
Synthesis and Characterization of Bis-Triazolyl-Pyridine Derivatives as Noncanonical DNA-Interacting Compounds

 ,
,  , , , , ,
, , , , ,  ,
,  and
and 
Abstract
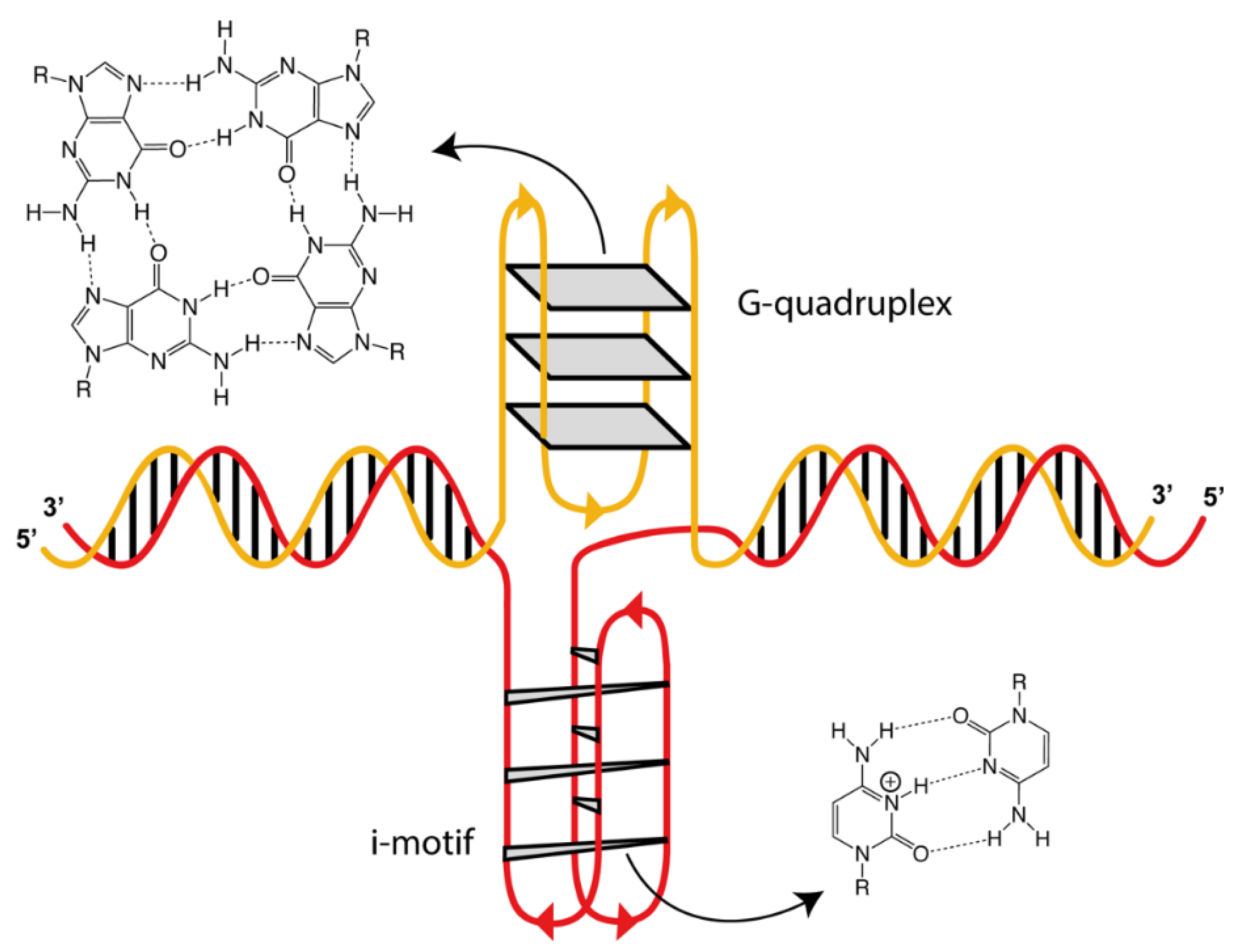
:1. Introduction
2. Results and Discussion
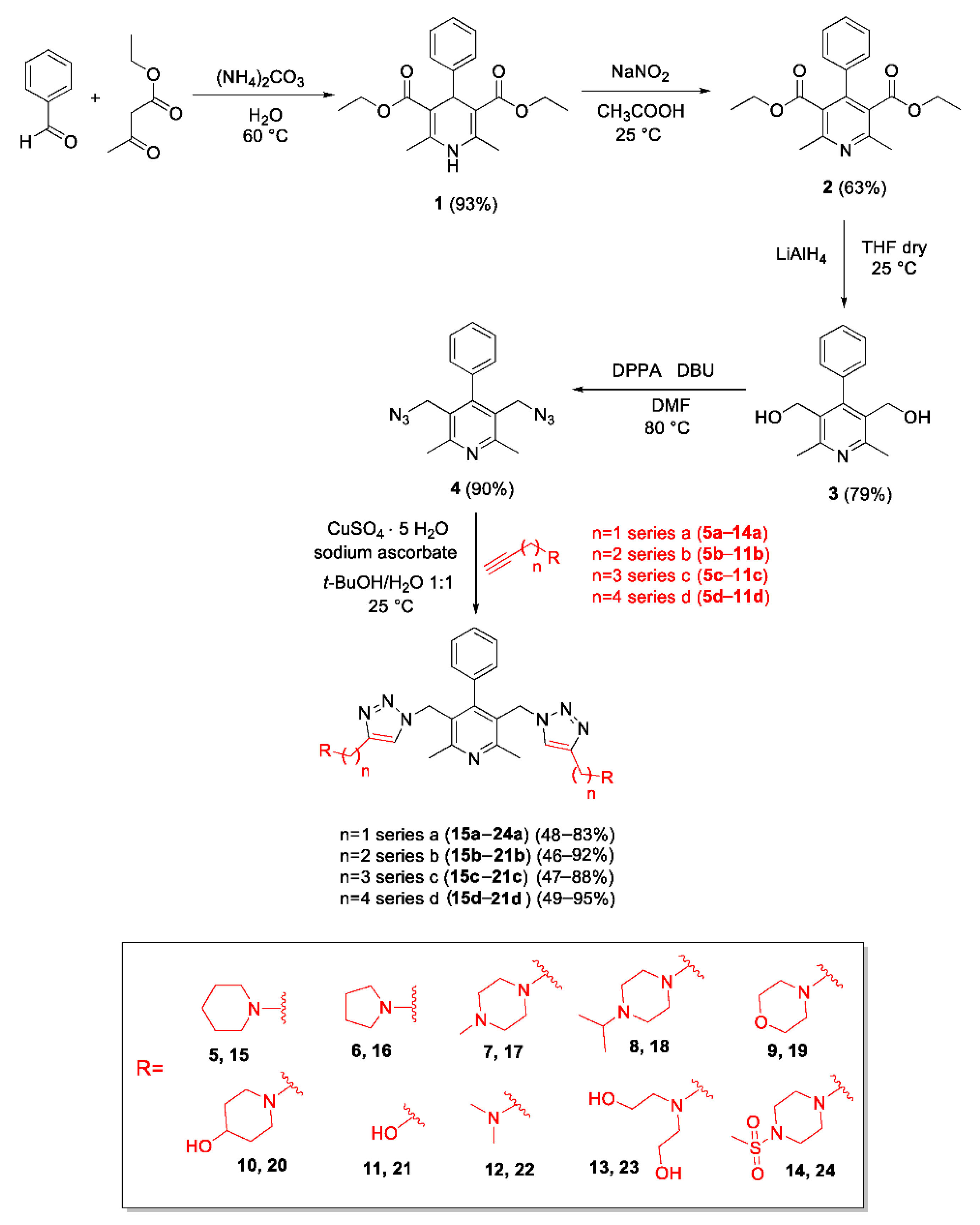
2.1. Chemistry
2.2. Circular Dichroism Screening and Selection of the Most Promising Compounds
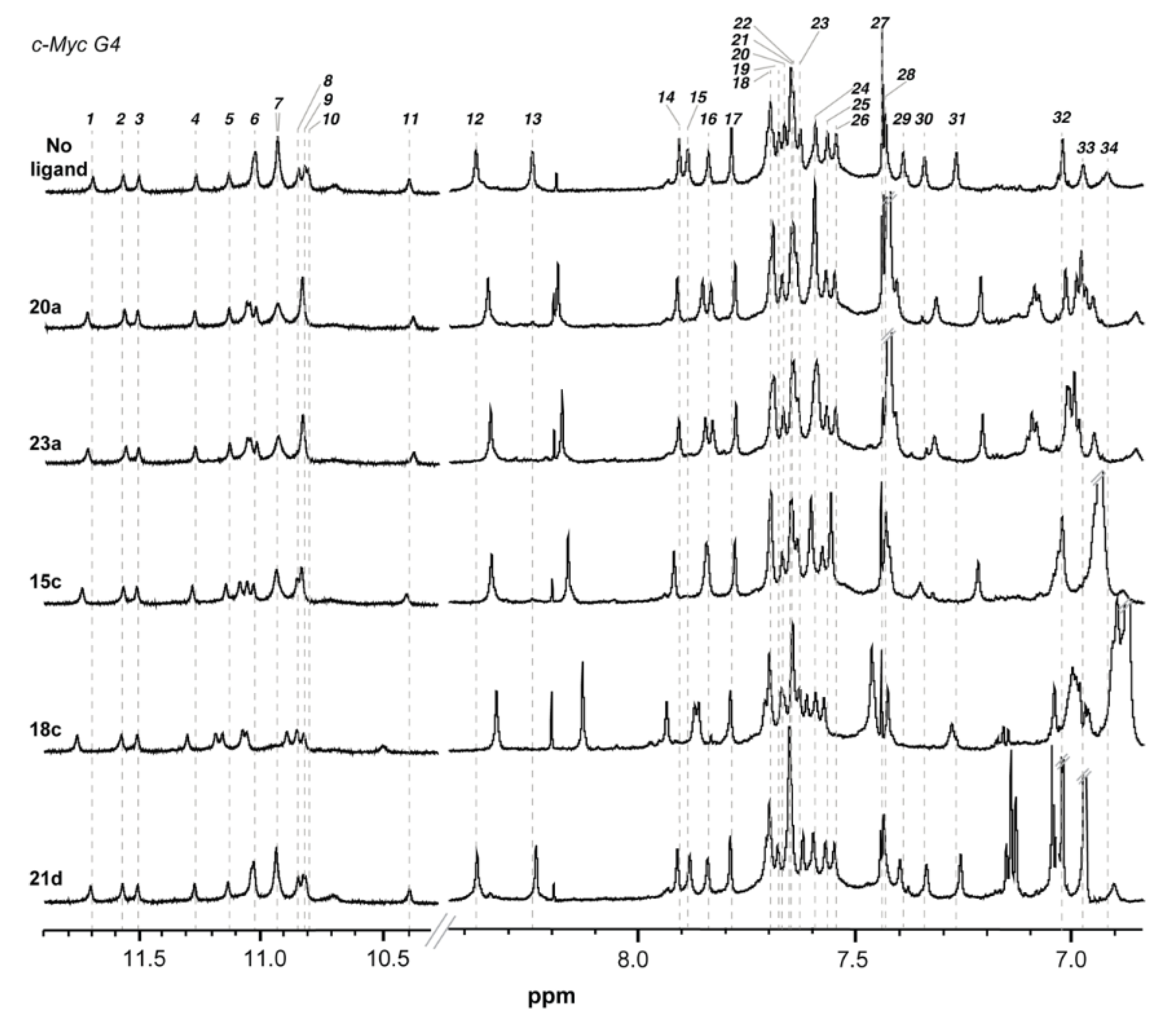
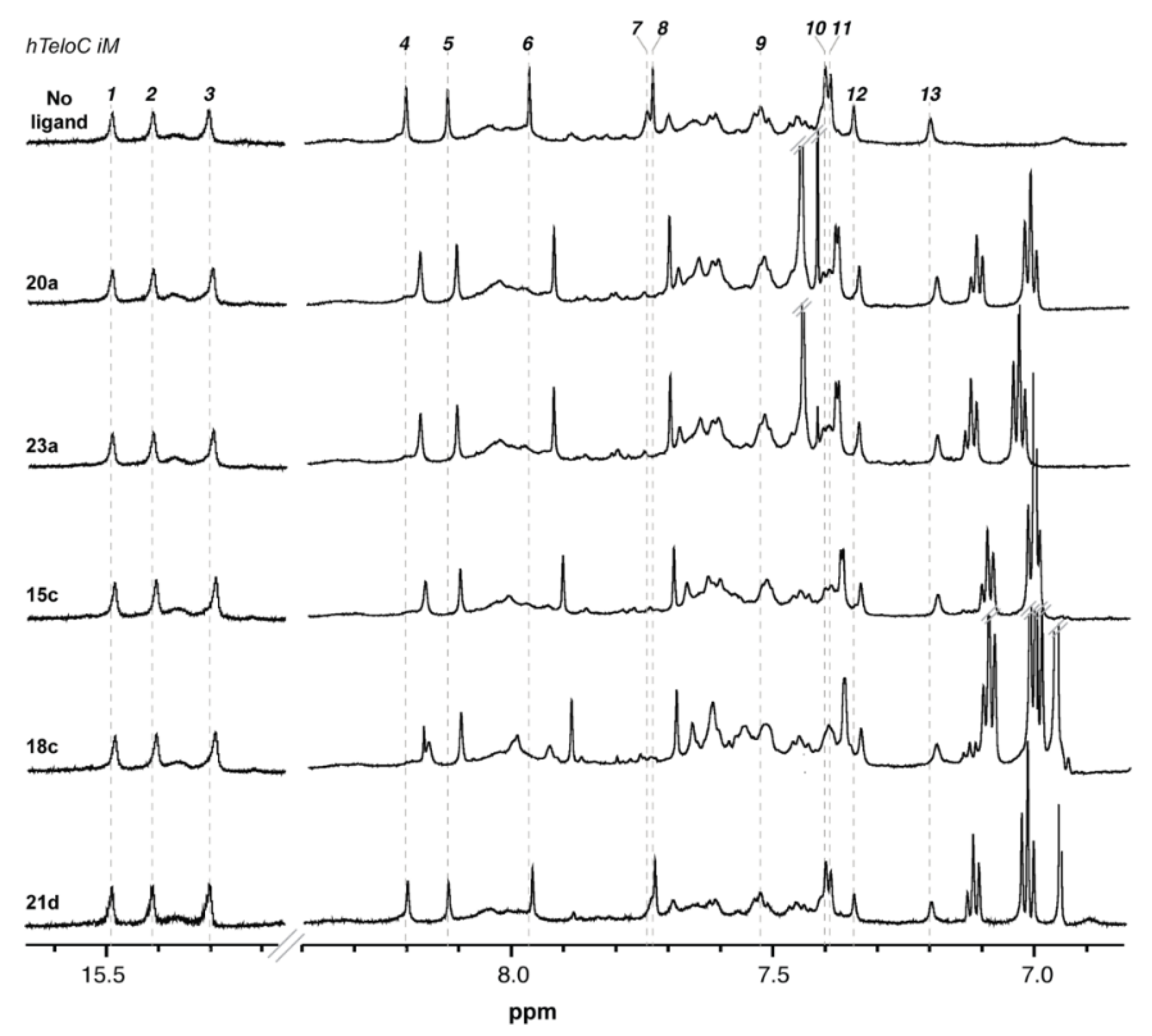
2.3. Nuclear Magnetic Resonance (NMR) Studies
2.4. Saturation-Transfer Difference (STD) NMR Analysis
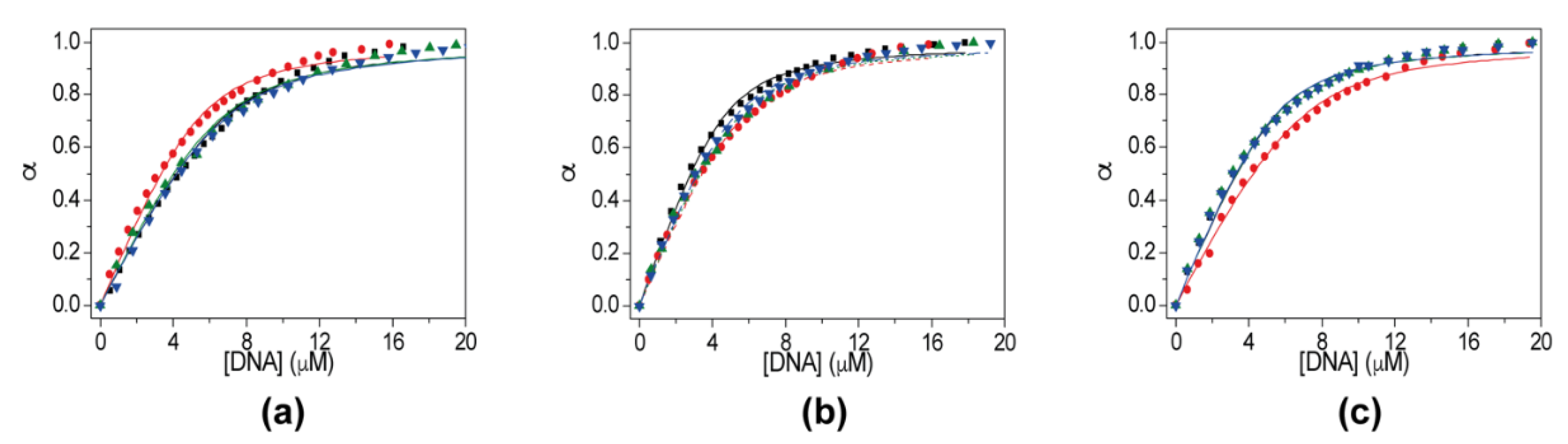
2.5. Evaluation of Ligand Affinity by Fluorescence Experiments
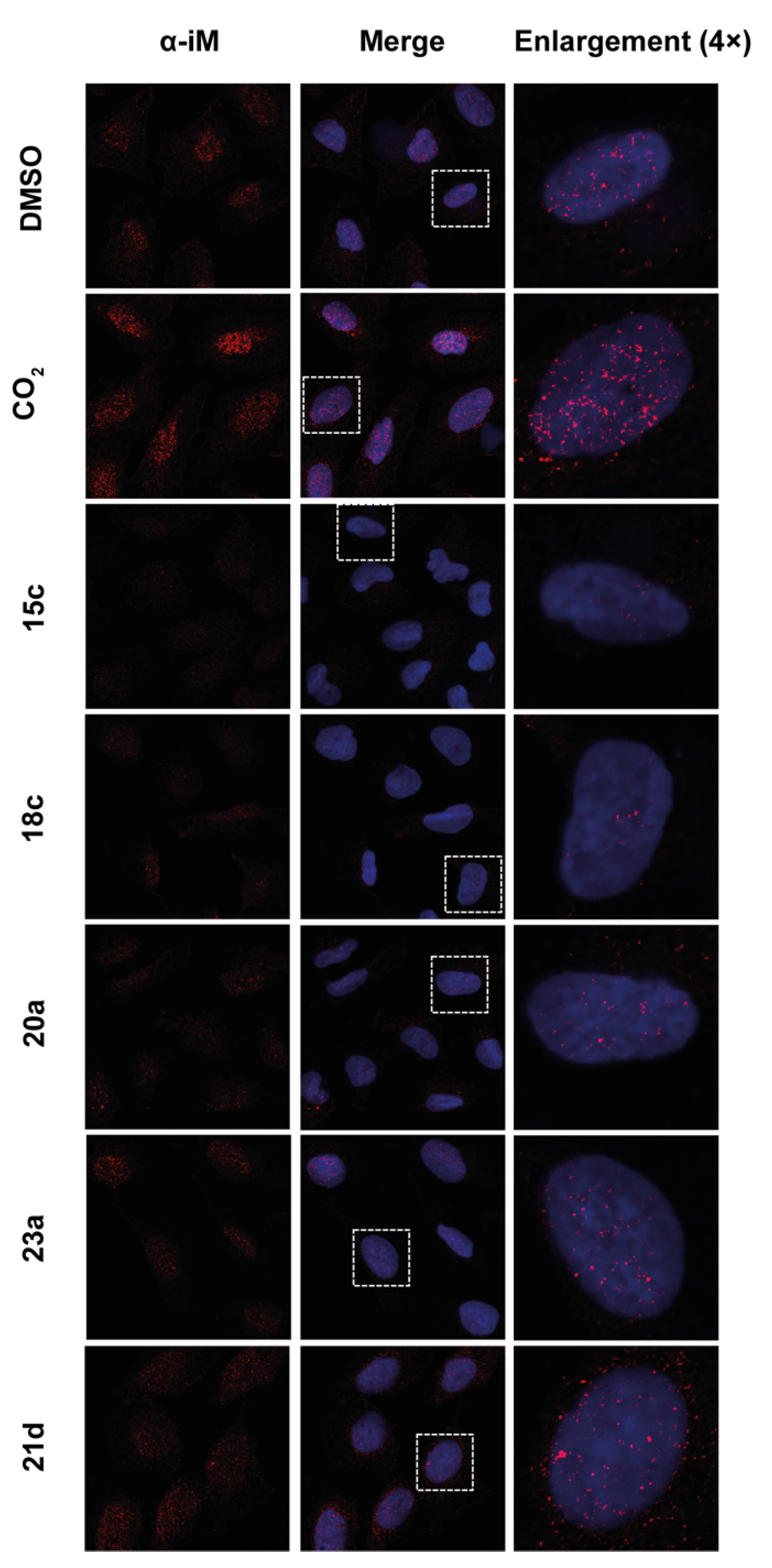
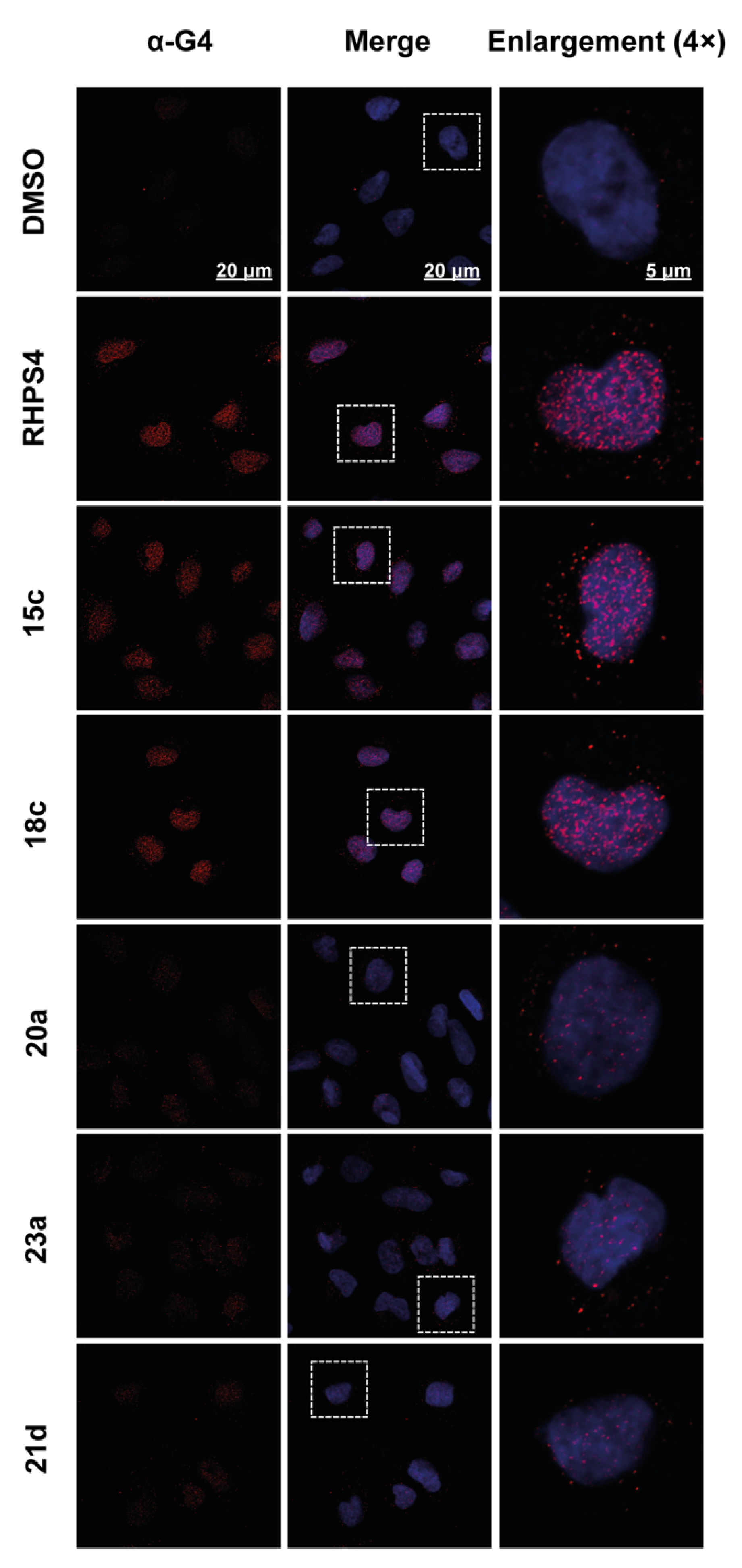
2.6. Effect of the Compounds on G4/iM Structures in Living Cells
3. Materials and Methods
3.1. Materials
3.2. Chemistry
3.2.1. Synthesis of Diethyl 2,6-Dimethyl-4-Phenyl-1,4-Dihydropyridine-3,5-Dicarboxylate (1)
3.2.2. Synthesis of Diethyl 2,6-Dimethyl-4-Phenylpyridine-3,5-Dicarboxylate (2)
3.2.3. Synthesis of (2,6-Dimethyl-4-Phenylpyridine-3,5-Diyl)Dimethanol (3)
3.2.4. Synthesis of 3,5-Bis(Azidomethyl)-2,6-Dimethyl-4-Phenylpyridine (4)
3.2.5. General Procedure for the Preparation of Bis-Triazolyl-Pyridines 15a–24a, 15b–21b, 15c–21c, 15d–21d
Series a: Bis-Triazolyl-Pyridines 15a–24a
Series b: Bis-Triazolyl-Pyridines 15b–21b
Series c: Bis-Triazolyl-Pyridines 15c–21c
Series d: Bis-Triazolyl-Pyridines 15d–21d
3.3. Oligonucleotide Synthesis and Sample Preparation
3.4. Circular Dichroism (CD) Experiments
3.5. Principal Component Analysis
3.6. Nuclear Magnetic Resonance Experiments
3.6.1. 1D 1H-NMR Experiments
3.6.2. STD NMR Experiments
3.7. Fluorescence Titrations
3.8. Biological Experiments
3.8.1. Cell Culture and Treatments
3.8.2. Immunofluorescence (IF) Microscopy Experiments
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| APT | Attached proton test |
| ATCC | American type culture collection |
| BCL-2 | B-cell lymphoma 2 |
| CD | Circular dichroism |
| c-MYC | Cellular MYC |
| CuAAC | Copper(I)-catalyzed alkyne-azide cycloaddition |
| DAPI | 4′,6-diamidino-2-phenylindole |
| DBU | 1,8-diazabicycloundec-7-ene |
| DMEM | Dulbecco’s modified eagle medium |
| DMF | N,N′-dimethylformamide |
| DMSO | Dimethyl sulfoxide |
| DPPA | Diphenylphosphoryl azide |
| ESI | Electrospray ionization |
| FBS | Fetal bovine serum |
| FT-IR | Fourier transform infrared |
| G4 | G-quadruplex |
| HPLC | High-performance liquid chromatography |
| IF | Immunofluorescence |
| iM | i-Motif |
| MS | Mass spectrometry |
| NMR | Nuclear magnetic resonance |
| NOE | Nuclear Overhauser effect |
| PBS | Phosphate buffered saline |
