
Neurotoxicity and Underlying Mechanisms of Endogenous Neurotoxins

 ,
,
Abstract
:1. Introduction
2. Endogenous Neurotoxins
2.1. Analogues of MPTP
2.1.1. Tetrahydroisoquinolines (TIQs)
Norsalsolinol and N-methyl-norsalsolinol
Salsolinol and N-methyl-salsolinol
THP
ADTIQ
1-BnTIQ
1-Methyl-TIQ
2.1.2. β-Carbolines (βCs)
Norharman and Harman
2-Me-βC+ and 2,9-DiMe-βC+
TaClo
2.2. Oxidation Products of Dopamine (DA)
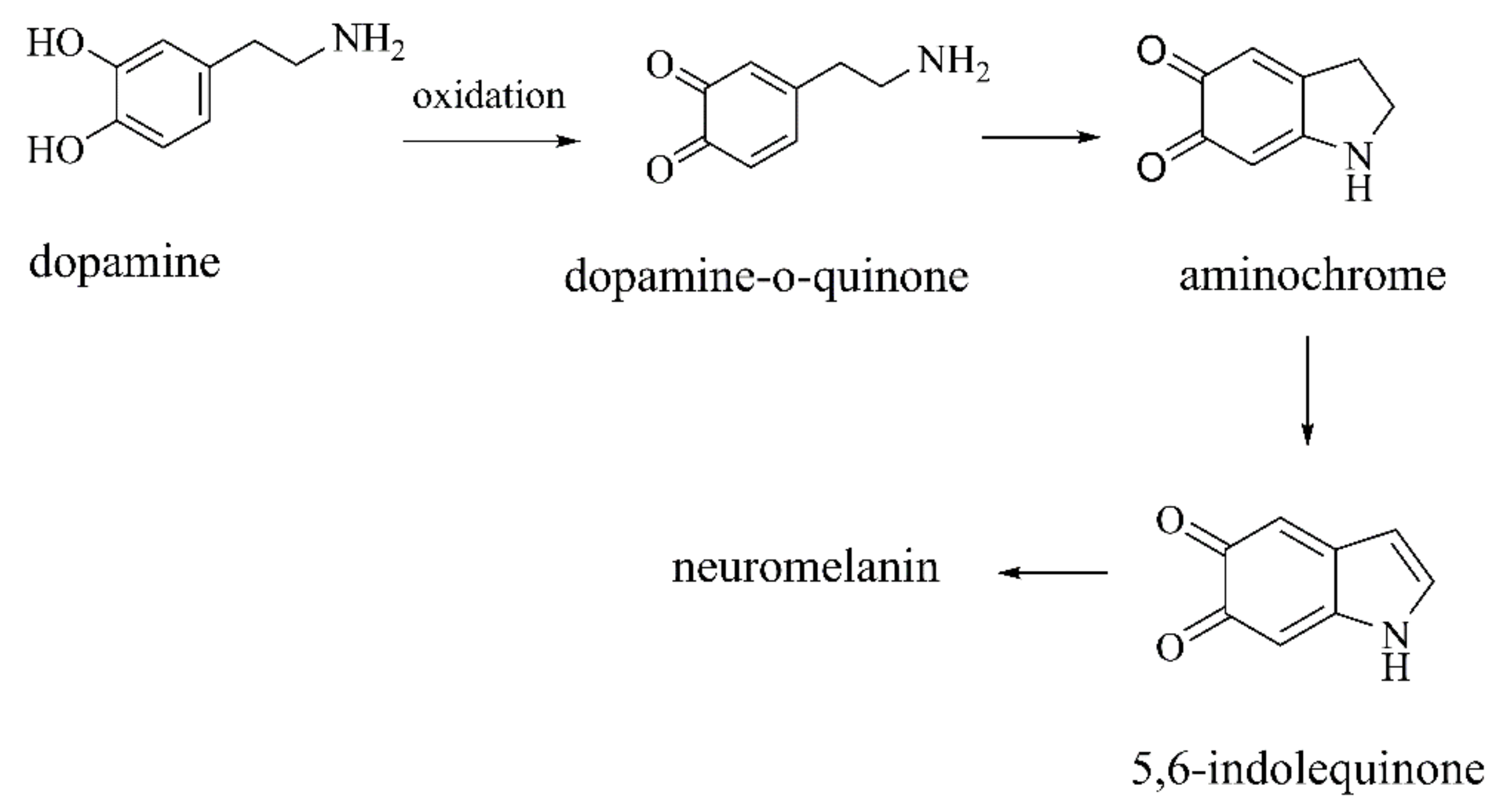
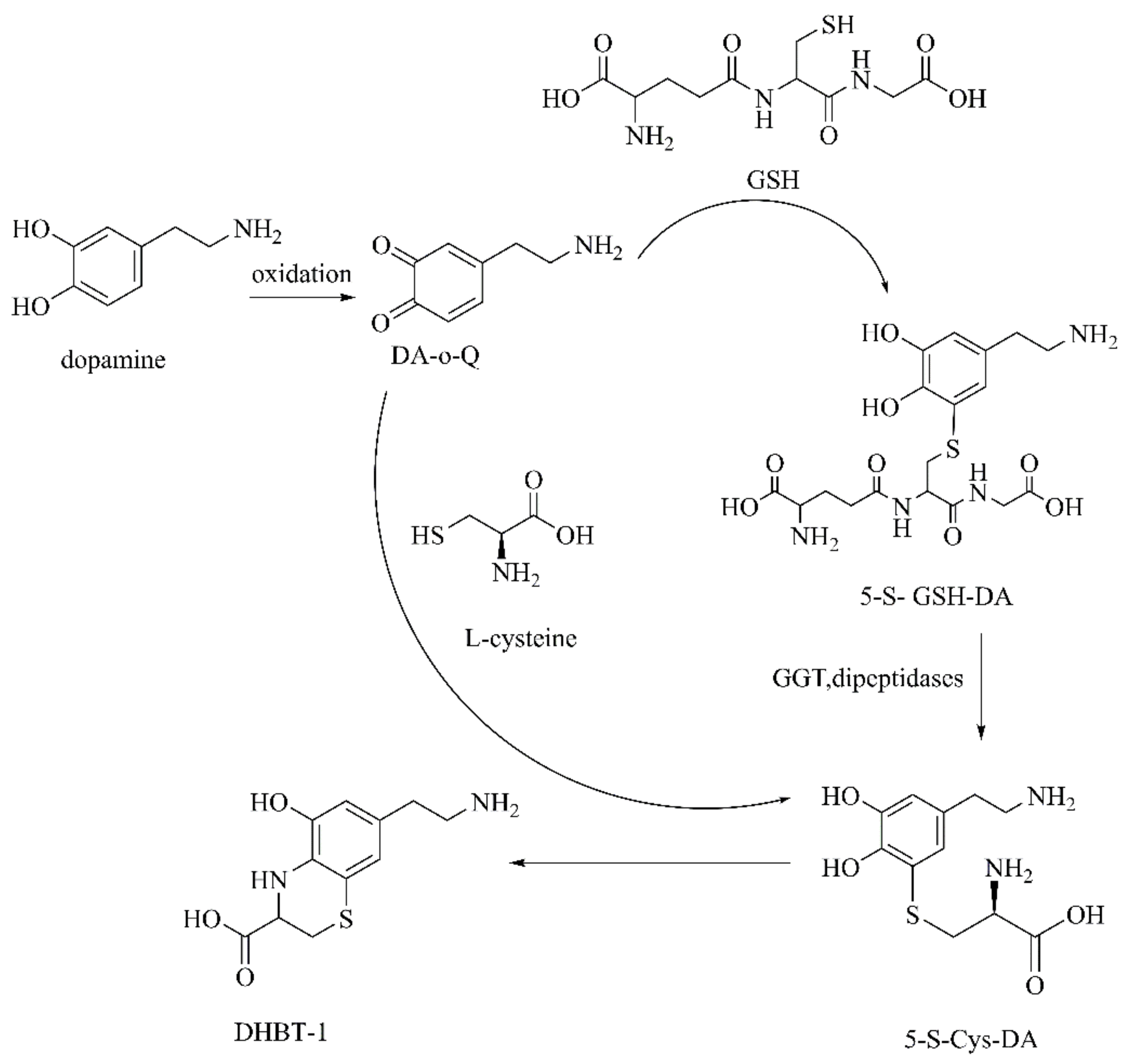
2.2.1. Autoxidation Products of DA
DA-o-Q
Aminochrome
2.2.2. Oxidation Products of Dopamine by Monoamine Oxidase (MAO)
DOPAL
DOPAC and HVA
2.3. Metabolites of Kynurenine Pathway
2.3.1. 3-HK
2.3.2. QUIN/QA
3. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| PD | Parkinson’s disease |
| DA | dopamine |
| AD | Alzheimer’s disease |
| ALS | amyotrophic lateral sclerosis |
| HD | Huntington’s disease |
| ALS-PDC | ALS-Parkinsonism Dementia Complex |
| CNS | central nervous system |
| TH | tyrosine hydroxylase |
| SNpc | substantia nigra pars compact |
| MAO | monoamine oxidase |
| DAT | dopamine transporter |
| ROS | reactive oxygen species |
| TRPM2 | transient receptor potential melastatin 2 |
| TIQs | tetrahydroisoquinolines |
| βCs | β-carbolines |
| norsalsolinol | 6,7-dihydroxy-1,2,3,4-tetrahydroisoquinoline |
| N-methyl-norsalsolinol | N-methyl-6,7-dihydroxy-1,2,3,4-tetrahydroisoquinoline |
| Sal | salsolinol |
| THP | tetrahydropapaveroline |
| ADTIQ | 1-acetyl-6,7-dihyroxy-1,2,3,4-tetrahydroisoquinoline |
| SN | substantia nigra |
| DOPAL | 3,4-dihydroxyphenylacetaldehyde |
| NGF | nerve growth factor |
| BDNF | brain-derived growth factor |
| SAM | S-adenosyl-L-methionine |
| norharman | 9H-pyrido[3,4-b]indole |
| harman | 1-methyl-9H-pyrido-[3,4-b]indole |
| 2-Me-βC+ | 2-methyl-β-carbolinium ion |
| 2,9-diMe-βC+ | 2,9-dimethyl-β-carbolinium ion |
| TaClo | 1-trichloromethyl-1,2,3,4-tetrahydro-carboline |
| 5-MTHF | 5-methylhydrofolate |
| ET | essential tremor |
| TCE | trichloroethylene |
| DA-o-Q | dopamine-o-quinone |
| DOPAL | 3,4-dihydroxyphenylacetaldehyde |
| DOPAC | 3,4-dihydroxyphenylacetic acid |
| HVA | homovanillic acid |
| 3-HK | hydroxykynurenine |
| QUIN | quinolinic acid |
| NMDA | N-methyl-D-aspartate |
| NOS | nitric oxide synthase |
| AMPK | 5′-adenosine monophosphate-activated protein kinase |
References
- Memon, A.A.; Coleman, J.J.; Amara, A.W. Effects of exercise on sleep in neurodegenerative disease. Neurobiol. Dis. 2020, 140, 104859. [Google Scholar] [CrossRef] [PubMed]
- Shaw, C.A.; Hoeglinger, G.U. Neurodegenerative diseases: Neurotoxins as sufficient etiologic agents? Neuromolecular Med. 2008, 10, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- A Kosenko, E.; NSolomadin, I.; ATikhonova, L.; Prakash Reddy, V.; Aliev, G.; G Kaminsky, Y. Pathogenesis of Alzheimer disease: Role of oxidative stress, amyloid-β peptides, systemic ammonia and erythrocyte energy metabolism. CNS Neurol. Disord. Drug Targets 2014, 13, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Calne, D. Neurotoxins and degeneration in the central nervous system. Neurotoxicology 1991, 12, 335–339. [Google Scholar]
- Wu, Y.-C.; Sonninen, T.-M.; Peltonen, S.; Koistinaho, J.; Lehtonen, Š. Blood–Brain Barrier and Neurodegenerative Diseases—Modeling with iPSC-Derived Brain Cells. Int. J. Mol. Sci. 2021, 22, 7710. [Google Scholar] [CrossRef]
- Soddu, E.; Rassu, G.; Giunchedi, P.; Sarmento, B.; Gavini, E. From naturally-occurring neurotoxic agents to CNS shuttles for drug delivery. Eur. J. Pharm. Sci. 2015, 74, 63–76. [Google Scholar] [CrossRef]
- Tipton, K.F.; Dajas, F. Neurotoxins in Neurobiology: Their actions and applications. Ellis Horwood Ser. Neurosci. Chichester 1994, 64, 1425. [Google Scholar]
- Silva, R.; Falcao, A.; Fernandes, A.; Gordo, A.; Brito, M.; Brites, D. Dissociated primary nerve cell cultures as models for assessment of neurotoxicity. Toxicol. Lett. 2006, 163, 1–9. [Google Scholar] [CrossRef]
