747–2758. [Google Scholar] [CrossRef]
- Kong, J.; Yu, S. Fourier Transform Infrared Spectroscopic Analysis of Protein Secondary Structures. Acta Biochim. Biophys. Sin. (Shanghai) 2007, 39, 549–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 [µM] | Cell Morphology in Culture | |
|---|---|---|
| 4a | Non-toxic | normal morphology for fibroblasts—elongated cells, single granular cells in 1 of 10 assessed fields of view |
| 5a | Non-toxic | normal morphology for fibroblasts—elongated cells, single granular cells in 1 of 10 assessed fields of view |
| 6a | Non-toxic | normal morphology for fibroblasts—elongated cells, single granular cells in 1 of 10 assessed fields of view |
| 7a | Non-toxic | normal morphology for fibroblasts—elongated cells, single granular cells in 1 of 10 assessed fields of view |
| 8a | 35.60 (4.46) | many granules, cells shrunken, cell lysis was observed |
| 9a | Non-toxic | normal morphology for fibroblasts—elongated cells, single granular cells in 1 of 10 assessed fields of view |
| 4b | Non-toxic | normal morphology for fibroblasts—elongated cells, single granular cells in 1 of 10 assessed fields of view |
| 5b | 200.00 (7.05) | granularities were observed in 3–5 fields of view from 10 analyzed fields, cells with an elongated shape characteristic of fibroblasts |
| 6b | 156.25 (6.70) | granularities were observed in 3–5 fields of view from 10 analyzed fields, cells with an elongated shape characteristic of fibroblasts |
| 7b | Non-toxic | normal morphology for fibroblasts—elongated cells, single granular cells in 1 of 10 assessed fields of view |
| 8b | 22.72 (2.25) | many granules, cells shrunken, cell lysis was observed |
| 9b | 30.80 (3.04) | many granules, cells shrunken, cell lysis was observed |
| 4c | Non-toxic | normal morphology for fibroblasts—elongated cells, single granular cells in 1 of 10 assessed fields of view |
| 5c | 166.67 (3.35) | granularities were observed in 3–5 fields of view from 10 analyzed fields, cells with an elongated shape characteristic of fibroblasts |
| 6c | 103.89 (2.75) | granularities were observed in 3–5 fields of view from 10 analyzed fields, cells with an elongated shape characteristic of fibroblasts |
| 7c | Non-toxic | normal morphology for fibroblasts—elongated cells, single granular cells in 1 of 10 assessed fields of view |
| 8c | 18.97 (1.84) | many granules, cells shrunken, cell lysis was observed |