| PCA | Principal component analysis |
| PE | Petroleum ether |
| RT | Room temperature |
| STD | Saturation transfer difference |
| THF | Tetrahydrofuran |
References
- Bacolla, A.; Wells, R.D. Non-B DNA Conformations as Determinants of Mutagenesis and Human Disease. Mol. Carcinog. 2009, 48, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Abou Assi, H.; Garavís, M.; González, C.; Damha, M.J. I-Motif DNA: Structural Features and Significance to Cell Biology. Nucleic Acids Res. 2018, 46, 8038–8056. [Google Scholar] [CrossRef] [Green Version]
- Gajarský, M.; Živković, M.L.; Stadlbauer, P.; Pagano, B.; Fiala, R.; Amato, J.; Tomáška, L.; Šponer, J.; Plavec, J.; Trantírek, L. Structure of a Stable G-Hairpin. J. Am. Chem. Soc. 2017, 139, 3591–3594. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, J.; Adhikari, S.; Balasubramanian, S. The Structure and Function of DNA G-Quadruplexes. Trends Chem. 2020, 2, 123–136. [Google Scholar] [CrossRef] [Green Version]
- Lane, A.N.; Chaires, J.B.; Gray, R.D.; Trent, J.O. Stability and Kinetics of G-Quadruplex Structures. Nucleic Acids Res. 2008, 36, 5482–5515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharyya, D.; Mirihana Arachchilage, G.; Basu, S. Metal Cations in G-Quadruplex Folding and Stability. Front. Chem. 2016, 4, 38. [Google Scholar] [CrossRef] [Green Version]
- Largy, E.; Mergny, J.-L.; Gabelica, V. Role of Alkali Metal Ions in G-Quadruplex Nucleic Acid Structure and Stability. In The Alkali Metal Ions: Their Role for Life. Metal Ions in Life Sciences; Sigel, A., Sigel, H., Sigel, R., Eds.; Springer: Berlin/Heidelberg, Germany, 2016; pp. 203–258. [Google Scholar]
- Pagano, B.; Mattia, C.A.; Cavallo, L.; Uesugi, S.; Giancola, C.; Fraternali, F. Stability and Cations Coordination of DNA and RNA 14-Mer G-Quadruplexes: A Multiscale Computational Approach. J. Phys. Chem. B 2008, 112, 12115–12123. [Google Scholar] [CrossRef]
- Guédin, A.; Gros, J.; Alberti, P.; Mergny, J.-L. How Long Is Too Long? Effects of Loop Size on G-Quadruplex Stability. Nucleic Acids Res. 2010, 38, 7858–7868. [Google Scholar] [CrossRef] [Green Version]
- Bugaut, A.; Balasubramanian, S. A Sequence-Independent Study of the Influence of Short Loop Lengths on the Stability and Topology of Intramolecular DNA G-Quadruplexes. Biochemistry 2008, 47, 689–697. [Google Scholar] [CrossRef] [Green Version]
- Marsico, G.; Chambers, V.S.; Sahakyan, A.B.; McCauley, P.; Boutell, J.M.; Di Antonio, M.; Balasubramanian, S. Whole Genome Experimental Maps of DNA G-Quadruplexes in Multiple Species. Nucleic Acids Res. 2019, 47, 3862–3874. [Google Scholar] [CrossRef] [Green Version]
