
iTRAQ-Based Proteomics Analysis of Response to Solanum tuberosum Leaves Treated with the Plant Phytotoxin Thaxtomin A

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
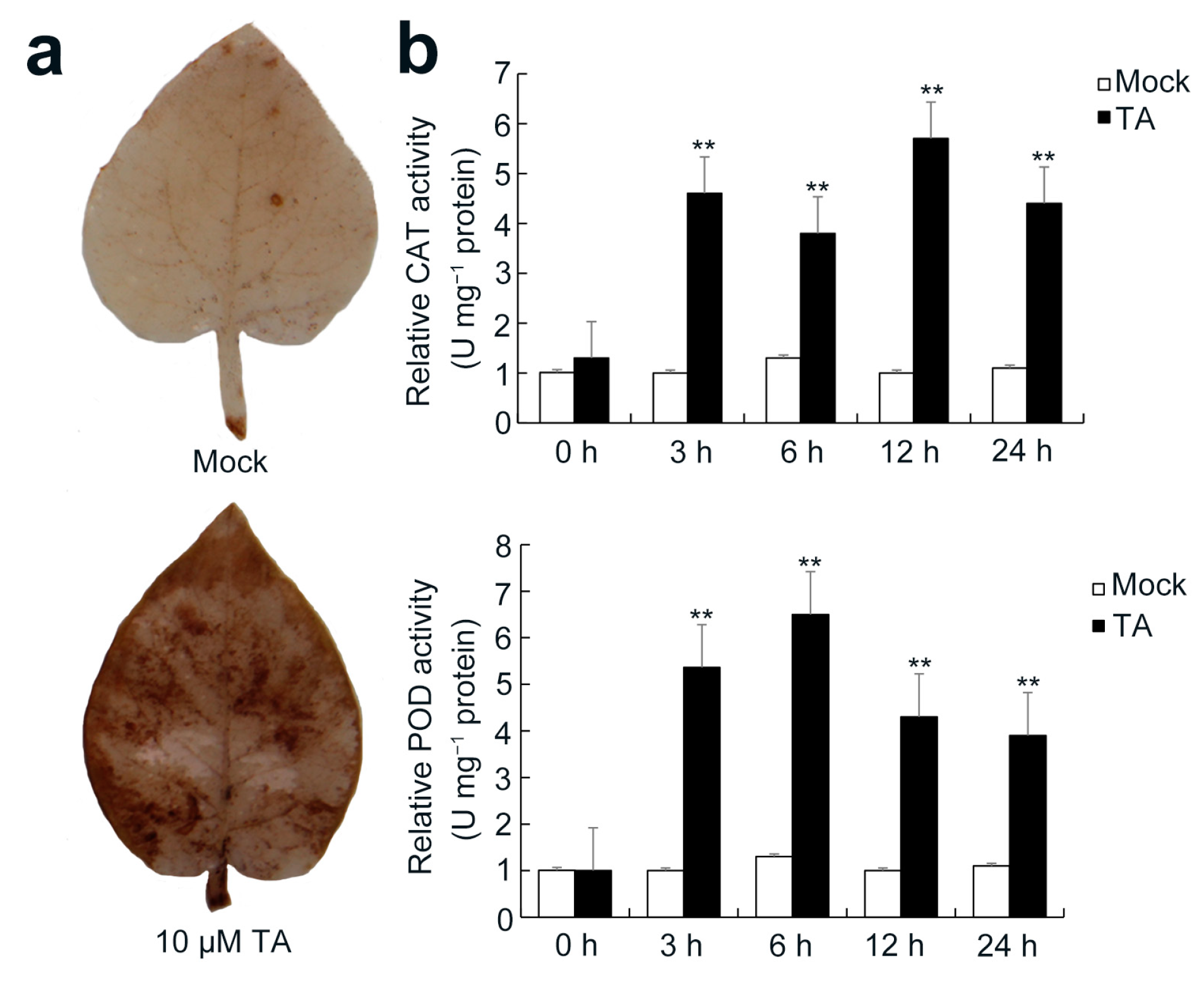
2.1. Effects of TA Treatment on Potato Leaves
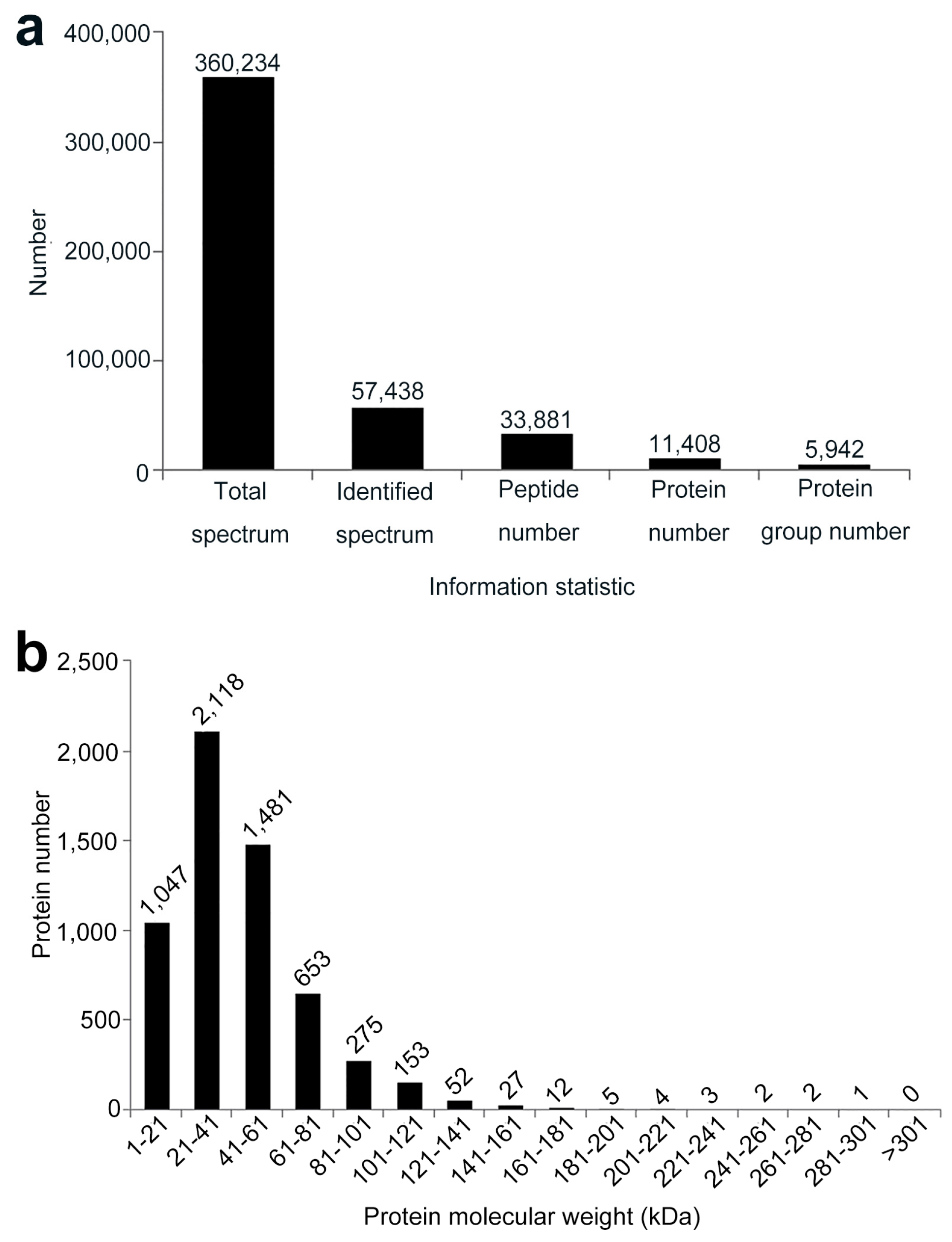
2.2. Quantitative Identification of Proteins
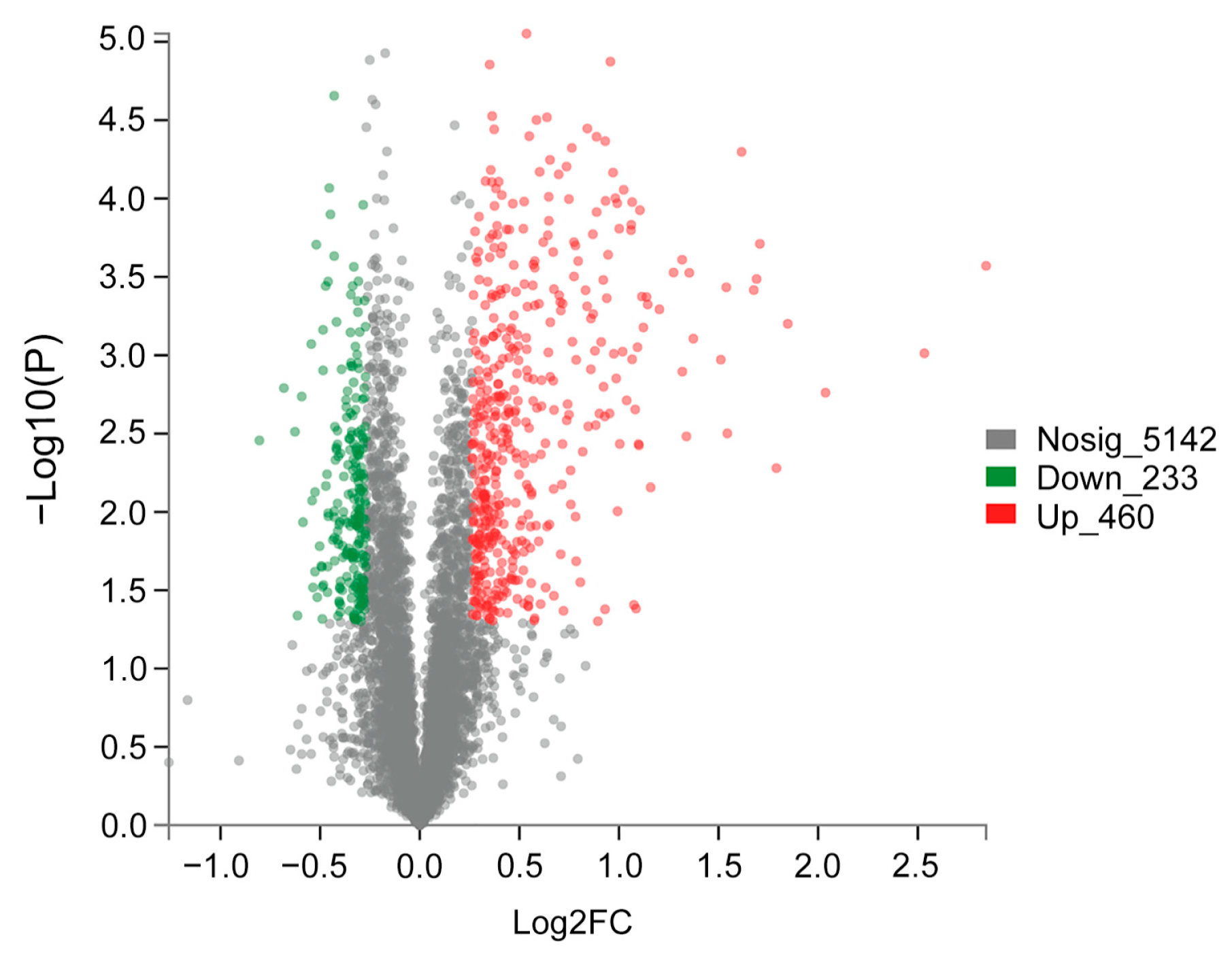
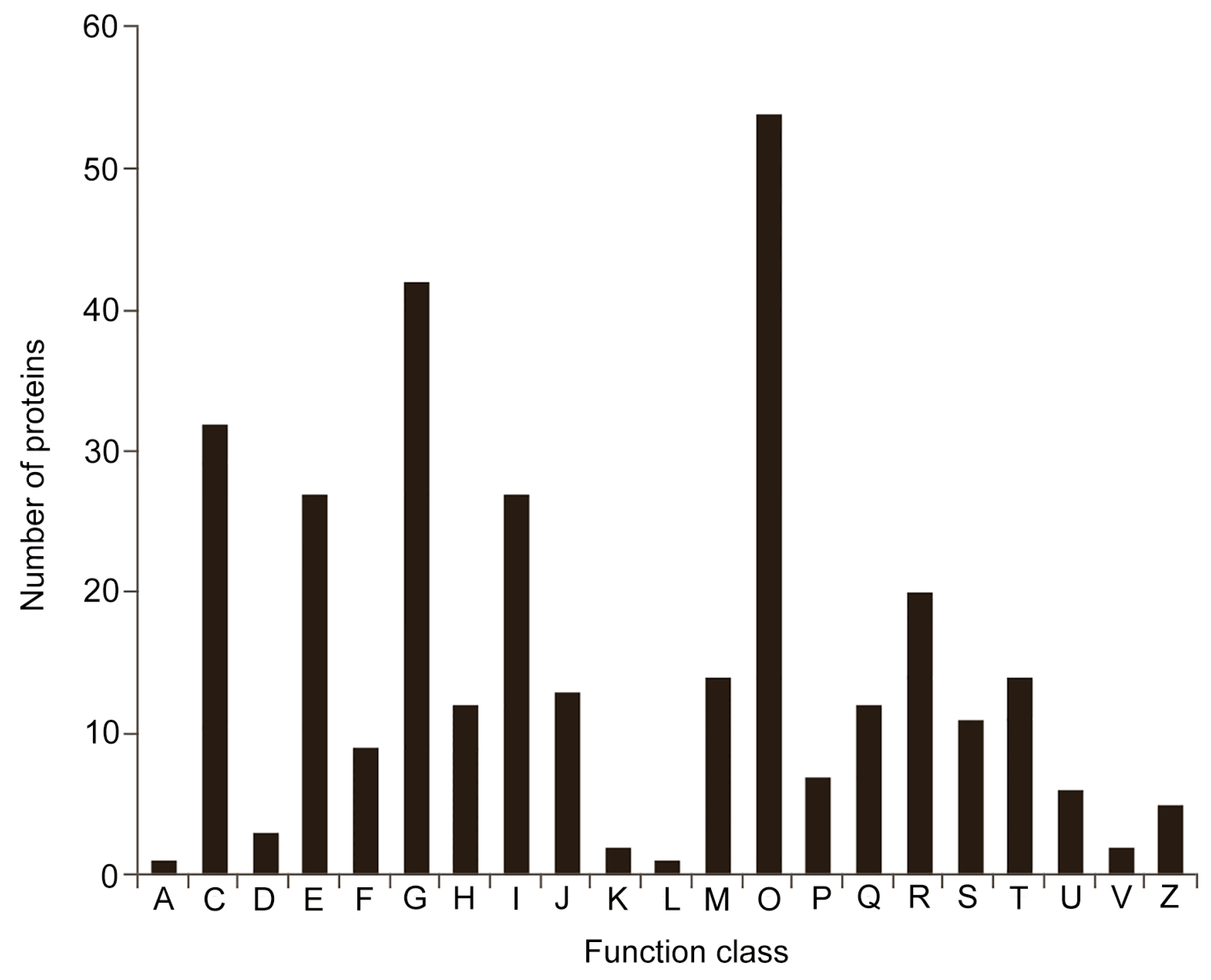
2.3. Analysis of DEPs
2.4. Validation of DEPs by qRT-PCR
2.5. Transcriptional Level Analysis of Selected DEPs
2.6. Detection of Antioxidant System Response after TA Treatment
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Thaxtomin A Preparation and Treatment
4.3. Protein Extraction and Quantification
4.4. Protein Digestion, iTRAQ Labeling, and Liquid Chromatography–Tandem Mass Spectrometry (LC–MS/MS)
4.5. Database Search and Proteomic Analysis
4.6. qRT-PCR Analysis
4.7. Histochemical Detection of Hydrogen Peroxide
4.8. POD and CAT Activities
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Knapp, S.; Vorontsova, M.S.; Särkinen, T. Dichotomous keys to the species of Solanum L. (Solanaceae) in continental Africa, Madagascar (incl. the Indian Ocean islands), Macaronesia and the Cape Verde Islands. PhytoKeys 2019, 127, 39–76. [Google Scholar] [CrossRef] [Green Version]
- Aliche, E.B.; Theeuwen, T.P.J.M.; Oortwijn, M.; Visser, R.G.F.; van der Linden, C.G. Carbon partitioning mechanisms in potato under drought stress. Plant Physiol. Biochem. 2020, 146, 211–219. [Google Scholar] [CrossRef]
- Braun, S.; Gevens, A.; Charkowski, A.; Allen, C.; Jansky, S. Potato common scab: A review of the causal pathogens, management practices, varietal resistance screening methods, and host resistance. Am. J. Potato Res. 2017, 94, 283–296. [Google Scholar] [CrossRef]
- Dees, M.W.; Lysøe, E.; Brurberg, M.B.; Somervuo, P.; Almvik, M.; Valkonen, J.P.T. Global gene expression in the common scab pathogen, Streptomyces scabies, exposed to potato microtubers. Ann. Appl. Biol. 2014, 165, 43–52. [Google Scholar] [CrossRef]
- Hiltunen, L.H.; Weckman, A.; Ylhainen, A.; Rita, H.; Richter, E.; Valkonen, J.P.T. Responses of potato cultivars to the common scab pathogens, Streptomyces scabies and S. turgidiscabies. Ann. Appl. Biol. 2005, 146, 395–403. [Google Scholar] [CrossRef]
- Loria, R.; Kers, J.; Joshi, M. Evolution of plant pathogenicity in Streptomyces. Annu. Rev. Phytopathol. 2006, 44, 469–487. [Google Scholar] [CrossRef] [PubMed]
- Goyer, C.; Vachon, J.; Beaulieu, C. Pathogenicity of Streptomyces scabies mutants altered in thaxtomin A production. Phytopathology 1998, 88, 442–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, R.R.; Lawrence, C.H.; Clark, M.C.; Calhoun, L.A. Isolation and characterization of phytotoxins associated with Streptomyces scabies. J. Chem. Soc. Chem. Commun. 1989, 13, 849–850. [Google Scholar] [CrossRef]
- Lawrence, C.H.; Clark, M.C.; King, R.R. Induction of common scab symptoms in aseptically cultured potato tubers by the vivotoxin, thaxtomin. Phytopathology 1990, 80, 606–608. [Google Scholar] [CrossRef]
