
Murine Oncostatin M Has Opposing Effects on the Proliferation of OP9 Bone Marrow Stromal Cells and NIH/3T3 Fibroblasts Signaling through the OSMR

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. OSM Has Differential Effects on the Proliferation of BM Stromal Cells and Fibroblasts
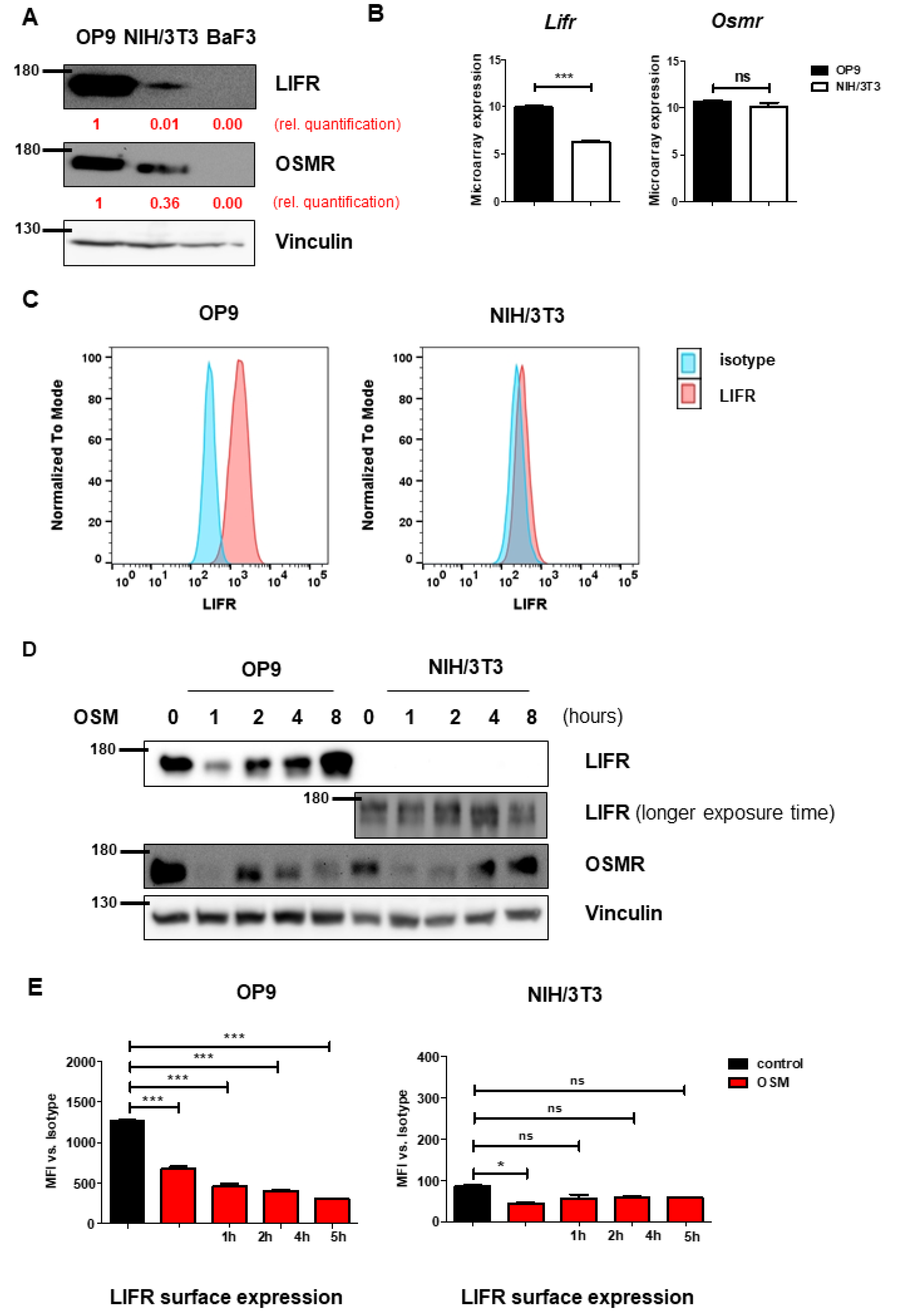
2.2. OP9 and NIH/3T3 Cells Show Differential Levels of OSMR and LIFR
2.3. OSMR Downregulation Attenuates OSM Effects on Proliferation
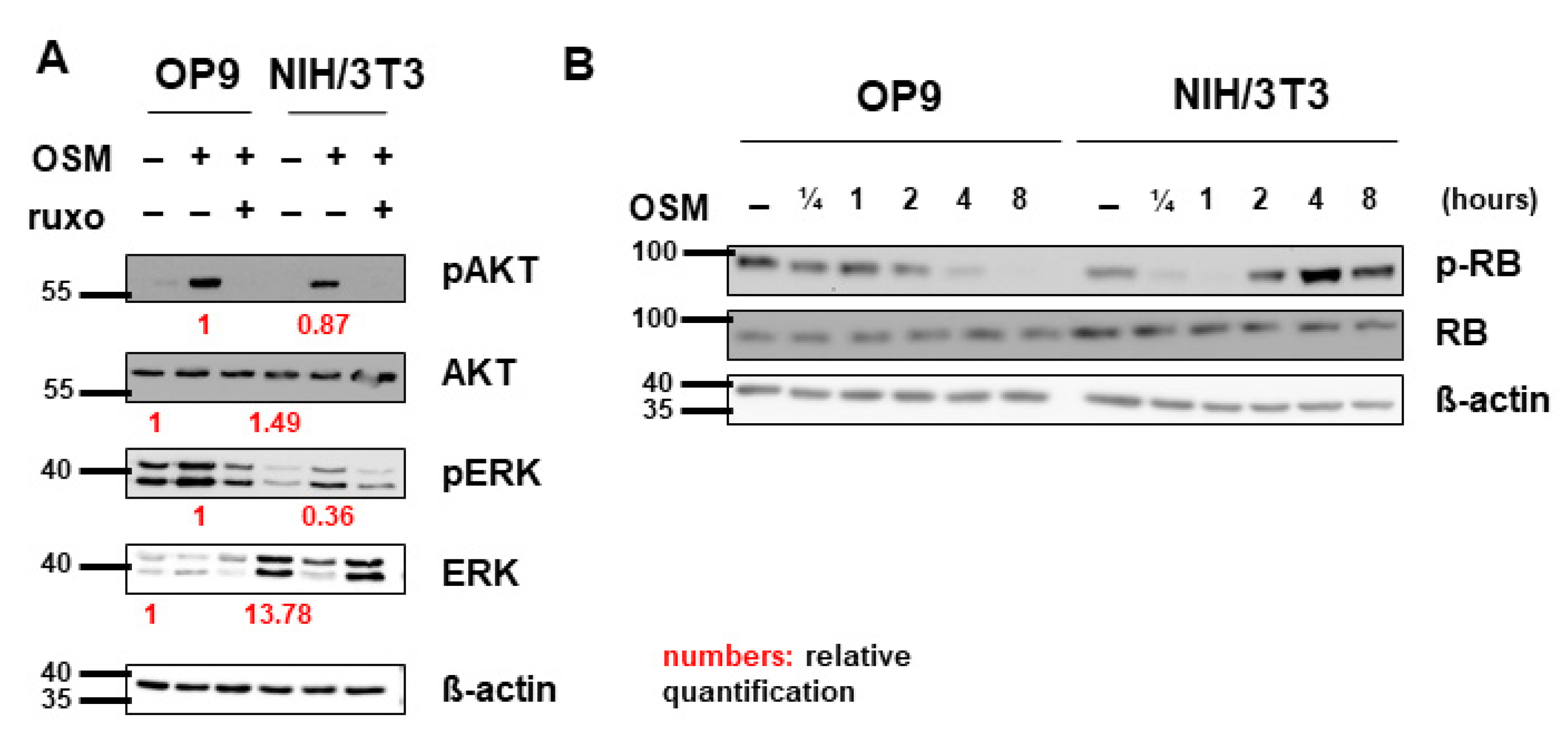
2.4. OSM Activates JAK-STAT, PI3K-AKT, and MAPK-ERK Pathways in OP9 and NIH/3T3 Cells
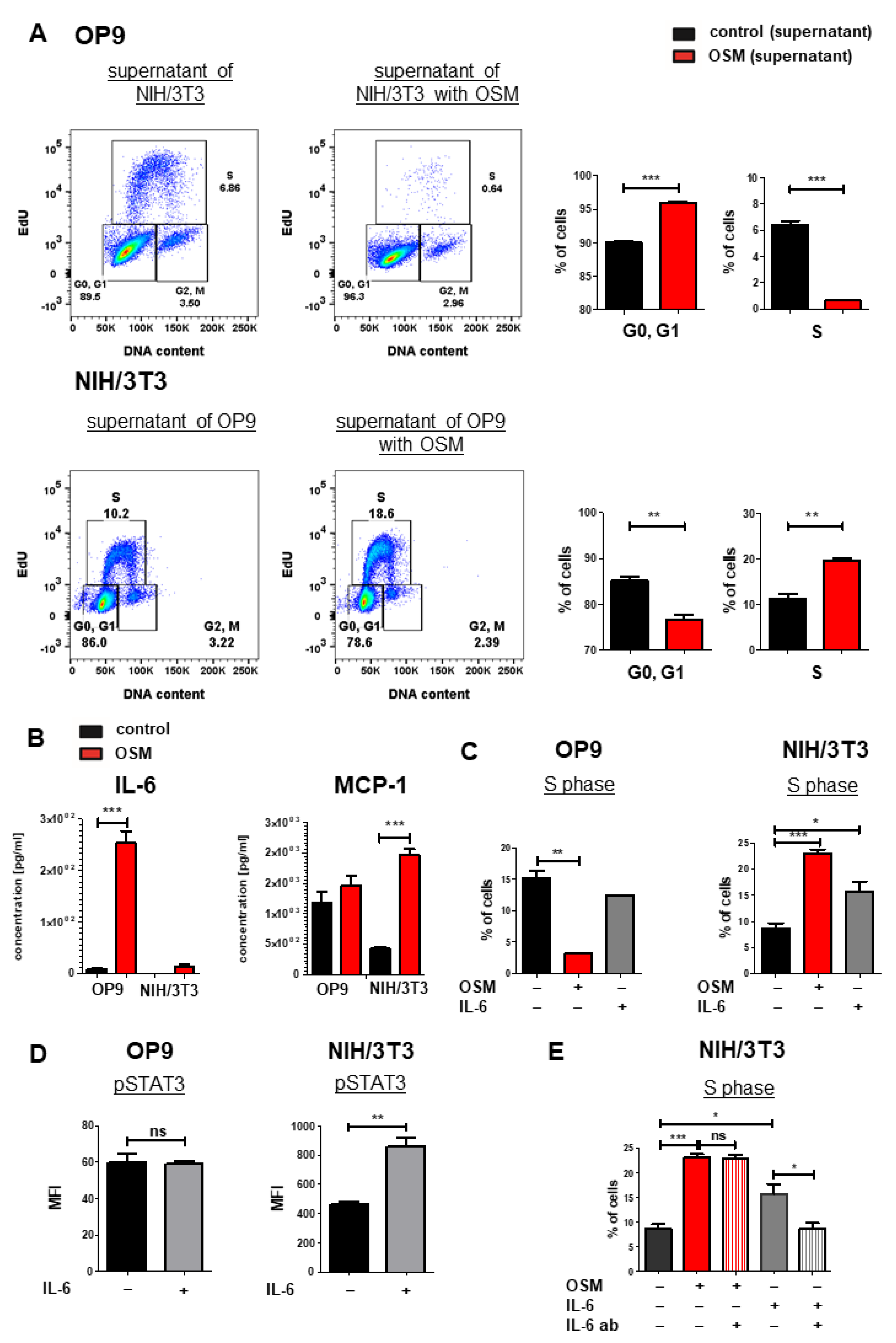
2.5. IL-6 Has Synergistic Effects to OSM on the Proliferation of NIH/3T3 Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Cell Culture Supplements
4.3. Retrovirus Production and Transduction
4.4. Proliferation Assays
4.5. DNA Constructs and Knockdown
4.6. Flow Cytometric Analysis and Cell Sorting
4.7. Immunoblotting
4.8. Cytokine Array
4.9. RNA Isolation and Microarray
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zarling, J.M.; Shoyab, M.; Marquardt, H.; Hanson, M.B.; Lioubin, M.N.; Todaro, G. Oncostatin M: A growth regulator produced by differentiated histiocytic lymphoma cells. Proc. Natl. Acad. Sci. USA 1986, 83, 9739–9743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyajima, A.; Kinoshita, T.; Tanaka, M.; Kamiya, A.; Mukouyama, Y.; Hara, T. Role of Oncostatin M in hematopoiesis and liver development. Cytokine Growth Factor Rev. 2000, 11, 177–183. [Google Scholar] [CrossRef]
- Malik, N.; Haugen, H.S.; Modrell, B.; Shoyab, M.; Clegg, C.H. Developmental abnormalities in mice transgenic for bovine oncostatin M. Mol. Cell Biol. 1995, 15, 2349–2358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, N.R.; Hegazy, A.N.; Owens, B.M.J.; Bullers, S.J.; Linggi, B.; Buonocore, S.; Coccia, M.; Görtz, D.; This, S.; Stockenhuber, K.; et al. Oncostatin M drives intestinal inflammation in mice and its abundance predicts response to tumor necrosis factor-neutralizing therapy in patients with inflammatory bowel disease. Nat. Med. 2017, 23, 579–589. [Google Scholar] [CrossRef]
- Minehata, K.; Takeuchi, M.; Hirabayashi, Y.; Inoue, T.; Donovan, P.J.; Tanaka, M.; Miyajima, A. Oncostatin m maintains the hematopoietic microenvironment and retains hematopoietic progenitors in the bone marrow. Int. J. Hematol. 2006, 84, 319–327. [Google Scholar] [CrossRef]
- Tanaka, M.; Hirabayashi, Y.; Sekiguchi, T.; Inoue, T.; Katsuki, M.; Miyajima, A. Targeted disruption of oncostatin M receptor results in altered hematopoiesis. Blood 2003, 102, 3154–3162. [Google Scholar] [CrossRef] [Green Version]
- Gurluler, E.; Tumay, L.V.; Guner, O.S.; Kucukmetin, N.T.; Hizli, B.; Zorluoglu, A. Oncostatin-M as a novel biomarker in colon cancer patients and its association with clinicopathologic variables. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 2042–2047. [Google Scholar]
- Torres, C.; Perales, S.; Alejandre, M.J.; Iglesias, J.; Palomino, R.J.; Martin, M.; Caba, O.; Prados, J.C.; Aránega, A.; Delgado, J.R.; et al. Serum cytokine profile in patients with pancreatic cancer. Pancreas 2014, 43, 1042–1049. [Google Scholar] [CrossRef]