- Zizza, P.; Cingolani, C.; Artuso, S.; Salvati, E.; Rizzo, A.; D’Angelo, C.; Porru, M.; Pagano, B.; Amato, J.; Randazzo, A.; et al. Intragenic G-Quadruplex Structure Formed in the Human CD133 and Its Biological and Translational Relevance. Nucleic Acids Res. 2016, 44, 1579–1590. [Google Scholar] [CrossRef]
- Kim, N. The Interplay between G-Quadruplex and Transcription. Curr. Med. Chem. 2019, 26, 2898–2917. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.L.; Kendrick, S. The I-Motif as a Molecular Target: More Than a Complementary DNA Secondary Structure. Pharmaceuticals 2021, 14, 96. [Google Scholar] [CrossRef] [PubMed]
- Dzatko, S.; Krafcikova, M.; Hänsel-Hertsch, R.; Fessl, T.; Fiala, R.; Loja, T.; Krafcik, D.; Mergny, J.-L.; Foldynova-Trantirkova, S.; Trantirek, L. Evaluation of the Stability of DNA I-Motifs in the Nuclei of Living Mammalian Cells. Angew. Chem. Int. Ed. 2018, 57, 2165–2169. [Google Scholar] [CrossRef] [Green Version]
- Zeraati, M.; Langley, D.B.; Schofield, P.; Moye, A.L.; Rouet, R.; Hughes, W.E.; Bryan, T.M.; Dinger, M.E.; Christ, D. I-Motif DNA Structures Are Formed in the Nuclei of Human Cells. Nat. Chem. 2018, 10, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Biffi, G.; Tannahill, D.; McCafferty, J.; Balasubramanian, S. Quantitative Visualization of DNA G-Quadruplex Structures in Human Cells. Nat. Chem. 2013, 5, 182–186. [Google Scholar] [CrossRef]
- Kosiol, N.; Juranek, S.; Brossart, P.; Heine, A.; Paeschke, K. G-Quadruplexes: A Promising Target for Cancer Therapy. Mol. Cancer 2021, 20, 40. [Google Scholar] [CrossRef] [PubMed]
- Amato, J.; Iaccarino, N.; Randazzo, A.; Novellino, E.; Pagano, B. Noncanonical DNA Secondary Structures as Drug Targets: The Prospect of the i-Motif. ChemMedChem 2014, 9, 2026–2030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, A.; Yadav, V.K.; Basundra, R.; Kumar, A.; Chowdhury, S. Evidence of Genome-Wide G4 DNA-Mediated Gene Expression in Human Cancer Cells. Nucleic Acids Res. 2009, 37, 4194–4204. [Google Scholar] [CrossRef] [Green Version]
- Shu, B.; Cao, J.; Kuang, G.; Qiu, J.; Zhang, M.; Zhang, Y.; Wang, M.; Li, X.; Kang, S.; Ou, T.-M.; et al. Syntheses and Evaluation of New Acridone Derivatives for Selective Binding of Oncogene C- Myc Promoter i-Motifs in Gene Transcriptional Regulation. Chem. Commun. 2018, 54, 2036–2039. [Google Scholar] [CrossRef]
- Brown, R.V.; Wang, T.; Chappeta, V.R.; Wu, G.; Onel, B.; Chawla, R.; Quijada, H.; Camp, S.M.; Chiang, E.T.; Lassiter, Q.R.; et al. The Consequences of Overlapping G-Quadruplexes and i-Motifs in the Platelet-Derived Growth Factor Receptor β Core Promoter Nuclease Hypersensitive Element Can Explain the Unexpected Effects of Mutations and Provide Opportunities for Selective Targeting Of. J. Am. Chem. Soc. 2017, 139, 7456–7475. [Google Scholar] [CrossRef]
