
Genetically Modified Cellular Therapies for Malignant Gliomas

 and
and
Abstract
:1. Introduction
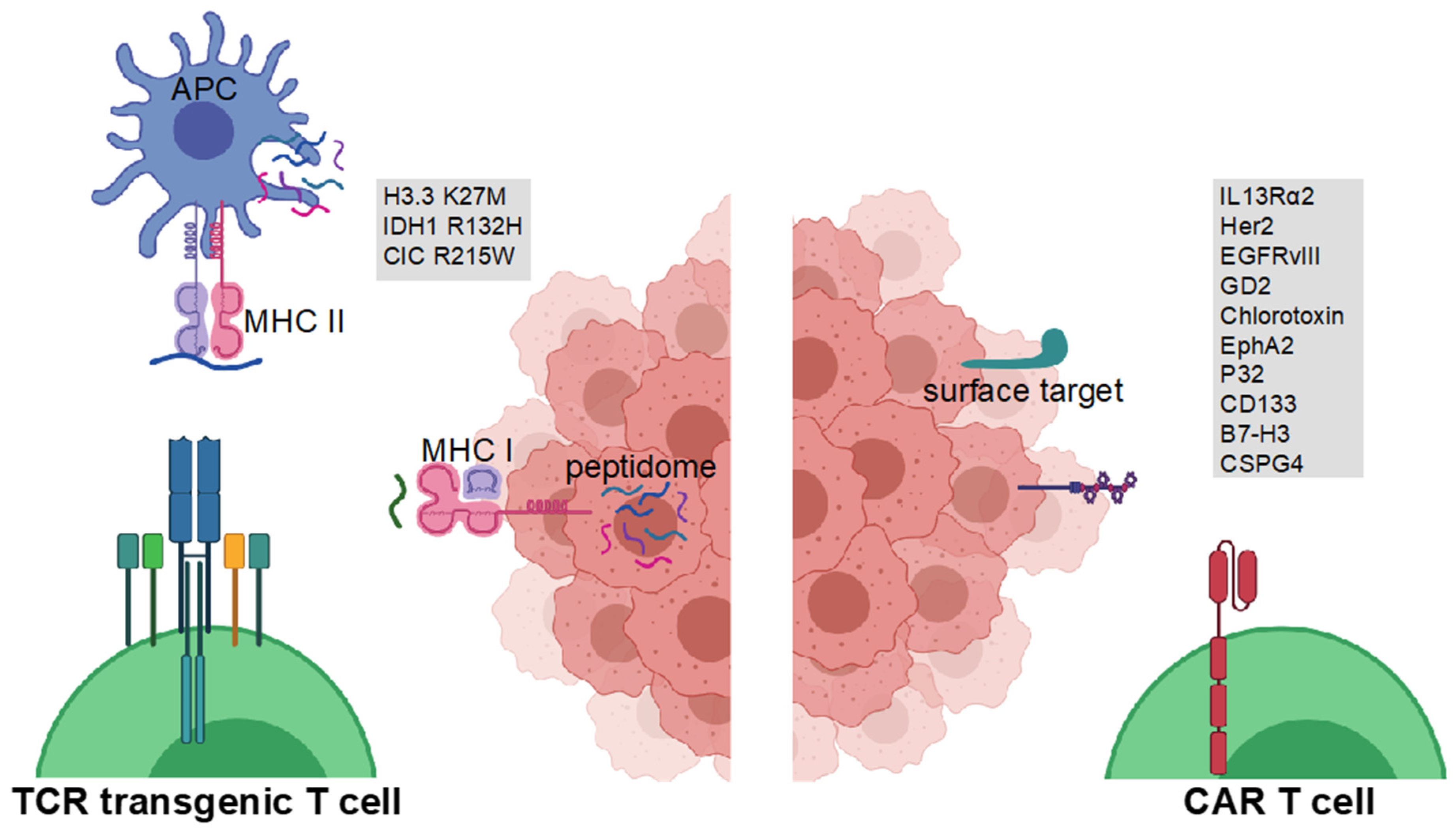
2. Glioma Antigens for CAR-Engineered T Cell Therapy
2.1. IL13Ra2
2.2. Her2
2.3. EGFRvIII
2.4. GD2
2.5. Chlorotoxin
2.6. EphA2
2.7. P32
2.8. CD133
2.9. CSPG4
2.10. B7-H3
3. Preclinical Strategies to Overcome Off-Target and On-Target Off-Tumor Side Effects
4. Overcoming Resistance to CAR T Cell Therapy
5. Application Routes for Genetically Modified T Cells in Preclinical Models
6. T Cell Receptor-Engineered T Cell Therapy
7. Perspective
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Aslan, K.; Turco, V.; Blobner, J.; Sonner, J.K.; Liuzzi, A.R.; Nunez, N.G.; De Feo, D.; Kickingereder, P.; Fischer, M.; Green, E.; et al. Heterogeneity of response to immune checkpoint blockade in hypermutated experimental gliomas. Nat. Commun. 2020, 11, 931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reardon, D.A.; Gokhale, P.C.; Klein, S.R.; Ligon, K.L.; Rodig, S.J.; Ramkissoon, S.H.; Jones, K.L.; Conway, A.S.; Liao, X.; Zhou, J.; et al. Glioblastoma Eradication Following Immune Checkpoint Blockade in an Orthotopic, Immunocompetent Model. Cancer Immunol. Res. 2016, 4, 124–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bahr, O.; et al. Effect of Nivolumab vs Bevacizumab in Patients with Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Omuro, A.; Brandes, A.A.; Rieger, J.; Wick, A.; Sepulveda, J.; Phuphanich, S.; de Souza, P.; Ahluwalia, M.S.; Lim, M.; et al. OS10.3 Randomized Phase 3 Study Evaluating the Efficacy and Safety of Nivolumab vs Bevacizumab in Patients with Recurrent Glioblastoma: CheckMate 143. Neuro Oncol. 2017, 19, iii21. [Google Scholar] [CrossRef] [Green Version]
- Maxwell, R.; Jackson, C.M.; Lim, M. Clinical Trials Investigating Immune Checkpoint Blockade in Glioblastoma. Curr. Treat. Options Oncol. 2017, 18, 51. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Schalper, K.A.; Rodriguez-Ruiz, M.E.; Diez-Valle, R.; Lopez-Janeiro, A.; Porciuncula, A.; Idoate, M.A.; Inoges, S.; de Andrea, C.; Lopez-Diaz de Cerio, A.; Tejada, S.; et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat. Med. 2019, 25, 470–476. [Google Scholar] [CrossRef]
- Zhao, J.; Chen, A.X.; Gartrell, R.D.; Silverman, A.M.; Aparicio, L.; Chu, T.; Bordbar, D.; Shan, D.; Samanamud, J.; Mahajan, A.; et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat. Med. 2019, 25, 462–469. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Lotze, M.T.; Muul, L.M.; Chang, A.E.; Avis, F.P.; Leitman, S.; Linehan, W.M.; Robertson, C.N.; Lee, R.E.; Rubin, J.T.; et al. A progress report on the treatment of 157 patients with advanced cancer using lymphokine-activated killer cells and interleukin-2 or high-dose interleukin-2 alone. N. Engl. J. Med. 1987, 316, 889–897. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Packard, B.S.; Aebersold, P.M.; Solomon, D.; Topalian, S.L.; Toy, S.T.; Simon, P.; Lotze, M.T.; Yang, J.C.; Seipp, C.A.; et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N. Engl. J. Med. 1988, 319, 1676–1680. [Google Scholar] [CrossRef]
- Topalian, S.L.; Solomon, D.; Avis, F.P.; Chang, A.E.; Freerksen, D.L.; Linehan, W.M.; Lotze, M.T.; Robertson, C.N.; Seipp, C.A.; Simon, P.; et al. Immunotherapy of patients with advanced cancer using tumor-infiltrating lymphocytes and recombinant interleukin-2: A pilot study. J. Clin. Oncol. 1988, 6, 839–853. [Google Scholar] [CrossRef]
- Dillman, R.O.; Duma, C.M.; Ellis, R.A.; Cornforth, A.N.; Schiltz, P.M.; Sharp, S.L.; DePriest, M.C. Intralesional lymphokine-activated killer cells as adjuvant therapy for primary glioblastoma. J. Immunother. 2009, 32, 914–919. [Google Scholar] [CrossRef]
- Dillman, R.O.; Duma, C.M.; Schiltz, P.M.; DePriest, C.; Ellis, R.A.; Okamoto, K.; Beutel, L.D.; De Leon, C.; Chico, S. Intracavitary placement of autologous lymphokine-activated killer (LAK) cells after resection of recurrent glioblastoma. J. Immunother. 2004, 27, 398–404. [Google Scholar] [CrossRef]
- Mehta, G.U.; Malekzadeh, P.; Shelton, T.; White, D.E.; Butman, J.A.; Yang, J.C.; Kammula, U.S.; Goff, S.L.; Rosenberg, S.A.; Sherry, R.M. Outcomes of Adoptive Cell Transfer with Tumor-infiltrating Lymphocytes for Metastatic Melanoma Patients with and Without Brain Metastases. J. Immunother. 2018, 41, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Meng, Q.; Bartek, J., Jr.; Poiret, T.; Persson, O.; Rane, L.; Rangelova, E.; Illies, C.; Peredo, I.H.; Luo, X.; et al. Tumor-infiltrating lymphocytes (TILs) from patients with glioma. Oncoimmunology 2017, 6, e1252894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reardon, D.A.; Wucherpfennig, K.W.; Freeman, G.; Wu, C.J.; Chiocca, E.A.; Wen, P.Y.; Curry, W.T., Jr.; Mitchell, D.A.; Fecci, P.E.; Sampson, J.H.; et al. An update on vaccine therapy and other immunotherapeutic approaches for glioblastoma. Expert Rev. Vaccines 2013, 12, 597–615. [Google Scholar] [CrossRef] [Green Version]