| 9c | 28.56 (4.59) | many granules, cells shrunken, cell lysis was observed |
| Compound | IC50 [µM] (SD) | COX-2/COX-1 Selectivity Ratio | |
|---|---|---|---|
| COX-1 | COX-2 | ||
| 4a | NA | 45.24 (0.018) | - |
| 5a | 95.75 (0.1) | 48.24 (0.04) | 0.50 |
| 6a | NA | 43.85 (0.035) | - |
| 7a | 70.96 (0.2) | 48.48 (0.037) | 0.68 |
| 9a | NA | 42.64 (0.015) | - |
| 4b | 79.47 (0.06) | 47.83 (0.039) | 0.60 |
| 5b | NA | NA | - |
| 6b | NA | NA | - |
| 7b | 86.30 (0.005) | 49.79 (0.001) | 0.58 |
| 4c | NA | 48.50 (0.027) | - |
| 5c | NA | NA | - |
| 6c | NA | NA | - |
| 7c | NA | NA | - |
| Meloxicam | 83.7 (0.03) | 59.2 (0.06) | 0.71 |
| Celecoxib | 56 (0.1) | 0.30 (0.08) | 0.005 |
| Diclofenac | 3.5 (0.04) | 16.6 (0.03) | 4.74 |
| ΔG° [kJmol−1] | ΔE1 [kJmol−1] | ΔE2 [kJmol−1] | ΔE3 [kJmol−1] | Ki [μM] | |
|---|---|---|---|---|---|
| 4a | −22.57 | −33.81 | −34.52 | 0.75 | 109 |
| 5a | −15.38 | −27.84 | −28.67 | 0.84 | 2010 |
| 6a | −23.28 | −34.48 | −35.44 | 0.96 | 83 |
| 7a | −11.32 | −23.83 | −23.83 | 0.00 | 10,250 |
| 9a | −21.02 | −33.48 | −33.41 | −0.16 | 206 |
| 4b | −20.40 | −32.85 | −34.36 | 1.50 | 226 |
| 5b | 11.60 | −2.17 | −3.89 | 1.76 | - |
| 6b | 13.29 | −0.83 | −0.25 | 1.08 | - |
| 7b | −5.93 | −19.64 | −19.10 | −0.84 | 90,800 |
| 4c | −8.07 | −21.82 | −23.24 | 1.42 | 38,200 |
| 5c | 29.63 | 14.67 | 11.87 | 2.71 | - |
| 6c | 52.36 | 36.65 | 35.44 | 1.21 | - |
| 7c | 6.18 | −8.78 | −8.66 | −0.12 | - |
| Meloxicam | −34.02 | −37.74 | −37.32 | −0.42 | 1.09 |
| Celecoxib | −30.17 | −36.40 | −36.11 | −0.08 | 5.13 |
| Diclofenac | −29.59 | −35.82 | −30.18 | −5.63 | 6.50 |
| Quenching | Binding | Thermodynamic | |||||||
|---|---|---|---|---|---|---|---|---|---|
| T [K] | Ksv × 105 [dm3mol−1] | kq × 1013 [dm3mol−1·s−1] | logKb | Kb × 104 [dm3mol−1] | n | ΔG° [kJmol−1] | ΔH° [kJmol−1] | ΔS° [Jmol−1K−1] | |
| 4a | 297 303 308 | 0.82 0.56 0.24 | 0.82 0.56 0.24 | 4.95 ± 0.09 4.65 ± 0.09 4.35 ± 0.27 | 8.91 4.47 2.24 | 1.00 ± 0.02 0.98 ± 0.02 0.99 ± 0.05 | −28.20 | −95.14 | −225.40 |
| 5a | 297 303 308 | 0.89 0.97 0.80 | 0.89 0.97 0.80 | 4.91 ± 0.14 4.74 ± 0.04 4.30 ± 0.07 | 8.13 5.50 2.00 | 0.99 ± 0.02 0.96 ± 0.01 0.89 ± 0.01 | −28.20 | −95.17 | −225.50 |
| 6a | 297 303 308 | 0.51 0.40 0.14 | 0.51 0.40 0.14 | 4.69 ± 0.13 4.27 ± 0.14 3.51 ± 0.22 | 4.90 1.86 0.33 | 0.99 ± 0.02 0.94 ± 0.02 0.89 ± 0.04 | −27.07 | −186.69 | −537.43 |
