
Mechanistic Insight from Preclinical Models of Parkinson’s Disease Could Help Redirect Clinical Trial Efforts in GDNF Therapy

 ,
, 
 , and
, and 
Abstract
:1. Introduction
2. The Role of α-Synuclein in Parkinson’s Disease
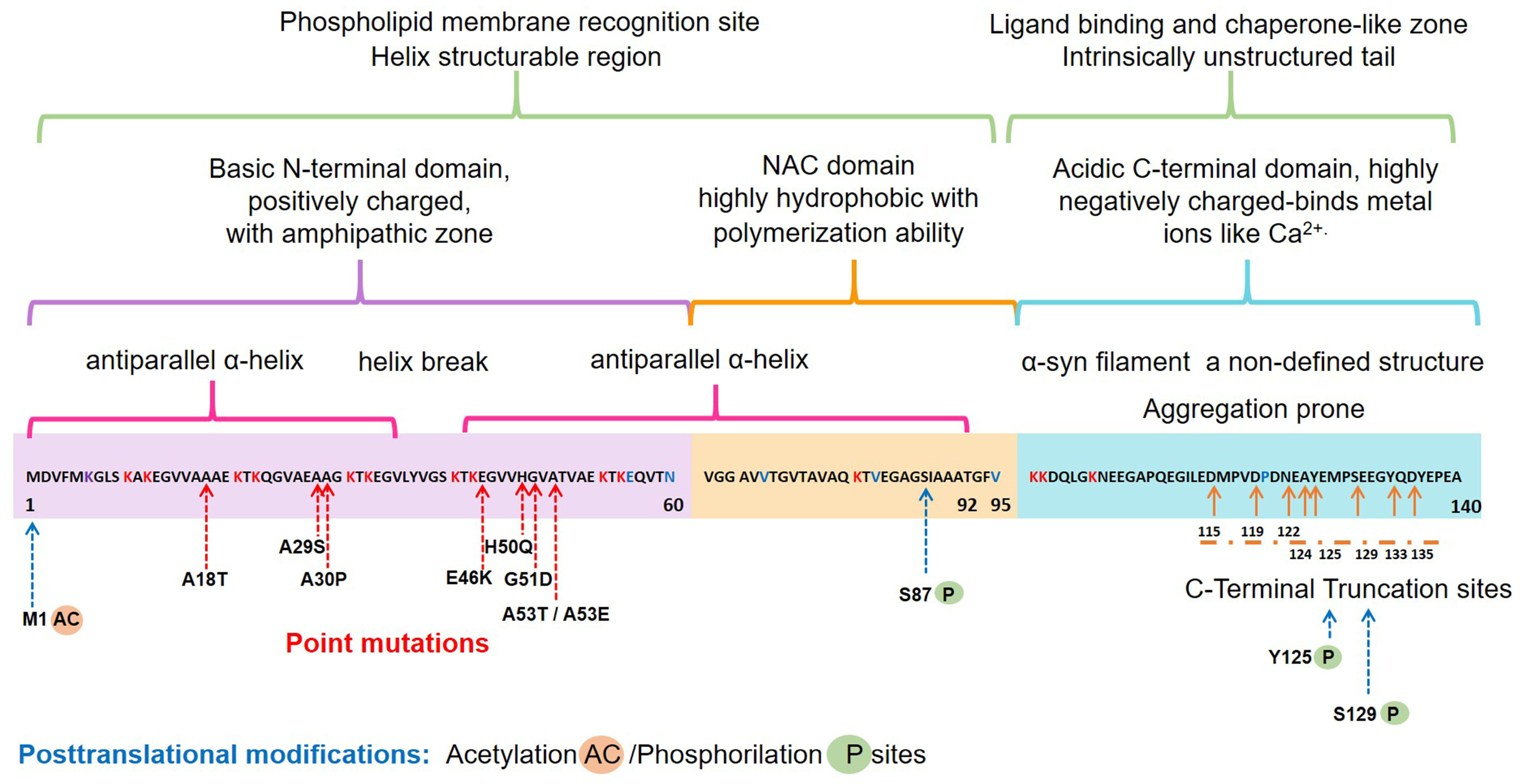
2.1. Structure and Physiology of α-Synuclein
2.2. Pathological α-Syn Aggregates and Prion-like Properties
3. The Misfolding of α-Synuclein and Its Association with Neuroinflammation in Parkinson’s Disease
4. Glial Cell-Derived Neurotrophic Factor in Parkinson’s Disease
4.1. Structure, Signaling Pathways, and Function
4.2. GDNF Alterations in Parkinson’s Disease
4.3. Neurotrophic Effects of GDNF in Preclinical Assays
4.4. Anti-α-Synuclein Effect of GDNF in Parkinson’s Disease Preclinical Trials
5. Anti-Inflammatory Effects of GDNF in Preclinical Assays
5.1. GDNF Anti-Inflammatory Effects in Neurodegenerative Disease Models
5.2. GDNF Anti-Inflammatory Effect in Other Neurological Disease Models
5.3. Anti-Inflammatory Effects of GDNF Therapy on Systemic Disease Models
6. Proof of Principle of GDNF in Clinical Trials for Parkinson’s Disease
7. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chmielarz, P.; Saarma, M. Neurotrophic factors for disease-modifying treatments of Parkinson’s disease: Gaps between basic science and clinical studies. Pharmacol. Rep. 2020, 72, 1195–1217. [Google Scholar] [CrossRef] [PubMed]
- Rocca, W.A. The burden of Parkinson’s disease: A worldwide perspective. Lancet Neurol. 2018, 17, 928–929. [Google Scholar] [CrossRef] [Green Version]
- Collaborators, G.B.D.P.s.D. Global, regional, and national burden of Parkinson’s disease, 1990-2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 939–953. [Google Scholar] [CrossRef] [Green Version]
- Parkinson, J. An essay on the shaking palsy. 1817. J. Neuropsychiatry Clin. Neurosci. 2002, 14, 223–236, discussion 222. [Google Scholar] [CrossRef]
- Ziemssen, T.; Reichmann, H. Non-motor dysfunction in Parkinson’s disease. Parkinsonism Relat. Disord. 2007, 13, 323–332. [Google Scholar] [CrossRef]
- Mahul-Mellier, A.L.; Burtscher, J.; Maharjan, N.; Weerens, L.; Croisier, M.; Kuttler, F.; Leleu, M.; Knott, G.W.; Lashuel, H.A. The process of Lewy body formation, rather than simply alpha-synuclein fibrillization, is one of the major drivers of neurodegeneration. Proc. Natl. Acad. Sci. USA 2020, 117, 4971–4982. [Google Scholar] [CrossRef] [Green Version]
- Goedert, M.; Jakes, R.; Spillantini, M.G. The Synucleinopathies: Twenty Years On. J. Parkinsons Dis. 2017, 7, S51–S69. [Google Scholar] [CrossRef] [Green Version]
- Sulzer, D.; Edwards, R.H. The physiological role of alpha-synuclein and its relationship to Parkinson’s Disease. J. Neurochem. 2019, 150, 475–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefanis, L. Alpha-Synuclein in Parkinson’s disease. Cold Spring Harb0 Perspect Med. 2012, 2, a009399. [Google Scholar] [CrossRef] [Green Version]
- Surguchov, A. Intracellular Dynamics of Synucleins: “Here, There and Everywhere”. Int. Rev. Cell. Mol. Biol. 2015, 320, 103–169. [Google Scholar] [CrossRef]
- Cascella, R.; Chen, S.W.; Bigi, A.; Camino, J.D.; Xu, C.K.; Dobson, C.M.; Chiti, F.; Cremades, N.; Cecchi, C. The release of toxic oligomers from alpha-synuclein fibrils induces dysfunction in neuronal cells. Nat. Commun. 2021, 12, 1814. [Google Scholar] [CrossRef]
- Lee, H.J.; Kim, C.; Lee, S.J. Alpha-synuclein stimulation of astrocytes: Potential role for neuroinflammation and neuroprotection. Oxid. Med. Cell Longev. 2010, 3, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Vivekanantham, S.; Shah, S.; Dewji, R.; Dewji, A.; Khatri, C.; Ologunde, R. Neuroinflammation in Parkinson’s disease: Role in neurodegeneration and tissue repair. Int. J. Neurosci. 2015, 125, 717–725. [Google Scholar] [CrossRef]
- Surguchev, A.A.; Emamzadeh, F.N.; Surguchov, A. Cell Responses to Extracellular alpha-Synuclein. Molecules 2019, 24, 305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, S.A.; Romero-Ramos, M. Microglia Response During Parkinson’s Disease: Alpha-Synuclein Intervention. Front. Cell. Neurosci. 2018, 12, 247. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.; Ho, D.H.; Suk, J.E.; You, S.; Michael, S.; Kang, J.; Joong Lee, S.; Masliah, E.; Hwang, D.; Lee, H.J.; et al. Neuron-released oligomeric alpha-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat. Commun. 2013, 4, 1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefanova, N.; Fellner, L.; Reindl, M.; Masliah, E.; Poewe, W.; Wenning, G.K. Toll-like receptor 4 promotes alpha-synuclein clearance and survival of nigral dopaminergic neurons. Am. J. Pathol. 2011, 179, 954–963. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Qin, L.; Wang, L.; Tan, J.; Zhang, H.; Tang, J.; Shen, X.; Tan, L.; Wang, C. alphasynuclein induces apoptosis of astrocytes by causing dysfunction of the endoplasmic reticulumGolgi compartment. Mol. Med. Rep. 2018, 18, 322–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, Y.; Kordower, J.H. GDNF signaling in subjects with minimal motor deficits and Parkinson’s disease. Neurobiol. Dis. 2021, 153, 105298. [Google Scholar] [CrossRef] [PubMed]
- Flores-Martinez, Y.M.; Fernandez-Parrilla, M.A.; Ayala-Davila, J.; Reyes-Corona, D.; Blanco-Alvarez, V.M.; Soto-Rojas, L.O.; Luna-Herrera, C.; Gonzalez-Barrios, J.A.; Leon-Chavez, B.A.; Gutierrez-Castillo, M.E.; et al. Acute Neuroinflammatory Response in the Substantia Nigra Pars Compacta of Rats after a Local Injection of Lipopolysaccharide. J. Immunol. Res. 2018, 2018, 1838921. [Google Scholar] [CrossRef]
- Gelders, G.; Baekelandt, V.; Van der Perren, A. Linking Neuroinflammation and Neurodegeneration in Parkinson’s Disease. J. Immunol. Res. 2018, 2018, 4784268. [Google Scholar] [CrossRef] [Green Version]
- Brochard, V.; Combadiere, B.; Prigent, A.; Laouar, Y.; Perrin, A.; Beray-Berthat, V.; Bonduelle, O.; Alvarez-Fischer, D.; Callebert, J.; Launay, J.M.; et al. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J. Clin. Investig. 2009, 119, 182–192. [Google Scholar] [CrossRef]
- Volpicelli-Daley, L.; Brundin, P. Prion-like propagation of pathology in Parkinson disease. Handb. Clin. Neurol. 2018, 153, 321–335. [Google Scholar] [CrossRef]
- Fahn, S.; Oakes, D.; Shoulson, I.; Kieburtz, K.; Rudolph, A.; Lang, A.; Olanow, C.W.; Tanner, C.; Marek, K.; Parkinson Study, G. Levodopa and the progression of Parkinson’s disease. N. Engl. J. Med. 2004, 351, 2498–2508. [Google Scholar] [CrossRef]
- Chmielarz, P.; Er, S.; Konovalova, J.; Bandres, L.; Hlushchuk, I.; Albert, K.; Panhelainen, A.; Luk, K.; Airavaara, M.; Domanskyi, A. GDNF/RET Signaling Pathway Activation Eliminates Lewy Body Pathology in Midbrain Dopamine Neurons. Mov. Disord. 2020, 35, 2279–2289. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Lin, Y.C.; Huang, C.Y.; Wu, S.R.; Chen, C.M.; Liu, H.L. Ultrasound-responsive neurotrophic factor-loaded microbubble- liposome complex: Preclinical investigation for Parkinson’s disease treatment. J. Control. Release 2020, 321, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Haney, M.J.; Jin, Y.S.; Uvarov, O.; Vinod, N.; Lee, Y.Z.; Langworthy, B.; Fine, J.P.; Rodriguez, M.; El-Hage, N.; et al. GDNF-expressing macrophages restore motor functions at a severe late-stage, and produce long-term neuroprotective effects at an early-stage of Parkinson’s disease in transgenic Parkin Q311X(A) mice. J. Control. Release 2019, 315, 139–149. [Google Scholar] [CrossRef]
