
miR-29b Regulates TGF-β1-Induced Epithelial–Mesenchymal Transition by Inhibiting Heat Shock Protein 47 Expression in Airway Epithelial Cells

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. HSP47 Expression, Targeted by miR-29b, Was Induced by TGF-β1 in A549 Cells
2.2. miR-29b Modulated mRNA and Protein Expression Levels of TGF-β1 in A549 Cells
2.3. Silencing the HSP47 Inhibited TGF-β1-Induced EMT in A549 Cells
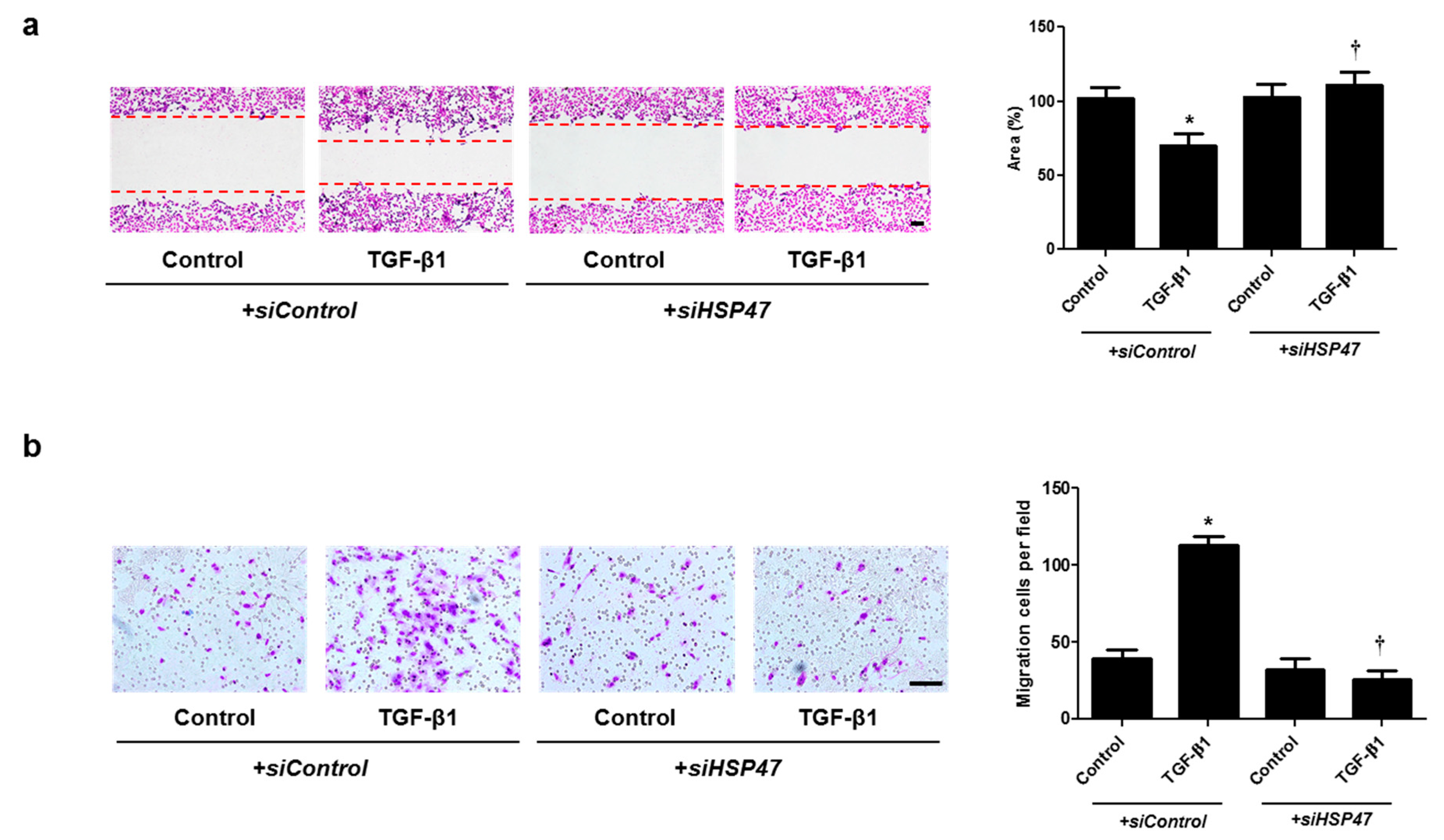
2.4. miR-29b Overexpression and HSP47 Silencing Inhibits TGF-β1-Induced Cell Migration in A549 Cells
2.5. Overexpression of miR-29b or Silencing of HSP47 Inhibits TGF-β1-Induced EMT in Primary Nasal Epithelial Cells
3. Discussion
4. Materials and Methods
4.1. Cell Cultures
4.2. Transfection with Small Interference (si)RNA Targeted against miR-29b and HSP47
4.3. qPCR
4.4. miR-29b Expression Analysis
4.5. Immunofluorescence Staining
4.6. Wound Scratch Migration Assay
4.7. Transwell Migration Assay
4.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fokkens, W.J.; Lund, V.J.; Mullol, J.; Bachert, C.; Alobid, I.; Baroody, F.; Cohen, N.; Cervin, A.; Douglas, R.; Gevaert, P.; et al. European Position Paper on Rhinosinusitis and Nasal Polyps 2012. Rhinol. Suppl. 2012, 23, 1–298. [Google Scholar]
- Georgalas, C.; Cornet, M.; Adriaensen, G.; Reinartz, S.; Holland, C.; Prokopakis, E.; Fokkens, W. Evidence-based Surgery for Chronic Rhinosinusitis with and without Nasal Polyps. Curr. Allergy Asthm. Rep. 2014, 14, 427. [Google Scholar] [CrossRef]
- Khalmuratova, R.; Shin, H.W. Crosstalk Between Mucosal Inflammation and Bone Metabolism in Chronic Rhinosinusitis. Clin. Exp. Otorhinolaryngol. 2021, 14, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Do, T.Q.; Barham, H.P.; Earls, P.; Sacks, R.; Christensen, J.M.; Rimmer, J.; Harvey, R.J. Clinical implications of mucosal remodeling from chronic rhinosinusitis. Int. Forum. Allergy Rhinol. 2016, 6, 835–840. [Google Scholar] [CrossRef] [PubMed]
- Barham, H.P.; Osborn, J.L.; Snidvongs, K.; Mrad, N.; Sacks, R.; Harvey, R.J. Remodeling changes of the upper airway with chronic rhinosinusitis. Int. Forum. Allergy Rhinol. 2015, 5, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Knight, D.A.; Grainge, C.L.; Stick, S.M.; Kicic, A.; Schuliga, M. Epithelial Mesenchymal Transition in Respiratory Disease: Fact or Fiction. Chest 2020, 157, 1591–1596. [Google Scholar] [CrossRef]