| 7a | 297 303 308 | 0.66 0.27 0.10 | 0.66 0.27 0.10 | 4.67 ± 0.07 4.09 ± 0.20 3.26 ± 0.21 | 4.68 1.23 0.99 | 0.97 ± 0.01 0.94 ± 0.03 0.87 ± 0.04 | −26.89 | −222.14 | −657.43 |
| 9a | 297 303 308 | 1.71 0.48 0.20 | 1.71 0.48 0.20 | 4.91 ± 0.05 4.52 ± 0.09 4.23 ± 0.18 | 4.90 1.86 0.33 | 0.94 ± 0.01 0.97 ± 0.02 0.98 ± 0.03 | −27.89 | −108.40 | −271.08 |
| 4b | 297 303 308 | 1.76 0.97 0.42 | 1.76 0.97 0.42 | 4.82 ± 0.20 4.47 ± 0.22 3.82 ± 0.20 | 6.61 2.95 0.61 | 0.92 ± 0.03 0.91 ± 0.03 0.86 ± 0.02 | −27.75 | −156.84 | −434.67 |
| 5b | 297 303 308 | 1.72 0.97 0.25 | 1.72 0.97 0.25 | 4.76 ± 0.12 4.22 ± 0.13 3.94 ± 0.17 | 5.75 1.66 0.87 | 0.92 ± 0.02 0.93 ± 0.02 0.92 ± 0.03 | −26.92 | −131.53 | −352.20 |
| 6b | 297 303 308 | 1.78 0.52 0.48 | 1.78 0.52 0.48 | 4.91 ± 0.15 4.32 ± 0.12 3.86 ± 0.06 | 8.13 2.09 0.72 | 0.95 ± 0.02 0.93 ± 0.02 0.90 ± 0.01 | −27.90 | −167.25 | −469.18 |
| 7b | 297 303 308 | 3.07 1.95 0.78 | 3.07 1.95 0.78 | 4.97 ± 0.17 4.53 ± 0.16 4.07 ± 0.10 | 9.33 3.39 1.17 | 0.91 ± 0.03 0.87 ± 0.02 0.86 ± 0.02 | −28.36 | −142.60 | −384.65 |
| 4c | 297 303 308 | 0.59 0.24 0.37 | 0.59 0.24 0.37 | 4.82 ± 0.19 4.37 ± 0.40 3.97 ± 0.12 | 5.61 2.34 0.93 | 1.01 ± 0.03 0.99 ± 0.08 0.90 ± 0.02 | −27.44 | −135.07 | −362.40 |
| 5c | 297 303 308 | 0.81 0.60 0.36 | 0.81 0.60 0.36 | 4.97 ± 0.18 4.51 ± 0.10 4.11 ± 0.13 | 9.33 3.24 1.29 | 1.01 ± 0.03 0.95 ± 0.02 0.92 ± 0.02 | −28.29 | −136.72 | −365.09 |
| 6c | 297 303 308 | 2.68 0.76 0.31 | 2.68 0.76 0.31 | 4.98 ± 0.12 4.61 ± 0.03 4.28 ± 0.12 | 9.55 4.07 1.91 | 0.92 ± 0.02 0.95 ± 0.01 0.96 ± 0.02 | −28.34 | −111.23 | −279.08 |
| 7c | 297 303 308 | 2.37 1.03 0.41 | 2.37 1.03 0.41 | 4.97 ± 0.20 4.53 ± 0.10 3.94 ± 0.18 | 9.33 3.39 0.87 | 0.93 ± 0.03 0.92 ± 0.02 0.88 ± 0.03 | −28.48 | −162.47 | −451.14 |
| BSA/Analyzed Compound Molar Ratio | α-Helix [%] | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 4a | 5a | 6a | 7a | 9a | 4b | 5b | 6b | 7b | 4c | 5c | 6c | 7c | |
| 1:0 | 51.83 | 52.15 | 51.93 | 51.59 | 51.90 | 51.08 | 52.80 | 52.44 | 52.87 | 52.70 | 53.10 | 53.49 | 52.77 |
| 1:0.5 | 51.35 | 51.52 | 51.28 | 50.49 | 50.79 | 51.40 | 51.05 | 51.89 | 51.78 | 52.64 | 52.24 | 52.41 | 52.07 |
| 1:1 | 51.16 | 50.62 | 50.74 | 49.99 | 50.74 | 50.48 | 50.86 | 49.81 | 51.75 | 51.66 | 52.49 | 51.81 | 51.22 |
| 1:5 | 49.80 | 50.26 | 49.59 | 48.79 | 49.76 | 49.74 | 51.21 | 49.15 | 49.92 | 51.10 | 50.36 | 50.74 | 50.31 |
| 1:10 | 48.09 | 47.56 | 47.45 | 48.87 | 46.94 | 47.00 | 48.53 | 47.66 | 46.99 | 47.45 | 50.12 | 48.01 | 48.19 |