- Abe, K.; Saitoh, T.; Horiguchi, Y.; Utsunomiya, I.; Taguchi, K. Synthesis and neurotoxicity of tetrahydroisoquinoline derivatives for studying Parkinson’s disease. Biol. Pharm. Bull. 2005, 28, 1355–1362. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Rasheed, M.; Deng, Y. The epigenetic mechanisms involved in mitochondrial dysfunction: Implication for Parkinson’s disease. Brain Pathol. 2021, e13012. [Google Scholar] [CrossRef]
- Wirdefeldt, K.; Adami, H.-O.; Cole, P.; Trichopoulos, D.; Mandel, J. Epidemiology and etiology of Parkinson’s disease: A review of the evidence. Eur. J. Epidemiol. 2011, 26, 1. [Google Scholar] [CrossRef]
- Peng, C.; Gathagan, R.J.; Covell, D.J.; Medellin, C.; Stieber, A.; Robinson, J.L.; Zhang, B.; Pitkin, R.M.; Olufemi, M.F.; Luk, K.C. Cellular milieu imparts distinct pathological α-synuclein strains in α-synucleinopathies. Nature 2018, 557, 558–563. [Google Scholar] [CrossRef]
- Ferreira, N.; Gram, H.; Sorrentino, Z.A.; Gregersen, E.; Schmidt, S.I.; Reimer, L.; Betzer, C.; Perez-Gozalbo, C.; Beltoja, M.; Nagaraj, M. Multiple system atrophy-associated oligodendroglial protein p25α stimulates formation of novel α-synuclein strain with enhanced neurodegenerative potential. Acta Neuropathol. 2021, 1–29. [Google Scholar] [CrossRef]
- Elfarrash, S.; Jensen, N.M.; Ferreira, N.; Betzer, C.; Thevathasan, J.V.; Diekmann, R.; Adel, M.; Omar, N.M.; Boraie, M.Z.; Gad, S. Organotypic slice culture model demonstrates inter-neuronal spreading of alpha-synuclein aggregates. Acta Neuropathol. Commun. 2019, 7, 1–16. [Google Scholar] [CrossRef]
- Ferreira, N.; Gonçalves, N.P.; Jan, A.; Jensen, N.M.; van der Laan, A.; Mohseni, S.; Vægter, C.B.; Jensen, P.H. Trans-synaptic spreading of alpha-synuclein pathology through sensory afferents leads to sensory nerve degeneration and neuropathic pain. Acta Neuropathol. Commun. 2021, 9, 1–17. [Google Scholar] [CrossRef]
- Van Den Berge, N.; Ferreira, N.; Gram, H.; Mikkelsen, T.W.; Alstrup, A.K.O.; Casadei, N.; Tsung-Pin, P.; Riess, O.; Nyengaard, J.R.; Tamgüney, G. Evidence for bidirectional and trans-synaptic parasympathetic and sympathetic propagation of alpha-synuclein in rats. Acta Neuropathol. 2019, 138, 535–550. [Google Scholar] [CrossRef] [Green Version]
- Sun, F.; Deng, Y.; Han, X.; Liu, Q.; Zhang, P.; Manzoor, R.; Ma, H. A secret that underlies Parkinson’s disease: The damaging cycle. Neurochem. Int. 2019, 129, 104484. [Google Scholar] [CrossRef]
- Zhang, Y.; Ma, H.; Xie, B.; Han, C.; Wang, C.; Qing, H.; Deng, Y. Alpha-synuclein overexpression induced mitochondrial damage by the generation of endogenous neurotoxins in PC12 cells. Neurosci. Lett. 2013, 547, 65–69. [Google Scholar] [CrossRef]
- Picklo Sr, M.J.; Montine, T.J. Mitochondrial effects of lipid-derived neurotoxins. J. Alzheimer’s Dis. 2007, 12, 185–193. [Google Scholar] [CrossRef]
- Fries, D.S.; Devries, J.; Hazelhoff, B.; Horn, A.S. Synthesis and Toxicity toward Nigrostriatal Dopamine Neurons of l-Methyl-4-phenyl-l,2,3,6-tetrahydropyridine (MPTP) Analogues. J. Med. Chem. 1986, 29, 424–427. [Google Scholar] [CrossRef]
- Maruyama, W.; Abe, T.; Tohgi, H.; Dostert, P.; Naoi, M. A dopaminergic neurotoxin, (R)-N-methylsalsolinol, increases in parkinsonian cerebrospinal fluid. Ann. Neurol. 1996, 40, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Casanova, Y.; Negro, S.; Barcia, E. Application of neurotoxin- and pesticide-induced animal models of Parkinson’s disease in the evaluation of new drug delivery systems. Acta Pharm. 2022, 72, 35–58. [Google Scholar] [CrossRef]
- Taib, C.N.M.; Mustapha, M. MPTP-induced mouse model of Parkinson’s disease: A promising direction of therapeutic strategies. Bosn. J. Basic Med Sci. 2021, 21, 422. [Google Scholar]
- Przedborski, S.; JacksonLewis, V.; Yokoyama, R.; Shibata, T.; Dawson, V.L.; Dawson, T.M. Role of neuronal nitric oxide in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced dopaminergic neurotoxicity. Proc. Natl. Acad. Sci. USA 1996, 93, 4565–4571. [Google Scholar] [CrossRef] [Green Version]
