
The Role of Thromboxane in the Course and Treatment of Ischemic Stroke: Review




{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Thromboxane and Stroke
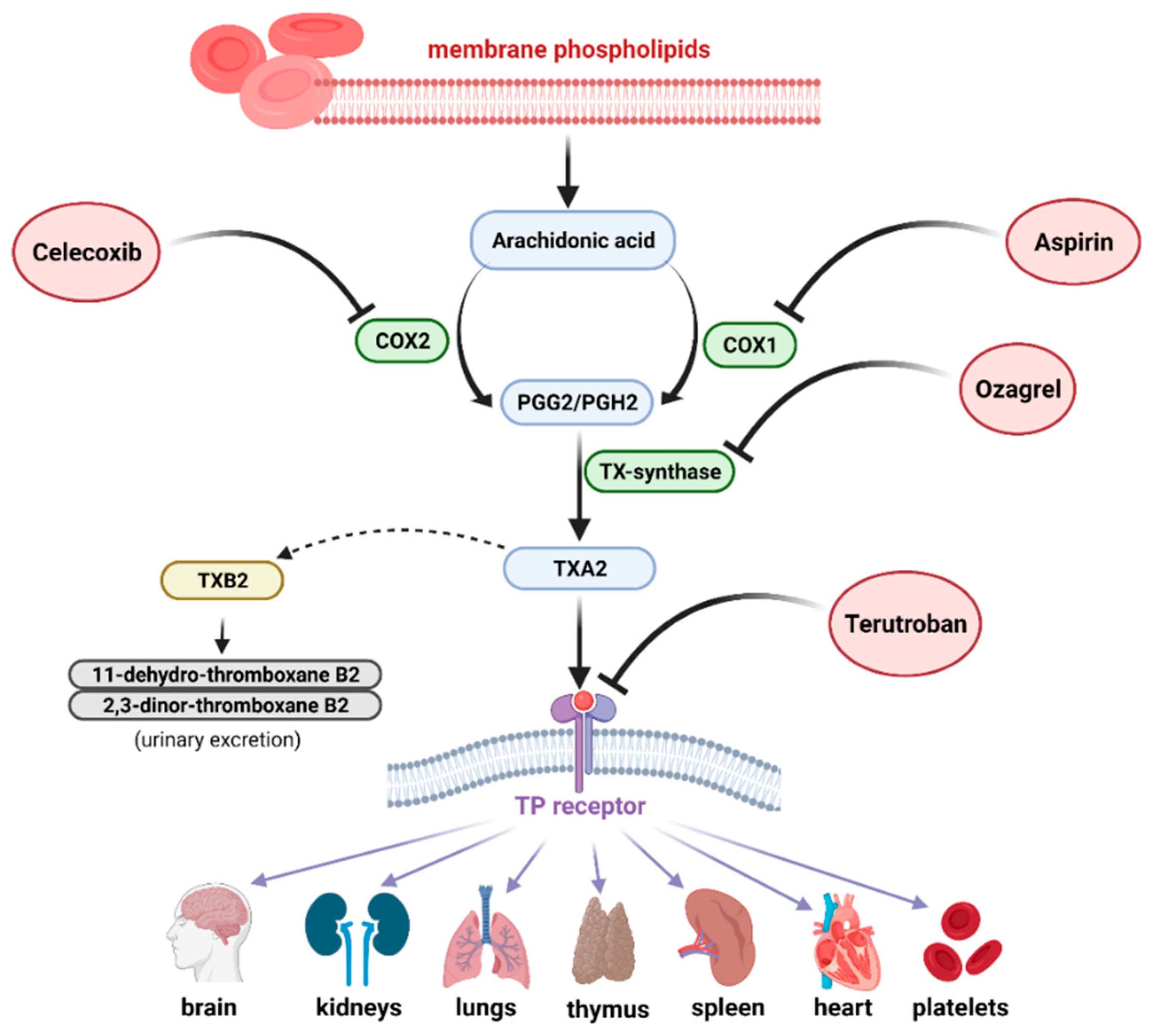
2.1. Thromboxane Synthesis and Its Mechanisms of Action
2.2. Natural Regulatory Substances Preventing Thromboxane Synthesis
2.3. Thromboxane, Inflammation, and Ischemic Stroke
2.4. Changes in Thromboxane Levels In Vitro and In Vivo
2.5. Changes in Thromboxane Levels in CVD
2.6. Application of Acetylsalicylic Acid
2.7. The Importance of ASA in the Treatment of Cardiovascular Diseases and Stroke
2.8. Incomplete Response to ASA Consumption
2.9. Other Methods of Blocking the TX Synthesis Pathway and Treating CVD
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Meschia, J.F.; Bushnell, C.; Boden-Albala, B.; Braun, L.T.; Bravata, D.M.; Chaturvedi, S.; Creager, M.A.; Eckel, R.H.; Elkind, M.S.V.; Fornage, M.; et al. Guidelines for the primary prevention of stroke: A statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2014, 45, 3754–3832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, A.; Baradaran, H.; Al-Dasuqi, K.; Knight-Greenfield, A.; Giambrone, A.E.; Delgado, D.; Wright, D.; Teng, Z.; Min, J.K.; Navi, B.B.; et al. Gadolinium Enhancement in Intracranial Atherosclerotic Plaque and Ischemic Stroke: A Systematic Review and Meta-Analysis. J. Am. Heart Assoc. 2016, 5, e003816. [Google Scholar] [CrossRef] [Green Version]
- Yan, A.; Zhang, T.; Yang, X.; Shao, J.; Fu, N.; Shen, F.; Fu, Y.; Xia, W. Thromboxane A2 Receptor Antagonist SQ29548 Reduces Ischemic Stroke-Induced Microglia/Macrophages Activation and Enrichment, and Ameliorates Brain Injury. Sci. Rep. 2016, 6, 35885. [Google Scholar] [CrossRef]
- Coupland, A.P.; Thapar, A.; Qureshi, M.I.; Jenkins, H.; Davies, A.H. The Definition of Stroke. J. R. Soc. Med. 2017, 110, 9–12. [Google Scholar] [CrossRef] [Green Version]
- Rucker, D.; Dhamoon, A.S. Physiology, Thromboxane A2. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Capra, V.; Bäck, M.; Barbieri, S.S.; Camera, M.; Tremoli, E.; Rovati, G.E. Eicosanoids and Their Drugs in Cardiovascular Diseases: Focus on Atherosclerosis and Stroke. Med. Res. Rev. 2013, 33, 364–438. [Google Scholar] [CrossRef] [PubMed]
- Sacerdoti, D.; Pesce, P.; Di Pascoli, M.; Brocco, S.; Cecchetto, L.; Bolognesi, M. Arachidonic Acid Metabolites and Endothelial Dysfunction of Portal Hypertension. Prostaglandins Other Lipid Mediat. 2015, 120, 80–90. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, T.; Tahara, Y.; Matsumoto, M.; Iguchi, M.; Sano, H.; Murayama, T.; Arai, H.; Oida, H.; Yurugi-Kobayashi, T.; Yamashita, J.K.; et al. Roles of Thromboxane A(2) and Prostacyclin in the Development of Atherosclerosis in ApoE-Deficient Mice. J. Clin. Investig. 2004, 114, 784–794. [Google Scholar] [CrossRef] [Green Version]