- Meng, J.; Zhou, P.; Liu, Y.; Liu, F.; Yi, X.; Liu, S.; Holtappels, G.; Bachert, C.; Zhang, N. The development of nasal polyp disease involves early nasal mucosal inflammation and remodelling. PLoS ONE 2013, 8, e82373. [Google Scholar] [CrossRef] [Green Version]
- Hupin, C.; Gohy, S.; Bouzin, C.; Lecocq, M.; Polette, M.; Pilette, C. Features of mesenchymal transition in the airway epithelium from chronic rhinosinusitis. Allergy 2014, 69, 1540–1549. [Google Scholar] [CrossRef]
- Xiao, J.; Meng, X.M.; Huang, X.R.; Chung, A.C.; Feng, Y.L.; Hui, D.S.; Yu, C.M.; Sung, J.J.; Lan, H.Y. miR-29 inhibits bleomycin-induced pulmonary fibrosis in mice. Mol. Ther. 2012, 20, 1251–1260. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Lin, Q.; Shao, Y.; Rong, L.; Zhang, D. miR-29b promotes skin wound healing and reduces excessive scar formation by inhibition of the TGF-β1/Smad/CTGF signaling pathway. Can. J. Physiol. Pharmacol. 2017, 95, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.P.; Li, Q.Y.; Lian, X.M.; Zhu, Z.H.; Chen, X.W.; Pei, W.Y.; Li, S.L.; Abbas, A.; Wang, Y.; Tian, L. MicroRNA-29b Mediates Lung Mesenchymal-Epithelial Transition and Prevents Lung Fibrosis in the Silicosis Model. Mol. Ther.-Nucl. Acids 2019, 14, 20–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.J.; Park, J.H.; Shin, J.M.; Yang, H.W.; Lee, H.M.; Park, I.H. TGF-β1-induced HSP47 regulates extracellular matrix accumulation via Smad2/3 signaling pathways in nasal fibroblasts. Sci. Rep. 2019, 9, 15563. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Li, Z.; Wang, Y.; Li, L.; Wang, D.; Zhang, W.; Liu, L.; Jiang, H.; Yang, J.; Cheng, J. Overexpression of miR-29b reduces collagen biosynthesis by inhibiting heat shock protein 47 during skin wound healing. Transl. Res. 2016, 178, 38–53.e6. [Google Scholar] [CrossRef] [PubMed]
- Akdis, C.A.; Bachert, C.; Cingi, C.; Dykewicz, M.S.; Hellings, P.W.; Naclerio, R.M.; Schleimer, R.P.; Ledford, D. Endotypes and phenotypes of chronic rhinosinusitis: A PRACTALL document of the European Academy of Allergy and Clinical Immunology and the American Academy of Allergy, Asthma & Immunology. J. Allergy Clin. Immunol. 2013, 131, 1479–1490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.; Tai, J.; Lee, S.H.; Kim, T.H. Advances in the Knowledge of the Underlying Airway Remodeling Mechanisms in Chronic Rhinosinusitis Based on the Endotypes: A Review. Int. J. Mol. Sci. 2021, 22, 910. [Google Scholar] [CrossRef]