- Okada, H.; Kohanbash, G.; Zhu, X.; Kastenhuber, E.R.; Hoji, A.; Ueda, R.; Fujita, M. Immunotherapeutic approaches for glioma. Crit. Rev. Immunol. 2009, 29, 1–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, A.J.; Chen, L.C.; Chen, Y.Y. Navigating CAR-T cells through the solid-tumour microenvironment. Nat. Rev. Drug Discov. 2021, 20, 531–550. [Google Scholar] [CrossRef]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [Green Version]
- Debinski, W.; Gibo, D.M.; Slagle, B.; Powers, S.K.; Gillespie, G.Y. Receptor for interleukin 13 is abundantly and specifically over-expressed in patients with glioblastoma multiforme. Int. J. Oncol. 1999, 15, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Joshi, B.H.; Plautz, G.E.; Puri, R.K. Interleukin-13 receptor α chain: A novel tumor-associated transmembrane protein in primary explants of human malignant gliomas. Cancer Res. 2000, 60, 1168–1172. [Google Scholar]
- Debinski, W.; Obiri, N.I.; Powers, S.K.; Pastan, I.; Puri, R.K. Human glioma cells overexpress receptors for interleukin 13 and are extremely sensitive to a novel chimeric protein composed of interleukin 13 and pseudomonas exotoxin. Clin. Cancer Res. 1995, 1, 1253–1258. [Google Scholar] [PubMed]
- Krebs, S.; Chow, K.K.; Yi, Z.; Rodriguez-Cruz, T.; Hegde, M.; Gerken, C.; Ahmed, N.; Gottschalk, S. T cells redirected to interleukin-13Ralpha2 with interleukin-13 mutein-chimeric antigen receptors have anti-glioma activity but also recognize interleukin-13Ralpha1. Cytotherapy 2014, 16, 1121–1131. [Google Scholar] [CrossRef] [Green Version]
- Debinski, W.; Gibo, D.M.; Obiri, N.I.; Kealiher, A.; Puri, R.K. Novel anti-brain tumor cytotoxins specific for cancer cells. Nat. Biotechnol. 1998, 16, 449–453. [Google Scholar] [CrossRef]
- Thaci, B.; Brown, C.E.; Binello, E.; Werbaneth, K.; Sampath, P.; Sengupta, S. Significance of interleukin-13 receptor alpha 2-targeted glioblastoma therapy. Neuro Oncol. 2014, 16, 1304–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahlon, K.S.; Brown, C.; Cooper, L.J.; Raubitschek, A.; Forman, S.J.; Jensen, M.C. Specific recognition and killing of glioblastoma multiforme by interleukin 13-zetakine redirected cytolytic T cells. Cancer Res. 2004, 64, 9160–9166. [Google Scholar] [CrossRef] [Green Version]
- Brown, C.E.; Aguilar, B.; Starr, R.; Yang, X.; Chang, W.C.; Weng, L.; Chang, B.; Sarkissian, A.; Brito, A.; Sanchez, J.F.; et al. Optimization of IL13Ralpha2-Targeted Chimeric Antigen Receptor T Cells for Improved Anti-tumor Efficacy against Glioblastoma. Mol. Ther. 2018, 26, 31–44. [Google Scholar] [CrossRef] [Green Version]
- Krenciute, G.; Prinzing, B.L.; Yi, Z.; Wu, M.F.; Liu, H.; Dotti, G.; Balyasnikova, I.V.; Gottschalk, S. Transgenic Expression of IL15 Improves Antiglioma Activity of IL13Ralpha2-CAR T Cells but Results in Antigen Loss Variants. Cancer Immunol. Res. 2017, 5, 571–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, C.E.; Badie, B.; Barish, M.E.; Weng, L.; Ostberg, J.R.; Chang, W.C.; Naranjo, A.; Starr, R.; Wagner, J.; Wright, C.; et al. Bioactivity and Safety of IL13Ralpha2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2015, 21, 4062–4072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef]
- Ahmed, N.; Salsman, V.S.; Kew, Y.; Shaffer, D.; Powell, S.; Zhang, Y.J.; Grossman, R.G.; Heslop, H.E.; Gottschalk, S. HER2-specific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clin. Cancer Res. 2010, 16, 474–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; Ying, H.; Zeng, G.; Wheeler, C.J.; Black, K.L.; Yu, J.S. HER-2, gp100, and MAGE-1 are expressed in human glioblastoma and recognized by cytotoxic T cells. Cancer Res. 2004, 64, 4980–4986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turatti, F.; Figini, M.; Alberti, P.; Willemsen, R.A.; Canevari, S.; Mezzanzanica, D. Highly efficient redirected anti-tumor activity of human lymphocytes transduced with a completely human chimeric immune receptor. J. Gene Med. 2005, 7, 158–170. [Google Scholar] [CrossRef]
- Toth, G.; Szollosi, J.; Abken, H.; Vereb, G.; Szoor, A. A Small Number of HER2 Redirected CAR T Cells Significantly Improves Immune Response of Adoptively Transferred Mouse Lymphocytes against Human Breast Cancer Xenografts. Int. J. Mol. Sci. 2020, 21, 1039. [Google Scholar] [CrossRef] [Green Version]
- Stancovski, I.; Schindler, D.G.; Waks, T.; Yarden, Y.; Sela, M.; Eshhar, Z. Targeting of T lymphocytes to Neu/HER2-expressing cells using chimeric single chain Fv receptors. J. Immunol. 1993, 151, 6577–6582. [Google Scholar]
- Li, S.; Yang, J.; Urban, F.A.; MacGregor, J.N.; Hughes, D.P.; Chang, A.E.; McDonagh, K.T.; Li, Q. Genetically engineered T cells expressing a HER2-specific chimeric receptor mediate antigen-specific tumor regression. Cancer Gene Ther. 2008, 15, 382–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajjar, A.; Hernan, R.; Kocak, M.; Fuller, C.; Lee, Y.; McKinnon, P.J.; Wallace, D.; Lau, C.; Chintagumpala, M.; Ashley, D.M.; et al. Clinical, histopathologic, and molecular markers of prognosis: Toward a new disease risk stratification system for medulloblastoma. J. Clin. Oncol. 2004, 22, 984–993. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Burger, M.C.; Jennewein, L.; Genssler, S.; Schonfeld, K.; Zeiner, P.; Hattingen, E.; Harter, P.N.; Mittelbronn, M.; Tonn, T.; et al. ErbB2/HER2-Specific NK Cells for Targeted Therapy of Glioblastoma. J. Natl. Cancer Inst. 2016, 108, djv375. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef]