- Smyth, E.M. Thromboxane and the Thromboxane Receptor in Cardiovascular Disease. Clin. Lipidol. 2010, 5, 209–219. [Google Scholar] [CrossRef] [Green Version]
- Nannoni, S.; de Groot, R.; Bell, S.; Markus, H.S. Stroke in COVID-19: A Systematic Review and Meta-Analysis. Int. J. Stroke Off. J. Int. Stroke Soc. 2021, 16, 137–149. [Google Scholar] [CrossRef]
- Hammock, B.D.; Wang, W.; Gilligan, M.M.; Panigrahy, D. Eicosanoids. Am. J. Pathol. 2020, 190, 1782–1788. [Google Scholar] [CrossRef]
- Yamakawa, M.; Kuno, T.; Mikami, T.; Takagi, H.; Gronseth, G. Clinical Characteristics of Stroke with COVID-19: A Systematic Review and Meta-Analysis. J. Stroke Cerebrovasc. Dis. 2020, 29, 105288. [Google Scholar] [CrossRef] [PubMed]
- Östling, J.; van Geest, M.; Schofield, J.P.R.; Jevnikar, Z.; Wilson, S.; Ward, J.; Lutter, R.; Shaw, D.E.; Bakke, P.S.; Caruso, M.; et al. IL-17-High Asthma with Features of a Psoriasis Immunophenotype. J. Allergy Clin. Immunol. 2019, 144, 1198–1213. [Google Scholar] [CrossRef] [Green Version]
- Szczeklik, W.; Stodółkiewicz, E.; Rzeszutko, M.; Tomala, M.; Chrustowicz, A.; Żmudka, K.; Sanak, M. Urinary 11-Dehydro-Thromboxane B2 as a Predictor of Acute Myocardial Infarction Outcomes: Results of Leukotrienes and Thromboxane In Myocardial Infarction (LTIMI) Study. J. Am. Heart Assoc. 2016, 5, e003702. [Google Scholar] [CrossRef] [PubMed]
- Nakano, Y.; Nakatani, Y.; Takami, M.; Taniyama, Y.; Arima, S. Diverse Associations between Oxidative Stress and Thromboxane A2 in Hypertensive Glomerular Injury. Hypertens. Res. Off. J. Jpn. Soc. Hypertens. 2019, 42, 450–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Chen, J.; Mao, J.; Li, H.; Wang, M.; Zhang, H.; Li, H.; Chen, W. Genistein Ameliorates Non-Alcoholic Fatty Liver Disease by Targeting the Thromboxane A2 Pathway. J. Agric. Food Chem. 2018, 66, 5853–5859. [Google Scholar] [CrossRef] [PubMed]
- Serhan, K.; Gartung, A.; Panigrahy, D. Drawing a Link between the Thromboxane A2 Pathway and the Role of Platelets and Tumor Cells in Ovarian Cancer. Prostaglandins Other Lipid Mediat. 2018, 137, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Szczuko, M.; Kotlęga, D.; Palma, J.; Zembroń-Łacny, A.; Tylutka, A.; Gołąb-Janowska, M.; Drozd, A. Lipoxins, RevD1 and 9, 13 HODE as the Most Important Derivatives after an Early Incident of Ischemic Stroke. Sci. Rep. 2020, 10, 12849. [Google Scholar] [CrossRef]