- Hernando, S.; Herran, E.; Figueiro-Silva, J.; Pedraz, J.L.; Igartua, M.; Carro, E.; Hernandez, R.M. Intranasal Administration of TAT-Conjugated Lipid Nanocarriers Loading GDNF for Parkinson’s Disease. Mol. Neurobiol. 2018, 55, 145–155. [Google Scholar] [CrossRef]
- Zhao, Y.; Haney, M.J.; Gupta, R.; Bohnsack, J.P.; He, Z.; Kabanov, A.V.; Batrakova, E.V. GDNF-transfected macrophages produce potent neuroprotective effects in Parkinson’s disease mouse model. PLoS ONE 2014, 9, e106867. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Cui, G.; Yang, X.; Zhang, Z.; Shi, H.; Zu, J.; Hua, F.; Shen, X. Intracerebral administration of ultrasound-induced dissolution of lipid-coated GDNF microbubbles provides neuroprotection in a rat model of Parkinson’s disease. Brain Res. Bull. 2014, 103, 60–65. [Google Scholar] [CrossRef]
- Rickert, U.; Grampp, S.; Wilms, H.; Spreu, J.; Knerlich-Lukoschus, F.; Held-Feindt, J.; Lucius, R. Glial Cell Line-Derived Neurotrophic Factor Family Members Reduce Microglial Activation via Inhibiting p38MAPKs-Mediated Inflammatory Responses. J. Neurodegener. Dis. 2014, 2014, 369468. [Google Scholar] [CrossRef] [PubMed]
- Littrell, O.M.; Granholm, A.C.; Gerhardt, G.A.; Boger, H.A. Glial cell-line derived neurotrophic factor (GDNF) replacement attenuates motor impairments and nigrostriatal dopamine deficits in 12-month-old mice with a partial deletion of GDNF. Pharmacol. Biochem. Behav. 2013, 104, 10–19. [Google Scholar] [CrossRef] [Green Version]
- Rocha, S.M.; Cristovao, A.C.; Campos, F.L.; Fonseca, C.P.; Baltazar, G. Astrocyte-derived GDNF is a potent inhibitor of microglial activation. Neurobiol. Dis. 2012, 47, 407–415. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.F.; Doherty, D.H.; Lile, J.D.; Bektesh, S.; Collins, F. GDNF: A glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science 1993, 260, 1130–1132. [Google Scholar] [CrossRef]
- Barker, R.A.; Bjorklund, A.; Gash, D.M.; Whone, A.; Van Laar, A.; Kordower, J.H.; Bankiewicz, K.; Kieburtz, K.; Saarma, M.; Booms, S.; et al. GDNF and Parkinson’s Disease: Where Next? A Summary from a Recent Workshop. J. Parkinsons Dis. 2020, 10, 875–891. [Google Scholar] [CrossRef]
- Nutt, J.G.; Burchiel, K.J.; Comella, C.L.; Jankovic, J.; Lang, A.E.; Laws, E.R., Jr.; Lozano, A.M.; Penn, R.D.; Simpson, R.K., Jr.; Stacy, M.; et al. Randomized, double-blind trial of glial cell line-derived neurotrophic factor (GDNF) in PD. Neurology 2003, 60, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Burre, J.; Sharma, M.; Sudhof, T.C. Cell Biology and Pathophysiology of alpha-Synuclein. Cold Spring Harb. Perspect. Med. 2018, 8, a024091. [Google Scholar] [CrossRef] [PubMed]
- Mori, F.; Tanji, K.; Yoshimoto, M.; Takahashi, H.; Wakabayashi, K. Demonstration of alpha-synuclein immunoreactivity in neuronal and glial cytoplasm in normal human brain tissue using proteinase K and formic acid pretreatment. Exp. Neurol. 2002, 176, 98–104. [Google Scholar] [CrossRef]
- Richter-Landsberg, C.; Gorath, M.; Trojanowski, J.Q.; Lee, V.M. alpha-synuclein is developmentally expressed in cultured rat brain oligodendrocytes. J. Neurosci. Res. 2000, 62, 9–14. [Google Scholar] [CrossRef]
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The many faces of alpha-synuclein: From structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 2013, 14, 38–48. [Google Scholar] [CrossRef] [Green Version]
- Emamzadeh, F.N. Alpha-synuclein structure, functions, and interactions. J. Res. Med. Sci. 2016, 21, 29. [Google Scholar] [CrossRef]
- Meade, R.M.; Fairlie, D.P.; Mason, J.M. Alpha-synuclein structure and Parkinson’s disease—Lessons and emerging principles. Mol. Neurodegener. 2019, 14, 29. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, N.; Arcos-Lopez, T.; Konig, A.; Quintanar, L.; Menacho Marquez, M.; Outeiro, T.F.; Fernandez, C.O. Effects of alpha-synuclein post-translational modifications on metal binding. J. Neurochem. 2019, 150, 507–521. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Zhao, C.; Li, D.; Tian, Z.; Lai, Y.; Diao, J.; Liu, C. Versatile Structures of alpha-Synuclein. Front. Mol. Neurosci. 2016, 9, 48. [Google Scholar] [CrossRef]
- Twohig, D.; Nielsen, H.M. alpha-synuclein in the pathophysiology of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Runfola, M.; De Simone, A.; Vendruscolo, M.; Dobson, C.M.; Fusco, G. The N-terminal Acetylation of alpha-Synuclein Changes the Affinity for Lipid Membranes but not the Structural Properties of the Bound State. Sci. Rep. 2020, 10, 204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carta, A.R.; Boi, L.; Pisanu, A.; Palmas, M.F.; Carboni, E.; De Simone, A. Advances in modelling alpha-synuclein-induced Parkinson’s diseases in rodents: Virus-based models versus inoculation of exogenous preformed toxic species. J. Neurosci. Methods 2020, 338, 108685. [Google Scholar] [CrossRef]
- Li, X.; Dong, C.; Hoffmann, M.; Garen, C.R.; Cortez, L.M.; Petersen, N.O.; Woodside, M.T. Early stages of aggregation of engineered alpha-synuclein monomers and oligomers in solution. Sci. Rep. 2019, 9, 1734. [Google Scholar] [CrossRef] [PubMed]
- Killinger, B.A.; Melki, R.; Brundin, P.; Kordower, J.H. Endogenous alpha-synuclein monomers, oligomers and resulting pathology: Let’s talk about the lipids in the room. NPJ Parkinsons Dis. 2019, 5, 23. [Google Scholar] [CrossRef] [Green Version]
- Alam, P.; Bousset, L.; Melki, R.; Otzen, D.E. alpha-synuclein oligomers and fibrils: A spectrum of species, a spectrum of toxicities. J. Neurochem. 2019, 150, 522–534. [Google Scholar] [CrossRef] [Green Version]
- Bridi, J.C.; Hirth, F. Mechanisms of alpha-Synuclein Induced Synaptopathy in Parkinson’s Disease. Front. Neurosci. 2018, 12, 80. [Google Scholar] [CrossRef] [Green Version]
- Vinueza-Gavilanes, R.; Inigo-Marco, I.; Larrea, L.; Lasa, M.; Carte, B.; Santamaria, E.; Fernandez-Irigoyen, J.; Bugallo, R.; Aragon, T.; Aldabe, R.; et al. N-terminal acetylation mutants affect alpha-synuclein stability, protein levels and neuronal toxicity. Neurobiol. Dis. 2020, 137, 104781. [Google Scholar] [CrossRef]
- Ma, M.R.; Hu, Z.W.; Zhao, Y.F.; Chen, Y.X.; Li, Y.M. Phosphorylation induces distinct alpha-synuclein strain formation. Sci. Rep. 2016, 6, 37130. [Google Scholar] [CrossRef] [PubMed]
- Dettmer, U. Rationally Designed Variants of alpha-Synuclein Illuminate Its in vivo Structural Properties in Health and Disease. Front. Neurosci. 2018, 12, 623. [Google Scholar] [CrossRef]
- Villar-Pique, A.; Lopes da Fonseca, T.; Outeiro, T.F. Structure, function and toxicity of alpha-synuclein: The Bermuda triangle in synucleinopathies. J. Neurochem. 2016, 139 (Suppl. 1), 240–255. [Google Scholar] [CrossRef] [PubMed]