- He, X.J.; Nakayama, H. Neurogenesis in Neurotoxin-induced Animal Models for Parkinson’s Disease-A Review of the Current Status. J. Toxicol. Pathol. 2009, 22, 101–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Sukumaran, P.; Selvaraj, S.; Cilz, N.I.; Schaar, A.; Lei, S.; Singh, B.B. TRPM2 Promotes Neurotoxin MPP+/MPTP-Induced Cell Death. Mol. Neurobiol. 2016, 55, 409–420. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.L.; Chen, J.L.; Mao, K.M.; She, H.; Ren, Y.X.; Gui, C.; Wu, X.; Zou, F.; Li, W.J. Mitochondrial calcium dysfunction contributes to autophagic cell death induced by MPP+ via AMPK pathway. Biochem. Biophys. Res. Commun. 2019, 509, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Segura-Aguilar, J. Neurotoxins as Preclinical Models for Parkinson’s Disease. Neurotox. Res. 2018, 34, 870–877. [Google Scholar] [CrossRef]
- Antkiewicz-Michaluk, L.; Wasik, A.; Michaluk, J. 1-Methyl-1,2,3,4-tetrahydroisoquinoline, an endogenous amine with unexpected mechanism of action: New vistas of therapeutic application. Neurotox. Res. 2014, 25, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Napolitano, A.; Manini, P.; d’Ischia, M. Oxidation Chemistry of Catecholamines and Neuronal Degeneration: An Update. Curr. Med. Chem. 2011, 18, 1832–1845. [Google Scholar] [CrossRef] [Green Version]
- Herraiz, T. N-methyltetrahydropyridines and pyridinium cations as toxins and comparison with naturally-occurring alkaloids. Food Chem. Toxicol. 2016, 97, 23–39. [Google Scholar] [CrossRef]
- DeCuypere, M.; Kalabokis, V.N.; Hao, R.; Schroeder, D.; Miller, D.D.; LeDoux, M.S. Localization of N-methyl-norsalsolinol within rodent and human brain. J. Neurosci. Res. 2008, 86, 2543–2552. [Google Scholar] [CrossRef]
- Scholz, J.; Toska, K.; Luborzewski, A.; Maass, A.; Schunemann, V.; Haavik, J.; Moser, A. Endogenous tetrahydroisoquinolines associated with Parkinson’s disease mimic the feedback inhibition of tyrosine hydroxylase by catecholamines. FEBS J. 2008, 275, 2109–2121. [Google Scholar] [CrossRef]
- DeCuypere, M.; Lu, Y.; Miller, D.D.; LeDoux, M.S. Regional distribution of tetrahydroisoquinoline derivatives in rodent, human, and Parkinson’s disease brain. J. Neurochem. 2008, 107, 1398–1413. [Google Scholar] [CrossRef] [Green Version]
- Maruyama, Y.; Teraoka, H.; Iwata, H.; Kazusaka, A.; Fujita, S. Inhibitory effects of endogenous dopaminergic neurotoxin, norsalsolinol on dopamine secretion in PC12 rat pheochromocytoma cells. Neurochem. Int. 2001, 38, 567–572. [Google Scholar] [CrossRef]
- Kobayashi, H.; Fukuhara, K.; Tada-Oikawa, S.; Yada, Y.; Hiraku, Y.; Murata, M.; Oikawa, S. The mechanisms of oxidative DNA damage and apoptosis induced by norsalsolinol, an endogenous tetrahydroisoquinoline derivative associated with Parkinson’s disease. J. Neurochem. 2009, 108, 397–407. [Google Scholar] [CrossRef]
- Scholz, J.; Bamberg, H.; Moser, A. N-methyl-norsalsolinol, an endogenous neurotoxin, inhibits tyrosine hydroxylase activity in the rat brain nucleus accumbens in vitro. Neurochem. Int. 1997, 31, 845–849. [Google Scholar] [CrossRef]
- Mravec, B. Salsolinol, a derivate of dopamine, is a possible modulator of catecholaminergic transmission: A review of recent developments. Physiological Research 2006, 55, 353–364. [Google Scholar]
- Deng, Y.L.; Maruyama, W.; Dostert, P.; Takahashi, T.; Kawai, M.; Naoi, M. Determination of the (R)- and (S)-enantiomers of salsolinol and N-methylsalsolinol by use of a chiral high-performance liquid chromatographic column. J. Chromatogr. B-Biomed. Appl. 1995, 670, 47–54. [Google Scholar] [CrossRef]
- Musshoff, F.; Schmidt, P.; Dettmeyer, R.; Priemer, F.; Jachau, K.; Madea, B. Determination of dopamine and dopamine-derived (R)-/(S)-salsolinol and norsalsolinol in various human brain areas using solid-phase extraction and gas chromatography/mass spectrometry. Forensic Sci. Int. 2000, 113, 359–366. [Google Scholar] [CrossRef]