- Simeone, P.; Liani, R.; Tripaldi, R.; Di Castelnuovo, A.; Guagnano, M.T.; Tartaro, A.; Bonadonna, R.C.; Federico, V.; Cipollone, F.; Consoli, A.; et al. Thromboxane-Dependent Platelet Activation in Obese Subjects with Prediabetes or Early Type 2 Diabetes: Effects of Liraglutide- or Lifestyle Changes-Induced Weight Loss. Nutrients 2018, 10, 1872. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.W.; Gardner, R.; Porter, D.L.; Louis, C.U.; Ahmed, N.; Jensen, M.; Grupp, S.A.; Mackall, C.L. Current Concepts in the Diagnosis and Management of Cytokine Release Syndrome. Blood 2014, 124, 188–195. [Google Scholar] [CrossRef] [Green Version]
- Russo, I.; Viretto, M.; Barale, C.; Mattiello, L.; Doronzo, G.; Pagliarino, A.; Cavalot, F.; Trovati, M.; Anfossi, G. High Glucose Inhibits the Aspirin-Induced Activation of the Nitric Oxide/CGMP/CGMP-Dependent Protein Kinase Pathway and Does Not Affect the Aspirin-Induced Inhibition of Thromboxane Synthesis in Human Platelets. Diabetes 2012, 61, 2913–2921. [Google Scholar] [CrossRef] [Green Version]
- Davì, G.; Santilli, F.; Vazzana, N. Thromboxane Receptors Antagonists and/or Synthase Inhibitors. Handb. Exp. Pharmacol. 2012, 210, 261–286. [Google Scholar] [CrossRef]
- Chang, M.-C.; Chang, H.-H.; Wang, T.-M.; Chan, C.-P.; Lin, B.-R.; Yeung, S.-Y.; Yeh, C.-Y.; Cheng, R.-H.; Jeng, J.-H. Antiplatelet Effect of Catechol Is Related to Inhibition of Cyclooxygenase, Reactive Oxygen Species, ERK/P38 Signaling and Thromboxane A2 Production. PLoS ONE 2014, 9, e104310. [Google Scholar] [CrossRef]
- Ed Nignpense, B.; Chinkwo, K.A.; Blanchard, C.L.; Santhakumar, A.B. Polyphenols: Modulators of Platelet Function and Platelet Microparticle Generation? Int. J. Mol. Sci. 2019, 21, 146. [Google Scholar] [CrossRef] [Green Version]
- Khan, H.; Jawad, M.; Kamal, M.A.; Baldi, A.; Xiao, J.; Nabavi, S.M.; Daglia, M. Evidence and Prospective of Plant Derived Flavonoids as Antiplatelet Agents: Strong Candidates to Be Drugs of Future. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2018, 119, 355–367. [Google Scholar] [CrossRef]
- Douglas, S.D.; Leeman, S.E. Neurokinin-1 Receptor: Functional Significance in the Immune System in Reference to Selected Infections and Inflammation. Ann. N. Y. Acad. Sci. 2011, 1217, 83–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corrigan, F.; Vink, R.; Turner, R.J. Inflammation in Acute CNS Injury: A Focus on the Role of Substance P. Br. J. Pharmacol. 2016, 173, 703–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arruzazabala, M.L.; Mas, R.; Molina, V.; Carbajal, D.; Fernández, L.; Illnait, J.; Castaño, G.; Fernández, J.; Mendoza, S. Effects of D-003, a New Substance Purified from Sugar Cane Wax, on Platelet Aggregation and Plasma Levels of Arachidonic Acid Metabolites in Healthy Volunteers. Int. J. Clin. Pharmacol. Res. 2004, 24, 55–63. [Google Scholar]