- Koskela, K.; Pelliniemi, T.-T.; Remes, K.; Rajamäki, A.; Pulkki, K. Serum oncostatin M in multiple myeloma: Association with prognostic factors. Br. J. Haematol. 1997, 96, 158–160. [Google Scholar] [CrossRef]
- Lilja, A.; Nordborg, C.; Brun, A.; Salford, L.; Aman, P. Expression of the IL-6 family cytokines in human brain tumors. Int. J. Oncol. 2001, 19, 495–499. [Google Scholar] [CrossRef]
- Robak, T.; Wierzbowska, A.; Błasińska-Morawiec, M.; Korycka, A.; Błoński, J.Z. Serum Levels of IL-6 Type Cytokines and Soluble IL-6 Receptors in Active B-Cell Chronic Lymphocytic Leukemia and in Cladribine Induced Remission. Mediat. Inflamm. 1999, 8, 277–286. [Google Scholar] [CrossRef] [Green Version]
- Liang, H.; Block, T.M.; Wang, M.; Nefsky, B.; Long, R.; Hafner, J.; Mehta, A.S.; Marrero, J.; Gish, R.; Norton, P.A. Interleukin-6 and oncostatin M are elevated in liver disease in conjunction with candidate hepatocellular carcinoma biomarker GP73. Cancer Biomark. 2012, 11, 161–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scotto, L.; Narayan, G.; Nandula, S.V.; Subramaniyam, S.; Kaufmann, A.M.; Wright, J.D.; Pothuri, B.; Mansukhani, M.; Schneider, A.; Arias-Pulido, H.; et al. Integrative genomics analysis of chromosome 5p gain in cervical cancer reveals target over-expressed genes, including Drosha. Mol. Cancer 2008, 7, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torossian, F.; Guerton, B.; Anginot, A.; Alexander, K.A.; Desterke, C.; Soave, S.; Tseng, H.-W.; Arouche, N.; Boutin, L.; Kulina, I.; et al. Macrophage-derived oncostatin M contributes to human and mouse neurogenic heterotopic ossifications. JCI Insight 2017, 2, e96034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guihard, P.; Danger, Y.; Brounais, B.; David, E.; Brion, R.; Delecrin, J.; Richards, C.D.; Chevalier, S.; Rédini, F.; Heymann, D.; et al. Induction of osteogenesis in mesenchymal stem cells by activated monocytes/macrophages depends on oncostatin M signaling. Stem Cells 2012, 30, 762–772. [Google Scholar] [CrossRef] [Green Version]
- Broxmeyer, H.E.; Bruns, H.A.; Zhang, S.; Cooper, S.; Hangoc, G.; McKenzie, A.N.; Dent, A.L.; Schindler, U.; Naeger, L.K.; Hoey, T.; et al. Th1 cells regulate hematopoietic progenitor cell homeostasis by production of oncostatin M. Immunity 2002, 16, 815–825. [Google Scholar] [CrossRef] [Green Version]
- Brown, T.J.; Lioubin, M.N.; Marquardt, H. Purification and characterization of cytostatic lymphokines produced by activated human T lymphocytes. Synergistic antiproliferative activity of transforming growth factor beta 1, interferon-gamma, and oncostatin M for human melanoma cells. J. Immunol. 1987, 139, 2977–2983. [Google Scholar]
- Suda, T.; Chida, K.; Todate, A.; Ide, K.; Asada, K.; Nakamura, Y.; Suzuki, K.; Kuwata, H.; Nakamura, H. Oncostatin M Production by Human Dendritic Cells In Response To Bacterial Products. Cytokine 2002, 17, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Goren, I.; Kämpfer, H.; Müller, E.; Schiefelbein, D.; Pfeilschifter, J.; Frank, S. Oncostatin M expression is functionally connected to neutrophils in the early inflammatory phase of skin repair: Implications for normal and diabetes-impaired wounds. J. Investig. Derm. 2006, 126, 628–637. [Google Scholar] [CrossRef] [Green Version]