- Loria, R.; Bukhalid, R.A.; Fry, B.A.; King, R.R. Plant pathogenicity in the genus Streptomyces. Plant Dis. 1997, 81, 836–846. [Google Scholar] [CrossRef] [Green Version]
- Healy, F.G.; Wach, M.; Krasnoff, S.B.; Gibson, D.M.; Loria, R. The txtAB genes of the plant pathogen Streptomyces acidiscabies encode a peptide synthetase required for phytotoxin thaxtomin A production and pathogenicity. Mol. Microbiol. 2000, 38, 794–804. [Google Scholar] [CrossRef] [PubMed]
- Healy, F.G.; Krasnoff, S.B.; Wach, M.; Gibson, D.M.; Loria, R. Involvement of a cytochrome P450 monooxygenase in thaxtomin A biosynthesis by Streptomyces acidiscabies. J. Bacteriol. 2002, 184, 2019–2029. [Google Scholar] [CrossRef] [Green Version]
- Kers, J.A.; Wach, M.J.; Krasnoff, S.B.; Widom, J.; Cameron, K.D.; Bukhalid, R.A.; Gibson, D.M.; Crane, B.R.; Loria, R. Nitration of a peptide phytotoxin by bacterial nitric oxide synthase. Nature 2004, 429, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Barry, S.M.; Kers, J.A.; Johnson, E.G.; Song, L.; Aston, P.R.; Patel, B.; Krasnoff, S.B.; Crane, B.R.; Gibson, D.M.; Loria, R.; et al. Cytochrome P450-catalyzed L-tryptophan nitration in thaxtomin phytotoxin biosynthesis. Nat. Chem. Biol. 2012, 8, 814–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Liu, J.; Adekunle, D.; Bown, L.; Tahlan, K.; Bignell, D.R.D. TxtH is a key component of the thaxtomin biosynthetic machinery in the potato common scab pathogen Streptomyces scabies. Mol. Plant Pathol. 2019, 20, 1379–1393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, D.; Grau, B.L.; Clark, C.A.; Taylor, C.M.; Loria, R.; Pettis, G.S. Evidence that thaxtomin C is an apathogenicity determinant of Streptomyces ipomoeae, the causative agent of Streptomyces soil rot disease of sweet potato. Mol. Plant-Microbe Interact. 2012, 25, 393–401. [Google Scholar] [CrossRef] [Green Version]
- Kers, J.A.; Cameron, K.D.; Joshi, M.V.; Bukhalid, R.A.; Morello, J.E.; Wach, M.J.; Gibson, D.M.; Loria, R. A large, mobile pathogenicity island confers plant pathogenicity on Streptomyces species. Mol. Microbiol. 2005, 55, 1025–1033. [Google Scholar] [CrossRef]
- Loria, R.; Bignell, D.R.D.; Moll, S.; Huguet-Tapia, J.C.; Joshi, M.V.; Johnson, E.G.; Seipke, R.F.; Gibson, D.M. Thaxtomin biosynthesis: The path to plant pathogenicity in the genus Streptomyces. Antonie van Leeuwenhoek 2008, 94, 3–10. [Google Scholar] [CrossRef]
- Joshi, M.V.; Bignell, D.R.D.; Johnson, E.G.; Sparks, J.P.; Gibson, D.M.; Loria, R. The AraC/XylS regulator TxtR modulates thaxtomin biosynthesis and virulence in Streptomyces scabies. Mol. Microbiol. 2007, 66, 633–642. [Google Scholar] [CrossRef]
- van Wezel, G.P.; McDowall, K.J. The regulation of the secondary metabolism of Streptomyces: New links and experimental advances. Nat. Prod. Rep. 2011, 28, 1311–1333. [Google Scholar] [CrossRef]