- Zhao, J.; Zheng, L.; Fei, Q.; Fu, Y.; Weng, Y.; Wu, H.; Li, H.; Jun, Q.; Shao, J.; Xu, Y. Association of Thromboxane A2 Receptor Gene Polymorphisms with Cerebral Infarction in a Chinese Population. Neurol. Sci. 2013, 34, 1791–1796. [Google Scholar] [CrossRef] [PubMed]
- Egan, K.M.; Wang, M.; Fries, S.; Lucitt, M.B.; Zukas, A.M.; Puré, E.; Lawson, J.A.; FitzGerald, G.A. Cyclooxygenases, Thromboxane, and Atherosclerosis: Plaque Destabilization by Cyclooxygenase-2 Inhibition Combined with Thromboxane Receptor Antagonism. Circulation 2005, 111, 334–342. [Google Scholar] [CrossRef]
- Li, L.; He, Z.; Wang, Y.; Liu, X.; Yuan, L. Associations between Thromboxane A Synthase 1 Gene Polymorphisms and the Risk of Ischemic Stroke in a Chinese Han Population. Neural Regen. Res. 2018, 13, 463–469. [Google Scholar] [CrossRef]
- Yi, X.; Lin, J.; Luo, H.; Wang, C.; Liu, Y. Genetic Variants of PTGS2, TXA2R and TXAS1 Are Associated with Carotid Plaque Vulnerability, Platelet Activation and TXA2 Levels in Ischemic Stroke Patients. PLoS ONE 2017, 12, e0180704. [Google Scholar] [CrossRef] [PubMed]
- Fanning, J.P.; Wong, A.A.; Fraser, J.F. The Epidemiology of Silent Brain Infarction: A Systematic Review of Population-Based Cohorts. BMC Med. 2014, 12, 119. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Hu, X.; Leak, R.K.; Shi, Y.; An, C.; Suenaga, J.; Chen, J.; Gao, Y. Demyelination as a Rational Therapeutic Target for Ischemic or Traumatic Brain Injury. Exp. Neurol. 2015, 272, 17–25. [Google Scholar] [CrossRef] [Green Version]
- An, C.; Shi, Y.; Li, P.; Hu, X.; Gan, Y.; Stetler, R.A.; Leak, R.K.; Gao, Y.; Sun, B.-L.; Zheng, P.; et al. Molecular Dialogs between the Ischemic Brain and the Peripheral Immune System: Dualistic Roles in Injury and Repair. Prog. Neurobiol. 2014, 115, 6–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conti, P.; Caraffa, A.; Gallenga, C.E.; Ross, R.; Kritas, S.K.; Frydas, I.; Younes, A.; Ronconi, G. Coronavirus-19 (SARS-CoV-2) Induces Acute Severe Lung Inflammation via IL-1 Causing Cytokine Storm in COVID-19: A Promising Inhibitory Strategy. J. Biol. Regul. Homeost. Agents 2020, 34, 1971–1975. [Google Scholar] [CrossRef] [PubMed]
- Conti, P.; Caraffa, A.; Gallenga, C.E.; Ross, R.; Kritas, S.K.; Frydas, I.; Younes, A.; Di Emidio, P.; Ronconi, G.; Toniato, E. IL-1 Induces Throboxane-A2 (TxA2) in COVID-19 Causing Inflammation and Micro-Thrombi: Inhibitory Effect of the IL-1 Receptor Antagonist (IL-1Ra). J. Biol. Regul. Homeost. Agents 2020, 34, 1623–1627. [Google Scholar] [CrossRef]
- Hladky, S.B.; Barrand, M.A. Elimination of Substances from the Brain Parenchyma: Efflux via Perivascular Pathways and via the Blood–Brain Barrier. Fluids Barriers CNS 2018, 15, 30. [Google Scholar] [CrossRef] [Green Version]