- Grenier, A.; Combaux, D.; Chastre, J.; Gougerot-Pocidalo, M.A.; Gibert, C.; Dehoux, M.; Chollet-Martin, S. Oncostatin M production by blood and alveolar neutrophils during acute lung injury. Lab. Investig. 2001, 81, 133–141. [Google Scholar] [CrossRef] [Green Version]
- Grenier, A.; Dehoux, M.; Boutten, A.; Arce-Vicioso, M.; Durand, G.; Gougerot-Pocidalo, M.A.; Chollet-Martin, S. Oncostatin M production and regulation by human polymorphonuclear neutrophils. Blood 1999, 93, 1413–1421. [Google Scholar] [CrossRef]
- Queen, M.M.; Ryan, R.E.; Holzer, R.G.; Keller-Peck, C.R.; Jorcyk, C.L. Breast cancer cells stimulate neutrophils to produce oncostatin M: Potential implications for tumor progression. Cancer Res. 2005, 65, 8896–8904. [Google Scholar] [CrossRef] [Green Version]
- Setiadi, H.; Yago, T.; Liu, Z.; McEver, R.P. Endothelial signaling by neutrophil-released oncostatin M enhances P-selectin–dependent inflammation and thrombosis. Blood Adv. 2019, 3, 168–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taga, T.; Hibi, M.; Hirata, Y.; Yamasaki, K.; Yasukawa, K.; Matsuda, T.; Hirano, T.; Kishimoto, T. Interleukin-6 triggers the association of its receptor with a possible signal transducer, gp130. Cell 1989, 58, 573–581. [Google Scholar] [CrossRef]
- Hirano, T.; Matsuda, T.; Nakajima, K. Signal transduction through gp130 that is shared among the receptors for the interleukin 6 related cytokine subfamily. Stem Cells 1994, 12, 262–277. [Google Scholar] [CrossRef] [PubMed]
- Gearing, D.P.; Comeau, M.R.; Friend, D.J.; Gimpel, S.D.; Thut, C.J.; McGourty, J.; Brasher, K.K.; King, J.A.; Gillis, S.; Mosley, B. The IL-6 signal transducer, gp130: An oncostatin M receptor and affinity converter for the LIF receptor. Science 1992, 255, 1434–1437. [Google Scholar] [CrossRef] [PubMed]
- Mosley, B.; Imus C de Friend, D.; Boiani, N.; Thoma, B.; Park, L.S.; Cosman, D. Dual oncostatin M (OSM) receptors. Cloning and characterization of an alternative signaling subunit conferring OSM-specific receptor activation. J. Biol. Chem. 1996, 271, 32635–32643. [Google Scholar] [CrossRef] [Green Version]
- Lindberg, R.A.; Juan, T.S.-C.; Welcher, A.A.; Sun, Y.; Cupples, R.; Guthrie, B.; Fletcher, F.A. Cloning and Characterization of a Specific Receptor for Mouse Oncostatin, M. Mol. Cell. Biol. 1998, 18, 3357–3367. [Google Scholar] [CrossRef] [Green Version]
- Ichihara, M.; Hara, T.; Kim, H.; Murate, T.; Miyajima, A. Oncostatin M and leukemia inhibitory factor do not use the same functional receptor in mice. Blood 1997, 90, 165–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, E.C.; McGregor, N.E.; Poulton, I.J.; Solano, M.; Pompolo, S.; Fernandes, T.J.; Constable, M.J.; Nicholson, G.C.; Zhang, J.G.; Nicola, N.A.; et al. Oncostatin M promotes bone formation independently of resorption when signaling through leukemia inhibitory factor receptor in mice. J. Clin. Investig. 2010, 120, 582–592. [Google Scholar] [CrossRef] [Green Version]