- Satpathi, S.; Sappati, S.; Das, K.; Hazra, P. Structural Characteristics Requisite for the Ligand-Based Selective Detection of i-Motif DNA. Org. Biomol. Chem. 2019, 17, 5392–5399. [Google Scholar] [CrossRef]
- Masoud, S.S.; Nagasawa, K. I-Motif-Binding Ligands and Their Effects on the Structure and Biological Functions of I-Motif. Chem. Pharm. Bull. 2018, 66, 1091–1103. [Google Scholar] [CrossRef] [Green Version]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Saha, P.; Panda, D.; Dash, J. The Application of Click Chemistry for Targeting Quadruplex Nucleic Acids. Chem. Commun. 2019, 55, 731–750. [Google Scholar] [CrossRef]
- Tron, G.C.; Pirali, T.; Billington, R.A.; Canonico, P.L.; Sorba, G.; Genazzani, A.A. Click Chemistry Reactions in Medicinal Chemistry: Applications of the 1,3-Dipolar Cycloaddition between Azides and Alkynes. Med. Res. Rev. 2008, 28, 278–308. [Google Scholar] [CrossRef] [PubMed]
- Bonandi, E.; Christodoulou, M.S.; Fumagalli, G.; Perdicchia, D.; Rastelli, G.; Passarella, D. The 1,2,3-Triazole Ring as a Bioisostere in Medicinal Chemistry. Drug Discov. Today 2017, 22, 1572–1581. [Google Scholar] [CrossRef] [PubMed]
- Moorhouse, A.D.; Santos, A.M.; Gunaratnam, M.; Moore, M.; Neidle, S.; Moses, J.E. Stabilization of G-Quadruplex DNA by Highly Selective Ligands via Click Chemistry. J. Am. Chem. Soc. 2006, 128, 15972–15973. [Google Scholar] [CrossRef]
- Ritson, D.J.; Moses, J.E. A Fragment Based Click Chemistry Approach towards Hybrid G-Quadruplex Ligands: Design, Synthesis and Biophysical Evaluation. Tetrahedron 2012, 68, 197–203. [Google Scholar] [CrossRef] [Green Version]
- Sparapani, S.; Haider, S.M.; Doria, F.; Gunaratnam, M.; Neidle, S. Rational Design of Acridine-Based Ligands with Selectivity for Human Telomeric Quadruplexes. J. Am. Chem. Soc. 2010, 132, 12263–12272. [Google Scholar] [CrossRef]
- Drewe, W.C.; Neidle, S. Click Chemistry Assembly of G-Quadruplex Ligands Incorporating a Diarylurea Scaffold and Triazole Linkers. Chem. Commun. 2008, 5295. [Google Scholar] [CrossRef]
- Rodriguez, R.; Müller, S.; Yeoman, J.A.; Trentesaux, C.; Riou, J.-F.; Balasubramanian, S. A Novel Small Molecule That Alters Shelterin Integrity and Triggers a DNA-Damage Response at Telomeres. J. Am. Chem. Soc. 2008, 130, 15758–15759. [Google Scholar] [CrossRef] [Green Version]
- Müller, S.; Sanders, D.A.; Di Antonio, M.; Matsis, S.; Riou, J.-F.; Rodriguez, R.; Balasubramanian, S. Pyridostatin Analogues Promote Telomere Dysfunction and Long-Term Growth Inhibition in Human Cancer Cells. Org. Biomol. Chem. 2012, 10, 6537–6546. [Google Scholar] [CrossRef]