- Hegde, M.; Joseph, S.K.; Pashankar, F.; DeRenzo, C.; Sanber, K.; Navai, S.; Byrd, T.T.; Hicks, J.; Xu, M.L.; Gerken, C.; et al. Tumor response and endogenous immune reactivity after administration of HER2 CAR T cells in a child with metastatic rhabdomyosarcoma. Nat. Commun. 2020, 11, 3549. [Google Scholar] [CrossRef]
- Ahmed, N.; Brawley, V.S.; Hegde, M.; Robertson, C.; Ghazi, A.; Gerken, C.; Liu, E.; Dakhova, O.; Ashoori, A.; Corder, A.; et al. Human Epidermal Growth Factor Receptor 2 (HER2) -Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J. Clin. Oncol. 2015, 33, 1688–1696. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2017, 3, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Vitanza, N.A.; Johnson, A.J.; Wilson, A.L.; Brown, C.; Yokoyama, J.K.; Kunkele, A.; Chang, C.A.; Rawlings-Rhea, S.; Huang, W.; Seidel, K.; et al. Locoregional infusion of HER2-specific CAR T cells in children and young adults with recurrent or refractory CNS tumors: An interim analysis. Nat. Med. 2021, 27, 1544–1552. [Google Scholar] [CrossRef] [PubMed]
- Ekstrand, A.J.; Longo, N.; Hamid, M.L.; Olson, J.J.; Liu, L.; Collins, V.P.; James, C.D. Functional characterization of an EGF receptor with a truncated extracellular domain expressed in glioblastomas with EGFR gene amplification. Oncogene 1994, 9, 2313–2320. [Google Scholar]
- Wikstrand, C.J.; McLendon, R.E.; Friedman, A.H.; Bigner, D.D. Cell surface localization and density of the tumor-associated variant of the epidermal growth factor receptor, EGFRvIII. Cancer Res. 1997, 57, 4130–4140. [Google Scholar]
- Heimberger, A.B.; Hlatky, R.; Suki, D.; Yang, D.; Weinberg, J.; Gilbert, M.; Sawaya, R.; Aldape, K. Prognostic effect of epidermal growth factor receptor and EGFRvIII in glioblastoma multiforme patients. Clin. Cancer Res. 2005, 11, 1462–1466. [Google Scholar] [CrossRef] [Green Version]
- Heimberger, A.B.; Crotty, L.E.; Archer, G.E.; Hess, K.R.; Wikstrand, C.J.; Friedman, A.H.; Friedman, H.S.; Bigner, D.D.; Sampson, J.H. Epidermal growth factor receptor VIII peptide vaccination is efficacious against established intracerebral tumors. Clin. Cancer Res. 2003, 9, 4247–4254. [Google Scholar]
- Heimberger, A.B.; Archer, G.E.; Crotty, L.E.; McLendon, R.E.; Friedman, A.H.; Friedman, H.S.; Bigner, D.D.; Sampson, J.H. Dendritic cells pulsed with a tumor-specific peptide induce long-lasting immunity and are effective against murine intracerebral melanoma. Neurosurgery 2002, 50, 158–164; discussion 156–164. [Google Scholar] [CrossRef]
- Choi, B.D.; Archer, G.E.; Mitchell, D.A.; Heimberger, A.B.; McLendon, R.E.; Bigner, D.D.; Sampson, J.H. EGFRvIII-targeted vaccination therapy of malignant glioma. Brain Pathol. 2009, 19, 713–723. [Google Scholar] [CrossRef] [Green Version]
- Morgan, R.A.; Johnson, L.A.; Davis, J.L.; Zheng, Z.; Woolard, K.D.; Reap, E.A.; Feldman, S.A.; Chinnasamy, N.; Kuan, C.T.; Song, H.; et al. Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Hum. Gene Ther. 2012, 23, 1043–1053. [Google Scholar] [CrossRef] [Green Version]
- Sampson, J.H.; Choi, B.D.; Sanchez-Perez, L.; Suryadevara, C.M.; Snyder, D.J.; Flores, C.T.; Schmittling, R.J.; Nair, S.K.; Reap, E.A.; Norberg, P.K.; et al. EGFRvIII mCAR-modified T-cell therapy cures mice with established intracerebral glioma and generates host immunity against tumor-antigen loss. Clin. Cancer Res. 2014, 20, 972–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohno, M.; Ohkuri, T.; Kosaka, A.; Tanahashi, K.; June, C.H.; Natsume, A.; Okada, H. Expression of miR-17-92 enhances anti-tumor activity of T-cells transduced with the anti-EGFRvIII chimeric antigen receptor in mice bearing human GBM xenografts. J. Immunother. Cancer 2013, 1, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravanpay, A.C.; Gust, J.; Johnson, A.J.; Rolczynski, L.S.; Cecchini, M.; Chang, C.A.; Hoglund, V.J.; Mukherjee, R.; Vitanza, N.A.; Orentas, R.J.; et al. EGFR806-CAR T cells selectively target a tumor-restricted EGFR epitope in glioblastoma. Oncotarget 2019, 10, 7080–7095. [Google Scholar] [CrossRef] [Green Version]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef] [Green Version]
- Johnson, L.A.; Scholler, J.; Ohkuri, T.; Kosaka, A.; Patel, P.R.; McGettigan, S.E.; Nace, A.K.; Dentchev, T.; Thekkat, P.; Loew, A.; et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci. Transl. Med. 2015, 7, 275ra222. [Google Scholar] [CrossRef] [Green Version]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; Goldlust, S.A.; Iwamoto, F.; et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): A randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017, 18, 1373–1385. [Google Scholar] [CrossRef] [Green Version]