- Eastwood, T.A.; Baker, K.; Brooker, H.R.; Frank, S.; Mulvihill, D.P. An enhanced recombinant amino-terminal acetylation system and novel in vivo high-throughput screen for molecules affecting alpha-synuclein oligomerisation. FEBS Lett. 2017, 591, 833–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mavroeidi, P.; Xilouri, M. Neurons and Glia Interplay in alpha-Synucleinopathies. Int. J. Mol. Sci. 2021, 22, 4994. [Google Scholar] [CrossRef]
- Du, X.Y.; Xie, X.X.; Liu, R.T. The Role of alpha-Synuclein Oligomers in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 8645. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K.; Bratzke, H.; Hamm-Clement, J.; Sandmann-Keil, D.; Rub, U. Staging of the intracerebral inclusion body pathology associated with idiopathic Parkinson’s disease (preclinical and clinical stages). J. Neurol. 2002, 249 (Suppl. 3), iii1–iii5. [Google Scholar] [CrossRef]
- Kim, S.; Kwon, S.H.; Kam, T.I.; Panicker, N.; Karuppagounder, S.S.; Lee, S.; Lee, J.H.; Kim, W.R.; Kook, M.; Foss, C.A.; et al. Transneuronal Propagation of Pathologic alpha-Synuclein from the Gut to the Brain Models Parkinson’s Disease. Neuron 2019, 103, 627–641. [Google Scholar] [CrossRef]
- Xia, Y.; Zhang, G.; Han, C.; Ma, K.; Guo, X.; Wan, F.; Kou, L.; Yin, S.; Liu, L.; Huang, J.; et al. Microglia as modulators of exosomal alpha-synuclein transmission. Cell Death Dis. 2019, 10, 174. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, Z.A.; Vijayaraghavan, N.; Gorion, K.M.; Riffe, C.J.; Strang, K.H.; Caldwell, J.; Giasson, B.I. Physiological C-terminal truncation of alpha-synuclein potentiates the prion-like formation of pathological inclusions. J. Biol. Chem. 2018, 293, 18914–18932. [Google Scholar] [CrossRef] [Green Version]
- Visanji, N.P.; Brooks, P.L.; Hazrati, L.N.; Lang, A.E. The prion hypothesis in Parkinson’s disease: Braak to the future. Acta Neuropathol. Commun. 2013, 1, 2. [Google Scholar] [CrossRef] [Green Version]
- Heras-Garvin, A.; Stefanova, N. From Synaptic Protein to Prion: The Long and Controversial Journey of alpha-Synuclein. Front. Synaptic Neurosci. 2020, 12, 584536. [Google Scholar] [CrossRef]
- Izco, M.; Blesa, J.; Verona, G.; Cooper, J.M.; Alvarez-Erviti, L. Glial activation precedes alpha-synuclein pathology in a mouse model of Parkinson’s disease. Neurosci. Res. 2021, 170, 330–340. [Google Scholar] [CrossRef]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef] [Green Version]
- Badanjak, K.; Fixemer, S.; Smajic, S.; Skupin, A.; Grunewald, A. The Contribution of Microglia to Neuroinflammation in Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 4676. [Google Scholar] [CrossRef] [PubMed]
- Doorn, K.J.; Moors, T.; Drukarch, B.; van de Berg, W.; Lucassen, P.J.; van Dam, A.M. Microglial phenotypes and toll-like receptor 2 in the substantia nigra and hippocampus of incidental Lewy body disease cases and Parkinson’s disease patients. Acta Neuropathol. Commun. 2014, 2, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halliday, G.M.; Stevens, C.H. Glia: Initiators and progressors of pathology in Parkinson’s disease. Mov. Disord. 2011, 26, 6–17. [Google Scholar] [CrossRef]
- Marques, O.; Outeiro, T.F. Alpha-synuclein: From secretion to dysfunction and death. Cell Death Dis. 2012, 3, e350. [Google Scholar] [CrossRef]
- Lee, S.B.; Park, S.M.; Ahn, K.J.; Chung, K.C.; Paik, S.R.; Kim, J. Identification of the amino acid sequence motif of alpha-synuclein responsible for macrophage activation. Biochem. Biophys. Res. Commun. 2009, 381, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Maguire-Zeiss, K.A.; Giuliano, R.; Prifti, L.; Venkatesh, K.; Federoff, H.J. Synuclein activates microglia in a model of Parkinson’s disease. Neurobiol. Aging 2008, 29, 1690–1701. [Google Scholar] [CrossRef] [Green Version]
- Couch, Y.; Alvarez-Erviti, L.; Sibson, N.R.; Wood, M.J.; Anthony, D.C. The acute inflammatory response to intranigral alpha-synuclein differs significantly from intranigral lipopolysaccharide and is exacerbated by peripheral inflammation. J. Neuroinflammation 2011, 8, 166. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.J.; Suk, J.E.; Patrick, C.; Bae, E.J.; Cho, J.H.; Rho, S.; Hwang, D.; Masliah, E.; Lee, S.J. Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J. Biol. Chem. 2010, 285, 9262–9272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynolds, A.D.; Stone, D.K.; Mosley, R.L.; Gendelman, H.E. Nitrated {alpha}-synuclein-induced alterations in microglial immunity are regulated by CD4+ T cell subsets. J. Immunol. 2009, 182, 4137–4149. [Google Scholar] [CrossRef] [Green Version]
- Benner, E.J.; Banerjee, R.; Reynolds, A.D.; Sherman, S.; Pisarev, V.M.; Tsiperson, V.; Nemachek, C.; Ciborowski, P.; Przedborski, S.; Mosley, R.L.; et al. Nitrated alpha-synuclein immunity accelerates degeneration of nigral dopaminergic neurons. PLoS ONE 2008, 3, e1376. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S.R.; Federoff, H.J. Targeting Microglial Activation States as a Therapeutic Avenue in Parkinson’s Disease. Front. Aging Neurosci. 2017, 9, 176. [Google Scholar] [CrossRef]
- Kim, S.; Cho, S.H.; Kim, K.Y.; Shin, K.Y.; Kim, H.S.; Park, C.H.; Chang, K.A.; Lee, S.H.; Cho, D.; Suh, Y.H. Alpha-synuclein induces migration of BV-2 microglial cells by up-regulation of CD44 and MT1-MMP. J. Neurochem. 2009, 109, 1483–1496. [Google Scholar] [CrossRef]
- Wang, S.; Chu, C.H.; Stewart, T.; Ginghina, C.; Wang, Y.; Nie, H.; Guo, M.; Wilson, B.; Hong, J.S.; Zhang, J. alpha-Synuclein, a chemoattractant, directs microglial migration via H2O2-dependent Lyn phosphorylation. Proc. Natl. Acad. Sci. USA 2015, 112, E1926–E1935. [Google Scholar] [CrossRef] [Green Version]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef] [Green Version]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [Green Version]
- Gordon, R.; Albornoz, E.A.; Christie, D.C.; Langley, M.R.; Kumar, V.; Mantovani, S.; Robertson, A.A.B.; Butler, M.S.; Rowe, D.B.; O’Neill, L.A.; et al. Inflammasome inhibition prevents alpha-synuclein pathology and dopaminergic neurodegeneration in mice. Sci. Transl. Med. 2018, 10, aah4066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pike, A.F.; Varanita, T.; Herrebout, M.A.C.; Plug, B.C.; Kole, J.; Musters, R.J.P.; Teunissen, C.E.; Hoozemans, J.J.M.; Bubacco, L.; Veerhuis, R. alpha-Synuclein evokes NLRP3 inflammasome-mediated IL-1beta secretion from primary human microglia. Glia 2021, 69, 1413–1428. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, J.; Wu, H.; Kagan, J.C. Gasdermin D activity in inflammation and host defense. Sci. Immunol. 2019, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, S.B.; Miao, E.A. Gasdermins: Effectors of Pyroptosis. Trends Cell Biol. 2017, 27, 673–684. [Google Scholar] [CrossRef]
- Wang, X.; Chi, J.; Huang, D.; Ding, L.; Zhao, X.; Jiang, L.; Yu, Y.; Gao, F. alpha-synuclein promotes progression of Parkinson’s disease by upregulating autophagy signaling pathway to activate NLRP3 inflammasome. Exp. Ther. Med. 2020, 19, 931–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dzamko, N.; Gysbers, A.; Perera, G.; Bahar, A.; Shankar, A.; Gao, J.; Fu, Y.; Halliday, G.M. Toll-like receptor 2 is increased in neurons in Parkinson’s disease brain and may contribute to alpha-synuclein pathology. Acta Neuropathol. 2017, 133, 303–319. [Google Scholar] [CrossRef] [Green Version]