- Di Fonzo, S.; Amato, J.; D’Aria, F.; Caterino, M.; D’Amico, F.; Gessini, A.; Brady, J.W.; Cesàro, A.; Pagano, B.; Giancola, C. Ligand Binding to G-Quadruplex DNA: New Insights from Ultraviolet Resonance Raman Spectroscopy. Phys. Chem. Chem. Phys. 2020, 22, 8128–8140. [Google Scholar] [CrossRef]
- Pagano, A.; Iaccarino, N.; Abdelhamid, M.A.S.; Brancaccio, D.; Garzarella, E.U.; Di Porzio, A.; Novellino, E.; Waller, Z.A.E.; Pagano, B.; Amato, J.; et al. Common G-Quadruplex Binding Agents Found to Interact With i-Motif-Forming DNA: Unexpected Multi-Target-Directed Compounds. Front. Chem. 2018, 6, 281. [Google Scholar] [CrossRef]
- Hantzsch, A. Condensationsprodukte Aus Aldehydammoniak Und Ketonartigen Verbindungen. Ber. Dtsch. Chem. Ges. 1881, 14, 1637–1638. [Google Scholar] [CrossRef] [Green Version]
- Hantzsch, A. Ueber Die Synthese Pyridinartiger Verbindungen Aus Acetessigäther Und Aldehydammoniak. Justus Liebig’s Ann. Chem. 1882, 215, 1–82. [Google Scholar] [CrossRef] [Green Version]
- Tamaddon, F.; Razmi, Z.; Jafari, A.A. Synthesis of 3,4-Dihydropyrimidin-2(1H)-Ones and 1,4-Dihydropyridines Using Ammonium Carbonate in Water. Tetrahedron Lett. 2010, 51, 1187–1189. [Google Scholar] [CrossRef]
- Pagano, B.; Cosconati, S.; Gabelica, V.; Petraccone, L.; De Tito, S.; Marinelli, L.; La Pietra, V.; Saverio di Leva, F.; Lauri, I.; Trotta, R.; et al. State-of-the-Art Methodologies for the Discovery and Characterization of DNA G-Quadruplex Binders. Curr. Pharm. Des. 2012, 18, 1880–1899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, J.; Chen, D.; Jones, R.A.; Hurley, L.H.; Yang, D. NMR Solution Structure of the Major G-Quadruplex Structure Formed in the Human BCL2 Promoter Region. Nucleic Acids Res. 2006, 34, 5133–5144. [Google Scholar] [CrossRef] [PubMed]
- Kendrick, S.; Akiyama, Y.; Hecht, S.M.; Hurley, L.H. The I-Motif in the Bcl-2 P1 Promoter Forms an Unexpectedly Stable Structure with a Unique 8:5:7 Loop Folding Pattern. J. Am. Chem. Soc. 2009, 131, 17667–17676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambrus, A.; Chen, D.; Dai, J.; Jones, R.A.; Yang, D. Solution Structure of the Biologically Relevant G-Quadruplex Element in the Human c-MYC Promoter. Implications for G-Quadruplex Stabilization. Biochemistry 2005, 44, 2048–2058. [Google Scholar] [CrossRef]
- Dai, J.; Hatzakis, E.; Hurley, L.H.; Yang, D. I-Motif Structures Formed in the Human c-MYC Promoter Are Highly Dynamic–Insights into Sequence Redundancy and I-Motif Stability. PLoS ONE 2010, 5, e11647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, J.; Carver, M.; Yang, D. Polymorphism of Human Telomeric Quadruplex Structures. Biochimie 2008, 90, 1172–1183. [Google Scholar] [CrossRef] [Green Version]