- Prapa, M.; Caldrer, S.; Spano, C.; Bestagno, M.; Golinelli, G.; Grisendi, G.; Petrachi, T.; Conte, P.; Horwitz, E.M.; Campana, D.; et al. A novel anti-GD2/4-1BB chimeric antigen receptor triggers neuroblastoma cell killing. Oncotarget 2015, 6, 24884–24894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mount, C.W.; Majzner, R.G.; Sundaresh, S.; Arnold, E.P.; Kadapakkam, M.; Haile, S.; Labanieh, L.; Hulleman, E.; Woo, P.J.; Rietberg, S.P.; et al. Potent antitumor efficacy of anti-GD2 CAR T cells in H3-K27M(+) diffuse midline gliomas. Nat. Med. 2018, 24, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Majzner, R.G.; Ramakrishna, S.; Mochizuki, A.; Patel, S.; Chinnasamy, H.; Yeom, K.; Schultz, L.; Richards, R.; Campen, C.; Reschke, A.; et al. Abstract CT031: GD2 CAR T cells mediate clinical activity and manageable toxicity in children and young adults with DIPG and H3K27M-mutated diffuse midline gliomas. Cancer Res. 2021, 81, CT031. [Google Scholar] [CrossRef]
- Soroceanu, L.; Gillespie, Y.; Khazaeli, M.B.; Sontheimer, H. Use of chlorotoxin for targeting of primary brain tumors. Cancer Res. 1998, 58, 4871–4879. [Google Scholar] [PubMed]
- Lyons, S.A.; O’Neal, J.; Sontheimer, H. Chlorotoxin, a scorpion-derived peptide, specifically binds to gliomas and tumors of neuroectodermal origin. Glia 2002, 39, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Starr, R.; Chang, W.C.; Aguilar, B.; Alizadeh, D.; Wright, S.L.; Yang, X.; Brito, A.; Sarkissian, A.; Ostberg, J.R.; et al. Chlorotoxin-directed CAR T cells for specific and effective targeting of glioblastoma. Sci. Transl. Med. 2020, 12, eaaw2672. [Google Scholar] [CrossRef]
- Ieguchi, K.; Maru, Y. Roles of EphA1/A2 and ephrin-A1 in cancer. Cancer Sci. 2019, 110, 841–848. [Google Scholar] [CrossRef]
- Miao, H.; Burnett, E.; Kinch, M.; Simon, E.; Wang, B. Activation of EphA2 kinase suppresses integrin function and causes focal-adhesion-kinase dephosphorylation. Nat. Cell Biol. 2000, 2, 62–69. [Google Scholar] [CrossRef]
- Kang, B.H.; Jensen, K.J.; Hatch, J.A.; Janes, K.A. Simultaneous profiling of 194 distinct receptor transcripts in human cells. Sci. Signal. 2013, 6, rs13. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.F.; Fokas, E.; Bieker, M.; Rose, F.; Rexin, P.; Zhu, Y.; Pagenstecher, A.; Engenhart-Cabillic, R.; An, H.X. Increased expression of EphA2 correlates with adverse outcome in primary and recurrent glioblastoma multiforme patients. Oncol. Rep. 2008, 19, 151–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, K.K.; Naik, S.; Kakarla, S.; Brawley, V.S.; Shaffer, D.R.; Yi, Z.; Rainusso, N.; Wu, M.F.; Liu, H.; Kew, Y.; et al. T cells redirected to EphA2 for the immunotherapy of glioblastoma. Mol. Ther. 2013, 21, 629–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, Z.; Prinzing, B.L.; Cao, F.; Gottschalk, S.; Krenciute, G. Optimizing EphA2-CAR T Cells for the Adoptive Immunotherapy of Glioma. Mol. Ther. Methods Clin. Dev. 2018, 9, 70–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donovan, L.K.; Delaidelli, A.; Joseph, S.K.; Bielamowicz, K.; Fousek, K.; Holgado, B.L.; Manno, A.; Srikanthan, D.; Gad, A.Z.; Van Ommeren, R.; et al. Locoregional delivery of CAR T cells to the cerebrospinal fluid for treatment of metastatic medulloblastoma and ependymoma. Nat. Med. 2020, 26, 720–731. [Google Scholar] [CrossRef]
- Agemy, L.; Kotamraju, V.R.; Friedmann-Morvinski, D.; Sharma, S.; Sugahara, K.N.; Ruoslahti, E. Proapoptotic peptide-mediated cancer therapy targeted to cell surface p32. Mol. Ther. 2013, 21, 2195–2204. [Google Scholar] [CrossRef] [Green Version]
- Rousso-Noori, L.; Mastandrea, I.; Talmor, S.; Waks, T.; Globerson Levin, A.; Haugas, M.; Teesalu, T.; Alvarez-Vallina, L.; Eshhar, Z.; Friedmann-Morvinski, D. P32-specific CAR T cells with dual antitumor and antiangiogenic therapeutic potential in gliomas. Nat. Commun. 2021, 12, 3615. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar]
- Vora, P.; Venugopal, C.; Salim, S.K.; Tatari, N.; Bakhshinyan, D.; Singh, M.; Seyfrid, M.; Upreti, D.; Rentas, S.; Wong, N.; et al. The Rational Development of CD133-Targeting Immunotherapies for Glioblastoma. Cell Stem Cell 2020, 26, 832–844.e6. [Google Scholar] [CrossRef]
- Uchida, N.; Buck, D.W.; He, D.; Reitsma, M.J.; Masek, M.; Phan, T.V.; Tsukamoto, A.S.; Gage, F.H.; Weissman, I.L. Direct isolation of human central nervous system stem cells. Proc. Natl. Acad. Sci. USA 2000, 97, 14720–14725. [Google Scholar] [CrossRef] [Green Version]
- Venugopal, C.; Hallett, R.; Vora, P.; Manoranjan, B.; Mahendram, S.; Qazi, M.A.; McFarlane, N.; Subapanditha, M.; Nolte, S.M.; Singh, M.; et al. Pyrvinium Targets CD133 in Human Glioblastoma Brain Tumor-Initiating Cells. Clin. Cancer Res. 2015, 21, 5324–5337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, C.A.; Pollett, A.; Gallinger, S.; Dick, J.E. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007, 445, 106–110. [Google Scholar] [CrossRef]