- Dees, M.W.; Wanner, L.A. In search of better management of potato common scab. Potato Res. 2012, 55, 249–268. [Google Scholar] [CrossRef]
- Duval, I.; Beaudoin, N. Transcriptional profiling in response to inhibition of cellulose synthesis by thaxtomin A and isoxaben in Arabidopsis thaliana suspension cells. Plant Cell Rep. 2009, 28, 811–830. [Google Scholar] [CrossRef] [PubMed]
- Errakhi, R.; Dauphin, A.; Meimoun, P.; Lehner, A.; Reboutier, D.; Vatsa, P.; Briand, J.; Madiona, K.; Rona, J.P.; Barakate, M.; et al. An early Ca2+ influx is a prerequisite to thaxtomin A-induced cell death in Arabidopsis thaliana cells. J. Exp. Bot. 2008, 59, 4259–4270. [Google Scholar] [CrossRef] [Green Version]
- Tegg, R.S.; Shabala, S.; Cuin, T.A.; Wilson, C.R. Mechanisms of thaxtomin A-induced root toxicity revealed by a thaxtomin A sensitive Arabidopsis mutant (ucu2-2/gi-2). Plant Cell Rep. 2016, 35, 347–356. [Google Scholar] [CrossRef]
- Francis, I.M.; Jourdan, S.; Fanara, S.; Loria, R.; Rigali, S. The cellobiose sensor CebR is the gatekeeper of Streptomyces scabies pathogenicity. mBio 2015, 6, e02018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jourdan, S.; Francis, I.M.; Kim, M.J.; Salazar, J.J.C.; Planckaert, S.; Frère, J.M.; Matagne, A.; Kerff, F.; Devreese, B.; Loria, R.; et al. The CebE/MsiK transporter is a doorway to the cello-oligosaccharide-mediated induction of Streptomyces scabies pathogenicity. Sci. Rep. 2016, 6, 27144. [Google Scholar] [CrossRef]
- Jourdan, S.; Francis, I.M.; Deflandre, B.; Tenconi, E.; Riley, J.; Planckaert, S.; Tocquin, P.; Martinet, L.; Devreese, B.; Loria, R.; et al. Contribution of the beta-glucosidase BglC to the onset of the pathogenic lifestyle of Streptomyces scabies. Mol. Plant Pathol. 2018, 19, 1480–1490. [Google Scholar] [CrossRef] [Green Version]
- Lauzier, A.; Simao-Beaunoir, A.M.; Bourassa, S.; Poirier, G.G.; Talbot, B.; Beaulieu, C. Effect of potato suberin on Streptomyces scabies proteome. Mol. Plant Pathol. 2008, 9, 753–762. [Google Scholar] [CrossRef]
- Joshi, M.V.; Mann, S.G.; Antelmann, H.; Widdick, D.A.; Fyans, J.K.; Chandra, G.; Hutchings, M.I.; Toth, I.; Hecker, M.; Loria, R.; et al. The twin arginine protein transport pathway exports multiple virulence proteins in the plant pathogen Streptomyces scabies. Mol. Microbiol. 2010, 77, 252–271. [Google Scholar] [CrossRef] [Green Version]
- Komeil, D.; Padilla-Reynaud, R.; Lerat, S.; Simao-Beaunoir, A.M.; Beaulieu, C. Comparative secretome analysis of Streptomyces scabiei during growth in the presence or absence of potato suberin. Proteome Sci. 2014, 12, 35. [Google Scholar] [CrossRef] [Green Version]