| Compound | α-Helix [%] | β-Sheet [%] | β-Turn [%] | β-Anti [%] | Random-Coil [%] |
|---|---|---|---|---|---|
| Free—BSA | 53.8 | 29.2 | 6.6 | 1.6 | 8.8 |
| 4a—BSA | 50.2 | 16.5 | 11.8 | 11.8 | 9.7 |
| 5a—BSA | 50.4 | 16.5 | 8.9 | 15.4 | 8.8 |
| 6a—BSA | 50.3 | 17.8 | 4.9 | 19.5 | 7.5 |
| 7a—BSA | 51.7 | 17.1 | 8.5 | 15.6 | 7.1 |
| 9a—BSA | 51.8 | 16.2 | 5.8 | 18.4 | 7.8 |
| 4b—BSA | 50.2 | 14.3 | 11.7 | 16.4 | 7.4 |
| 5b—BSA | 49.7 | 13.8 | 13.7 | 12.5 | 10.3 |
| 6b—BSA | 49.8 | 12.4 | 12.3 | 13.4 | 12.1 |
| 7b—BSA | 51.5 | 14.2 | 15.6 | 9.9 | 8.8 |
| 4c—BSA | 50.1 | 17.3 | 13.2 | 8.3 | 11.1 |
| 5c—BSA | 49.1 | 6.1 | 16.4 | 3.6 | 24.8 |
| 6c—BSA | 49.9 | 5.6 | 18.8 | 4.1 | 21.6 |
| 7c—BSA | 48.3 | 6.6 | 18.6 | 4.2 | 22.3 |
| Site Marker | logKb | Site Marker | logKb | Site Marker | logKb | |||
|---|---|---|---|---|---|---|---|---|
| 4a | - BSA + IBP BSA + PHP | 4.95 ± 0.09 3.50 ± 0.07 4.54 ± 0.05 | 4b | - BSA + IBP BSA + PHP | 4.82 ± 0.20 3.55 ± 0.19 4.55 ± 0.07 | 4c | - BSA + IBP BSA + PHP | 4.82 ± 0.19 3.30 ± 0.08 4.57 ± 0.12 |
| 5a | - BSA + IBP BSA + PHP | 4.91 ± 0.14 3.60 ± 0.09 4.36 ± 0.07 | 5b | - BSA + IBP BSA + PHP | 4.76 ± 0.12 3.45 ± 0.05 4.25 ± 0.13 | 5c | - BSA + IBP BSA + PHP | 4.97 ± 0.18 3.27 ± 0.09 4.58 ± 0.05 |
| 6a | - BSA + IBP BSA + PHP | 4.69 ± 0.13 3.40 ± 0.11 4.10 ± 0.09 | 6b | - BSA + IBP BSA + PHP | 4.91 ± 0.15 3.60 ± 0.12 4.46 ± 0.15 | 6c | - BSA + IBP BSA + PHP | 4.98 ± 0.12 3.68 ± 0.06 4.63 ± 0.05 |
| 7a | - BSA + IBP BSA + PHP | 4.67 ± 0.07 3.37 ± 0.09 4.20 ± 0.11 | 7b | - BSA + IBP BSA + PHP | 4.97 ± 0.17 3.20 ± 0.08 4.39 ± 0.07 | 7c | - BSA + IBP BSA + PHP | 4.97 ± 0.20 3.50 ± 0.14 4.58 ± 0.09 |
| 9a | - BSA + IBP BSA + PHP | 4.91 ± 0.05 3.50 ± 0.09 4.55 ± 0.10 |
| Binding Site | ΔG° [kJmol−1] | ΔE1 [kJmol−1] | ΔE2 [kJmol−1] | ΔE3 [kJmol−1] | |
|---|---|---|---|---|---|
| 4a | site I site II(m) site II(l) | −31.02 −40.08 −27.42 | −42.22 −51.33 −38.66 | −37.66 −46.73 −39.88 | −4.56 −4.60 1.21 |
| 5a | site I site II(m) site II(l) | −29.93 −38.22 −28.88 | −42.38 −50.74 −41.38 | −39.00 −46.89 −42.38 | −3.38 −3.85 1.00 |
| 6a | site I site II(m) site II(l) | −30.18 −38.03 −31.30 | −42.79 −49.28 −42.55 | −38.67 −46.48 −43.72 | −4.10 −2.80 1.17 |
| 7a | site I site II(m) site II(l) | −30.51 −35.69 −28.55 | −43.01 −48.20 −41.04 | −42.93 −47.36 −40.38 | −0.04 −0.84 −0.67 |
| 9a | site I site II(m) site II(l) | −31.06 −35.44 −25.16 | −43.55 −47.95 −37.62 | −43.35 −47.19 −37.12 | −0.21 −0.76 −0.50 |