- Patrono, C.; Rocca, B. Measurement of Thromboxane Biosynthesis in Health and Disease. Front. Pharmacol. 2019, 10. [Google Scholar] [CrossRef]
- Lopez, L.R.; Guyer, K.E.; Torre, I.G.D.L.; Pitts, K.R.; Matsuura, E.; Ames, P.R. Platelet Thromboxane (11-Dehydro-Thromboxane B2) and Aspirin Response in Patients with Diabetes and Coronary Artery Disease. World J. Diabetes 2014, 5, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Parker, W.A.E.; Orme, R.C.; Hanson, J.; Stokes, H.M.; Bridge, C.M.; Shaw, P.A.; Sumaya, W.; Thorneycroft, K.; Petrucci, G.; Porro, B.; et al. Very-Low-Dose Twice-Daily Aspirin Maintains Platelet Inhibition and Improves Haemostasis during Dual-Antiplatelet Therapy for Acute Coronary Syndrome. Platelets 2019, 30, 148–157. [Google Scholar] [CrossRef]
- Tucker, K.L.; Sheppard, J.P.; Stevens, R.; Bosworth, H.B.; Bove, A.; Bray, E.P.; Earle, K.; George, J.; Godwin, M.; Green, B.B.; et al. Self-Monitoring of Blood Pressure in Hypertension: A Systematic Review and Individual Patient Data Meta-Analysis. PLoS Med. 2017, 14, 8532. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Hu, J.; Gao, X.; Liang, H.; Yu, H.; Liu, S.; Liu, Z. Hyperglycemia via Activation of Thromboxane A2 Receptor Impairs the Integrity and Function of Blood-Brain Barrier in Microvascular Endothelial Cells. Oncotarget 2017, 8, 30030–30038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothwell, P.M.; Cook, N.R.; Gaziano, J.M.; Price, J.F.; Belch, J.F.F.; Roncaglioni, M.C.; Morimoto, T.; Mehta, Z. Effects of Aspirin on Risks of Vascular Events and Cancer According to Bodyweight and Dose: Analysis of Individual Patient Data from Randomised Trials. Lancet 2018, 392, 387–399. [Google Scholar] [CrossRef] [Green Version]
- Effects of Aspirin for Primary Prevention in Persons with Diabetes Mellitus. N. Engl. J. Med. 2018, 379, 1529–1539. [CrossRef] [PubMed]
- Woods, R.L.; Polekhina, G.; Wolfe, R.; Nelson, M.R.; Ernst, M.E.; Reid, C.M.; Shah, R.C.; Lockery, J.E.; Orchard, S.G.; Murray, A.M.; et al. No Modulation of the Effect of Aspirin by Body Weight in Healthy Older Men and Women. Circulation 2020, 141, 1110–1112. [Google Scholar] [CrossRef]
- Peace, A.; McCall, M.; Tedesco, T.; Kenny, D.; Conroy, R.M.; Foley, D.; Cox, D. The Role of Weight and Enteric Coating on Aspirin Response in Cardiovascular Patients. J. Thromb. Haemost. JTH 2010, 8, 2323–2325. [Google Scholar] [CrossRef]
- Filipiak, K.J. Jaką Dawkę Kwasu Acetylosalicylowego Należy Stosować w Codziennej Praktyce Klinicznej? Wielodyscyplinarne Stanowisko Ekspertów. Chor. Serca Naczyń 2016, 13, 147–158. [Google Scholar]