- Planckaert, S.; Jourdan, S.; Francis, I.M.; Deflandre, B.; Rigali, S.; Devreese, B. Proteomic response to thaxtomin phytotoxin elicitor cellobiose and to deletion of cellulose utilization regulator CebR in Streptomyces scabies. J. Proteome Res. 2018, 17, 3837–3852. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Bi, X.; An, M.; Xia, Z.; Wu, Y. iTRAQ-based proteomic analysis of watermelon fruits in response to Cucumber green mottle mosaic virus infection. Int. J. Mol. Sci. 2020, 21, 2541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, X.; Bandow, J.; Sherman, J.; Baker, J.D.; Brown, P.W.; McDowell, M.T.; Molloy, M.P. iTRAQ experimental design for plasma biomarker discovery. J. Proteome Res. 2008, 7, 2952–2958. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.W.; Wang, G.; Baek, S.J.; Shen, R.F. Comparative study of three proteomic quantitative methods, DIGE, cICAT, and iTRAQ, using 2D gel or LC-MALDITOF/TOF. J. Proteome Res. 2006, 5, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Wang, Z.; Guan, X.; Hu, Z.; Zhang, Z.; Zheng, J.; Lu, Y. Proteomic analysis of Potentilla fruticosa L. leaves by iTRAQ reveals responses to heat stress. PLoS ONE 2017, 12, e0182917. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Chen, Z.; Leng, P.; Hu, Z. iTRAQ-based quantitative proteomic analysis of the response of Hylotelephium erythrostictum leaves to salt stress. Sci. Hortic. 2020, 264, 109190. [Google Scholar] [CrossRef]
- Alvarez, S.; Roy Choudhury, S.; Pandey, S. Comparative quantitative proteomics analysis of the ABA response of roots of drought-sensitive and drought-tolerant wheat varieties identifies proteomic signatures of drought adaptability. J. Proteome Res. 2014, 13, 1688–1701. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Wang, L.; Zhao, M.; Gu, S.; Wang, C.; Zhao, J.; Tang, Z.; Gao, H.; Zhang, L.; Fu, L.; et al. iTRAQ proteomics reveals the regulatory response to Magnaporthe oryzae in durable resistant vs. susceptible rice genotypes. PLoS ONE 2020, 15, e0227470. [Google Scholar] [CrossRef]
- Zhang, M.; Cheng, S.T.; Wang, H.Y.; Wu, J.H.; Luo, Y.M.; Wang, Q.; Wang, F.X.; Xia, G.X. iTRAQ-based proteomic analysis of defence responses triggered by the necrotrophic pathogen Rhizoctonia solani in cotton. J. Proteom. 2017, 152, 226–235. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.X.; Luo, Y.M.; Ye, Z.Q.; Cao, X.; Liang, J.N.; Wang, Q.; Wu, Y.; Wu, J.H.; Wang, H.Y.; Zhang, M.; et al. iTRAQ-based proteomics analysis of autophagy-mediated immune responses against the vascular fungal pathogen Verticillium dahliae in Arabidopsis. Autophagy 2018, 14, 598–618. [Google Scholar] [CrossRef] [Green Version]
- Nejat, N.; Mantri, N. Plant immune system: Crosstalk between responses to biotic and abiotic stresses the missing link in understanding plant defence. Curr. Issues Mol. Biol. 2017, 23, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodds, P.N.; Rathjen, J.P. Plant immunity: Towards an integrated view of plant-pathogen interactions. Nat. Rev. Genet. 2010, 11, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, K.; Katagiri, F. Comparing signaling mechanisms engaged in pattern-triggered and effector-triggered immunity. Curr. Opin. Plant Biol. 2010, 13, 459–465. [Google Scholar] [CrossRef]
- Robert-Seilaniantz, A.; Grant, M.; Jones, J.D.G. Hormone crosstalk in plant disease and defense: More than just jasmonate-salicylate antagonism. Annu. Rev. Phytopathol. 2011, 49, 317–343. [Google Scholar] [CrossRef] [PubMed]