- Rannikko, E.H.; Weber, S.S.; Kahle, P.J. Exogenous alpha-synuclein induces toll-like receptor 4 dependent inflammatory responses in astrocytes. BMC Neurosci. 2015, 16, 57. [Google Scholar] [CrossRef] [Green Version]
- Fellner, L.; Irschick, R.; Schanda, K.; Reindl, M.; Klimaschewski, L.; Poewe, W.; Wenning, G.K.; Stefanova, N. Toll-like receptor 4 is required for alpha-synuclein dependent activation of microglia and astroglia. Glia 2013, 61, 349–360. [Google Scholar] [CrossRef] [Green Version]
- Chavarria, C.; Rodriguez-Bottero, S.; Quijano, C.; Cassina, P.; Souza, J.M. Impact of monomeric, oligomeric and fibrillar alpha-synuclein on astrocyte reactivity and toxicity to neurons. Biochem. J. 2018, 475, 3153–3169. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Standaert, D.G.; Harms, A.S. The gamma chain subunit of Fc receptors is required for alpha-synuclein-induced pro-inflammatory signaling in microglia. J. Neuroinflamm. 2012, 9, 259. [Google Scholar] [CrossRef] [Green Version]
- Cao, S.; Theodore, S.; Standaert, D.G. Fcgamma receptors are required for NF-kappaB signaling, microglial activation and dopaminergic neurodegeneration in an AAV-synuclein mouse model of Parkinson’s disease. Mol. Neurodegener. 2010, 5, 42. [Google Scholar] [CrossRef] [Green Version]
- Hou, L.; Bao, X.; Zang, C.; Yang, H.; Sun, F.; Che, Y.; Wu, X.; Li, S.; Zhang, D.; Wang, Q. Integrin CD11b mediates alpha-synuclein-induced activation of NADPH oxidase through a Rho-dependent pathway. Redox Biol. 2018, 14, 600–608. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, T.; Pei, Z.; Miller, D.S.; Wu, X.; Block, M.L.; Wilson, B.; Zhang, W.; Zhou, Y.; Hong, J.S.; et al. Aggregated alpha-synuclein activates microglia: A process leading to disease progression in Parkinson’s disease. FASEB J. 2005, 19, 533–542. [Google Scholar] [CrossRef]
- Jin, J.; Shie, F.S.; Liu, J.; Wang, Y.; Davis, J.; Schantz, A.M.; Montine, K.S.; Montine, T.J.; Zhang, J. Prostaglandin E2 receptor subtype 2 (EP2) regulates microglial activation and associated neurotoxicity induced by aggregated alpha-synuclein. J. Neuroinflammation 2007, 4, 2. [Google Scholar] [CrossRef] [PubMed]
- Johansson, J.U.; Woodling, N.S.; Wang, Q.; Panchal, M.; Liang, X.; Trueba-Saiz, A.; Brown, H.D.; Mhatre, S.D.; Loui, T.; Andreasson, K.I. Prostaglandin signaling suppresses beneficial microglial function in Alzheimer’s disease models. J. Clin. Investig. 2015, 125, 350–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, T.; Hoekstra, J.; Heng, X.; Kang, W.; Ding, J.; Liu, J.; Chen, S.; Zhang, J. P2X7 receptor is critical in alpha-synuclein–mediated microglial NADPH oxidase activation. Neurobiol. Aging 2015, 36, 2304–2318. [Google Scholar] [CrossRef]
- Russ, K.; Teku, G.; Bousset, L.; Redeker, V.; Piel, S.; Savchenko, E.; Pomeshchik, Y.; Savistchenko, J.; Stummann, T.C.; Azevedo, C.; et al. TNF-alpha and alpha-synuclein fibrils differently regulate human astrocyte immune reactivity and impair mitochondrial respiration. Cell Rep. 2021, 34, 108895. [Google Scholar] [CrossRef]
- Lindstrom, V.; Gustafsson, G.; Sanders, L.H.; Howlett, E.H.; Sigvardson, J.; Kasrayan, A.; Ingelsson, M.; Bergstrom, J.; Erlandsson, A. Extensive uptake of alpha-synuclein oligomers in astrocytes results in sustained intracellular deposits and mitochondrial damage. Mol. Cell. Neurosci. 2017, 82, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Dohgu, S.; Takata, F.; Matsumoto, J.; Kimura, I.; Yamauchi, A.; Kataoka, Y. Monomeric alpha-synuclein induces blood-brain barrier dysfunction through activated brain pericytes releasing inflammatory mediators in vitro. Microvasc. Res. 2019, 124, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Sommer, A.; Marxreiter, F.; Krach, F.; Fadler, T.; Grosch, J.; Maroni, M.; Graef, D.; Eberhardt, E.; Riemenschneider, M.J.; Yeo, G.W.; et al. Th17 Lymphocytes Induce Neuronal Cell Death in a Human iPSC-Based Model of Parkinson’s Disease. Cell Stem Cell 2018, 23, 123–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subbarayan, M.S.; Hudson, C.; Moss, L.D.; Nash, K.R.; Bickford, P.C. T cell infiltration and upregulation of MHCII in microglia leads to accelerated neuronal loss in an alpha-synuclein rat model of Parkinson’s disease. J. Neuroinflamm. 2020, 17, 242. [Google Scholar] [CrossRef] [PubMed]
- Kustrimovic, N.; Comi, C.; Magistrelli, L.; Rasini, E.; Legnaro, M.; Bombelli, R.; Aleksic, I.; Blandini, F.; Minafra, B.; Riboldazzi, G.; et al. Parkinson’s disease patients have a complex phenotypic and functional Th1 bias: Cross-sectional studies of CD4+ Th1/Th2/T17 and Treg in drug-naive and drug-treated patients. J. Neuroinflamm. 2018, 15, 205. [Google Scholar] [CrossRef]
- Kotzbauer, P.T.; Lampe, P.A.; Heuckeroth, R.O.; Golden, J.P.; Creedon, D.J.; Johnson, E.M., Jr.; Milbrandt, J. Neurturin, a relative of glial-cell-line-derived neurotrophic factor. Nature 1996, 384, 467–470. [Google Scholar] [CrossRef] [PubMed]
- Cintron-Colon, A.F.; Almeida-Alves, G.; Boynton, A.M.; Spitsbergen, J.M. GDNF synthesis, signaling, and retrograde transport in motor neurons. Cell Tissue Res. 2020, 382, 47–56. [Google Scholar] [CrossRef]
- Parkash, V.; Leppanen, V.M.; Virtanen, H.; Jurvansuu, J.M.; Bespalov, M.M.; Sidorova, Y.A.; Runeberg-Roos, P.; Saarma, M.; Goldman, A. The structure of the glial cell line-derived neurotrophic factor-coreceptor complex: Insights into RET signaling and heparin binding. J. Biol. Chem. 2008, 283, 35164–35172. [Google Scholar] [CrossRef] [Green Version]
- Airaksinen, M.S.; Saarma, M. The GDNF family: Signalling, biological functions and therapeutic value. Nat. Rev. Neurosci. 2002, 3, 383–394. [Google Scholar] [CrossRef]
- Paratcha, G.; Ledda, F.; Ibanez, C.F. The neural cell adhesion molecule NCAM is an alternative signaling receptor for GDNF family ligands. Cell 2003, 113, 867–879. [Google Scholar] [CrossRef] [Green Version]
- Kawai, K.; Takahashi, M. Intracellular RET signaling pathways activated by GDNF. Cell Tissue Res. 2020, 382, 113–123. [Google Scholar] [CrossRef]
- Coulpier, M.; Anders, J.; Ibanez, C.F. Coordinated activation of autophosphorylation sites in the RET receptor tyrosine kinase: Importance of tyrosine 1062 for GDNF mediated neuronal differentiation and survival. J. Biol. Chem. 2002, 277, 1991–1999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bespalov, M.M.; Sidorova, Y.A.; Tumova, S.; Ahonen-Bishopp, A.; Magalhaes, A.C.; Kulesskiy, E.; Paveliev, M.; Rivera, C.; Rauvala, H.; Saarma, M. Heparan sulfate proteoglycan syndecan-3 is a novel receptor for GDNF, neurturin, and artemin. J. Cell Biol. 2011, 192, 153–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The GTEx Portal. Available online: https://gtexportal.org/home/gene/GDNF (accessed on 29 August 2021).
- Hellmich, H.L.; Kos, L.; Cho, E.S.; Mahon, K.A.; Zimmer, A. Embryonic expression of glial cell-line derived neurotrophic factor (GDNF) suggests multiple developmental roles in neural differentiation and epithelial-mesenchymal interactions. Mech. Dev. 1996, 54, 95–105. [Google Scholar] [CrossRef]
- The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000168621-GDNF/tissue (accessed on 29 August 2021).