- Quintanilla, M.E.; Rivera-Meza, M.; Berrios-Carcamo, P.; Cassels, B.K.; Herrera-Marschitz, M.; Israel, Y. (R)-Salsolinol, a product of ethanol metabolism, stereospecifically induces behavioral sensitization and leads to excessive alcohol intake. Addict. Biol. 2016, 21, 1063–1071. [Google Scholar] [CrossRef]
- Hashizume, T.; Watanabe, R.; Inaba, Y.; Sawai, K.; Fulop, F.; Nagy, G.M. Hypothalamic dopamine is required for salsolinol-induced prolactin secretion in goats. Anim. Sci. J. 2017, 88, 1588–1594. [Google Scholar] [CrossRef]
- Gorski, K.; Marciniak, E.; Zielinska-Gorska, M.; Misztal, T. Salsolinol Up-Regulates Oxytocin Expression and Release During Lactation in Sheep. J. Neuroendocrinol. 2016, 28. [Google Scholar] [CrossRef]
- Kurnik-Lucka, M.; Latacz, G.; Martyniak, A.; Bugajski, A.; Kiec-Kononowicz, K.; Gil, K. Salsolinol-neurotoxic or Neuroprotective? Neurotox. Res. 2020, 37, 286–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurnik-Lucka, M.; Panula, P.; Bugajski, A.; Gil, K. Salsolinol: An Unintelligible and Double-Faced Molecule-Lessons Learned from In Vivo and In Vitro Experiments. Neurotox. Res. 2018, 33, 485–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villageliu, D.N.; Borts, D.J.; Lyte, M. Production of the Neurotoxin Salsolinol by a Gut-Associated Bacterium and Its Modulation by Alcohol. Front. Microbiol. 2018, 9, 3092. [Google Scholar] [CrossRef]
- Zheng, X.; Chen, X.; Guo, M.; Ali, S.; Huang, Y.; Sun, F.; Liu, K.; Chen, Z.; Deng, Y.; Zhong, R. Changes in salsolinol production and salsolinol synthase activity in Parkinson’s disease model. Neurosci. Lett. 2018, 673, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Naoi, M.; Maruyama, W.; Takahashi, T.; Akao, Y.; Nakagawa, Y. Involvement of endogenous N-methyl(R) salsolinol in Parkinson’s disease: Induction of apoptosis and protection by (-)deprenyl. In Advances in Research on Neurodegeneration; Springer: Berlin/Heidelberg, Germany, 2000; pp. 111–121. [Google Scholar]
- Naoi, M.; Maruyama, W.; Zhang, J.H.; Takahashi, T.; Deng, Y.L.; Dostert, P. Enzymatic oxidation of the dopaminergic neurotoxin, 1(R), 2(N)-dimethyl-6,7-dihydroxy-1,2,3,4-tetrahydroisoquinoline, into 1,2(N)-dimethyl-6,7-dihydroxyisoquinolinium ion. Life Sci. 1995, 57, 1061–1066. [Google Scholar] [CrossRef]
- Maruyama, W.; Dostert, P.; Matsubara, K.; Naoi, M. N-methyl(R)salsolinol produces hydroxyl radicals: Involvement to neurotoxicity. Free. Radic. Biol. Med. 1995, 19, 67–75. [Google Scholar] [CrossRef]
- Weitz, C.J.; Faull, K.F.; Goldstein, A. Synthesis of the skeleton of the morphine molecule by mammalian liver. Nature 1987, 330, 674–677. [Google Scholar] [CrossRef]
- Soto-Otero, R.; Sanmartin-Suarez, C.; Sanchez-Iglesias, S.; Hermida-Ameijeiras, A.; Sanchez-Sellero, I.; Mendez-Alvarez, E. Study on the ability of 1,2,3,4-tetrahydropapaveroline to cause oxidative stress: Mechanisms and potential implications in relation to Parkinson’s disease. J. Biochem. Mol. Toxicol. 2006, 20, 209–220. [Google Scholar] [CrossRef]
- Cashaw, J.L.; Geraghty, C.A. Tetrahydropapaveroline and the blood-brain barrier in rats. Alcohol 1991, 8, 317–319. [Google Scholar] [CrossRef]
- Kobayashi, H.; Oikawa, S.; Kawanishi, S. Mechanism of DNA damage and apoptosis induced by tetrahydropapaveroline, a metabolite of dopamine. Neurochem. Res. 2006, 31, 523–532. [Google Scholar] [CrossRef]
- Sango, K.; Maruyama, W.; Matsubara, K.; Dostert, P.; Naoi, M. Enantio-selective occurrence of (S)-tetrahydropapaveroline in human brain. Neurosci. Lett. 2000, 283, 224–226. [Google Scholar] [CrossRef]
- Kyeong, I.G.; Eum, W.S.; Choi, S.Y.; Kang, J.H. Oxidative modification of neurofilament-L and neuronal cell death induced by the catechol neurotoxin, tetrahydropapaveroline. Toxicol. Lett. 2013, 217, 59–66. [Google Scholar] [CrossRef]
- Lee, J.J.; Yu, M.K.; Shou, Y.Y.; Park, H.D.; Min, H.K.; Jin, T.H.; Lee, M.K. Aggravation of L-DOPA-induced neurotoxicity by tetrahydropapaveroline in PC12 cells. Biochem. Pharmacol. 2003, 66, 1787–1795. [Google Scholar] [CrossRef]