- Piasecka, A.; Jedrzejczak-Rey, N.; Bednarek, P. Secondary metabolites in plant innate immunity: Conserved function of divergent chemicals. New Phytol. 2015, 206, 948–964. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhou, J.M. Plant immunity triggered by microbial molecular signatures. Mol. Plant 2010, 3, 783–793. [Google Scholar] [CrossRef] [Green Version]
- Bigeard, J.; Colcombet, J.; Hirt, H. Signaling mechanisms in pattern-triggered immunity (PTI). Mol. Plant 2015, 8, 521–539. [Google Scholar] [CrossRef] [Green Version]
- Ramirez-Prado, J.S.; Abulfaraj, A.A.; Rayapuram, N.; Benhamed, M.; Hirt, H. Plant immunity: From signaling to epigenetic control of defense. Trends Plant Sci. 2018, 23, 833–844. [Google Scholar] [CrossRef]
- Van Breusegem, F.; Dat, J.F. Reactive oxygen species in plant cell death. Plant Physiol. 2006, 141, 384–390. [Google Scholar] [CrossRef] [Green Version]
- del Río, L.A. ROS and RNS in plant physiology: An overview. J. Exp. Bot. 2015, 66, 2827–2837. [Google Scholar] [CrossRef] [Green Version]
- Camejo, D.; Guzmán-Cedeño, Á.; Moreno, A. Reactive oxygen species, essential molecules, during plant-pathogen interactions. Plant Physiol. Biochem. 2016, 103, 10–23. [Google Scholar] [CrossRef]
- Vivancos, P.D.; Dong, Y.; Ziegler, K.; Markovic, J.; Pallardó, F.V.; Pellny, T.K.; Verrier, P.J.; Foyer, C.H. Recruitment of glutathione into the nucleus during cell proliferation adjusts whole-cell redox homeostasis in Arabidopsis thaliana and lowers the oxidative defence shield. Plant J. 2010, 64, 825–838. [Google Scholar] [CrossRef] [PubMed]
- Camejo, D.; Martí, M.C.; Jiménez, A.; Cabrera, J.C.; Olmos, E.; Sevilla, F. Effect of oligogalacturonides on root length, extracellular alkalinization and O2−-accumulation in alfalfa. J. Plant Physiol. 2011, 168, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Waszczak, C.; Carmody, M.; Kangasjärvi, J. Reactive oxygen species in plant signaling. Annu. Rev. Plant Biol. 2018, 69, 209–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gill, S.S.; Tuteja, N. Reactive oxygen species and antioxidant machinery in abiotic stress tolerance in crop plants. Plant Physiol. Biochem. 2010, 48, 909–930. [Google Scholar] [CrossRef] [PubMed]
- Nouman, W.; Anwar, F.; Gull, T.; Newton, A.; Rosa, E.; Domínguez-Perles, R. Profiling of polyphenolics, nutrients and antioxidant potential of germplasm’s leaves from seven cultivars of Moringa oleifera Lam. Ind. Crops Prod. 2016, 83, 166–176. [Google Scholar] [CrossRef]
- Gilroy, S.; Białasek, M.; Suzuki, N.; Górecka, M.; Devireddy, A.R.; Karpinski, S.; Mittler, R. ROS, calcium, and electric signals: Key mediators of rapid systemic signaling in plants. Plant Physiol. 2016, 171, 1606–1615. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Song, C.P.; Wang, B.; Zhou, J.; Kangasjärvi, J.; Zhu, J.K.; Gong, Z. Reactive oxygen species signaling and stomatal movement in plant responses to drought stress and pathogen attack. J. Integr. Plant Biol. 2018, 60, 805–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kao, C.H. Mechanisms of salt tolerance in rice plants: Cell wall-related genes and expansins. J. Taiwan Agric. Res. 2017, 66, 87–93. [Google Scholar]