- Mogi, M.; Togari, A.; Kondo, T.; Mizuno, Y.; Kogure, O.; Kuno, S.; Ichinose, H.; Nagatsu, T. Glial cell line-derived neurotrophic factor in the substantia nigra from control and parkinsonian brains. Neurosci. Lett. 2001, 300, 179–181. [Google Scholar] [CrossRef]
- Michos, O.; Cebrian, C.; Hyink, D.; Grieshammer, U.; Williams, L.; D’Agati, V.; Licht, J.D.; Martin, G.R.; Costantini, F. Kidney development in the absence of Gdnf and Spry1 requires Fgf10. PLoS Genet 2010, 6, e1000809. [Google Scholar] [CrossRef] [Green Version]
- Gianino, S.; Grider, J.R.; Cresswell, J.; Enomoto, H.; Heuckeroth, R.O. GDNF availability determines enteric neuron number by controlling precursor proliferation. Development 2003, 130, 2187–2198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enomoto, H.; Heuckeroth, R.O.; Golden, J.P.; Johnson, E.M.; Milbrandt, J. Development of cranial parasympathetic ganglia requires sequential actions of GDNF and neurturin. Development 2000, 127, 4877–4889. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, N.B.; Siegel, G.J.; Lee, J.M. Depletion of glial cell line-derived neurotrophic factor in substantia nigra neurons of Parkinson’s disease brain. J. Chem. Neuroanat. 2001, 21, 277–288. [Google Scholar] [CrossRef]
- Virachit, S.; Mathews, K.J.; Cottam, V.; Werry, E.; Galli, E.; Rappou, E.; Lindholm, P.; Saarma, M.; Halliday, G.M.; Shannon Weickert, C.; et al. Levels of glial cell line-derived neurotrophic factor are decreased, but fibroblast growth factor 2 and cerebral dopamine neurotrophic factor are increased in the hippocampus in Parkinson’s disease. Brain Pathol. 2019, 29, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Griego, E.; Herrera-Lopez, G.; Gomez-Lira, G.; Barrionuevo, G.; Gutierrez, R.; Galvan, E.J. Functional expression of TrkB receptors on interneurones and pyramidal cells of area CA3 of the rat hippocampus. Neuropharmacology 2021, 182, 108379. [Google Scholar] [CrossRef]
- Backman, C.M.; Shan, L.; Zhang, Y.J.; Hoffer, B.J.; Leonard, S.; Troncoso, J.C.; Vonsatel, P.; Tomac, A.C. Gene expression patterns for GDNF and its receptors in the human putamen affected by Parkinson’s disease: A real-time PCR study. Mol. Cell. Endocrinol. 2006, 252, 160–166. [Google Scholar] [CrossRef]
- Su, X.; Fischer, D.L.; Li, X.; Bankiewicz, K.; Sortwell, C.E.; Federoff, H.J. Alpha-Synuclein mRNA Is Not Increased in Sporadic PD and Alpha-Synuclein Accumulation Does Not Block GDNF Signaling in Parkinson’s Disease and Disease Models. Mol. Ther. 2017, 25, 2231–2235. [Google Scholar] [CrossRef] [Green Version]
- Migliore, M.M.; Ortiz, R.; Dye, S.; Campbell, R.B.; Amiji, M.M.; Waszczak, B.L. Neurotrophic and neuroprotective efficacy of intranasal GDNF in a rat model of Parkinson’s disease. Neuroscience 2014, 274, 11–23. [Google Scholar] [CrossRef]
- Kordower, J.H.; Bjorklund, A. Trophic factor gene therapy for Parkinson’s disease. Mov. Disord. 2013, 28, 96–109. [Google Scholar] [CrossRef]
- Sullivan, A.M.; Toulouse, A. Neurotrophic factors for the treatment of Parkinson’s disease. Cytokine Growth Factor Rev. 2011, 22, 157–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, F.; Luan, L.; Ai, Y.; Walton, A.; Gerhardt, G.A.; Gash, D.M.; Grondin, R.; Zhang, Z. Development of a stable, early stage unilateral model of Parkinson’s disease in middle-aged rhesus monkeys. Exp. Neurol. 2008, 212, 431–439. [Google Scholar] [CrossRef] [Green Version]
- Grondin, R.; Zhang, Z.; Yi, A.; Cass, W.A.; Maswood, N.; Andersen, A.H.; Elsberry, D.D.; Klein, M.C.; Gerhardt, G.A.; Gash, D.M. Chronic, controlled GDNF infusion promotes structural and functional recovery in advanced parkinsonian monkeys. Brain 2002, 125, 2191–2201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gash, D.M.; Zhang, Z.; Ovadia, A.; Cass, W.A.; Yi, A.; Simmerman, L.; Russell, D.; Martin, D.; Lapchak, P.A.; Collins, F.; et al. Functional recovery in parkinsonian monkeys treated with GDNF. Nature 1996, 380, 252–255. [Google Scholar] [CrossRef]
- Hirsch, E.C. Animal models in neurodegenerative diseases. J. Neural. Transm. Suppl. 2007, 72, 87–90. [Google Scholar] [CrossRef] [Green Version]
- Luz, M.; Whone, A.; Bassani, N.; Wyse, R.K.; Stebbins, G.T.; Mohr, E. The Parkinson’s Disease Comprehensive Response (PDCORE): A composite approach integrating three standard outcome measures. Brain Commun. 2020, 2, fcaa046. [Google Scholar] [CrossRef]
- Sidorova, Y.A.; Saarma, M. Can Growth Factors Cure Parkinson’s Disease? Trends Pharmacol. Sci. 2020, 41, 909–922. [Google Scholar] [CrossRef]
- Huttunen, H.J.; Saarma, M. CDNF Protein Therapy in Parkinson’s Disease. Cell Transplant. 2019, 28, 349–366. [Google Scholar] [CrossRef] [Green Version]
- Whone, A.; Luz, M.; Boca, M.; Woolley, M.; Mooney, L.; Dharia, S.; Broadfoot, J.; Cronin, D.; Schroers, C.; Barua, N.U.; et al. Randomized trial of intermittent intraputamenal glial cell line-derived neurotrophic factor in Parkinson’s disease. Brain 2019, 142, 512–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidorova, Y.A.; Volcho, K.P.; Salakhutdinov, N.F. Neuroregeneration in Parkinson’s Disease: From Proteins to Small Molecules. Curr. Neuropharmacol. 2019, 17, 268–287. [Google Scholar] [CrossRef]
- Allen, S.J.; Watson, J.J.; Shoemark, D.K.; Barua, N.U.; Patel, N.K. GDNF, NGF and BDNF as therapeutic options for neurodegeneration. Pharmacol. Ther. 2013, 138, 155–175. [Google Scholar] [CrossRef]
- Salvatore, M.F.; Ai, Y.; Fischer, B.; Zhang, A.M.; Grondin, R.C.; Zhang, Z.; Gerhardt, G.A.; Gash, D.M. Point source concentration of GDNF may explain failure of phase II clinical trial. Exp. Neurol. 2006, 202, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Kearns, C.M.; Cass, W.A.; Smoot, K.; Kryscio, R.; Gash, D.M. GDNF protection against 6-OHDA: Time dependence and requirement for protein synthesis. J. Neurosci. 1997, 17, 7111–7118. [Google Scholar] [CrossRef]
- Kirik, D.; Rosenblad, C.; Bjorklund, A. Preservation of a functional nigrostriatal dopamine pathway by GDNF in the intrastriatal 6-OHDA lesion model depends on the site of administration of the trophic factor. Eur. J. Neurosci. 2000, 12, 3871–3882. [Google Scholar] [CrossRef]
- Blum, D.; Torch, S.; Lambeng, N.; Nissou, M.; Benabid, A.L.; Sadoul, R.; Verna, J.M. Molecular pathways involved in the neurotoxicity of 6-OHDA, dopamine and MPTP: Contribution to the apoptotic theory in Parkinson’s disease. Prog. Neurobiol. 2001, 65, 135–172. [Google Scholar] [CrossRef]
- Van Kampen, J.M.; Robertson, H.A. The BSSG rat model of Parkinson’s disease: Progressing towards a valid, predictive model of disease. EPMA J. 2017, 8, 261–271. [Google Scholar] [CrossRef] [Green Version]
- Soto-Rojas, L.O.; Garces-Ramirez, L.; Luna-Herrera, C.; Flores-Martinez, Y.M.; Soto-Rodriguez, G.; Gatica-Garcia, B.; Lopez-Salas, F.E.; Ayala-Davila, J.; Gutierrez-Castillo, M.E.; Padilla-Viveros, A.; et al. A single intranigral administration of beta-sitosterol beta-d-glucoside elicits bilateral sensorimotor and non-motor alterations in the rat. Behav. Brain Res. 2020, 378, 112279. [Google Scholar] [CrossRef] [PubMed]
- Soto-Rojas, L.O.; Martinez-Davila, I.A.; Luna-Herrera, C.; Gutierrez-Castillo, M.E.; Lopez-Salas, F.E.; Gatica-Garcia, B.; Soto-Rodriguez, G.; Bringas Tobon, M.E.; Flores, G.; Padilla-Viveros, A.; et al. Unilateral intranigral administration of beta-sitosterol beta-D-glucoside triggers pathological alpha-synuclein spreading and bilateral nigrostriatal dopaminergic neurodegeneration in the rat. Acta Neuropathol. Commun. 2020, 8, 56. [Google Scholar] [CrossRef]
- Luna-Herrera, C.; Martinez-Davila, I.A.; Soto-Rojas, L.O.; Flores-Martinez, Y.M.; Fernandez-Parrilla, M.A.; Ayala-Davila, J.; Leon-Chavez, B.A.; Soto-Rodriguez, G.; Blanco-Alvarez, V.M.; Lopez-Salas, F.E.; et al. Intranigral Administration of beta-Sitosterol-beta-D-Glucoside Elicits Neurotoxic A1 Astrocyte Reactivity and Chronic Neuroinflammation in the Rat Substantia Nigra. J. Immunol. Res. 2020, 2020, 5907591. [Google Scholar] [CrossRef]
- Gash, D.M.; Gerhardt, G.A.; Bradley, L.H.; Wagner, R.; Slevin, J.T. GDNF clinical trials for Parkinson’s disease: A critical human dimension. Cell Tissue Res. 2020, 382, 65–70. [Google Scholar] [CrossRef]
- Yasuhara, T.; Kameda, M.; Agari, T.; Date, I. Regenerative medicine for Parkinson’s disease. Neurol. Med. Chir. 2015, 55, 113–123. [Google Scholar] [CrossRef] [Green Version]
- Lapchak, P.A.; Jiao, S.; Collins, F.; Miller, P.J. Glial cell line-derived neurotrophic factor: Distribution and pharmacology in the rat following a bolus intraventricular injection. Brain Res. 1997, 747, 92–102. [Google Scholar] [CrossRef]
- Martin, D.; Miller, G.; Fischer, N.; Diz, D.; Cullen, T.; Russell, D. Glial cell line-derived neurotrophic factor: The lateral cerebral ventricle as a site of administration for stimulation of the substantia nigra dopamine system in rats. Eur. J. Neurosci. 1996, 8, 1249–1255. [Google Scholar] [CrossRef] [PubMed]
- Bowenkamp, K.E.; Lapchak, P.A.; Hoffer, B.J.; Miller, P.J.; Bickford, P.C. Intracerebroventricular glial cell line-derived neurotrophic factor improves motor function and supports nigrostriatal dopamine neurons in bilaterally 6-hydroxydopamine lesioned rats. Exp. Neurol. 1997, 145, 104–117. [Google Scholar] [CrossRef]
- Sullivan, A.M.; Opacka-Juffry, J.; Blunt, S.B. Long-term protection of the rat nigrostriatal dopaminergic system by glial cell line-derived neurotrophic factor against 6-hydroxydopamine in vivo. Eur. J. Neurosci. 1998, 10, 57–63. [Google Scholar] [CrossRef]
- Winkler, C.; Sauer, H.; Lee, C.S.; Bjorklund, A. Short-term GDNF treatment provides long-term rescue of lesioned nigral dopaminergic neurons in a rat model of Parkinson’s disease. J. Neurosci. 1996, 16, 7206–7215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Barrios, J.A.; Lindahl, M.; Bannon, M.J.; Anaya-Martinez, V.; Flores, G.; Navarro-Quiroga, I.; Trudeau, L.E.; Aceves, J.; Martinez-Arguelles, D.B.; Garcia-Villegas, R.; et al. Neurotensin polyplex as an efficient carrier for delivering the human GDNF gene into nigral dopamine neurons of hemiparkinsonian rats. Mol. Ther. 2006, 14, 857–865. [Google Scholar] [CrossRef] [PubMed]