- Deng, Y.; Zhang, Y.; Li, Y.; Xiao, S.; Song, D.; Qing, H.; Li, Q.; Rajput, A.H. Occurrence and distribution of salsolinol-like compound, 1-acetyl-6,7-dihydroxy-1,2,3,4-tetrahydroisoquinoline (ADTIQ) in parkinsonian brains. J. Neural. Transm. 2012, 119, 435–441. [Google Scholar] [CrossRef]
- Xie, B.; Lin, F.; Ullah, K.; Peng, L.; Ding, W.; Dai, R.; Qing, H.; Deng, Y. A newly discovered neurotoxin ADTIQ associated with hyperglycemia and Parkinson’s disease. Biochem. Biophys. Res. Commun. 2015, 459, 361–366. [Google Scholar] [CrossRef]
- Morgan, P.E.; Dean, R.T.; Davies, M.J. Inactivation of cellular enzymes by carbonyls and protein-bound glycation/glycoxidation products. Arch. Biochem. Biophys. 2002, 403, 259–269. [Google Scholar] [CrossRef]
- Arriba, S.G.D.; Stuchbury, G.; Yarin, J.; Burnell, J.; Münch, G. Methylglyoxal impairs glucose metabolism and leads to energy depletion in neuronal cells--protection by carbonyl scavengers. Neurobiol. Aging 2007, 28, 1044–1050. [Google Scholar] [CrossRef]
- Shavali, S.; Ebadi, M. 1-Benzyl-1,2,3,4-tetrahydroisoquinoline (1-BnTIQ), an endogenous neurotoxin, induces dopaminergic cell death through apoptosis. Neurotoxicology 2003, 24, 417–424. [Google Scholar] [CrossRef]
- Wsik, A.; Romańska, I.; Antkiewicz-Michaluk, L. 1-Benzyl-1,2,3,4-Tetrahydroisoquinoline, an Endogenous Parkinsonism-Inducing Toxin, Strongly Potentiates MAO-Dependent Dopamine Oxidation and Impairs Dopamine Release: Ex vivo and In vivo Neurochemical Studies. Neurotox. Res. 2009, 15, 15. [Google Scholar] [CrossRef]
- Wąsik, A.; Kajta, M.G.; Lenda, T.; Antkiewicz-Michaluk, L. Concentration-Dependent Opposite Effects of 1-Benzyl-1,2,3,4-tetrahydroisoquinoline on Markers of Apoptosis: In Vitro and Ex Vivo Studies. Neurotox. Res. 2014, 25, 90–99. [Google Scholar] [CrossRef] [Green Version]
- Kotake, Y.; Sekiya, Y.; Okuda, K.; Ohta, S. Detection of a novel neurotoxic metabolite of Parkinson’s disease-related neurotoxin, 1-benzyl-1,2,3,4-tetrahydroisoquinoline. J. Toxicol. Sci. 2014, 39, 749–754. [Google Scholar] [CrossRef] [Green Version]
- Makino, Y.; Ohta, S.; Tachikawa, O.; Hirobe, M. Presence of tetrahydroisoquinoline and 1-methyl-tetrahydro-isoquinoline in foods: Compounds related to Parkinson’s disease. Life Sci. 1988, 43, 373–378. [Google Scholar] [CrossRef]
- Yamakawa, T.; Ohta, S. Isolation of 1-methyl-1,2,3,4-tetrahydroisoquinoline-synthesizing enzyme from rat brain: A possible Parkinson’s disease-preventing enzyme. Biochem. Biophys. Res. Commun. 1997, 236, 676–681. [Google Scholar] [CrossRef]
- Wasik, A.; Antkiewicz-Michaluk, L. The mechanism of neuroprotective action of natural compounds. Pharmacol. Rep. 2017, 69, 851–860. [Google Scholar] [CrossRef]
- Yamakawa, T.; Kotake, Y.; Fujitani, M.; Shintani, H.; Makino, Y.; Ohta, S. Regional distribution of parkinsonism-preventing endogenous tetrahydroisoquinoline derivatives and an endogenous parkinsonism-preventing substance-synthesizing enzyme in monkey brain. Neurosci. Lett. 1999, 276, 68–70. [Google Scholar] [CrossRef]
- Wasik, A.; Romanska, I.; Zelek-Molik, A.; Antkiewicz-Michaluk, L. Multiple Administration of Endogenous Amines TIQ and 1MeTIQ Protects Against a 6-OHDA-Induced Essential Fall of Dopamine Release in the Rat Striatum: In Vivo Microdialysis Study. Neurotox. Res. 2018, 33, 523–531. [Google Scholar] [CrossRef] [Green Version]
- Abe, A.; Kokuba, H. Harmol induces autophagy and subsequent apoptosis in U251MG human glioma cells through the downregulation of survivin. Oncol. Rep. 2013, 29, 1333–1342. [Google Scholar] [CrossRef] [Green Version]
- Piechowska, P.; Zawirska-Wojtasiak, R.; Mildner-Szkudlarz, S. Bioactive beta-Carbolines in Food: A Review. Nutrients 2019, 11, 814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsubara, K.; Collins, M.A.; Akane, A.; Ikebuchi, J.; Neafsey, E.J.; Kagawa, M.; Shiono, H. Potential bioactivated neurotoxicants, N-methylated beta-carbolinium ions, are present in human brain. Brain Res. 1993, 610, 90–96. [Google Scholar] [CrossRef]