- Hauck, P.; Thilmony, R.; He, S.Y. A Pseudomonas syringae type III effector suppresses cell wall-based extracellular defense in susceptible Arabidopsis plants. Proc. Natl. Acad. Sci. USA 2003, 100, 8577–8582. [Google Scholar] [CrossRef] [Green Version]
- Vellosillo, T.; Vicente, J.; Kulasekaran, S.; Hamberg, M.; Castresana, C. Emerging complexity in reactive oxygen species production and signaling during the response of plants to pathogens. Plant Physiol. 2010, 154, 444–448. [Google Scholar] [CrossRef] [Green Version]
- López-Cruz, J.; Crespo-Salvador, Ó.; Fernández-Crespo, E.; García-Agustín, P.; González-Bosch, C. Absence of Cu-Zn superoxide dismutase BCSOD1 reduces Botrytis cinerea virulence in Arabidopsis and in tomato plants, which reveals interplay among ROS, callose and signaling pathways. Mol. Plant Pathol. 2017, 18, 16–31. [Google Scholar] [CrossRef] [PubMed]
- Awwad, F.; Bertrand, G.; Grandbois, M.; Beaudoin, N. Reactive oxygen species alleviate cell death induced by thaxtomin A in Arabidopsis thaliana cell cultures. Plants 2019, 8, 332. [Google Scholar] [CrossRef] [Green Version]
- Goyer, C.; Charest, P.M.; Toussaint, V.; Beaulieu, C. Ultrastructural effects of thaxtomin A produced by Streptomyces scabies on mature potato tuber tissues. Can. J. Bot. 2000, 78, 374–380. [Google Scholar]
- King, R.R.; Calhoun, L.A. The thaxtomin phytotoxins: Sources, synthesis, biosynthesis, biotransformation and biological activity. Phytochemistry 2009, 70, 833–841. [Google Scholar] [CrossRef]
- Scheible, W.R.; Fry, B.; Kochevenko, A.; Schindelasch, D.; Zimmerli, L.; Somerville, S.; Loria, R.; Somerville, C.R. An Arabidopsis mutant resistant to thaxtomin A, a cellulose synthesis inhibitor from Streptomyces species. Plant Cell 2003, 15, 1781–1794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tegg, R.S.; Shabala, S.N.; Cuin, T.A.; Davies, N.W.; Wilson, C.R. Enhanced resistance to the cellulose biosynthetic inhibitors, thaxtomin A and isoxaben in Arabidopsis thaliana mutants, also provides specific co-resistance to the auxin transport inhibitor, 1-NPA. BMC Plant Biol. 2013, 13, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bischoff, V.; Cookson, S.J.; Wu, S.; Scheible, W.R. Thaxtomin A affects CESA-complex density, expression of cell wall genes, cell wall composition, and causes ectopic lignification in Arabidopsis thaliana seedlings. J. Exp. Bot. 2009, 60, 955–965. [Google Scholar] [CrossRef] [Green Version]
- Caretto, S.; Linsalata, V.; Colella, G.; Mita, G.; Lattanzio, V. Carbon fluxes between primary metabolism and phenolic pathway in plant tissues under stress. Int. J. Mol. Sci. 2015, 16, 26378–26394. [Google Scholar] [CrossRef] [Green Version]