- Walker, E.C.; Johnson, R.W.; Hu, Y.; Brennan, H.J.; Poulton, I.J.; Zhang, J.-G.; Jenkins, B.J.; Smyth, G.K.; Nicola, N.A.; Sims, N. Murine Oncostatin M Acts via Leukemia Inhibitory Factor Receptor to Phosphorylate Signal Transducer and Activator of Transcription 3 (STAT3) but Not STAT1, an Effect That Protects Bone Mass. J. Biol. Chem. 2016, 291, 21703–21716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N.; et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef]
- Auguste, P.; Guillet, C.; Fourcin, M.; Olivier, C.; Veziers, J.; Pouplard-Barthelaix, A.; Gascan, H. Signaling of type II oncostatin M receptor. J. Biol. Chem. 1997, 272, 15760–15764. [Google Scholar] [CrossRef] [Green Version]
- Hermanns, H.M.; Radtke, S.; Schaper, F.; Heinrich, P.C.; Behrmann, I. Non-redundant signal transduction of interleukin-6-type cytokines. The adapter protein Shc is specifically recruited to rhe oncostatin M receptor. J. Biol. Chem. 2000, 275, 40742–40748. [Google Scholar] [CrossRef] [Green Version]
- Hermanns, H.M.; Radtke, S.; Haan, C.; Schmitz-Van de Leur, H.; Tavernier, J.; Heinrich, P.C.; Behrmann, I. Contributions of leukemia inhibitory factor receptor and oncostatin M receptor to signal transduction in heterodimeric complexes with glycoprotein 130. J. Immunol. 1999, 163, 6651–6658. [Google Scholar]
- Böing, I.; Stross, C.; Radtke, S.; Lippok, B.E.; Heinrich, P.C.; Hermanns, H.M. Oncostatin M-induced activation of stress-activated MAP kinases depends on tyrosine 861 in the OSM receptor and requires Jak1 but not Src kinases. Cell. Signal. 2006, 18, 50–61. [Google Scholar] [CrossRef]
- David, C.; Smyth, C.K.; Richards, D.C. Oncostatin M-Induced IL-6 Expression in Murine Fibroblasts Requires the Activation of Protein Kinase Cδ. J. Immunol. 2006, 177, 8740–8747. [Google Scholar] [CrossRef] [Green Version]
- Smyth, D.C.; Takenaka, S.; Yeung, C.; Richards, C.D. Oncostatin M regulates osteogenic differentiation of murine adipose-derived mesenchymal progenitor cells through a PKCdelta-dependent mechanism. Cell Tissue Res. 2015, 360, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Schnittker, D.; Kwofie, K.; Ashkar, A.; Trigatti, B.; Richards, C.D. Oncostatin M and TLR-4 ligand synergize to induce MCP-1, IL-6, and VEGF in human aortic adventitial fibroblasts and smooth muscle cells. Mediat. Inflamm. 2013, 2013, 317503. [Google Scholar] [CrossRef]
- Li, C.; Ahlborn, T.E.; Kraemer, F.B.; Liu, J. Oncostatin M-induced growth inhibition and morphological changes of MDA-MB231 breast cancer cells are abolished by blocking the MEK/ERK signaling pathway. Breast Cancer Res. Treat. 2001, 66, 111–121. [Google Scholar] [CrossRef] [PubMed]
- David, E.; Guihard, P.; Brounais, B.; Riet, A.; Charrier, C.; Battaglia, S.; Gouin, F.; Ponsolle, S.; Le Bot, R.; Richards, C.D.; et al. Direct anti-cancer effect of oncostatin M on chondrosarcoma. Int. J. Cancer 2011, 128, 1822–1835. [Google Scholar] [CrossRef]
- Wang, M.-L.; Pan, C.-M.; Chiou, S.-H.; Chen, W.-H.; Chang, H.-Y.; Lee, O.K.-S.; Hsu, H.-S.; Wu, C.-W. Oncostatin m modulates the mesenchymal-epithelial transition of lung adenocarcinoma cells by a mesenchymal stem cell-mediated paracrine effect. Cancer Res. 2012, 72, 6051–6064. [Google Scholar] [CrossRef] [Green Version]