- Lawana, V.; Um, S.Y.; Rochet, J.-C.; Turesky, R.J.; Shannahan, J.H.; Cannon, J.R. Neuromelanin Modulates Heterocyclic Aromatic Amine-Induced Dopaminergic Neurotoxicity. Toxicol. Sci. 2020, 173, 171–188. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, K.; Aoyama, K.; Suno, M.; Awaya, T. N-methylation underlying Parkinson’s disease. Neurotoxicology Teratol. 2002, 24, 593–598. [Google Scholar] [CrossRef]
- Pfau, W.; Skog, K.J. Exposure to β-carbolines norharman and harman. J. Chromatogr. B 2004, 802, 115–126. [Google Scholar] [CrossRef]
- Rommelspacher, H.; May, T.; Susilo, R. β-Carbolines and Tetrahydroisoquinolines: Detection and Function in Mammals. Planta Med. 1991, 57, S85–S92. [Google Scholar] [CrossRef]
- Kuhn, W.; Muller, T.; Grosse, H.; Rommelspacher, H. Elevated levels of harman and norharman in cerebrospinal fluid of parkinsonian patients. J. Neural Transm. 1996, 103, 1435–1440. [Google Scholar] [CrossRef]
- Sammi, S.R.; Agim, Z.S.; Cannon, J.R. Harmane-Induced Selective Dopaminergic Neurotoxicity in Caenorhabditis elegans. Toxicol. Sci. 2018, 161, 335–348. [Google Scholar] [CrossRef]
- Louis, E.D.; Jiang, W.; Pellegrino, K.M.; Rios, E.; Factorlitvak, P.; Henchcliffe, C.; Zheng, W. Elevated blood harmane (1-methyl-9H-pyrido[3,4-b]indole) concentrations in essential tremor. NeuroToxicology 2008, 29, 294–300. [Google Scholar] [CrossRef] [Green Version]
- Louis, E.D.; Eliasen, E.H.; Ferrer, M.; Hernandez, D.; Gaini, S.; Jiang, W.; Zheng, W.; Nielsen, F.; Petersen, M.S. Blood Harmane (1-Methyl-9H-Pyrido 3,4-b indole) and Mercury in Essential Tremor: A Population-Based, Environmental Epidemiology Study in the Faroe Islands. Neuroepidemiology 2020, 54, 272–280. [Google Scholar] [CrossRef]
- Yang, Y.J.; Lim, S.C.; Lee, M.K. The harman and norharman reduced dopamine content and induced cytotoxicity in PC12 cells. Biomol. Ther. 2008, 16, 106–112. [Google Scholar] [CrossRef] [Green Version]
- Rasse-Suriani, F.A.O.; García-Einschlag, F.S.; Rafti, M.; Schmidt De León, T.; David Gara, P.M.; Erra-Balsells, R.; Cabrerizo, F.M. Photophysical and Photochemical Properties of Naturally Occurring normelinonine F and Melinonine F Alkaloids and Structurally Related N(2)- and/or N(9)-methyl-β-carboline Derivatives. Photochem. Photobiol. 2018, 94, 36–51. [Google Scholar] [CrossRef]
- Hamann, J.; Wernicke, C.; Lehmann, J.; Reichmann, H.; Rommelspacher, H.; Gille, G. 9-Methyl-β-carboline up-regulates the appearance of differentiated dopaminergic neurones in primary mesencephalic culture. Neurochem. Int. 2008, 52, 688–700. [Google Scholar] [CrossRef]
- Hamann, J.; Rommelspacher, H.; Storch, A.; Reichmann, H.; Gille, G. Neurotoxic mechanisms of 2,9-dimethyl-beta-carbolinium ion in primary dopaminergic culture. J. Neurochem. 2006, 98, 1185–1199. [Google Scholar] [CrossRef]
- Keane, P.C.; Hanson, P.S.; Patterson, L.; Blain, P.G.; Hepplewhite, P.; Khundakar, A.A.; Judge, S.J.; Kahle, P.J.; LeBeau, F.E.N.; Morris, C.M. Trichloroethylene and its metabolite TaClo lead to degeneration of substantia nigra dopaminergic neurones: Effects in wild type and human A30P mutant alpha-synuclein mice. Neurosci. Lett. 2019, 711, 134437. [Google Scholar] [CrossRef]
- Liu, M.; Shin, E.-J.; Dang, D.-K.; Jin, C.-H.; Lee, P.H.; Jeong, J.H.; Park, S.-J.; Kim, Y.-S.; Xing, B.; Xin, T.; et al. Trichloroethylene and Parkinson’s Disease: Risk Assessment. Mol. Neurobiol. 2018, 55, 6201–6214. [Google Scholar] [CrossRef]
- Riederer, P.; Foley, P.; Bringmann, G.; Feineis, D.; Brückner, R.; Gerlach, M. Biochemical and pharmacological characterization of 1-trichloromethyl-1,2,3,4-tetrahydro-β-carboline: A biologically relevant neurotoxin? Eur. J. Pharmacol. 2002, 442, 1–16. [Google Scholar] [CrossRef]
- Sharma, R.K.; Candelario-Jalil, E.; Feineis, D.; Bringmann, G.; Fiebich, B.L.; Akundi, R.S. 1-Trichloromethyl-1, 2, 3, 4-tetrahydro-beta-carboline (TaClo) alters cell cycle progression in human neuroblastoma cell lines. Neurotox. Res. 2017, 32, 649–660. [Google Scholar] [CrossRef]