- Wach, M.J.; Krasnoff, S.B.; Loria, R.; Gibson, D.M. Effect of carbohydrates on the production of thaxtomin A by Streptomyces acidiscabies. Arch. Microbiol. 2007, 188, 81–88. [Google Scholar] [CrossRef]
- Pratelli, R.; Pilot, G. Regulation of amino acid metabolic enzymes and transporters in plants. J. Exp. Bot. 2014, 65, 5535–5556. [Google Scholar] [CrossRef]
- Jia, M.; Wu, H.; Clay, K.L.; Jung, R.; Larkins, B.A.; Gibbon, B.C. Identification and characterization of lysine-rich proteins and starch biosynthesis genes in the opaque2 mutant by transcriptional and proteomic analysis. BMC Plant Biol. 2013, 13, 60. [Google Scholar] [CrossRef] [Green Version]
- Rocha, M.; Licausi, F.; Araújom, W.L.; Nunes-Nesim, A.; Sodek, L.; Fernie, A.R.; van Dongen, J.T. Glycolysis and the tricarboxylic acid cycle are linked by alanine aminotransferase during hypoxia induced by waterlogging of Lotus japonicus. Plant Physiol. 2010, 152, 1501–1513. [Google Scholar] [CrossRef] [Green Version]
- Zhong, M.; Liu, X.; Liu, F.; Ren, Y.; Wang, Y.; Zhu, J.; Teng, X.; Duan, E.; Wang, F.; Zhang, H.; et al. FLOURY ENDOSPERM12 encoding alanine aminotransferase 1 regulates carbon and nitrogen metabolism in rice. J. Plant Biol. 2019, 62, 61–73. [Google Scholar] [CrossRef]
- Wang, W.; Xu, M.; Wang, G.; Galili, G. New insights into the metabolism of aspartate-family amino acids in plant seeds. Plant Reprod. 2018, 31, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Lauzier, A.; Goyer, C.; Ruest, L.; Brzezinski, R.; Crawford, D.L.; Beaulieu, C. Effect of amino acids on thaxtomin A biosynthesis by Streptomyces scabies. Can. J. Microbiol. 2002, 48, 359–364. [Google Scholar] [CrossRef]
- Kiyota, E.; Pena, I.A.; Arruda, P. The saccharopine pathway in seed development and stress response of maize. Plant Cell Environ. 2015, 38, 2450–2461. [Google Scholar] [CrossRef] [PubMed]
- Bernsdorff, F.; Döring, A.C.; Gruner, K.; Schuck, S.; Bräutigam, A.; Zeier, J. Pipecolic acid orchestrates plant systemic acquired resistance and defense priming via salicylic acid-dependent and -independent pathways. Plant Cell 2016, 28, 102–129. [Google Scholar] [CrossRef] [Green Version]
- Arruda, P.; Barreto, P. Lysine catabolism through the saccharopine pathway: Enzymes and intermediates involved in plant responses to abiotic and biotic stress. Front. Plant Sci. 2020, 11, 587. [Google Scholar] [CrossRef] [PubMed]
- Zeier, J. New insights into the regulation of plant immunity by amino acid metabolic pathways. Plant Cell Environ. 2013, 36, 2085–2103. [Google Scholar] [CrossRef]
- Wilson, C.R.; Luckman, G.A.; Tegg, R.S.; Yuan, Z.Q.; Wilson, A.J.; Eyles, A.; Conner, A.J. Enhanced resistance to common scab of potato through somatic cell selection in cv. Iwa with the phytotoxin thaxtomin A. Plant Pathol. 2009, 58, 137–144. [Google Scholar] [CrossRef]
- Brochu, V.; Girard-Martel, M.; Duval, I.; Lerat, S.; Grondin, G.; Domingue, O.; Beaulieu, C.; Beaudoin, N. Habituation to thaxtomin A in hybrid poplar cell suspensions provides enhanced and durable resistance to inhibitors of cellulose synthesis. BMC Plant Biol. 2010, 10, 272. [Google Scholar] [CrossRef] [Green Version]