- Lapchak, P.A.; Miller, P.J.; Collins, F.; Jiao, S. Glial cell line-derived neurotrophic factor attenuates behavioural deficits and regulates nigrostriatal dopaminergic and peptidergic markers in 6-hydroxydopamine-lesioned adult rats: Comparison of intraventricular and intranigral delivery. Neuroscience 1997, 78, 61–72. [Google Scholar] [CrossRef]
- Kirik, D.; Georgievska, B.; Rosenblad, C.; Bjorklund, A. Delayed infusion of GDNF promotes recovery of motor function in the partial lesion model of Parkinson’s disease. Eur. J. Neurosci. 2001, 13, 1589–1599. [Google Scholar] [CrossRef] [PubMed]
- Garbayo, E.; Montero-Menei, C.N.; Ansorena, E.; Lanciego, J.L.; Aymerich, M.S.; Blanco-Prieto, M.J. Effective GDNF brain delivery using microspheres—A promising strategy for Parkinson’s disease. J. Control. Release 2009, 135, 119–126. [Google Scholar] [CrossRef]
- Fletcher, A.M.; Kowalczyk, T.H.; Padegimas, L.; Cooper, M.J.; Yurek, D.M. Transgene expression in the striatum following intracerebral injections of DNA nanoparticles encoding for human glial cell line-derived neurotrophic factor. Neuroscience 2011, 194, 220–226. [Google Scholar] [CrossRef] [Green Version]
- Yurek, D.M.; Flectcher, A.M.; Kowalczyk, T.H.; Padegimas, L.; Cooper, M.J. Compacted DNA nanoparticle gene transfer of GDNF to the rat striatum enhances the survival of grafted fetal dopamine neurons. Cell Transplant. 2009, 18, 1183–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grandoso, L.; Ponce, S.; Manuel, I.; Arrue, A.; Ruiz-Ortega, J.A.; Ulibarri, I.; Orive, G.; Hernandez, R.M.; Rodriguez, A.; Rodriguez-Puertas, R.; et al. Long-term survival of encapsulated GDNF secreting cells implanted within the striatum of parkinsonized rats. Int. J. Pharm. 2007, 343, 69–78. [Google Scholar] [CrossRef]
- Garbayo, E.; Ansorena, E.; Lanciego, J.L.; Blanco-Prieto, M.J.; Aymerich, M.S. Long-term neuroprotection and neurorestoration by glial cell-derived neurotrophic factor microspheres for the treatment of Parkinson’s disease. Mov. Disord. 2011, 26, 1943–1947. [Google Scholar] [CrossRef] [Green Version]
- Yue, P.; Miao, W.; Gao, L.; Zhao, X.; Teng, J. Ultrasound-Triggered Effects of the Microbubbles Coupled to GDNF Plasmid-Loaded PEGylated Liposomes in a Rat Model of Parkinson’s Disease. Front. Neurosci. 2018, 12, 222. [Google Scholar] [CrossRef] [Green Version]
- Iravani, M.M.; Costa, S.; Jackson, M.J.; Tel, B.C.; Cannizzaro, C.; Pearce, R.K.; Jenner, P. GDNF reverses priming for dyskinesia in MPTP-treated, L-DOPA-primed common marmosets. Eur. J. Neurosci. 2001, 13, 597–608. [Google Scholar] [CrossRef]
- Costa, S.; Iravani, M.M.; Pearce, R.K.; Jenner, P. Glial cell line-derived neurotrophic factor concentration dependently improves disability and motor activity in MPTP-treated common marmosets. Eur. J. Pharmacol. 2001, 412, 45–50. [Google Scholar] [CrossRef]
- Gerhardt, G.A.; Cass, W.A.; Huettl, P.; Brock, S.; Zhang, Z.; Gash, D.M. GDNF improves dopamine function in the substantia nigra but not the putamen of unilateral MPTP-lesioned rhesus monkeys. Brain. Res. 1999, 817, 163–171. [Google Scholar] [CrossRef]
- Zhang, Z.; Miyoshi, Y.; Lapchak, P.A.; Collins, F.; Hilt, D.; Lebel, C.; Kryscio, R.; Gash, D.M. Dose response to intraventricular glial cell line-derived neurotrophic factor administration in parkinsonian monkeys. J. Pharmacol. Exp. Ther. 1997, 282, 1396–1401. [Google Scholar] [PubMed]
- Ai, Y.; Markesbery, W.; Zhang, Z.; Grondin, R.; Elseberry, D.; Gerhardt, G.A.; Gash, D.M. Intraputamenal infusion of GDNF in aged rhesus monkeys: Distribution and dopaminergic effects. J. Comp. Neurol. 2003, 461, 250–261. [Google Scholar] [CrossRef]
- Grondin, R.; Cass, W.A.; Zhang, Z.; Stanford, J.A.; Gash, D.M.; Gerhardt, G.A. Glial cell line-derived neurotrophic factor increases stimulus-evoked dopamine release and motor speed in aged rhesus monkeys. J. Neurosci. 2003, 23, 1974–1980. [Google Scholar] [CrossRef] [PubMed]
- Maswood, N.; Grondin, R.; Zhang, Z.; Stanford, J.A.; Surgener, S.P.; Gash, D.M.; Gerhardt, G.A. Effects of chronic intraputamenal infusion of glial cell line-derived neurotrophic factor (GDNF) in aged Rhesus monkeys. Neurobiol. Aging 2002, 23, 881–889. [Google Scholar] [CrossRef]
- Kells, A.P.; Eberling, J.; Su, X.; Pivirotto, P.; Bringas, J.; Hadaczek, P.; Narrow, W.C.; Bowers, W.J.; Federoff, H.J.; Forsayeth, J.; et al. Regeneration of the MPTP-lesioned dopaminergic system after convection-enhanced delivery of AAV2-GDNF. J. Neurosci. 2010, 30, 9567–9577. [Google Scholar] [CrossRef] [PubMed]
- Lo Bianco, C.; Deglon, N.; Pralong, W.; Aebischer, P. Lentiviral nigral delivery of GDNF does not prevent neurodegeneration in a genetic rat model of Parkinson’s disease. Neurobiol. Dis. 2004, 17, 283–289. [Google Scholar] [CrossRef]
- Decressac, M.; Ulusoy, A.; Mattsson, B.; Georgievska, B.; Romero-Ramos, M.; Kirik, D.; Bjorklund, A. GDNF fails to exert neuroprotection in a rat alpha-synuclein model of Parkinson’s disease. Brain 2011, 134, 2302–2311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decressac, M.; Kadkhodaei, B.; Mattsson, B.; Laguna, A.; Perlmann, T.; Bjorklund, A. alpha-Synuclein-induced down-regulation of Nurr1 disrupts GDNF signaling in nigral dopamine neurons. Sci. Transl. Med. 2012, 4, 163ra156. [Google Scholar] [CrossRef]
- Quintino, L.; Avallone, M.; Brannstrom, E.; Kavanagh, P.; Lockowandt, M.; Garcia Jareno, P.; Breger, L.S.; Lundberg, C. GDNF-mediated rescue of the nigrostriatal system depends on the degree of degeneration. Gene Ther. 2019, 26, 57–64. [Google Scholar] [CrossRef] [Green Version]
- Kwon, H.S.; Koh, S.H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Qing, J.; Liu, X.; Wu, Q.; Zhou, M.; Zhang, Y.; Mazhar, M.; Huang, X.; Wang, L.; He, F. Hippo/YAP Pathway Plays a Critical Role in Effect of GDNF Against Abeta-Induced Inflammation in Microglial Cells. DNA Cell Biol. 2020, 39, 1064–1071. [Google Scholar] [CrossRef]
- Van Dyke, J.M.; Smit-Oistad, I.M.; Macrander, C.; Krakora, D.; Meyer, M.G.; Suzuki, M. Macrophage-mediated inflammation and glial response in the skeletal muscle of a rat model of familial amyotrophic lateral sclerosis (ALS). Exp. Neurol. 2016, 277, 275–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Deng, L.; Wang, Y.; Yin, L.; Yang, C.; Du, J.; Yuan, Q. GDNF Enhances Therapeutic Efficiency of Neural Stem Cells-Based Therapy in Chronic Experimental Allergic Encephalomyelitis in Rat. Stem Cells Int. 2016, 2016, 1431349. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Liu, Y.; Zhou, J. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl. Neurodegener. 2015, 4, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Z.; Chen, A.; Fa, Z.; Ding, Z.; Xie, J.; Sun, Y.; Zhang, R.; Wang, Q. Adipose-Derived Stem Cells Modulate BV2 Microglial M1/M2 Polarization by Producing GDNF. Stem Cells Dev. 2020, 29, 714–727. [Google Scholar] [CrossRef]
- Chou, A.K.; Yang, M.C.; Tsai, H.P.; Chai, C.Y.; Tai, M.H.; Kwan, A.L.; Hong, Y.R. Adenoviral-mediated glial cell line-derived neurotrophic factor gene transfer has a protective effect on sciatic nerve following constriction-induced spinal cord injury. PLoS ONE 2014, 9, e92264. [Google Scholar] [CrossRef]
- Lin, C.H.; Wang, C.H.; Hsu, S.L.; Liao, L.Y.; Lin, T.A.; Hsueh, C.M. Molecular Mechanisms Responsible for Neuron-Derived Conditioned Medium (NCM)-Mediated Protection of Ischemic Brain. PLoS ONE 2016, 11, e0146692. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Li, S.; Wang, Y.; Zhang, X.; Chen, L.; Sun, D. GDNF enhances the anti-inflammatory effect of human adipose-derived mesenchymal stem cell-based therapy in renal interstitial fibrosis. Stem Cell Res. 2019, 41, 101605. [Google Scholar] [CrossRef]
- Zhang, D.K.; He, F.Q.; Li, T.K.; Pang, X.H.; Cui, D.J.; Xie, Q.; Huang, X.L.; Gan, H.T. Glial-derived neurotrophic factor regulates intestinal epithelial barrier function and inflammation and is therapeutic for murine colitis. J. Pathol. 2010, 222, 213–222. [Google Scholar] [CrossRef]
- Shinoda, M.; Fukuoka, T.; Takeda, M.; Iwata, K.; Noguchi, K. Spinal glial cell line-derived neurotrophic factor infusion reverses reduction of Kv4.1-mediated A-type potassium currents of injured myelinated primary afferent neurons in a neuropathic pain model. Mol. Pain 2019, 15, 1744806919841196. [Google Scholar] [CrossRef]
- Wang, Y.; Lin, S.Z.; Chiou, A.L.; Williams, L.R.; Hoffer, B.J. Glial cell line-derived neurotrophic factor protects against ischemia-induced injury in the cerebral cortex. J. Neurosci. 1997, 17, 4341–4348. [Google Scholar] [CrossRef]
- Hayward, N.J.; Harding, M.; Lloyd, S.A.; McKnight, A.T.; Hughes, J.; Woodruff, G.N. The effect of CCKB/gastrin antagonists on stimulated gastric acid secretion in the anaesthetized rat. Br. J. Pharmacol. 1991, 104, 973–977. [Google Scholar] [CrossRef] [Green Version]
- Neurath, M.F. Cytokines in inflammatory bowel disease. Nat. Rev. Immunol. 2014, 14, 329–342. [Google Scholar] [CrossRef]
- Steinkamp, M.; Geerling, I.; Seufferlein, T.; von Boyen, G.; Egger, B.; Grossmann, J.; Ludwig, L.; Adler, G.; Reinshagen, M. Glial-derived neurotrophic factor regulates apoptosis in colonic epithelial cells. Gastroenterology 2003, 124, 1748–1757. [Google Scholar] [CrossRef]
- Lang, A.E.; Gill, S.; Patel, N.K.; Lozano, A.; Nutt, J.G.; Penn, R.; Brooks, D.J.; Hotton, G.; Moro, E.; Heywood, P.; et al. Randomized controlled trial of intraputamenal glial cell line-derived neurotrophic factor infusion in Parkinson disease. Ann. Neurol. 2006, 59, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.K.; Bunnage, M.; Plaha, P.; Svendsen, C.N.; Heywood, P.; Gill, S.S. Intraputamenal infusion of glial cell line-derived neurotrophic factor in PD: A two-year outcome study. Ann. Neurol. 2005, 57, 298–302. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/history/NCT04167540?V_3=View#StudyPageTop (accessed on 29 August 2021).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03652363 (accessed on 30 August 2021).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT01621581 (accessed on 28 August 2021).