- Friedrich, M.; Höss, N.; Stögbauer, F.; Senner, V.; Paulus, W.; Ringelstein, E.B.; Halfter, H. Complete inhibition of in vivo glioma growth by oncostatin M. J. Neurochem. 2001, 76, 1589–1592. [Google Scholar] [CrossRef] [Green Version]
- Mori, S.; Murakami-Mori, K.; Bonavida, B. Oncostatin M (OM) promotes the growth of DU 145 human prostate cancer cells, but not PC-3 or LNCaP, through the signaling of the OM specific receptor. Anticancer Res. 1999, 19, 1011–1015. [Google Scholar]
- Godoy-Tundidor, S.; Cavarretta, I.T.R.; Fuchs, D.; Fiechtl, M.; Steiner, H.; Friedbichler, K.; Bartsch, G.; Hobisch, A.; Culig, Z. Interleukin-6 and oncostatin M stimulation of proliferation of prostate cancer 22Rv1 cells through the signaling pathways of p38 mitogen-activated protein kinase and phosphatidylinositol 3-kinase. Prostate 2005, 64, 209–216. [Google Scholar] [CrossRef]
- Lee, M.J.; Heo, S.C.; Shin, S.H.; Kwon, Y.W.; Do, E.K.; Suh, D.-S.; Yoon, M.-S.; Kim, J.H. Oncostatin M promotes mesenchymal stem cell-stimulated tumor growth through a paracrine mechanism involving periostin and TGFBI. Int. J. Biochem. Cell Biol. 2013, 45, 1869–1877. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhu, J.; Sun, F.; Liu, L.; Liu, X.; Yue, Y. Oncostatin M promotes proliferation of ovarian cancer cells through signal transducer and activator of transcription 3. Int. J. Mol. Med. 2011, 28, 101–108. [Google Scholar] [CrossRef] [PubMed]
- David, E.; Tirode, F.; Baud’huin, M.; Guihard, P.; Laud, K.; Delattre, O.; Heymann, M.F.; Heymann, D.; Redini, F.; Blanchard, F. Oncostatin M is a growth factor for Ewing sarcoma. Am. J. Pathol. 2012, 181, 1782–1795. [Google Scholar] [CrossRef]
- Wang, H.; Lei, L.; Hu, J.; Li, Y. Oncostatin M upregulates Livin to promote keratinocyte proliferation and survival via ERK and STAT3 signalling pathways. Exp. Physiol. 2020, 105, 1151–1158. [Google Scholar] [CrossRef]
- Schwaller, J.; Parganas, E.; Wang, D.; Cain, D.; Aster, J.C.; Williams, I.R.; Lee, C.-K.; Gerthner, R.; Kitamura, T.; Frantsve, J.; et al. Stat5 Is Essential for the Myelo- and Lymphoproliferative Disease Induced by TEL/JAK2. Mol. Cell 2000, 6, 693–704. [Google Scholar] [CrossRef]
- Müller, T.A.; Grundler, R.; Istvanffy, R.; Rudelius, M.; Hennighausen, L.; Illert, A.L.; Duyster, J. Lineage-specific STAT5 target gene activation in hematopoietic progenitor cells predicts the FLT3(+)-mediated leukemic phenotype. Leukemia 2016, 30, 1725–1733. [Google Scholar] [CrossRef] [PubMed]
- Bisht, K.; McGirr, C.; Lee, S.-Y.; Tseng, H.-W.; Fleming, W.; Alexander, K.A.; Matsumoto, T.; Barbier, V.; Sims, N.A.; Müller-Newen, G.; et al. Oncostatin M regulates hematopoietic stem cell (HSC) niches in the bone marrow to restrict HSC mobilization. Leukemia 2021. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Bian, S.; Zhou, X.; Cui, Y.; Wang, W.; Wen, L.; Guo, L.; Fu, W.; Tang, F. Single-Cell Multiomics Sequencing Reveals Prevalent Genomic Alterations in Tumor Stromal Cells of Human Colorectal Cancer. Cancer Cell 2020, 38, 818–828.e5. [Google Scholar] [CrossRef] [PubMed]