- Shibahara, I.; Sonoda, Y.; Saito, R.; Kanamori, M.; Yamashita, Y.; Kumabe, T.; Watanabe, M.; Suzuki, H.; Watanabe, T.; Ishioka, C.; et al. The expression status of CD133 is associated with the pattern and timing of primary glioblastoma recurrence. Neuro Oncol. 2013, 15, 1151–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellegatta, S.; Savoldo, B.; Di Ianni, N.; Corbetta, C.; Chen, Y.; Patane, M.; Sun, C.; Pollo, B.; Ferrone, S.; DiMeco, F.; et al. Constitutive and TNFalpha-inducible expression of chondroitin sulfate proteoglycan 4 in glioblastoma and neurospheres: Implications for CAR-T cell therapy. Sci. Transl. Med. 2018, 10, eaao2731. [Google Scholar] [CrossRef] [Green Version]
- Nehama, D.; Di Ianni, N.; Musio, S.; Du, H.; Patane, M.; Pollo, B.; Finocchiaro, G.; Park, J.J.H.; Dunn, D.E.; Edwards, D.S.; et al. B7-H3-redirected chimeric antigen receptor T cells target glioblastoma and neurospheres. EBioMedicine 2019, 47, 33–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, X.; Zhao, S.; Zhang, Y.; Wang, Y.; Zhang, Z.; Yang, M.; Zhu, Y.; Zhang, G.; Guo, G.; Tong, A.; et al. B7-H3 as a Novel CAR-T Therapeutic Target for Glioblastoma. Mol. Ther. Oncolytics 2019, 14, 279–287. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Tang, X.; Zhang, Z.; Gu, L.; Wei, H.; Zhao, S.; Zhong, K.; Mu, M.; Huang, C.; Jiang, C.; et al. Tandem CAR-T cells targeting CD70 and B7-H3 exhibit potent preclinical activity against multiple solid tumors. Theranostics 2020, 10, 7622–7634. [Google Scholar] [CrossRef]
- Parker, K.R.; Migliorini, D.; Perkey, E.; Yost, K.E.; Bhaduri, A.; Bagga, P.; Haris, M.; Wilson, N.E.; Liu, F.; Gabunia, K.; et al. Single-Cell Analyses Identify Brain Mural Cells Expressing CD19 as Potential Off-Tumor Targets for CAR-T Immunotherapies. Cell 2020, 183, 126–142.e17. [Google Scholar] [CrossRef]
- Yu, S.; Yi, M.; Qin, S.; Wu, K. Next generation chimeric antigen receptor T cells: Safety strategies to overcome toxicity. Mol. Cancer 2019, 18, 125. [Google Scholar] [CrossRef]
- Bonifant, C.L.; Szoor, A.; Torres, D.; Joseph, N.; Velasquez, M.P.; Iwahori, K.; Gaikwad, A.; Nguyen, P.; Arber, C.; Song, X.T.; et al. CD123-Engager T Cells as a Novel Immunotherapeutic for Acute Myeloid Leukemia. Mol. Ther. 2016, 24, 1615–1626. [Google Scholar] [CrossRef] [Green Version]
- Roybal, K.T.; Rupp, L.J.; Morsut, L.; Walker, W.J.; McNally, K.A.; Park, J.S.; Lim, W.A. Precision Tumor Recognition by T Cells with Combinatorial Antigen-Sensing Circuits. Cell 2016, 164, 770–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choe, J.H.; Watchmaker, P.B.; Simic, M.S.; Gilbert, R.D.; Li, A.W.; Krasnow, N.A.; Downey, K.M.; Yu, W.; Carrera, D.A.; Celli, A.; et al. SynNotch-CAR T cells overcome challenges of specificity, heterogeneity, and persistence in treating glioblastoma. Sci. Transl. Med. 2021, 13, eabe7378. [Google Scholar] [CrossRef] [PubMed]
- Van de Donk, N.; Themeli, M.; Usmani, S.Z. Determinants of response and mechanisms of resistance of CAR T-cell therapy in multiple myeloma. Blood Cancer Discov. 2021, 2, 302–318. [Google Scholar] [CrossRef] [PubMed]
- Song, M.K.; Park, B.B.; Uhm, J.E. Resistance Mechanisms to CAR T-Cell Therapy and Overcoming Strategy in B-Cell Hematologic Malignancies. Int. J. Mol. Sci. 2019, 20, 5010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, N.N.; Fry, T.J. Mechanisms of resistance to CAR T cell therapy. Nat. Rev. Clin. Oncol. 2019, 16, 372–385. [Google Scholar] [CrossRef]
- Cheng, J.; Zhao, L.; Zhang, Y.; Qin, Y.; Guan, Y.; Zhang, T.; Liu, C.; Zhou, J. Understanding the Mechanisms of Resistance to CAR T-Cell Therapy in Malignancies. Front. Oncol. 2019, 9, 1237. [Google Scholar] [CrossRef] [Green Version]
- Akhavan, D.; Alizadeh, D.; Wang, D.; Weist, M.R.; Shepphird, J.K.; Brown, C.E. CAR T cells for brain tumors: Lessons learned and road ahead. Immunol. Rev. 2019, 290, 60–84. [Google Scholar] [CrossRef] [Green Version]
- Agliardi, G.; Liuzzi, A.R.; Hotblack, A.; De Feo, D.; Nunez, N.; Stowe, C.L.; Friebel, E.; Nannini, F.; Rindlisbacher, L.; Roberts, T.A.; et al. Intratumoral IL-12 delivery empowers CAR-T cell immunotherapy in a pre-clinical model of glioblastoma. Nat. Commun. 2021, 12, 444. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Sun, C.; Landoni, E.; Metelitsa, L.; Dotti, G.; Savoldo, B. Eradication of Neuroblastoma by T Cells Redirected with an Optimized GD2-Specific Chimeric Antigen Receptor and Interleukin-15. Clin. Cancer Res. 2019, 25, 2915–2924. [Google Scholar] [CrossRef] [Green Version]
- Suryadevara, C.M.; Desai, R.; Abel, M.L.; Riccione, K.A.; Batich, K.A.; Shen, S.H.; Chongsathidkiet, P.; Gedeon, P.C.; Elsamadicy, A.A.; Snyder, D.J.; et al. Temozolomide lymphodepletion enhances CAR abundance and correlates with antitumor efficacy against established glioblastoma. Oncoimmunology 2018, 7, e1434464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegde, M.; Corder, A.; Chow, K.K.; Mukherjee, M.; Ashoori, A.; Kew, Y.; Zhang, Y.J.; Baskin, D.S.; Merchant, F.A.; Brawley, V.S.; et al. Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Mol. Ther. 2013, 21, 2087–2101. [Google Scholar] [CrossRef] [Green Version]