- Kao, S.S.; Bassiouni, A.; Ramezanpour, M.; Finnie, J.; Chegeni, N.; Colella, A.D.; Chataway, T.K.; Wormald, P.J.; Vreugde, S.; Psaltis, A.J. Proteomic analysis of nasal mucus samples of healthy patients and patients with chronic rhinosinusitis. J. Allergy Clin. Immunol. 2021, 147, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Ryu, G.; Mo, J.H.; Shin, H.W. Epithelial-to-mesenchymal transition in neutrophilic chronic rhinosinusitis. Curr. Opin Allergy Clin. Immunol. 2021, 21, 30–37. [Google Scholar] [CrossRef]
- Radajewski, K.; Wierzchowska, M.; Grzanka, D.; Antosik, P.; Zdrenka, M.; Burduk, P. Tissue remodelling in chronic rhinosinusitis—Review of literature. Otolaryngologia polska. Polish Otolaryngol. 2019, 73, 8–11. [Google Scholar] [CrossRef]
- Hagiwara, S.; Iwasaka, H.; Shingu, C.; Matumoto, S.; Hasegawa, A.; Noguchi, T. Heat Shock Protein 47 (HSP47) Antisense Oligonucleotides Reduce Cardiac Remodeling and Improve Cardiac Function in a Rat Model of Myocardial Infarction. Thorac. Cardiov. Surg. 2011, 59, 386–392. [Google Scholar] [CrossRef]
- Nakayama, S.; Mukae, H.; Sakamoto, N.; Kakugawa, T.; Yoshioka, S.; Soda, H.; Oku, H.; Urata, Y.; Kondo, T.; Kubota, H.; et al. Pirfenidone inhibits the expression of HSP47 in TGF-beta1-stimulated human lung fibroblasts. Life Sci. 2008, 82, 210–217. [Google Scholar] [CrossRef] [Green Version]
- Samitas, K.; Carter, A.; Kariyawasam, H.H.; Xanthou, G. Upper and lower airway remodelling mechanisms in asthma, allergic rhinitis and chronic rhinosinusitis: The one airway concept revisited. Allergy 2018, 73, 993–1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, H.; Fabbri, M.; Calin, G.A. MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nat. Rev. Drug Discov. 2013, 12, 847–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Zhang, X.H.; Callejas-Díaz, B.; Mullol, J. MicroRNA in United Airway Diseases. Int. J. Mol. Sci. 2016, 17, 716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Zhang, J.; Sun, X.; Su, Q.; You, C. Down-regulation of miR-29b in carcinoma associated fibroblasts promotes cell growth and metastasis of breast cancer. Oncotarget 2017, 8, 39559–39570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Yi, N.; Chen, R.; Meng, Y.; Wang, Y.; Liu, H.; Cao, W.; Hu, Y.; Gu, Y.; Tong, C.; et al. miR-29b-3p protects cardiomyocytes against endotoxin-induced apoptosis and inflammatory response through targeting FOXO3A. Cell. Signal. 