- Eikelboom, J.W.; Hirsh, J.; Spencer, F.A.; Baglin, T.P.; Weitz, J.I. Antiplatelet Drugs - Antithrombotic Therapy and Prevention of Thrombosis, 9th Ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012, 141, e89S–e119S. [Google Scholar] [CrossRef] [Green Version]
- Mancia, G. Short- and Long-Term Blood Pressure Variability: Present and Future. Hypertension 2012, 60, 512–517. [Google Scholar] [CrossRef] [Green Version]
- Paniccia, R.; Priora, R.; Liotta, A.A.; Abbate, R. Platelet Function Tests: A Comparative Review. Vasc. Health Risk Manag. 2015, 11, 133–148. [Google Scholar] [CrossRef] [Green Version]
- Racca, C.; van Diemen, J.J.K.; Fuijkschot, W.W.; Spit, K.; Bonten, T.N.; Numans, M.E.; van der Bom, J.G.; Smulders, Y.M.; Thijs, A. Aspirin Intake in the Morning Is Associated with Suboptimal Platelet Inhibition, as Measured by Serum Thromboxane B2, during Infarct-Prone Early-Morning Hours. Platelets 2019, 30, 871–877. [Google Scholar] [CrossRef] [PubMed]
- Vandvik, P.O.; Lincoff, A.M.; Gore, J.M.; Gutterman, D.D.; Sonnenberg, F.A.; Alonso-Coello, P.; Akl, E.A.; Lansberg, M.G.; Guyatt, G.H.; Spencer, F.A. Primary and Secondary Prevention of Cardiovascular Disease: Antithrombotic Therapy and Prevention of Thrombosis, 9th Ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012, 141, e637S–e668S. [Google Scholar] [CrossRef] [Green Version]
- Smith, S.M.; Soubhi, H.; Fortin, M.; Hudon, C.; O’Dowd, T. Managing Patients with Multimorbidity: Systematic Review of Interventions in Primary Care and Community Settings. BMJ 2012, 345, e5205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baggen, V.J.M.; Leiner, T.; Post, M.C.; van Dijk, A.P.; Roos-Hesselink, J.W.; Boersma, E.; Habets, J.; Sieswerda, G.T. Cardiac Magnetic Resonance Findings Predicting Mortality in Patients with Pulmonary Arterial Hypertension: A Systematic Review and Meta-Analysis. Eur. Radiol. 2016, 26, 3771–3780. [Google Scholar] [CrossRef] [Green Version]
- Szałek, E. The pleiotropic effect of acetylsalicylic acid. Farmacja Współczesna 2015, 8, 52–58. [Google Scholar]
- Bliden, K.P.; Tantry, U.S.; Gesheff, M.G.; Franzese, C.J.; Pandya, S.; Toth, P.P.; Mathew, D.P.; Chaudhary, R.; Gurbel, P.A. Thrombin-Induced Platelet-Fibrin Clot Strength Identified by Thrombelastography. J. Intervent. Cardiol. 2016, 29, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Bliden, K.P.; Tantry, U.S.; Chaudhary, R.; Byun, S.; Gurbel, P.A. Extended-Release Acetylsalicylic Acid for Secondary Prevention of Stroke and Cardiovascular Events. Expert Rev. Cardiovasc. Ther. 2016, 14, 779–791. [Google Scholar] [CrossRef]
- Vasudeva, K.; Chaurasia, P.; Singh, S.; Munshi, A. Genetic Signatures in Ischemic Stroke: Focus on Aspirin Resistance. CNS Neurol. Disord. Drug Targets 2017, 16, 974–982. [Google Scholar] [CrossRef]