- Yang, Y.H.; Pang, B.; Liu, Z.H.; Li, J.; Zhang, Z.; Zhang, R.; Hou, X.Z.; Guo, H.; Chi, L.Y.; Pang, Q.; et al. 1-Trichloromethyl-1,2,3,4-tetrahydro-beta-carboline (TaClo) Induces the Apoptosis of Dopaminergic Neurons via Oxidative Stress and Neuroinflammation. Oxidative Med. Cell. Longev. 2019, 2019, 1292891. [Google Scholar] [CrossRef] [Green Version]
- Sontag, T.A.; Lange, K.W.; Heim, C.; Kolasiewicz, W.; Tucha, O.; Sontag, K.-H. Alterations of nocturnal activity in rats following subchronic oral administration of the neurotoxin 1-trichloromethyl-1, 2, 3, 4-tetrahydro-β-carboline. J. Neural Transm. 2009, 116, 1267–1271. [Google Scholar] [CrossRef] [Green Version]
- Segura-Aguilar, J.; Paris, I.; Muoz, P.; Ferrari, E.; Zucca, F. Protective and toxic roles of dopamine in Parkinson’s disease. J. Neurochem. 2014, 129, 898–915. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, R.; Wang, G. Impact of Dopamine Oxidation on Dopaminergic Neurodegeneration. ACS Chem. Neurosci. 2019, 10, 945–953. [Google Scholar] [CrossRef]
- Antkiewicz-Michaluk, L. Endogenous risk factors in Parkinson’s disease: Dopamine and tetrahydroisoquinolines. Pol. J. Pharmacol. 2002, 54, 567–572. [Google Scholar]
- Segura-Aguilar, J. On the role of endogenous neurotoxins and neuroprotection in Parkinson’s disease. Neural Regen. Res. 2017, 12, 897–901. [Google Scholar] [CrossRef]
- Jameson, G.N.L.; Zhang, J.; Jameson, R.F.; Linert, W. Kinetic evidence that cysteine reacts with dopaminoquinone via reversible adduct formation to yield 5-cysteinyl-dopamine: An important precursor of neuromelanin. Org. Biomol. Chem. 2004, 2, 777–782. [Google Scholar] [CrossRef]
- Badillo-Ramirez, I.; Saniger, J.M.; Rivas-Arancibia, S. 5-S-cysteinyl-dopamine, a neurotoxic endogenous metabolite of dopamine: Implications for Parkinson’s disease. Neurochem. Int. 2019, 129, 104514. [Google Scholar] [CrossRef]
- Segura-Aguilar, J. Aminochrome as preclinical model for Parkinson’s disease. Oncotarget 2017, 8, 45036–45037. [Google Scholar] [CrossRef]
- Silva, V.; Segura-Aguilar, J. State and perspectives on flavonoid neuroprotection against aminochrome-induced neurotoxicity. Neural Regen. Res. 2021, 16, 1797–1798. [Google Scholar]
- Meiser, J.; Weindl, D.; Hiller, K. Complexity of dopamine metabolism. Cell Commun. Signal. 2013, 11, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Masato, A.; Plotegher, N.; Boassa, D.; Bubacco, L. Impaired dopamine metabolism in Parkinson’s disease pathogenesis. Mol. Neurodegener. 2019, 14, 35. [Google Scholar] [CrossRef] [Green Version]
- Vanle, B.C.; Florang, V.R.; Murry, D.J.; Aguirre, A.L.; Doorn, J.A. Inactivation of glyceraldehyde-3-phosphate dehydrogenase by the dopamine metabolite, 3,4-dihydroxyphenylacetaldehyde. Biochem. Biophys. Res. Commun. 2017, 492, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Nunes, C.; Almeida, L.; Laranjinha, J. 3,4-Dihydroxyphenylacetic acid (DOPAC) modulates the toxicity induced by nitric oxide in PC-12 cells via mitochondrial dysfunctioning. NeuroToxicology 2008, 29, 998–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morimoto, S.; Takao, M.; Hatsuta, H.; Nishina, Y.; Murayama, S. Homovanillic acid and 5-hydroxyindole acetic acid as biomarkers for dementia with Lewy bodies and coincident Alzheimer’s disease: An autopsy-confirmed study. PLoS ONE 2017, 12, e0171524. [Google Scholar]
- Stefani, A.; Pierantozzi, M.; Olivola, E.; Galati, S.; Cerroni, R.; D’Angelo, V.; Hainsworth, A.H.; Saviozzi, V.; Fedele, E.; Liguori, C. Homovanillic acid in CSF of mild stage Parkinson’s disease patients correlates with motor impairment. Neurochem. Int. 2017, 105, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Markianos, M.; Panas, M.; Kalfakis, N.; Vassilopoulos, D. Plasma Homovanillic Acid and Prolactin in Huntington’s Disease. Neurochem. Res. 2009, 34, 917–922. [Google Scholar] [CrossRef] [PubMed]