- Whone, A.L.; Boca, M.; Luz, M.; Woolley, M.; Mooney, L.; Dharia, S.; Broadfoot, J.; Cronin, D.; Schroers, C.; Barua, N.U.; et al. Extended Treatment with Glial Cell Line-Derived Neurotrophic Factor in Parkinson’s Disease. J. Parkinsons Dis. 2019, 9, 301–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT00006488 (accessed on 31 August 2021).




{kind=link}
{kind=link}
{kind=link}
| α-Syn Aggregation Pattern | Glial Receptor/Mechanism | Signaling Pathway | Neuroinflammatory/Neurotoxic Effects | Ref. |
|---|---|---|---|---|
| Fibrils | NLRP3 a | α-syn acts as DAMP and activates the NLRP3 inflammasome. | Synthesis and release of IL-1β and cleaved caspase-1 that triggers pyroptosis *. | [15,83,84,85,86,87] |
| Monomers, oligomers, and fibrils | TLRs a,b,† | TLRs sense DAMPs, including α-syn, leading to nuclear translocation of NF-κB. | Release of pro-inflammatory cytokines (TNF-α and IL-6). Dual effect on the astrocyte: secretion of pro-inflammatory and/or neuroprotective factors. | [12,15,16,17,88,89,90,91] |
| Fibrils | FcγR a | Internalization in phagosomes and nuclear translocation of NF-kB p65. | Clearance of α-syn, triggering the release of pro-inflammatory molecules and neurodegeneration. | [15,92,93] |
| Oligomers and fibrils | CD11b a | NOX2 activation through a RhoA-dependent pathway. | NOX2 activation mediates the chemoattractant ability of α-syn. Induction of superoxide production. | [15,79,94] |
| Oligomers | EP2 a | The cyclooxygenase/prostaglandin E2 (COX/PGE2) pathway. | Activation of PHOX NADPH and increase in prostaglandin E2 levels, leading to neuronal toxicity. | [15,95,96,97] |
| Monomers | CD36 a | Phosphorylation of ERK2, a downstream kinase activated by CD36 ligation. | Neuronal death through the release of TNF-α and ROS and up-regulation of COX2, NOX2, and iNOS. | [15,72] |
| Oligomers | P2X7R a | Activation of the PI3K/AKT pathway. | Increase of oxidative stress by p47phox translocation and PHOX activation. | [15,98] |
| Fibrils | MHC b | Changes in the expression of HLA genes encoding MHC class I and II proteins. | Impairment of ATP-generating mitochondrial respiration. | [99] |
| Oligomers | Endocytosis b | Dysfunction in mitochondrial dynamics. | Neuronal death is mediated by cytokines release. | [100] |
| GDNF Therapy and Lesion Models | Neurodegenerative or Neuroprotective Effects of GDNF Therapy | Ref. |
|---|---|---|
| (a) Murine models | ||
| ICV infusion of rhGDNF or 125Iodine-labelled GDNF in unlesioned rats. | Significantly increased striatal and nigral DA levels. | [148,149] |
| Bilateral ICV GDNF peptide infusion in aged rats with 6-OHDA lesion. | Improved locomotor performance and increased striatal DA turnover. | [150] |
| Intraventricular infusion of rhGDNF in rats with 6-OHDA lesion. | AIRB prevention. PET analysis showed DA reuptake reduction in the ipsilateral STR. Reduced loss of TH positive neurons in the SNpc and VTA. | [151] |
| Intranigral GDNF peptide administration in rats with 6-OHDA intrastriatal lesion. | No protection of STR terminals, absence MD recovery, but prevention of cell death in SN. | [152] |
| Intranigral hGDNF gene transfection in 6-OHDA lesioned rats. | Improved locomotor performance resulting from the regeneration of the nigrostriatal dopaminergic system. | [153] |
| Intranigral or ICV GDNF peptide administration in rats with 6-OHDA lesion. | The intranigral therapy prevented AIRB and increased TH activity in SN but not in the STR. The ICV therapy transiently reduced AIRB and increased TH only in the ipsilateral SN. | [154] |
| Intraventricular or intrastriatal rhGDNF infusion via an osmotic minipump over 4 wk in rats with 2 wk-lesion of 6-OHDA. | The ICV therapy successfully blocked the late neuron degeneration in the SN and caused long-lasting relief of MD. Intrastriatal therapy transiently improved MD only during the infusion period, and DA cells’ rescue was less prominent. | [155] |
| Intrastriatal administration of microspheres of N-glycosylated rhGDNF in rats with 6-OHDA lesion. | Relieved the AIRB and increased the density of TH+ fibers at the striatal level. The therapy proved to be suitable to release biologically active GDNF over up to 5 wk. | [156] |
| Intrastriatal administration of hGDNF gene-loaded nanoparticles in rats with 6-OHDA lesion. | Promoted survival of grafted fetal DA neurons, increased the survival of TH + cells, and significantly improved motor behavior. | [157,158] |
| Intrastriatal grafts of genetically modified fibroblasts to produce GDNF in 6-OHDA rats. | Behavioral improvements and, 6 months after grafting, strong GDNF immunoreactivity in the STR. No changes in DA levels and its metabolites neither TH immunoreactivity in the STR. | [159] |
| Intraestriatal administration of GDNF-loaded microspheres in animals with 2- wk 6-OHDA lesion. | Increased DA striatal terminals and neuroprotection of DA neurons, long-term improvements of behavior until the end of the study (wk 30). | [160] |
| FUS *-facilitated the delivery of rGDNF-PLs-MBs to the rat brain with 6-OHDA-lesion. | Neuroprotection in effects on TH+ cell number and levels of DA and its metabolites. Also, it prevented the progression of motor-related behavioral abnormalities. | [161] |
| (b) Non-human primate models | ||
| Intraventricular rhGDNF administration in rhesus monkeys and marmosets with MPTP-lesion. | Relief of MD correlates with increased DA levels and its metabolites in the SN, but not in the Pu of rhesus monkeys. Promising improvements were observed in MD in the marmosets. | [162,163,164,165] |
| Intrastriatal continuous hGDNF administration in rhesus monkeys with MPTP-lesion. | It increased the DA cell number in the SN and fiber density in the CN, Pu, and GP and increased DA and its metabolite levels. Improvement of the PD rating scale. | [129,166,167,168] |
| Intraputamenal CED delivery of AAV2-GDNF in rhesus monkeys with MPTP-lesion. | Led to GDNF expression in the Pu, anterograde transport to the SN and rescue of DA neurons via retrograde striatonigral transport, and reversion of neuroregeneration. | [169] |
| Experimental Model | Type of Therapy with GDNF | Featured Results | Proposed Mechanism/Concluding Remarks | Ref. |
|---|---|---|---|---|
| Genetic rat model of PD (human A30P mutant α-synuclein overexpression by Lenti-A30P). | Intranigral transduction of GDNF-LV (200,000 ng of p24/mL), 2 wk before the intranigral injection Lenti-A30P). | GDNF did not prevent the loss of DA neurons and nerve terminals induced by α-syn toxicity, despite promoting the sprouting of DA axons. | The lack of GDNF neuroprotective effects may be caused by α-syn-mediated blockade of the GDNF/GFRα/RET signaling pathway. | [170] |
| Genetic rat model of PD (human wild-type α-syn overexpression by AAV-α-syn). | Intrastriatal (2 wk before AAV-α-syn) and intranigral (3 wk before AAV-α-syn) injection of GDNF-LV (1 × 107 transduction units/mL) and AAV-GDNF (1 × 1012 genome copies/mL). | Both GDNF therapies did not protect nigral neurons and striatal DA innervation against α-syn-induced toxicity. Also, GDNF did not affect the process of α-syn aggregation. | The α-syn-based rat model can cause poor axonal transport, which may interfere with antegrade or retrograde transport of GDNF in the nigrostriatal system. | [171] |
| Genetic: human WT α-syn overexpression by AAV-α-syn. | Intrastriatal or intranigral injection of rhGDNF (1 μg/3 μL) 2 wk after administration of AAV- α-syn. | GDNF failed to activate the AKT and MAPK pathways in the genetic model. It also reduced the expression of the RET and NR4A2 (Nurr1) genes. | Therapeutic failure was caused by the toxicity of α-syn that blockades the GDNF/RET/Nurr1 signaling pathway. | [172] |
| Parkin Q311X(A) transgenic mice. | GDNF-producing macrophage injections (i.v. 2 × 106 cells/100 μL/mouse) every wk for 3 wks. | GDNF reduced the formation of α-syn aggregates. It also restored the impaired locomotor functions. | The result is attributed to the anti-inflammatory subtype of M2 macrophages. | [27] |
| Primary embryonic midbrain cultures with α-syn PFFs added on culture day 8. Intrastriatal injection of α-syn PFFs in adult C57Bl/6NCrI mice. | pCDH-hSYN-hGDNF LV (MOI ≈ 5, on days 0, 5, and 9). Intrastriatal injection of AAV1-hGDNF (2.14 × 1012 vg/m) one wk before α-syn PFFs injection. | In vitro, GDNF overexpression reduced misfolded α-syn and phosphorylated α-syn. In vivo, GDNF overexpression prevents the α-syn aggregates in the SN and then spreads to the brain. | Activation of the GDNF/RET pathway prevents Lewy pathology. The inhibition of the PI3K/AKT signaling increased the phospho α-syn. The inhibition of SRC blocks the effect of GDNF on α-syn accumulation. | [25] |