- Ren, G.; Zhao, X.; Wang, Y.; Zhang, X.; Chen, X.; Xu, C.; Yuan, Z.-R.; Roberts, A.I.; Zhang, L.; Zheng, B.; et al. CCR2-Dependent Recruitment of Macrophages by Tumor-Educated Mesenchymal Stromal Cells Promotes Tumor Development and Is Mimicked by TNFα. Cell Stem Cell 2012, 11, 812–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horn, D.; Fitzpatrick, W.C.; Gompper, P.T.; Ochs, V.; Bolton-Hansen, M.; Zarling, J.; Malik, N.; Todaro, G.J.; Linsley, P.S. Regulation of cell growth by recombinant oncostatin M. Growth Factors 1990, 2, 157–165. [Google Scholar] [CrossRef]
- Wang, E.C.E.; Dai, Z.; Ferrante, A.W.; Drake, C.G.; Christiano, A.M. A Subset of TREM2+ Dermal Macrophages Secretes Oncostatin M to Maintain Hair Follicle Stem Cell Quiescence and Inhibit Hair Growth. Cell Stem Cell 2019, 24, 654–669.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tvorogov, D.; Thomas, D.; Liau, N.P.D.; Dottore, M.; Barry, E.F.; Lathi, M.; Kan, W.L.; Hercus, T.R.; Stomski, F.; Hughes, T.P.; et al. Accumulation of JAK activation loop phosphorylation is linked to type I JAK inhibitor withdrawal syndrome in myelofibrosis. Sci. Adv. 2018, 4, eaat3834. [Google Scholar] [CrossRef] [Green Version]
- Su, C.-M.; Lee, W.-L.; Hsu, C.-J.; Lu, T.-T.; Wang, L.-H.; Xu, G.-H.; Tang, C.-H. Adiponectin Induces Oncostatin M Expression in Osteoblasts through the PI3K/Akt Signaling Pathway. Int. J. Mol. Sci. 2015, 17, 29. [Google Scholar] [CrossRef] [Green Version]
- West, N.R.; Murray, J.I.; Watson, P.H. Oncostatin-M promotes phenotypic changes associated with mesenchymal and stem cell-like differentiation in breast cancer. Oncogene 2014, 33, 1485–1494. [Google Scholar] [CrossRef]
- Brown, T.J.; Rowe, J.M.; Liu, J.W.; Shoyab, M. Regulation of IL-6 expression by oncostatin M. J. Immunol. 1991, 147, 2175–2180. [Google Scholar]
- Yanai, N.; Obinata, M. Oncostatin m regulates mesenchymal cell differentiation and enhances hematopoietic supportive activity of bone marrow stromal cell lines. In vitro cellular & developmental biology. Animal 2001, 37, 698–704. [Google Scholar] [CrossRef]
- Guo, L.; Chen, C.; Shi, M.; Wang, F.; Chen, X.; Diao, D.; Hu, M.; Yu, M.; Qian, L.; Guo, N. Stat3-coordinated Lin-28-let-7-HMGA2 and miR-200-ZEB1 circuits initiate and maintain oncostatin M-driven epithelial-mesenchymal transition. Oncogene 2013, 32, 5272–5282. [Google Scholar] [CrossRef] [PubMed]
- Gavin, E.; Jarvis, A.J. Thompson Evidence for an effect of receptor density on ligand occupancy and agonist EC 50. Sci. Rep. 2013, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Levy, R.S.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.; Miller, C.; Silver, R.T.; et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N. Engl. J. Med. 2012, 366, 799–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vannucchi, A.M.; Kiladjian, J.J.; Griesshammer, M.; Masszi, T.; Durrant, S.; Passamonti, F.; Harrison, C.N.; Pane, F.; Zachee, P.; Mesa, R.; et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N. Engl. J. Med. 2015, 372, 426–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeiser, R.; Bubnoff, N.; von Butler, J.; Mohty, M.; Niederwieser, D.; Or, R.; Szer, J.; Wagner, E.M.; Zuckerman, T.; Mahuzier, B.; et al. Ruxolitinib for Glucocorticoid-Refractory Acute Graft-versus-Host Disease. N. Engl. J. Med. 2020, 382, 1800–1810. [Google Scholar] [CrossRef]