- Pagano, B.; Amato, J.; Iaccarino, N.; Cingolani, C.; Zizza, P.; Biroccio, A.; Novellino, E.; Randazzo, A. Looking for Efficient G-Quadruplex Ligands: Evidence for Selective Stabilizing Properties and Telomere Damage by Drug-like Molecules. ChemMedChem 2015, 10, 640–649. [Google Scholar] [CrossRef]
- Phan, A.T.; Guéron, M.; Leroy, J.-L. The Solution Structure and Internal Motions of a Fragment of the Cytidine-Rich Strand of the Human Telomere. J. Mol. Biol. 2000, 299, 123–144. [Google Scholar] [CrossRef]
- Iaccarino, N.; Cheng, M.; Qiu, D.; Pagano, B.; Amato, J.; Di Porzio, A.; Zhou, J.; Randazzo, A.; Mergny, J.-L. Effects of Sequence and Base Composition on the CD and TDS Profiles of I-DNA. Angew. Chem. Int. Ed. 2021, 60, 10295–10303. [Google Scholar] [CrossRef]
- Wold, S.; Esbensen, K.; Geladi, P. Principal Component Analysis. Chemom. Intell. Lab. Syst. 1987, 2, 37–52. [Google Scholar] [CrossRef]
- Amato, J.; Morigi, R.; Pagano, B.; Pagano, A.; Ohnmacht, S.; De Magis, A.; Tiang, Y.-P.; Capranico, G.; Locatelli, A.; Graziadio, A.; et al. Toward the Development of Specific G-Quadruplex Binders: Synthesis, Biophysical, and Biological Studies of New Hydrazone Derivatives. J. Med. Chem. 2016, 59, 5706–5720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amato, J.; Miglietta, G.; Morigi, R.; Iaccarino, N.; Locatelli, A.; Leoni, A.; Novellino, E.; Pagano, B.; Capranico, G.; Randazzo, A. Monohydrazone Based G-Quadruplex Selective Ligands Induce DNA Damage and Genome Instability in Human Cancer Cells. J. Med. Chem. 2020, 63, 3090–3103. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, C.E.; Van Ert, N.A.; Agrawal, P.; Chawla, R.; Yang, D.; Hurley, L.H. Insight into the Complexity of the I-Motif and G-Quadruplex DNA Structures Formed in the KRAS Promoter and Subsequent Drug-Induced Gene Repression. J. Am. Chem. Soc. 2017, 139, 8522–8536. [Google Scholar] [CrossRef]
- Mayer, M.; Meyer, B. Characterization of Ligand Binding by Saturation Transfer Difference NMR Spectroscopy. Angew. Chem. Int. Ed. 1999, 38, 1784–1788. [Google Scholar] [CrossRef]
- Giancola, C.; Pagano, B. Energetics of Ligand Binding to G-Quadruplexes. Top. Curr. Chem. 2013, 330, 211–242. [Google Scholar] [PubMed]
- King, J.J.; Irving, K.L.; Evans, C.W.; Chikhale, R.V.; Becker, R.; Morris, C.J.; Peña Martinez, C.D.; Schofield, P.; Christ, D.; Hurley, L.H.; et al. DNA G-Quadruplex and i-Motif Structure Formation Is Interdependent in Human Cells. J. Am. Chem. Soc. 2020, 142, 20600–20604. [Google Scholar] [CrossRef] [PubMed]
- Böcker, R.H.; Guengerich, F.P. Oxidation of 4-Aryl- and 4-Alkyl-Substituted 2,6-Dimethyl-3,5-Bis(Alkoxycarbonyl)-1,4-Dihydropyridines by Human Liver Microsomes and Immunochemical Evidence for the Involvement of a Form of Cytochrome P-450. J. Med. Chem. 1986, 29, 1596–1603. [Google Scholar] [CrossRef]