| Experimental Model/GDNF Therapeutic Approach | GDNF Relevant Effects | Proposed Mechanisms | Ref. |
|---|---|---|---|
| (a) Neurodegenerative disease | |||
| Parkinson’s disease | |||
| Midbrain microglial cultures activated by Zymosan A/Therapy with astrocyte-derived GDNF. | GDNF decreases microglial activation in a model of neuroinflammation in vitro. | Activation of the GFRα1-RET complex and inhibition of the FAK pathway. | [33] |
| 12-month-old GDNF+/- knockdown mice/Bilateral intrastriatal injection of GDNF (10 μg per hemisphere). | Attenuation of motor impairments and nigrostriatal dopamine levels. | Reduction of the COX-2 expression and increase in SOD-2 levels in the SN suggests reduced microglial activation. | [32] |
| Rat microglial culture activated by LPS/Therapy with rHGDNF (50 ng/mL, before LPS administration). | Prevention of NO synthesis and iNOS COX-2, IL-6, IL-1β, and TNF-α expression. | Inhibition of microglial activation by reducing phosphorylation of p38 *. | [31] |
| 6-OHDA-induced PD rat model/Intrastriatal GDNF released by LCM (1.5 mg/kg, 3d after 6-OHDA injection). | Reduction of caspase-3 and TNF-α levels and activation of microglia. | Reduced microglial activation by lowering MMP-9 and MHC II expression, which consequently retains DA. | [30] |
| Intracutaneous 6-OHDA injection in mice/GDNF transfected macrophages (i.v. 1 × 106 cells/100 μL, 2d after 6-OHDA). | It decreased activated microglia in SNpc. | The macrophages migrate to inflammation sites and provide neuroprotection modulating glial cells activation. | [27] |
| Intraperitoneal MPTP injection in mouse/IN GDNF delivery by peptide-conjugated lipid nanocarriers (2.5 μg, 2.5 μL per nostril, 4 sessions). | It decreased activated microglia. | It targets microglia activation, modulating the neuroinflammation. | [28] |
| Transgenic Parkin Q311X(A) mice (4-month-old)/Administration of GDNF-macrophages (i.v. 2 × 106 cells/100 μL/mouse, once a wk for 3wk). | Disminution of microgliosis and astrogliosis. | Anti-neuroinflammatory effect by modulating glial activation. | [27] |
| Intraperitoneal MPTP injection in mouse twice a wk for 3wk/GDNF gene delivery via the UTMD system (3.6 × 108 MBs/mL, twice a wk for 3wk, 1d after MPTP). | It decreased apoptosis and astrogliosis. | Reduced expression of calcium ions and the apoptotic protein caspase 3. | [26] |
| Alzheimer’s disease | |||
| Murine microglial cell line BV2 stimulated with Aβ-protein/GDNF (100 and 500 ng/mL, after Aβ-protein administration). | Decreased levels of TNF-α, TGF-β, IL-1β, and IL-12β, in a dose-dependent manner. | Inhibition of microglial activation by reducing YAP phosphorylation (Hippo/YAP pathway). | [175] |
| Amyotrophic lateral sclerosis | |||
| Transgenic SOD1G93A rat (90d old)/i.m. grafts of hMSC-GDNF (150,000 cells in 50 μL, at 24h, 1 and 2wk). | Reduced inflammation markers and promoted TSC survival. Delayed onset of ALS symptom | Delayed ALS symptom onset by preserving NMJ integrity. | [176] |
| Multiple sclerosis | |||
| CEAE animal model for multiple sclerosis: Transplantation of GDNF/NSCs in each lateral ventricle (5 × 105 in 10 μL, 10d after the CEAE induction). | Reduced inflammatory infiltrates in the STR and the astrocyte differentiation of NSCs. | Possible activation of myeloid dendritic cells and subsequent restriction T cell expansions. | [177] |
| (b) Other neurological diseases | |||
| Murine microglial cell line BV2 with ADSCs-GDNF (500 ng/L). | Inhibition of the M1 phenotype and promoting the M2 phenotype. | Microglial activation and polarization via PI3K/AKT signaling pathway. | [179] |
| Neuropathic pain model by CCI in rats/rAV-GDNF [2 × 109 pfu/100 mL PBS] injection in the triceps brachii muscle. | Down-regulated protein expression of MMP9, p38, IL6, IL1β, and iNOS. | Inhibition of microglial activation and cytokine production via p38 and PKC signaling. | [180] |
| FCI rat animal model and neuronal culture GOSD/NCM with TGFβ1, GDNF, and NT-3 (1 ng/mL for each brain cell in plates; 6.4 × 106/plate) and incubated under GOSD conditions (2 h). | Reduction of the brain infarction and motor deficit. | Anti-apoptotic, anti-oxidant, and potentially anti-glutamate activities. | [181] |
| (c) Systemic diseases | |||
| UUO mice model/GDNF-AMSCs (i.v. 150 μl cell of suspension containing 5 × 105 cells via the tail vein). | Activation of M2 macrophages and reduced renal fibrosis by suppressing inflammation. | Down-regulated TNF-α and iNOS; upregulated IL-4 and IL10. | [182] |
| Experimental colitis mouse model induced by DSS/rAV-GDNF (intracolonically 1 × 1010 pfu/mouse) | Reduction in enhanced permeability by restoring epithelial barrier function. | Inhibition of TNF-α, IL-1β, MPO, caspase-3, and NF-kB. Increase in ZO-1 and AKT. | [183] |
| CT Identifier. Clinical Phase and Status. | Characteristics of Subjects Recruited | Aims of the Study | Outcome Measures/Preliminary Results | Ref. |
|---|---|---|---|---|
| NCT04167540 Clinical Phase: I Clinical status: Active, not recruiting. | 25 individuals, males, and females at least 18 years of age (recent/long-standing diagnosis of PD) who are not well controlled by medications (follow up for 5 years). | Evaluate the safety and clinical effect of AAV2-GDNF delivered to the Pu. | Access to disease improvement: MRI, serum and CSF tests, movement and behavioral tests. No preliminary results | [191,192] |
| NCT01621581 Clinical Phase: I Clinical status: Completed. | 25 individuals, males, and females at least 18 years of age diagnosed with idiopathic and advanced PD (follow up for 5 years). | Evaluate the safety and effectiveness of AAV2-GDNF gene transfer to the Pu via CED. | Good drug tolerability based on clinical and neuroimaging tests. The PET showed increased F-DOPA uptake in the infused areas at 6 and 18 months in 10/13 and 12/13 patients, respectively. | [135,192,193] |
| NCT03652363 Clinical Phase: II Clinical status: Completed. | 42 individuals, females and males between 35 to 75 years diagnosed with idiopathic PD (followup for 5 years). | Analyze the safety and efficacy of intermittent bilateral intraputamenal GDNF via CED. | Periodic evaluation with specific clinical and blood tests or an MRI scan. Disappointing final results, with no significant clinical improvement between GDNF and placebo group. | [135,192,194] |
| NCT00006488 Clinical Phase: I Clinical status: Completed. | Females and males between 18 and 75 years of age were diagnosed with idiopathic PD. The number of individuals is not specified. | Determine the benefits and TEAE of continuous infusion of r-metHuGDNF in Pu. | Subjects were evaluated using clinical neurological tests, computerized gait assessment, and neurological imaging. The results of this study were not specified. | [192,195] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Delgado-Minjares, K.M.; Martinez-Fong, D.; Martínez-Dávila, I.A.; Bañuelos, C.; Gutierrez-Castillo, M.E.; Blanco-Alvarez, V.M.; Cardenas-Aguayo, M.-d.-C.; Luna-Muñoz, J.; Pacheco-Herrero, M.; Soto-Rojas, L.O. Mechanistic Insight from Preclinical Models of Parkinson’s Disease Could Help Redirect Clinical Trial Efforts in GDNF Therapy. Int. J. Mol. Sci. 2021, 22, 11702. https://doi.org/10.3390/ijms222111702
Delgado-Minjares KM, Martinez-Fong D, Martínez-Dávila IA, Bañuelos C, Gutierrez-Castillo ME, Blanco-Alvarez VM, Cardenas-Aguayo M-d-C, Luna-Muñoz J, Pacheco-Herrero M, Soto-Rojas LO. Mechanistic Insight from Preclinical Models of Parkinson’s Disease Could Help Redirect Clinical Trial Efforts in GDNF Therapy. International Journal of Molecular Sciences. 2021; 22(21):11702. https://doi.org/10.3390/ijms222111702
Chicago/Turabian StyleDelgado-Minjares, Karen M., Daniel Martinez-Fong, Irma A. Martínez-Dávila, Cecilia Bañuelos, M. E. Gutierrez-Castillo, Víctor Manuel Blanco-Alvarez, Maria-del-Carmen Cardenas-Aguayo, José Luna-Muñoz, Mar Pacheco-Herrero, and Luis O. Soto-Rojas. 2021. "Mechanistic Insight from Preclinical Models of Parkinson’s Disease Could Help Redirect Clinical Trial Efforts in GDNF Therapy" International Journal of Molecular Sciences 22, no. 21: 11702. https://doi.org/10.3390/ijms222111702
APA StyleDelgado-Minjares, K. M., Martinez-Fong, D., Martínez-Dávila, I. A., Bañuelos, C., Gutierrez-Castillo, M. E., Blanco-Alvarez, V. M., Cardenas-Aguayo, M. -d. -C., Luna-Muñoz, J., Pacheco-Herrero, M., & Soto-Rojas, L. O. (2021). Mechanistic Insight from Preclinical Models of Parkinson’s Disease Could Help Redirect Clinical Trial Efforts in GDNF Therapy. International Journal of Molecular Sciences, 22(21), 11702. https://doi.org/10.3390/ijms222111702