- Harel, S.; Higgins, C.A.; Cerise, J.E.; Dai, Z.; Chen, J.C.; Clynes, R.; Christiano, A.M. Pharmacologic inhibition of JAK-STAT signaling promotes hair growth. Sci. Adv. 2015, 1, e1500973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araujo ED de Keserű, G.M.; Gunning, P.T.; Moriggl, R. Targeting STAT3 and STAT5 in Cancer. Cancers 2020, 12, 2002. [Google Scholar] [CrossRef]
- Walz, C.; Ahmed, W.; Lazarides, K.; Betancur, M.; Patel, N.; Hennighausen, L.; Zaleskas, V.M.; van Etten, R.A. Essential role for Stat5a/b in myeloproliferative neoplasms induced by BCR-ABL1 and JAK2(V617F) in mice. Blood 2012, 119, 3550–3560. [Google Scholar] [CrossRef]
- Swoboda, A.; Soukup, R.; Eckel, O.; Kinslechner, K.; Wingelhofer, B.; Schörghofer, D.; Sternberg, C.; Pham, H.T.T.; Vallianou, M.; Horvath, J.; et al. STAT3 promotes melanoma metastasis by CEBP-induced repression of the MITF pathway. Oncogene 2021, 40, 1091–1105. [Google Scholar] [CrossRef]
- Liu, X.; Robinson, G.W.; Wagner, K.U.; Garrett, L.; Wynshaw-Boris, A.; Hennighausen, L. Stat5a is mandatory for adult mammary gland development and lactogenesis. Genes Dev. 1997, 11, 179–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, Y.E.; Kitagawa, M.; Su, W.C.; You, Z.H.; Iwamoto, Y.; Fu, X.Y. Cell growth arrest and induction of cyclin-dependent kinase inhibitor p21 WAF1/CIP1 mediated by STAT1. Science 1996, 272, 719–722. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, J.F.; Horvath, C.M.; Wen, Z.; Schreiber, R.D.; Darnell, J.E. Transcriptionally active Stat1 is required for the antiproliferative effects of both interferon alpha and interferon gamma. Proc. Natl. Acad. Sci. USA 1996, 93, 7673–7678. [Google Scholar] [CrossRef] [Green Version]
- Dimberg, A.; Karlberg, I.; Nilsson, K.; Oberg, F. Ser727/Tyr701-phosphorylated Stat1 is required for the regulation of c-Myc, cyclins, and p27Kip1 associated with ATRA-induced G0/G1 arrest of U-937 cells. Blood 2003, 102, 254–261. [Google Scholar] [CrossRef]
- Nivarthi, H.; Gordziel, C.; Themanns, M.; Kramer, N.; Eberl, M.; Rabe, B.; Schlederer, M.; Rose-John, S.; Knösel, T.; Kenner, L.; et al. The ratio of STAT1 to STAT3 expression is a determinant of colorectal cancer growth. Oncotarget 2016, 7, 51096–51106. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.-K.; Raz, R.; Gimeno, R.; Gertner, R.; Wistinghausen, B.; Takeshita, K.; DePinho, R.A.; Levy, D.E. STAT3 Is a Negative Regulator of Granulopoiesis but Is Not Required for G-CSF-Dependent Differentiation. Immunity 2002, 17, 63–72. [Google Scholar] [CrossRef] [Green Version]
- Oostendorp, R.A.J.; Gilfillan, S.; Parmar, A.; Schiemann, M.; Marz, S.; Niemeyer, M.; Schill, S.; Hammerschmid, E.; Jacobs, V.R.; Peschel, C.; et al. Oncostatin M-mediated regulation of KIT-ligand-induced extracellular signal-regulated kinase signaling maintains hematopoietic repopulating activity of Lin-CD34+CD133+ cord blood cells. Stem Cells 2008, 26, 2164–2172. [Google Scholar] [CrossRef]
- Kim, H.; Jo, C.; Jang, B.G.; Oh, U.; Jo, S.A. Oncostatin M induces growth arrest of skeletal muscle cells in G1 phase by regulating cyclin D1 protein level. Cell. Signal. 2008, 20, 120–129. [Google Scholar] [CrossRef]