- Genssler, S.; Burger, M.C.; Zhang, C.; Oelsner, S.; Mildenberger, I.; Wagner, M.; Steinbach, J.P.; Wels, W.S. Dual targeting of glioblastoma with chimeric antigen receptor-engineered natural killer cells overcomes heterogeneity of target antigen expression and enhances antitumor activity and survival. Oncoimmunology 2016, 5, e1119354. [Google Scholar] [CrossRef] [PubMed]
- Bielamowicz, K.; Fousek, K.; Byrd, T.T.; Samaha, H.; Mukherjee, M.; Aware, N.; Wu, M.F.; Orange, J.S.; Sumazin, P.; Man, T.K.; et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro Oncol. 2018, 20, 506–518. [Google Scholar] [CrossRef]
- El Khawanky, N.; Hughes, A.; Yu, W.; Taromi, S.; Clarson, J.; Lopez, A.F.; Brown, M.P.; Duyster, J.; Hughes, T.P.; White, D.L.; et al. Azacytidine Sensitizes AML Cells for Effective Elimination by CD123 CAR T-Cells. Blood 2019, 134, 3904. [Google Scholar] [CrossRef]
- Terracina, K.P.; Graham, L.J.; Payne, K.K.; Manjili, M.H.; Baek, A.; Damle, S.R.; Bear, H.D. DNA methyltransferase inhibition increases efficacy of adoptive cellular immunotherapy of murine breast cancer. Cancer Immunol. Immunother. 2016, 65, 1061–1073. [Google Scholar] [CrossRef]
- Wang, Y.; Tong, C.; Dai, H.; Wu, Z.; Han, X.; Guo, Y.; Chen, D.; Wei, J.; Ti, D.; Liu, Z.; et al. Low-Dose decitabine priming endows CAR T cells with enhanced and persistent antitumour potential via epigenetic reprogramming. Nat. Commun. 2021, 12, 409. [Google Scholar] [CrossRef]
- You, L.; Han, Q.; Zhu, L.; Zhu, Y.; Bao, C.; Yang, C.; Lei, W.; Qian, W. Decitabine-Mediated Epigenetic Reprograming Enhances Anti-leukemia Efficacy of CD123-Targeted Chimeric Antigen Receptor T-Cells. Front. Immunol. 2020, 11, 1787. [Google Scholar] [CrossRef]
- Priceman, S.J.; Tilakawardane, D.; Jeang, B.; Aguilar, B.; Murad, J.P.; Park, A.K.; Chang, W.C.; Ostberg, J.R.; Neman, J.; Jandial, R.; et al. Regional Delivery of Chimeric Antigen Receptor-Engineered T Cells Effectively Targets HER2(+) Breast Cancer Metastasis to the Brain. Clin. Cancer Res. 2018, 24, 95–105. [Google Scholar] [CrossRef] [Green Version]
- Majzner, R.G.; Theruvath, J.L.; Nellan, A.; Heitzeneder, S.; Cui, Y.; Mount, C.W.; Rietberg, S.P.; Linde, M.H.; Xu, P.; Rota, C.; et al. CAR T Cells Targeting B7-H3, a Pan-Cancer Antigen, Demonstrate Potent Preclinical Activity Against Pediatric Solid Tumors and Brain Tumors. Clin. Cancer Res. 2019, 25, 2560–2574. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Hirabayashi, K.; Ahn, S.; Kren, N.P.; Montgomery, S.A.; Wang, X.; Tiruthani, K.; Mirlekar, B.; Michaud, D.; Greene, K.; et al. Antitumor Responses in the Absence of Toxicity in Solid Tumors by Targeting B7-H3 via Chimeric Antigen Receptor T Cells. Cancer Cell 2019, 35, 221–237.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, C.E.; Alizadeh, D.; Jonsson, V.; Hibbard, J.; Yahn, S.; Wong, R.A.; Yang, X.; Ng, R.; Dullerud, N.; Maker, M.; et al. Abstract 59: CAR T cell therapy reshapes the tumor microenvironment to promote host antitumor immune repsonses in glioblastoma. Cancer Res. 2021, 81, 59. [Google Scholar] [CrossRef]
- Skog, J. Glioma-Specific antigens for immune tumor therapy. Expert Rev. Vaccines 2006, 5, 793–802. [Google Scholar] [CrossRef]
- Rimoldi, D.; Romero, P.; Carrel, S. The human melanoma antigen-encoding gene, MAGE-1, is expressed by other tumour cells of neuroectodermal origin such as glioblastomas and neuroblastomas. Int. J. Cancer 1993, 54, 527–528. [Google Scholar] [CrossRef] [PubMed]
- Imaizumi, T.; Kuramoto, T.; Matsunaga, K.; Shichijo, S.; Yutani, S.; Shigemori, M.; Oizumi, K.; Itoh, K. Expression of the tumor-rejection antigen SART1 in brain tumors. Int. J. Cancer 1999, 83, 760–764. [Google Scholar] [CrossRef]
- Morgan, R.A.; Dudley, M.E.; Wunderlich, J.R.; Hughes, M.S.; Yang, J.C.; Sherry, R.M.; Royal, R.E.; Topalian, S.L.; Kammula, U.S.; Restifo, N.P.; et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 2006, 314, 126–129. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, S.A.; Restifo, N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015, 348, 62–68. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Li, X.; Zhou, W.L.; Huang, Y.; Liang, X.; Jiang, L.; Yang, X.; Sun, J.; Li, Z.; Han, W.D.; et al. Genetically engineered T cells for cancer immunotherapy. Signal. Transduct. Target. Ther. 2019, 4, 35. [Google Scholar] [CrossRef]
- Johnson, L.A.; Morgan, R.A.; Dudley, M.E.; Cassard, L.; Yang, J.C.; Hughes, M.S.; Kammula, U.S.; Royal, R.E.; Sherry, R.M.; Wunderlich, J.R.; et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 2009, 114, 535–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parkhurst, M.R.; Yang, J.C.; Langan, R.C.; Dudley, M.E.; Nathan, D.A.; Feldman, S.A.; Davis, J.L.; Morgan, R.A.; Merino, M.J.; Sherry, R.M.; et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol. Ther. 2011, 19, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Robbins, P.F.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; Dudley, M.E.; Wunderlich, J.R.; Nahvi, A.V.; Helman, L.J.; Mackall, C.L.; et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J. Clin. Oncol. 2011, 29, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Linette, G.P.; Stadtmauer, E.A.; Maus, M.V.; Rapoport, A.P.; Levine, B.L.; Emery, L.; Litzky, L.; Bagg, A.; Carreno, B.M.; Cimino, P.J.; et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood 2013, 122, 863–871. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Sampson, J.H.; Gunn, M.D.; Fecci, P.E.; Ashley, D.M. Brain immunology and immunotherapy in brain tumours. Nat. Rev. Cancer 2020, 20, 12–25. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Hilf, N.; Kuttruff-Coqui, S.; Frenzel, K.; Bukur, V.; Stevanovic, S.; Gouttefangeas, C.; Platten, M.; Tabatabai, G.; Dutoit, V.; van der Burg, S.H.; et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature 2019, 565, 240–245. [Google Scholar] [CrossRef]