- Lugo-Huitron, R.; Ugalde Muniz, P.; Pineda, B.; Pedraza-Chaverri, J.; Rios, C.; Perez-de la Cruz, V. Quinolinic acid: An endogenous neurotoxin with multiple targets. Oxid. Med. Cell. Longev. 2013, 2013, 104024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarcz, R.; Pellicciari, R. Manipulation of brain kynurenines: Glial targets, neuronal effects, and clinical opportunities. J. Pharmacol. Exp. Ther. 2002, 303, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Sas, K.; Robotka, H.; Toldi, J.; Vécsei, L. Mitochondria, metabolic disturbances, oxidative stress and the kynurenine system, with focus on neurodegenerative disorders. J. Neurol. Sci. 2007, 257, 221–239. [Google Scholar] [CrossRef]
- Ramírez-Ortega, D.; Ramiro-Salazar, A.; González-Esquivel, D.; Ríos, C.; Pineda, B.; Pérez de la Cruz, V. 3-Hydroxykynurenine and 3-hydroxyanthranilic acid enhance the toxicity induced by copper in rat astrocyte culture. Oxidative Med. Cell. Longev. 2017, 2017, 2371895. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.R.; Jamie, J.F.; Guillemin, G.J. Kynurenine-3-monooxygenase: A review of structure, mechanism, and inhibitors. Drug Discov. Today 2016, 21, 315–324. [Google Scholar] [CrossRef]
- Guilarte, T.R.; Eastman, C.L. Is 3-hydroxykynurenine an endogenous neurotoxin in Huntington’s disease? J. Neurol. Sci. 1993, 116, 227–228. [Google Scholar] [CrossRef]
- Okuda, S.; Nishiyama, N.; Saito, H.; Katsuki, H. Hydrogen peroxide-mediated neuronal cell death induced by an endogenous neurotoxin, 3-hydroxykynurenine. Proc. Natl. Acad. Sci. USA 1996, 93, 12553–12558. [Google Scholar] [CrossRef] [Green Version]
- Stone, T.W. Endogenous neurotoxins from tryptophan. Toxicon 2001, 39, 61–73. [Google Scholar] [CrossRef]
- Dehhaghi, M.; Tan, V.; Heng, B.; Braidy, N.; Mohammadipanah, F.; Guillemin, G. Neuroprotective Effect of Myxobacterial Extracts on Quinolinic Acid-Induced Toxicity in Primary Human Neurons. Neurotox. Res. 2019, 35, 281–290. [Google Scholar] [CrossRef]
- Esmaeili, S.; Ghobadi, N.; Akbari, V.; Moradi, S.; Shahlaie, M.; Ghobadi, S.; Jalalvand, A.R.; Amani, M.; Khodarahmi, R. Pyridine-2,3-dicarboxylate, quinolinic acid, induces 1N4R Tau amyloid aggregation in vitro: Another evidence for the detrimental effect of the inescapable endogenous neurotoxin. Chem. Biol. Interact. 2020, 315, 108884. [Google Scholar] [CrossRef]
- Zhang, Y.; Buckmaster, P.S.; Qiu, L.; Wang, J.; Keunen, O.; Ghobadi, S.N.; Huang, A.; Hou, Q.; Li, N.; Narang, S. Non-invasive, neurotoxic surgery reduces seizures in a rat model of temporal lobe epilepsy. Exp. Neurol. 2021, 343, 113761. [Google Scholar] [CrossRef]
- Blesa, J.; Trigo-Damas, I.; Quiroga-Varela, A.; Jackson-Lewis, V.R. Oxidative stress and Parkinson’s disease. Front. Neuroanat. 2015, 9, 91. [Google Scholar] [CrossRef] [Green Version]















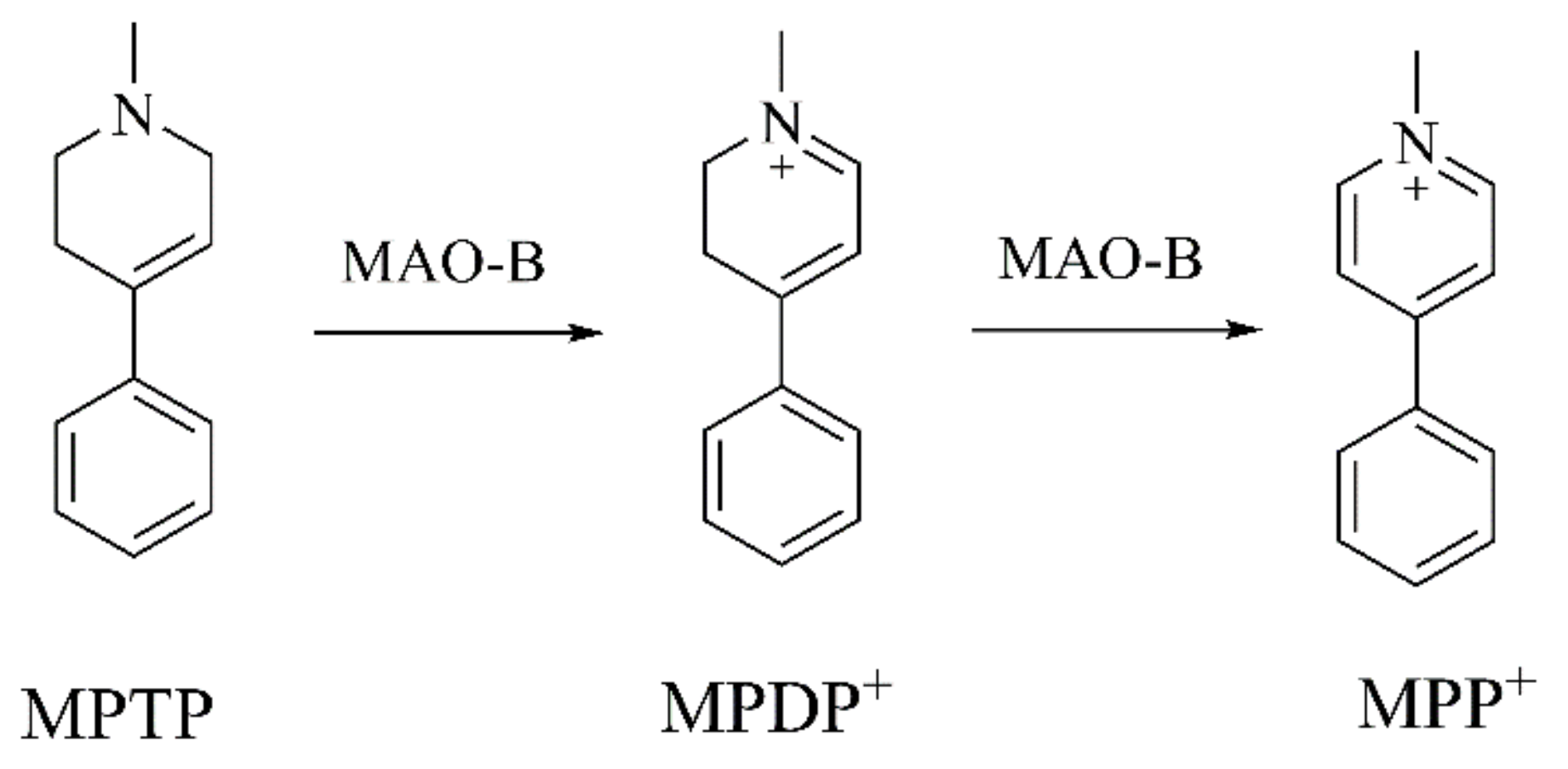{kind=link}
{kind=link}
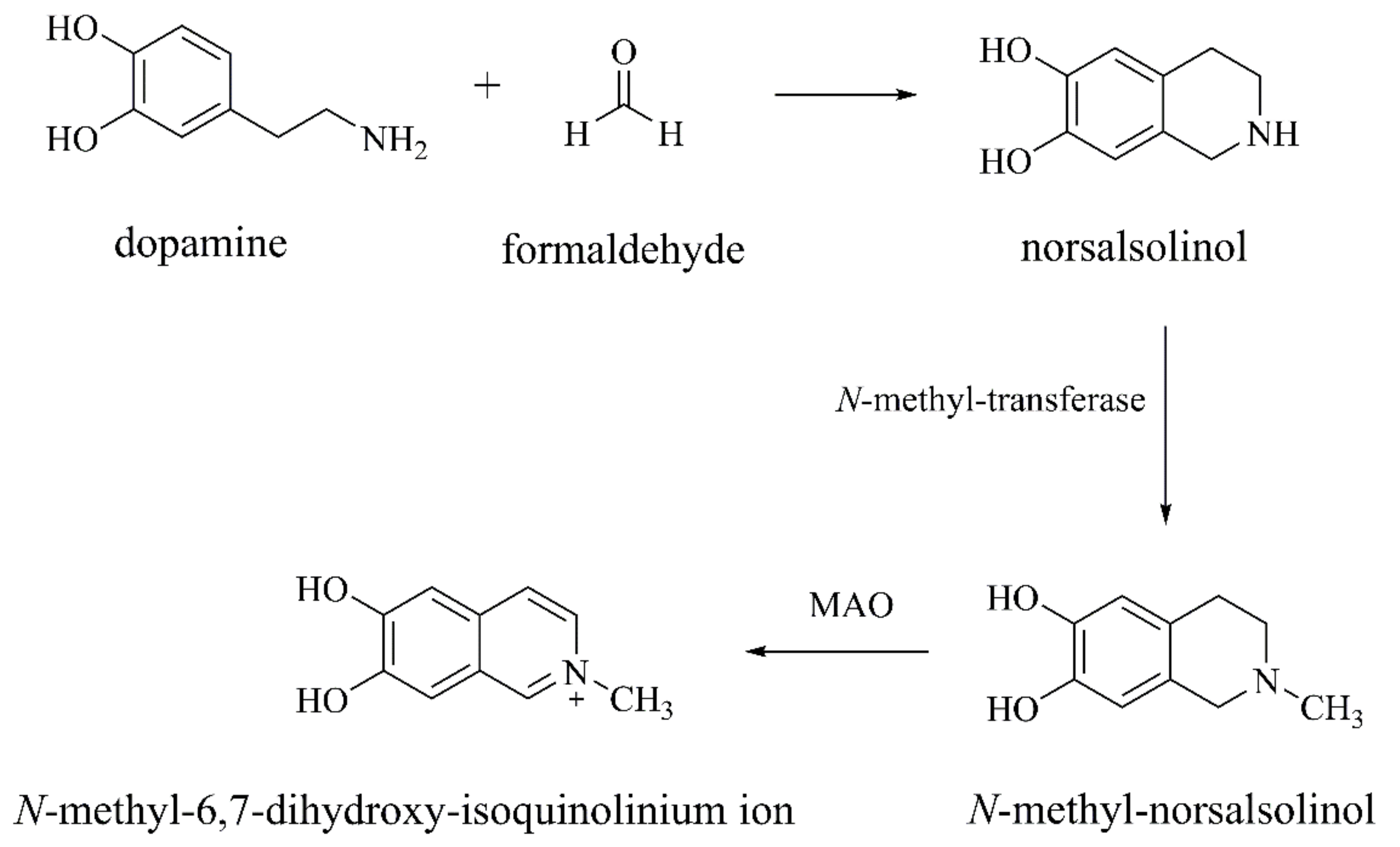{kind=link}
{kind=link}
{kind=link}
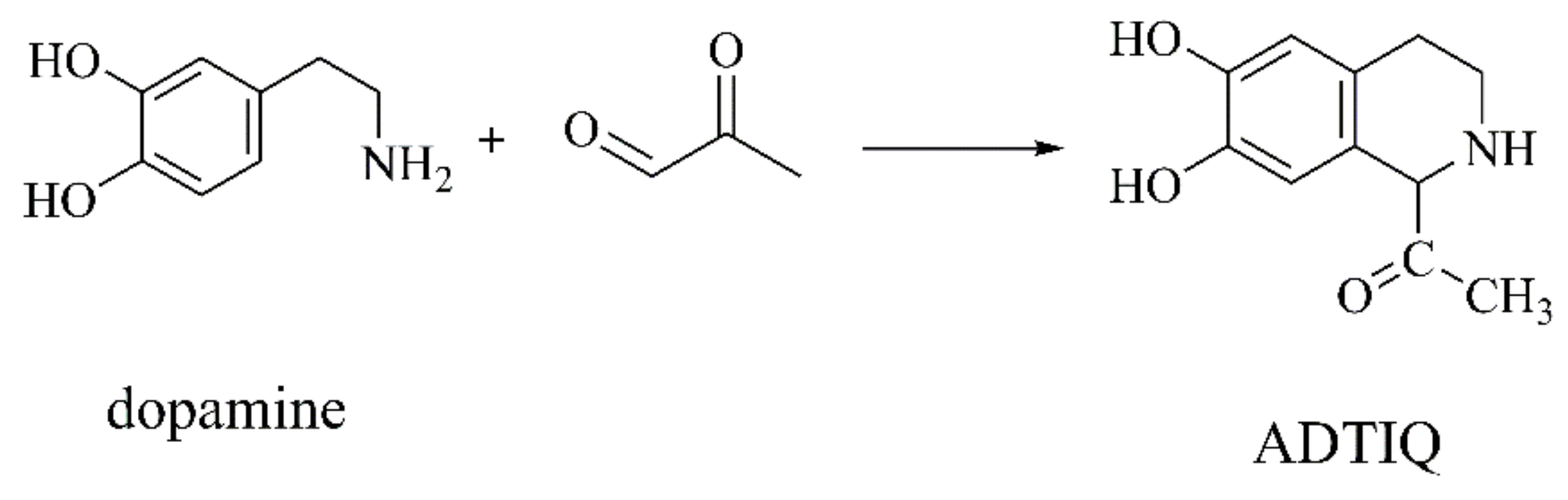{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
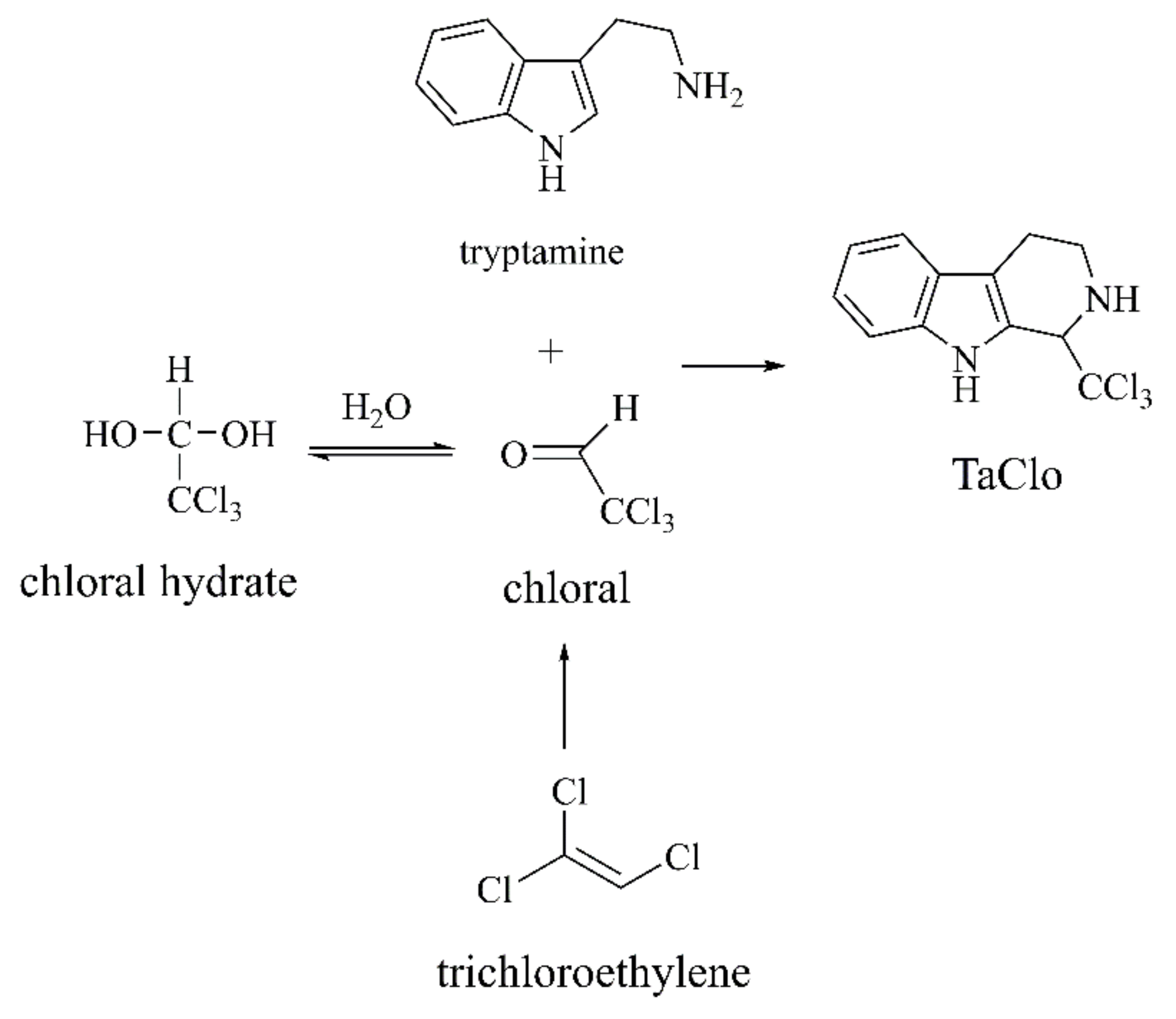{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, Y.; Li, B.; Ismail, N.; Smith, K.; Li, T.; Dai, R.; Deng, Y. Neurotoxicity and Underlying Mechanisms of Endogenous Neurotoxins. Int. J. Mol. Sci. 2021, 22, 12805. https://doi.org/10.3390/ijms222312805
Cao Y, Li B, Ismail N, Smith K, Li T, Dai R, Deng Y. Neurotoxicity and Underlying Mechanisms of Endogenous Neurotoxins. International Journal of Molecular Sciences. 2021; 22(23):12805. https://doi.org/10.3390/ijms222312805
Chicago/Turabian StyleCao, Yanlu, Bo Li, Nafissa Ismail, Kevin Smith, Tianmei Li, Rongji Dai, and Yulin Deng. 2021. "Neurotoxicity and Underlying Mechanisms of Endogenous Neurotoxins" International Journal of Molecular Sciences 22, no. 23: 12805. https://doi.org/10.3390/ijms222312805
APA StyleCao, Y., Li, B., Ismail, N., Smith, K., Li, T., Dai, R., & Deng, Y. (2021). Neurotoxicity and Underlying Mechanisms of Endogenous Neurotoxins. International Journal of Molecular Sciences, 22(23), 12805. https://doi.org/10.3390/ijms222312805