- Dharmasaroja, P.A.; Muengtaweepongsa, S.; Sae-Lim, S. Aspirin Nonresponders in Patients with Ischaemic Stroke. Blood Coagul. Fibrinolysis Int. J. Haemost. Thromb. 2013, 24, 361–364. [Google Scholar] [CrossRef]
- Abraham, N.S.; Singh, S.; Caleb Alexander, G.; Heien, H.; Haas, L.R.; Crown, W.; Shah, N.D. Comparative Risk of Gastrointestinal Bleeding with Dabigatran, Rivaroxaban, and Warfarin: Population Based Cohort Study. BMJ 2015, 350, h1857. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.-F.; Hsiao, F.-Y.; Wen, Y.-W.; Tsai, Y.-W. Cardiovascular Events Associated with the Use of Four Nonselective NSAIDs (Etodolac, Nabumetone, Ibuprofen, or Naproxen) versus a Cyclooxygenase-2 Inhibitor (Celecoxib): A Population-Based Analysis in Taiwanese Adults. Clin. Ther. 2006, 28, 1827–1836. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, J.; Chang, X.; Zhang, C.; Zhou, H.; Liu, M. Ozagrel for Acute Ischemic Stroke: A Meta-Analysis of Data from Randomized Controlled Trials. Neurol. Res. 2012, 34, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Sathler, P.C.; Santana, M.; Lourenço, A.L.; Rodrigues, C.R.; Abreu, P.; Cabral, L.M.; Castro, H.C. Human Thromboxane Synthase: Comparative Modeling and Docking Evaluation with the Competitive Inhibitors Dazoxiben and Ozagrel. J. Enzyme Inhib. Med. Chem. 2014, 29, 527–531. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.R.; Fredette, N.C.; Barton, M.; Prossnitz, E.R. G Protein-Coupled Estrogen Receptor Inhibits Vascular Prostanoid Production and Activity. J. Endocrinol. 2015, 227, 61–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szczuko, M.; Kozioł, I.; Kotlęga, D.; Brodowski, J.; Drozd, A. The Role of Thromboxane in the Course and Treatment of Ischemic Stroke: Review. Int. J. Mol. Sci. 2021, 22, 11644. https://doi.org/10.3390/ijms222111644
Szczuko M, Kozioł I, Kotlęga D, Brodowski J, Drozd A. The Role of Thromboxane in the Course and Treatment of Ischemic Stroke: Review. International Journal of Molecular Sciences. 2021; 22(21):11644. https://doi.org/10.3390/ijms222111644
Chicago/Turabian StyleSzczuko, Małgorzata, Igor Kozioł, Dariusz Kotlęga, Jacek Brodowski, and Arleta Drozd. 2021. "The Role of Thromboxane in the Course and Treatment of Ischemic Stroke: Review" International Journal of Molecular Sciences 22, no. 21: 11644. https://doi.org/10.3390/ijms222111644
APA StyleSzczuko, M., Kozioł, I., Kotlęga, D., Brodowski, J., & Drozd, A. (2021). The Role of Thromboxane in the Course and Treatment of Ischemic Stroke: Review. International Journal of Molecular Sciences, 22(21), 11644. https://doi.org/10.3390/ijms222111644