- Loev, B.; Snader, K.M. The Hantzsch Reaction. I. Oxidative Dealkylation of Certain Dihydropyridines. J. Org. Chem. 1965, 30, 1914–1916. [Google Scholar] [CrossRef]
- Shahabi, D.; Amrollahi, M.A. Synthesis of Some Novel and Water-Soluble 2,4,6-Substituted 3,5-Dihydroxymethylpyridines. Bulg. Chem. Commun. 2014, 46, 264–268. [Google Scholar]
- Amato, J.; Pagano, A.; Cosconati, S.; Amendola, G.; Fotticchia, I.; Iaccarino, N.; Marinello, J.; De Magis, A.; Capranico, G.; Novellino, E.; et al. Discovery of the First Dual G-Triplex/G-Quadruplex Stabilizing Compound: A New Opportunity in the Targeting of G-Rich DNA Structures? Biochim. Biophys. Acta—Gen. Subj. 2017, 1861, 1271–1280. [Google Scholar] [CrossRef] [PubMed]
- Cantor, C.R.; Warshaw, M.M.; Shapiro, H. Oligonucleotide Interactions. III. Circular Dichroism Studies of the Conformation of Deoxyoligonucleolides. Biopolymers 1970, 9, 1059–1077. [Google Scholar] [CrossRef]
- Iaccarino, N.; Di Porzio, A.; Amato, J.; Pagano, B.; Brancaccio, D.; Novellino, E.; Leardi, R.; Randazzo, A. Assessing the Influence of pH and Cationic Strength on I-Motif DNA Structure. Anal. Bioanal. Chem. 2019, 411, 7473–7479. [Google Scholar] [CrossRef]
- Musumeci, D.; Amato, J.; Zizza, P.; Platella, C.; Cosconati, S.; Cingolani, C.; Biroccio, A.; Novellino, E.; Randazzo, A.; Giancola, C.; et al. Tandem Application of Ligand-Based Virtual Screening and G4-OAS Assay to Identify Novel G-Quadruplex-Targeting Chemotypes. Biochim. Biophys. Acta—Gen. Subj. 2017, 1861, 1341–1352. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.Y.; Chang, E.Y.C.; Lim, J.; Kwan, H.H.; Monchaud, D.; Yip, S.; Stirling, P.C.; Wong, J.M.Y. G-Quadruplexes Mark Alternative Lengthening of Telomeres. NAR Cancer 2021, 3, zcab031. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | ΔT1/2 (°C) 1 | ||||||
|---|---|---|---|---|---|---|---|
| Hairpin | Bcl-2 G4 | c-Myc G4 | Tel23 G4 | Bcl-2 iM | c-Myc iM | hTeloC iM | |
| 15a | 0 | 5.0 | 7.0 | 1.1 | −7.0 | −18.4 | −12 |
| 16a | −0.2 | 4.5 | 5.0 | 1.5 | −11.0 | −27.5 | −25.0 |
| 17a | −1.0 | 4.0 | 5.0 | 0 | −10.5 | −27.0 | −24.5 |
| 18a | 0.2 | 4.5 | 7.5 | 3.5 | −13.5 | −24.5 | −21.0 |
| 19a | 1.7 | 0.5 | 2.0 | 1.0 | −4.0 | −5.5 | −4.0 |
| 20a | 3.9 | 0.5 | 1.0 | 0 | −8.0 | −10.4 | −18.0 |
| 21a | 2.9 | 0 | 0 | 0 | 0 | −0.8 | 2.5 |
| 22a | −0.2 | 2.7 | 4.5 | 0.5 | −11.0 | −19.0 | −25.5 |
| 23a | 2.9 | 0 | 0.5 | 0.5 | −6.0 | −17.0 | −12.5 |
| 24a | −0.7 | 1.5 | 2.0 | −0.5 | −2.0 | −2.8 | 1.5 |
| 15b | −0.1 | 5.1 | 7.5 | 1.6 | −6.0 | −20 | −26.5 |
| 16b | −0.1 | 4.5 | 8.0 | 1.6 | −13.0 | −19.4 | −24.5 |
| 17b | −0.1 | 4.0 | 5.5 | 1.0 | −10 | −17.4 | −23.5 |