| 4b | site I site II(m) site II(l) | −35.53 −41.17 −25.37 | −47.98 −53.62 −37.87 | −44.02 −50.20 −38.03 | −4.01 −3.43 0.20 |
| 5b | site I site II(m) site II(l) | −32.02 −39.54 −30.47 | −45.73 −53.30 −44.18 | −41.05 −50.95 −45.06 | −4.68 −2.30 0.84 |
| 6b | site I site II(m) site II(l) | −33.56 −41.17 −28.30 | −46.02 −53.63 −40.76 | −41.08 −51.00 −39.67 | −4.22 −2.63 −1.08 |
| 7b | site I site II(m) site II(l) | −29.30 −39.67 −23.57 | −43.01 −53.38 −37.28 | −42.64 −52.83 −37.57 | −0.37 −0.55 0.29 |
| 4c | site I site II(m) site II(l) | −34.48 −38.79 −28.34 | −48.19 −52.50 −45.01 | −43.76 −49.87 −43.09 | −4.43 −2.63 1.08 |
| 5c | site I site II(m) site II(l) | −31.51 −42.48 −22.40 | −46.48 −57.81 −37.37 | −42.18 −54.88 −39.17 | −4.26 −2.93 1.80 |
| 6c | site I site II(m) site II(l) | −31.77 −37.20 −24.03 | −45.48 −50.91 −37.75 | −42.39 −47.82 −36.12 | −3.09 −3.09 −1.63 |
| 7c | site I site II(m) site II(l) | −27.21 −38.12 −33.89 | −42.17 −53.08 −48.86 | −41.08 −52.37 −48.91 | −1.09 −0.71 0.05 |
| Compound | Physicochemical Properties—Lipinski’s Rule of Five (Ro5) | ||||
|---|---|---|---|---|---|
| #H-Bond Acceptors | #H-Bond Donors | Log Po/w (MLOGP) | MW [g/mol] | #Violations | |
| 4a | 5 | 0 | 2.88 | 550.72 | 1 |
| 5a | 8 | 0 | 3.36 | 618.72 | 1 |
| 6a | 5 | 0 | 3.07 | 564.75 | 1 |
| 7a | 6 | 0 | 2.99 | 578.73 | 2 |
| 9a | 6 | 0 | 3.18 | 592.76 | 2 |
| 4b | 5 | 0 | 3.89 | 612.79 | 1 |
| 5b | 8 | 0 | 4.35 | 680.79 | 2 |
| 6b | 5 | 0 | 4.08 | 626.81 | 1 |
| 7b | 5 | 0 | 3.99 | 640.80 | 2 |
| 4c | 6 | 0 | 3.58 | 642.81 | 2 |
| 5c | 9 | 0 | 4.03 | 710.81 | 2 |
| 6c | 6 | 0 | 3.76 | 656.84 | 2 |
| 7c | 7 | 0 | 3.68 | 670.82 | 2 |
| Compound | Pharmacokinetics | |||
|---|---|---|---|---|
| GI Absorption | BBB Permeant | P-gp Substrate | Water Solubility | |
| 4a | High | No | Yes | Moderately soluble |
| 5a | High | No | Yes | Poorly soluble |
| 6a | High | No | Yes | Moderately soluble |
| 7a | High | No | Yes | Poorly soluble |
| 9a | High | No | Yes | Poorly soluble |
| 4b | High | No | Yes | Poorly soluble |
| 5b | Low | No | Yes | Poorly soluble |
| 6b | High | No | Yes | Poorly soluble |
| 7b | Low | No | Yes | Poorly soluble |
| 4c | High | No | Yes | Poorly soluble |
| 5c | Low | No | Yes | Poorly soluble |
| 6c | High | No | Yes | Poorly soluble |
| 7c | Low | No | Yes | Poorly soluble |
| Compound | Drug-Likeness | |||
|---|---|---|---|---|
| Lipinski | Veber | Bioavailability Score | TPSA [Å2] | |