2020, 74, 109716. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, R.L.; Yu, G.; Latimer, P.A.; Stack, C.; Robinson, K.; Dalby, C.M.; Kaminski, N.; van Rooij, E. MicroRNA mimicry blocks pulmonary fibrosis. EMBO Mol. Med. 2014, 6, 1347–1356. [Google Scholar] [CrossRef]
- Li, X.; Li, C.; Zhu, G.; Yuan, W.; Xiao, Z.A. TGF-β1 Induces Epithelial-Mesenchymal Transition of Chronic Sinusitis with Nasal Polyps through MicroRNA-21. Int. Arch. Allergy Immunol. 2019, 179, 304–319. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Chung, A.C.; Huang, X.R.; Meng, X.M.; Hui, D.S.; Yu, C.M.; Sung, J.J.; Lan, H.Y. TGF-β/Smad3 signaling promotes renal fibrosis by inhibiting miR-29. J. Am. Soc. Nephrol. JASN 2011, 22, 1462–1474. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shin, J.-M.; Park, J.-H.; Yang, H.-W.; Moon, J.W.; Lee, H.-M.; Park, I.-H. miR-29b Regulates TGF-β1-Induced Epithelial–Mesenchymal Transition by Inhibiting Heat Shock Protein 47 Expression in Airway Epithelial Cells. Int. J. Mol. Sci. 2021, 22, 11535. https://doi.org/10.3390/ijms222111535
Shin J-M, Park J-H, Yang H-W, Moon JW, Lee H-M, Park I-H. miR-29b Regulates TGF-β1-Induced Epithelial–Mesenchymal Transition by Inhibiting Heat Shock Protein 47 Expression in Airway Epithelial Cells. International Journal of Molecular Sciences. 2021; 22(21):11535. https://doi.org/10.3390/ijms222111535
Chicago/Turabian StyleShin, Jae-Min, Joo-Hoo Park, Hyun-Woo Yang, Jee Won Moon, Heung-Man Lee, and Il-Ho Park. 2021. "miR-29b Regulates TGF-β1-Induced Epithelial–Mesenchymal Transition by Inhibiting Heat Shock Protein 47 Expression in Airway Epithelial Cells" International Journal of Molecular Sciences 22, no. 21: 11535. https://doi.org/10.3390/ijms222111535
APA StyleShin, J. -M., Park, J. -H., Yang, H. -W., Moon, J. W., Lee, H. -M., & Park, I. -H. (2021). miR-29b Regulates TGF-β1-Induced Epithelial–Mesenchymal Transition by Inhibiting Heat Shock Protein 47 Expression in Airway Epithelial Cells. International Journal of Molecular Sciences, 22(21), 11535. https://doi.org/10.3390/ijms222111535