- Bult, C.J.; Blake, J.A.; Smith, C.L.; Kadin, J.A.; Richardson, J.E. Mouse Genome Database (MGD) 2019. Nucleic Acids Res. 2019, 47, D801–D806. [Google Scholar] [CrossRef] [Green Version]
- Masjedi, A.; Hajizadeh, F.; Beigi Dargani, F.; Beyzai, B.; Aksoun, M.; Hojjat-Farsangi, M.; Zekiy, A.; Jadidi-Niaragh, F. Oncostatin M: A mysterious cytokine in cancers. Int. Immunopharmacol. 2021, 90, 107158. [Google Scholar] [CrossRef] [PubMed]
- Pear, W.S.; Miller, J.P.; Xu, L.; Pui, J.C.; Soffer, B.; Quackenbush, R.C.; Pendergast, A.M.; Bronson, R.; Aster, J.C.; Scott, M.L.; et al. Efficient and rapid induction of a chronic myelogenous leukemia-like myeloproliferative disease in mice receiving P210 bcr/abl-transduced bone marrow. Blood 1998, 92, 3780–3792. [Google Scholar] [CrossRef]
- Klein, C.; Zwick, A.; Kissel, S.; Forster, C.U.; Pfeifer, D.; Follo, M.; Illert, A.L.; Decker, S.; Benkler, T.; Pahl, H.; et al. Ptch2 loss drives myeloproliferation and myeloproliferative neoplasm progression. J. Exp. Med. 2016, 213, 273–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renström, J.; Kröger, M.; Peschel, C.; Oostendorp, R.A.J. How the niche regulates hematopoietic stem cells. Chem. Biol. Interact. 2010, 184, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Grundler, R.; Thiede, C.; Miething, C.; Steudel, C.; Peschel, C.; Duyster, J. Sensitivity toward tyrosine kinase inhibitors varies between different activating mutations of the FLT3 receptor. Blood 2003, 102, 646–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jakob, L.; Müller, T.A.; Rassner, M.; Kleinfelder, H.; Veratti, P.; Mitschke, J.; Miething, C.; Oostendorp, R.A.J.; Pfeifer, D.; Waterhouse, M.; et al. Murine Oncostatin M Has Opposing Effects on the Proliferation of OP9 Bone Marrow Stromal Cells and NIH/3T3 Fibroblasts Signaling through the OSMR. Int. J. Mol. Sci. 2021, 22, 11649. https://doi.org/10.3390/ijms222111649
Jakob L, Müller TA, Rassner M, Kleinfelder H, Veratti P, Mitschke J, Miething C, Oostendorp RAJ, Pfeifer D, Waterhouse M, et al. Murine Oncostatin M Has Opposing Effects on the Proliferation of OP9 Bone Marrow Stromal Cells and NIH/3T3 Fibroblasts Signaling through the OSMR. International Journal of Molecular Sciences. 2021; 22(21):11649. https://doi.org/10.3390/ijms222111649
Chicago/Turabian StyleJakob, Lena, Tony Andreas Müller, Michael Rassner, Helen Kleinfelder, Pia Veratti, Jan Mitschke, Cornelius Miething, Robert A. J. Oostendorp, Dietmar Pfeifer, Miguel Waterhouse, and et al. 2021. "Murine Oncostatin M Has Opposing Effects on the Proliferation of OP9 Bone Marrow Stromal Cells and NIH/3T3 Fibroblasts Signaling through the OSMR" International Journal of Molecular Sciences 22, no. 21: 11649. https://doi.org/10.3390/ijms222111649
APA StyleJakob, L., Müller, T. A., Rassner, M., Kleinfelder, H., Veratti, P., Mitschke, J., Miething, C., Oostendorp, R. A. J., Pfeifer, D., Waterhouse, M., & Duyster, J. (2021). Murine Oncostatin M Has Opposing Effects on the Proliferation of OP9 Bone Marrow Stromal Cells and NIH/3T3 Fibroblasts Signaling through the OSMR. International Journal of Molecular Sciences, 22(21), 11649. https://doi.org/10.3390/ijms222111649