| 4a | Yes, 1 violation | Yes | 0.55 | 110.37 |
| 5a | Yes, 1 violation | Yes | 0.55 | 110.37 |
| 6a | Yes, 1 violation | Yes | 0.55 | 110.37 |
| 7a | No, 2 violations | No, 1 violation | 0.17 | 128.61 |
| 9a | No, 2 violations | No, 1 violation | 0.17 | 128.61 |
| 4b | Yes, 1 violation | Yes | 0.55 | 110.37 |
| 5b | No, 2 violations | No, 1 violation | 0.17 | 110.37 |
| 6b | Yes, 1 violation | Yes | 0.55 | 110.37 |
| 7b | No, 2 violations | No, 1 violation | 0.17 | 128.61 |
| 4c | No, 2 violations | No, 1 violation | 0.17 | 119.60 |
| 5c | No, 2 violations | No, 1 violation | 0.17 | 119.60 |
| 6c | No, 2 violations | No, 1 violation | 0.17 | 119.60 |
| 7c | No, 2 violations | No, 1 violation | 0.17 | 137.84 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szczukowski, Ł.; Krzyżak, E.; Wiatrak, B.; Jawień, P.; Marciniak, A.; Kotynia, A.; Świątek, P. New N-Substituted-1,2,4-triazole Derivatives of Pyrrolo[3,4-d]pyridazinone with Significant Anti-Inflammatory Activity—Design, Synthesis and Complementary In Vitro, Computational and Spectroscopic Studies. Int. J. Mol. Sci. 2021, 22, 11235. https://doi.org/10.3390/ijms222011235
Szczukowski Ł, Krzyżak E, Wiatrak B, Jawień P, Marciniak A, Kotynia A, Świątek P. New N-Substituted-1,2,4-triazole Derivatives of Pyrrolo[3,4-d]pyridazinone with Significant Anti-Inflammatory Activity—Design, Synthesis and Complementary In Vitro, Computational and Spectroscopic Studies. International Journal of Molecular Sciences. 2021; 22(20):11235. https://doi.org/10.3390/ijms222011235
Chicago/Turabian StyleSzczukowski, Łukasz, Edward Krzyżak, Benita Wiatrak, Paulina Jawień, Aleksandra Marciniak, Aleksandra Kotynia, and Piotr Świątek. 2021. "New N-Substituted-1,2,4-triazole Derivatives of Pyrrolo[3,4-d]pyridazinone with Significant Anti-Inflammatory Activity—Design, Synthesis and Complementary In Vitro, Computational and Spectroscopic Studies" International Journal of Molecular Sciences 22, no. 20: 11235. https://doi.org/10.3390/ijms222011235
APA StyleSzczukowski, Ł., Krzyżak, E., Wiatrak, B., Jawień, P., Marciniak, A., Kotynia, A., & Świątek, P. (2021). New N-Substituted-1,2,4-triazole Derivatives of Pyrrolo[3,4-d]pyridazinone with Significant Anti-Inflammatory Activity—Design, Synthesis and Complementary In Vitro, Computational and Spectroscopic Studies. International Journal of Molecular Sciences, 22(20), 11235. https://doi.org/10.3390/ijms222011235