| 18b | −0.1 | 5.5 | 8.5 | 1.5 | −6.0 | −11.4 | −15.5 |
| 19b | 0 | 3.0 | 2.0 | 1.2 | −5.0 | −4.0 | −7.0 |
| 20b | 2.2 | 5.9 | 4.0 | 0.1 | −8.0 | −6.0 | −9.0 |
| 21b | 1.9 | 1.0 | 1.0 | 2.0 | 0 | −0.4 | 2.0 |
| 15c | −0.1 | 6.1 | 10.6 | 0 | −11.0 | −24 | −29 |
| 16c | −0.1 | 5.6 | 9.5 | 0.5 | −10.0 | −17.4 | −24.1 |
| 17c | −0.1 | 5.6 | 6.5 | 2.0 | −12.0 | −21.1 | −27.4 |
| 18c | 1.1 | 7.6 | 10.5 | 1.6 | −10.5 | −20.0 | −25.5 |
| 19c | −1.1 | 5.1 | 4.2 | −1.0 | −3.0 | −3.0 | −10.0 |
| 20c | 3.0 | 7.1 | 6.8 | 0 | −9.0 | −2.9 | −9.0 |
| 21c | 1.9 | 0 | 0 | 2.0 | 0 | −0.9 | 1.5 |
| 15d | 0 | 8.8 | 9.8 | −1.0 | −5.0 | −4.9 | −7.0 |
| 16d | −0.3 | 8.0 | 8.1 | 2.0 | −5.0 | −4.9 | −7.0 |
| 17d | 1.0 | 9.0 | 6.9 | 2.0 | −8.0 | −7.0 | −8.9 |
| 18d | 0 | 10.1 | 9.8 | −1.0 | −8.0 | −8.0 | −9.0 |
| 19d | 0.9 | 4.2 | 2.8 | −2.0 | −5.0 | −5.0 | −7.0 |
| 20d | 2.9 | 8.0 | 8.2 | 0 | −8.0 | −5.0 | −9.0 |
| 21d | 1.9 | 2.0 | 0.8 | −0.8 | −1.0 | 0 | −1.0 |
| Compound | Kb (×106 M−1) 1 | ||
|---|---|---|---|
| c-Myc G4 | c-Myc iM | hTeloC iM | |
| 20a | 2.3 | 3.1 | 3.1 |
| 23a | 2.2 | 3.2 | 3.1 |
| 15c | 2.5 | 3.4 | 3.2 |
| 18c | 3.1 | 2.8 | 2.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Porzio, A.; Galli, U.; Amato, J.; Zizza, P.; Iachettini, S.; Iaccarino, N.; Marzano, S.; Santoro, F.; Brancaccio, D.; Carotenuto, A.; et al. Synthesis and Characterization of Bis-Triazolyl-Pyridine Derivatives as Noncanonical DNA-Interacting Compounds. Int. J. Mol. Sci. 2021, 22, 11959. https://doi.org/10.3390/ijms222111959
Di Porzio A, Galli U, Amato J, Zizza P, Iachettini S, Iaccarino N, Marzano S, Santoro F, Brancaccio D, Carotenuto A, et al. Synthesis and Characterization of Bis-Triazolyl-Pyridine Derivatives as Noncanonical DNA-Interacting Compounds. International Journal of Molecular Sciences. 2021; 22(21):11959. https://doi.org/10.3390/ijms222111959
Chicago/Turabian StyleDi Porzio, Anna, Ubaldina Galli, Jussara Amato, Pasquale Zizza, Sara Iachettini, Nunzia Iaccarino, Simona Marzano, Federica Santoro, Diego Brancaccio, Alfonso Carotenuto, and et al. 2021. "Synthesis and Characterization of Bis-Triazolyl-Pyridine Derivatives as Noncanonical DNA-Interacting Compounds" International Journal of Molecular Sciences 22, no. 21: 11959. https://doi.org/10.3390/ijms222111959
APA StyleDi Porzio, A., Galli, U., Amato, J., Zizza, P., Iachettini, S., Iaccarino, N., Marzano, S., Santoro, F., Brancaccio, D., Carotenuto, A., De Tito, S., Biroccio, A., Pagano, B., Tron, G. C., & Randazzo, A. (2021). Synthesis and Characterization of Bis-Triazolyl-Pyridine Derivatives as Noncanonical DNA-Interacting Compounds. International Journal of Molecular Sciences, 22(21), 11959. https://doi.org/10.3390/ijms222111959