- Keskin, D.B.; Anandappa, A.J.; Sun, J.; Tirosh, I.; Mathewson, N.D.; Li, S.; Oliveira, G.; Giobbie-Hurder, A.; Felt, K.; Gjini, E.; et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 2019, 565, 234–239. [Google Scholar] [CrossRef]
- Kilian, M.; Friedrich, M.; Sanghvi, K.; Green, E.; Pusch, S.; Kawauchi, D.; Löwer, M.; Sonner, J.K.; Krämer, C.; Zaman, J.; et al. T cell receptor therapy targeting mutant capicua transcriptional repressor in experimental gliomas. Clin. Cancer Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Platten, M.; Bunse, L.; Wick, A.; Bunse, T.; Le Cornet, L.; Harting, I.; Sahm, F.; Sanghvi, K.; Tan, C.L.; Poschke, I.; et al. A vaccine targeting mutant IDH1 in newly diagnosed glioma. Nature 2021, 592, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Bunse, L.; Pusch, S.; Bunse, T.; Sahm, F.; Sanghvi, K.; Friedrich, M.; Alansary, D.; Sonner, J.K.; Green, E.; Deumelandt, K.; et al. Suppression of antitumor T cell immunity by the oncometabolite (R)-2-hydroxyglutarate. Nat. Med. 2018, 24, 1192–1203. [Google Scholar] [CrossRef]
- Bunse, L.; Schumacher, T.; Sahm, F.; Pusch, S.; Oezen, I.; Rauschenbach, K.; Gonzalez, M.; Solecki, G.; Osswald, M.; Capper, D.; et al. Proximity ligation assay evaluates IDH1R132H presentation in gliomas. J. Clin. Investig. 2015, 125, 593–606. [Google Scholar] [CrossRef] [Green Version]
- Ochs, K.; Ott, M.; Bunse, T.; Sahm, F.; Bunse, L.; Deumelandt, K.; Sonner, J.K.; Keil, M.; von Deimling, A.; Wick, W.; et al. K27M-mutant histone-3 as a novel target for glioma immunotherapy. Oncoimmunology 2017, 6, e1328340. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, T.; Bunse, L.; Wick, W.; Platten, M. Mutant IDH1: An immunotherapeutic target in tumors. Oncoimmunology 2014, 3, e974392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellegatta, S.; Valletta, L.; Corbetta, C.; Patane, M.; Zucca, I.; Riccardi Sirtori, F.; Bruzzone, M.G.; Fogliatto, G.; Isacchi, A.; Pollo, B.; et al. Effective immuno-targeting of the IDH1 mutation R132H in a murine model of intracranial glioma. Acta Neuropathol. Commun. 2015, 3, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cairns, R.A.; Mak, T.W. Oncogenic isocitrate dehydrogenase mutations: Mechanisms, models, and clinical opportunities. Cancer Discov. 2013, 3, 730–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sturm, D.; Witt, H.; Hovestadt, V.; Khuong-Quang, D.A.; Jones, D.T.; Konermann, C.; Pfaff, E.; Tonjes, M.; Sill, M.; Bender, S.; et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 2012, 22, 425–437. [Google Scholar] [CrossRef] [Green Version]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.; Tonjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef]
- Wu, G.; Broniscer, A.; McEachron, T.A.; Lu, C.; Paugh, B.S.; Becksfort, J.; Qu, C.; Ding, L.; Huether, R.; Parker, M.; et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 2012, 44, 251–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bender, S.; Tang, Y.; Lindroth, A.M.; Hovestadt, V.; Jones, D.T.; Kool, M.; Zapatka, M.; Northcott, P.A.; Sturm, D.; Wang, W.; et al. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell 2013, 24, 660–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klemm, F.; Maas, R.R.; Bowman, R.L.; Kornete, M.; Soukup, K.; Nassiri, S.; Brouland, J.P.; Iacobuzio-Donahue, C.A.; Brennan, C.; Tabar, V.; et al. Interrogation of the Microenvironmental Landscape in Brain Tumors Reveals Disease-Specific Alterations of Immune Cells. Cell 2020, 181, 1643–1660.e17. [Google Scholar] [CrossRef]
- Friebel, E.; Kapolou, K.; Unger, S.; Nunez, N.G.; Utz, S.; Rushing, E.J.; Regli, L.; Weller, M.; Greter, M.; Tugues, S.; et al. Single-Cell Mapping of Human Brain Cancer Reveals Tumor-Specific Instruction of Tissue-Invading Leukocytes. Cell 2020, 181, 1626–1642.e20. [Google Scholar] [CrossRef]
- Friedrich, M.; Sankowski, R.; Bunse, L.; Kilian, M.; Green, E.; Ramallo Guevara, C.; Pusch, S.; Poschet, G.; Sanghvi, K.; Hahn, M.; et al. Tryptophan metabolism drives dynamic immunosuppressive myeloid states in IDH-mutant gliomas. Nat. Cancer 2021, 2, 723–740. [Google Scholar] [CrossRef]
- Kadiyala, P.; Carney, S.V.; Gauss, J.C.; Garcia-Fabiani, M.B.; Haase, S.; Alghamri, M.S.; Nunez, F.J.; Liu, Y.; Yu, M.; Taher, A.; et al. Inhibition of 2-hydroxyglutarate elicits metabolic reprogramming and mutant IDH1 glioma immunity in mice. J. Clin. Investig. 2021, 131, e139542. [Google Scholar] [CrossRef] [PubMed]