- Hiltunen, L.H.; Alanen, M.; Laakso, I.; Kangas, A.; Virtanen, E.; Valkonen, J.P.T. Elimination of common scab sensitive progeny from a potato breeding population using thaxtomin A as a selective agent. Plant Pathol. 2011, 60, 426–435. [Google Scholar] [CrossRef]
- Murashige, T.; Skoog, F. A revised medium for rapid growth and bioassays with tobacco tissue culture. Physiol. Plant 1962, 15, 473–497. [Google Scholar] [CrossRef]
- Mo, S.; Zhang, Y.; Wang, X.; Yang, J.; Sun, Z.; Zhang, D.; Chen, B.; Wang, G.; Ke, H.; Liu, Z.; et al. Cotton GhSSI2 isoforms from the stearoyl acyl carrier protein fatty acid desaturase family regulate Verticillium wilt resistance. Mol. Plant Pathol. 2021, 22, 1041–1056. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, X.; Deng, X.; Han, H.; Shi, W.; Li, Y. A protein extraction method compatible with proteomic analysis for the euhalophyte Salicornia europaea. Electrophoresis 2007, 28, 3976–3987. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Daudi, A.; O’Brien, J.A. Detection of hydrogen peroxide by DAB staining in Arabidopsis leaves. Bio. Protoc. 2012, 2, e263. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.Q.; Wang, Y.S.; Lou, Z.P.; Dong, J.D. Effect of heavy metal stress on antioxidative enzymes and lipid peroxidation in leaves and roots of two mangrove plant seedlings (Kandelia candel and Bruguiera gymnorrhiza). Chemosphere 2007, 67, 44–50. [Google Scholar] [CrossRef]
- Hanif, A.; Ahmad, S.; Jaskani, M.J.; Ahmad, R. Papaya treatment with putrescine maintained the overall quality and promoted the antioxidative enzyme activities of the stored fruit. Sci. Hortic. 2020, 268, 109367. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, L.; Hao, L.; Liu, N.; Zhao, Y.; Zhong, N.; Zhao, P. iTRAQ-Based Proteomics Analysis of Response to Solanum tuberosum Leaves Treated with the Plant Phytotoxin Thaxtomin A. Int. J. Mol. Sci. 2021, 22, 12036. https://doi.org/10.3390/ijms222112036
Liu L, Hao L, Liu N, Zhao Y, Zhong N, Zhao P. iTRAQ-Based Proteomics Analysis of Response to Solanum tuberosum Leaves Treated with the Plant Phytotoxin Thaxtomin A. International Journal of Molecular Sciences. 2021; 22(21):12036. https://doi.org/10.3390/ijms222112036
Chicago/Turabian StyleLiu, Lu, Liaoyang Hao, Ning Liu, Yonglong Zhao, Naiqin Zhong, and Pan Zhao. 2021. "iTRAQ-Based Proteomics Analysis of Response to Solanum tuberosum Leaves Treated with the Plant Phytotoxin Thaxtomin A" International Journal of Molecular Sciences 22, no. 21: 12036. https://doi.org/10.3390/ijms222112036
APA StyleLiu, L., Hao, L., Liu, N., Zhao, Y., Zhong, N., & Zhao, P. (2021). iTRAQ-Based Proteomics Analysis of Response to Solanum tuberosum Leaves Treated with the Plant Phytotoxin Thaxtomin A. International Journal of Molecular Sciences, 22(21), 12036. https://doi.org/10.3390/ijms222112036