- Kohanbash, G.; Carrera, D.A.; Shrivastav, S.; Ahn, B.J.; Jahan, N.; Mazor, T.; Chheda, Z.S.; Downey, K.M.; Watchmaker, P.B.; Beppler, C.; et al. Isocitrate dehydrogenase mutations suppress STAT1 and CD8+ T cell accumulation in gliomas. J. Clin. Investig. 2017, 127, 1425–1437. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, N.; Rao, A.; Sandlesh, P.; Yerneni, S.S.; Swain, A.D.; Bullock, K.M.; Hansen, K.M.; Zhang, X.; Jaman, E.; Allen, J.; et al. Characterization of systemic immunosuppression by IDH mutant glioma small extracellular vesicles. Neuro Oncol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Chheda, Z.S.; Kohanbash, G.; Okada, K.; Jahan, N.; Sidney, J.; Pecoraro, M.; Yang, X.; Carrera, D.A.; Downey, K.M.; Shrivastav, S.; et al. Novel and shared neoantigen derived from histone 3 variant H3.3K27M mutation for glioma T cell therapy. J. Exp. Med. 2018, 215, 141–157. [Google Scholar] [CrossRef] [PubMed]
- Green, E.W.; Bunse, L.; Bozza, M.; Sanghvi, K.; Platten, M. TCR validation toward gene therapy for cancer. Methods Enzym. 2019, 629, 419–441. [Google Scholar] [CrossRef]
- Bunse, L.; Green, E.W.; Platten, M. High-throughput discovery of cancer-targeting TCRs. Methods Enzym. 2019, 629, 401–417. [Google Scholar] [CrossRef]
- Behnan, J.; Finocchiaro, G.; Hanna, G. The landscape of the mesenchymal signature in brain tumours. Brain 2019, 142, 847–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337–341. [Google Scholar] [CrossRef]
- Louveau, A.; Herz, J.; Alme, M.N.; Salvador, A.F.; Dong, M.Q.; Viar, K.E.; Herod, S.G.; Knopp, J.; Setliff, J.C.; Lupi, A.L.; et al. CNS lymphatic drainage and neuroinflammation are regulated by meningeal lymphatic vasculature. Nat. Neurosci 2018, 21, 1380–1391. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Dichwalkar, T.; Chang, J.Y.H.; Cossette, B.; Garafola, D.; Zhang, A.Q.; Fichter, M.; Wang, C.; Liang, S.; Silva, M.; et al. Enhanced CAR-T cell activity against solid tumors by vaccine boosting through the chimeric receptor. Science 2019, 365, 162–168. [Google Scholar] [CrossRef]


{kind=link}
| Clinical Trial | Entity | Target | Start | Phase | Combination |
|---|---|---|---|---|---|
| NCT04196413 | Diffuse intrinsic pontine gliomas (DIPG) + Spinal diffuse midline glioma (DMG) | GD2 | Dec 19 | 1 | Fludara, Cyclo |
| NCT04003649 | Recurrent or refractory GBM | IL13Ra2 + Ipi | Jul 19 | 1 | Nivo + Ipi |
| NCT02442297 | HER2-positive CNS tumors | Her2 | May 15 | 1 | |
| NCT04510051 | Recurrent or refractory brain tumors in children | IL13Ra2 | Aug 20 | 1 | Fludara, Cyclo |
| NCT04099797 | GD2-positive brain tumors | GD2 | Sep 19 | 1 | Fludara, Cyclo |
| NCT04661384 | Leptomeningeal GBM, ependymoma, or medulloblastoma | IL13a2 | Dec 20 | 1 | |
| NCT04185038 | DIPG/DMG and recurrent or refractory pediatric CNS tumors | B7-H3 | Dec 29 | 1 | |
| NCT03638167 | EGFR-positive recurrent or refractory pediatric CNS tumors | EGFR806 | Aug 18 | 1 | |
| NCT03500991 | HER2-positive recurrent or refractory pediatric CNS tumors | HER2 | Apr 18 | 1 | |
| NCT03696030 | Recurrent brain or leptomeningeal metastases | HER2 | Oct 18 | 1 | |
| NCT04903795 * | Grade 4 malignant glioma | EGFRvIII | May 21 | 1 | autologous activated T cells |
| NCT04385173 | Recurrent and refractory GBM | B7-H3 | May 20 | 1 | TMZ |
| NCT04077866 | Recurrent or refractory GBM | B7-H3 | Sep 19 | 1/2 | TMZ |
| NCT04214392 | MPP2-positive recurrent or progressive GBMa | Chlorotoxin-derived CAR | Jan 20 | 1 | |
| NCT04045847 | Recurrent malignant glioma | CD147 | Aug 19 | 1 | |
| NCT03389230 | Recurrent or refractory grade 3–4 glioma | Her2 | Jan 18 | 1 | |
| NCT02208362 | Recurrent or refractory malignant glioma | IL13Ra2 | Aug 14 | 1 | |
| NCT02575261 # | EphA2-positive malignant glioma | EphA2 | Oct 15 | 1/2 | |
| NCT03726515 $ | Newly diagnosed MGMT-unmethylated GBM | EGFRvIII | Oct 18 | 1 | Pembro |
| NCT03170141 ~ | GBM | several | May 17 | 1 | Fludara, Cyclo |
| NCT03423992 | Recurrent malignant gliomas | several | Feb 18 | 1 | |
| NCT03383978 | Recurrent HER2-positive GBM | HER2 | Dec 17 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kilian, M.; Bunse, T.; Wick, W.; Platten, M.; Bunse, L. Genetically Modified Cellular Therapies for Malignant Gliomas. Int. J. Mol. Sci. 2021, 22, 12810. https://doi.org/10.3390/ijms222312810
Kilian M, Bunse T, Wick W, Platten M, Bunse L. Genetically Modified Cellular Therapies for Malignant Gliomas. International Journal of Molecular Sciences. 2021; 22(23):12810. https://doi.org/10.3390/ijms222312810
Chicago/Turabian StyleKilian, Michael, Theresa Bunse, Wolfgang Wick, Michael Platten, and Lukas Bunse. 2021. "Genetically Modified Cellular Therapies for Malignant Gliomas" International Journal of Molecular Sciences 22, no. 23: 12810. https://doi.org/10.3390/ijms222312810
APA StyleKilian, M., Bunse, T., Wick, W., Platten, M., & Bunse, L. (2021). Genetically Modified Cellular Therapies for Malignant Gliomas. International Journal of Molecular Sciences, 22(23), 12810. https://doi.org/10.3390/ijms222312810