
Comparative Transcriptome Analysis Revealed the Key Genes Regulating Ascorbic Acid Synthesis in Actinidia

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Transcriptome of Actinidia Fruits
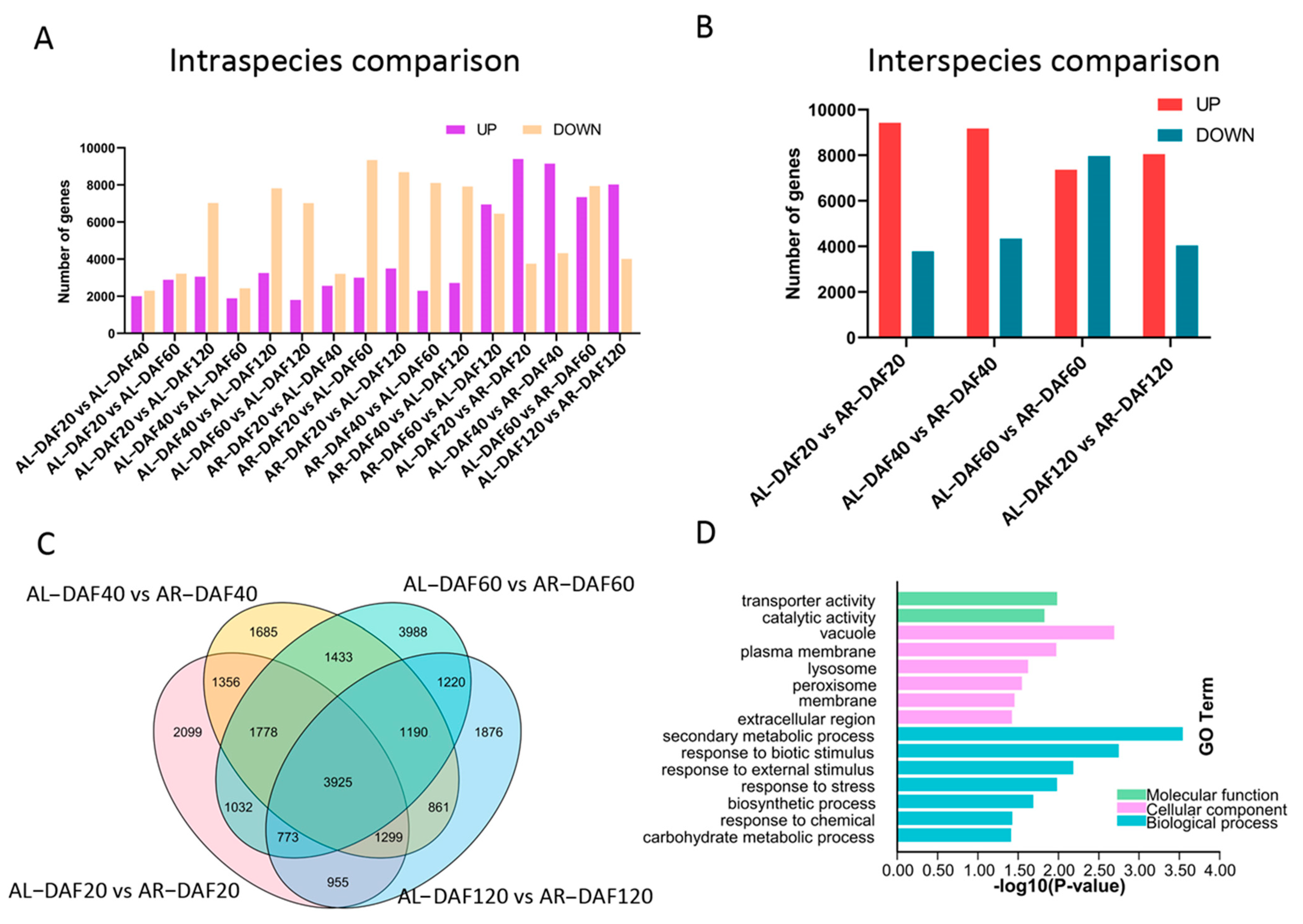
2.2. Differential Expression Analysis
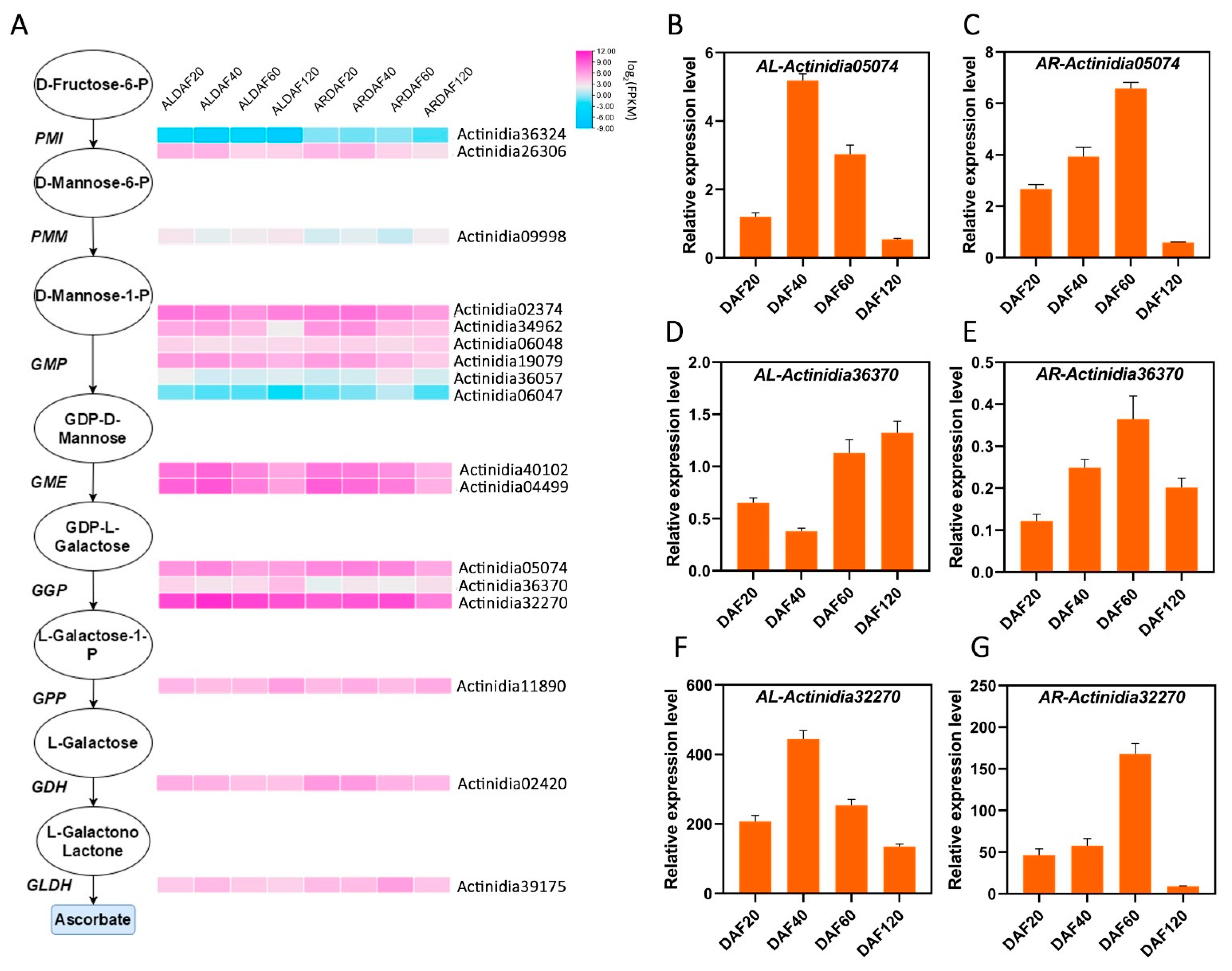
2.3. AsA Synthesis Pathway Analysis in Kiwifruit
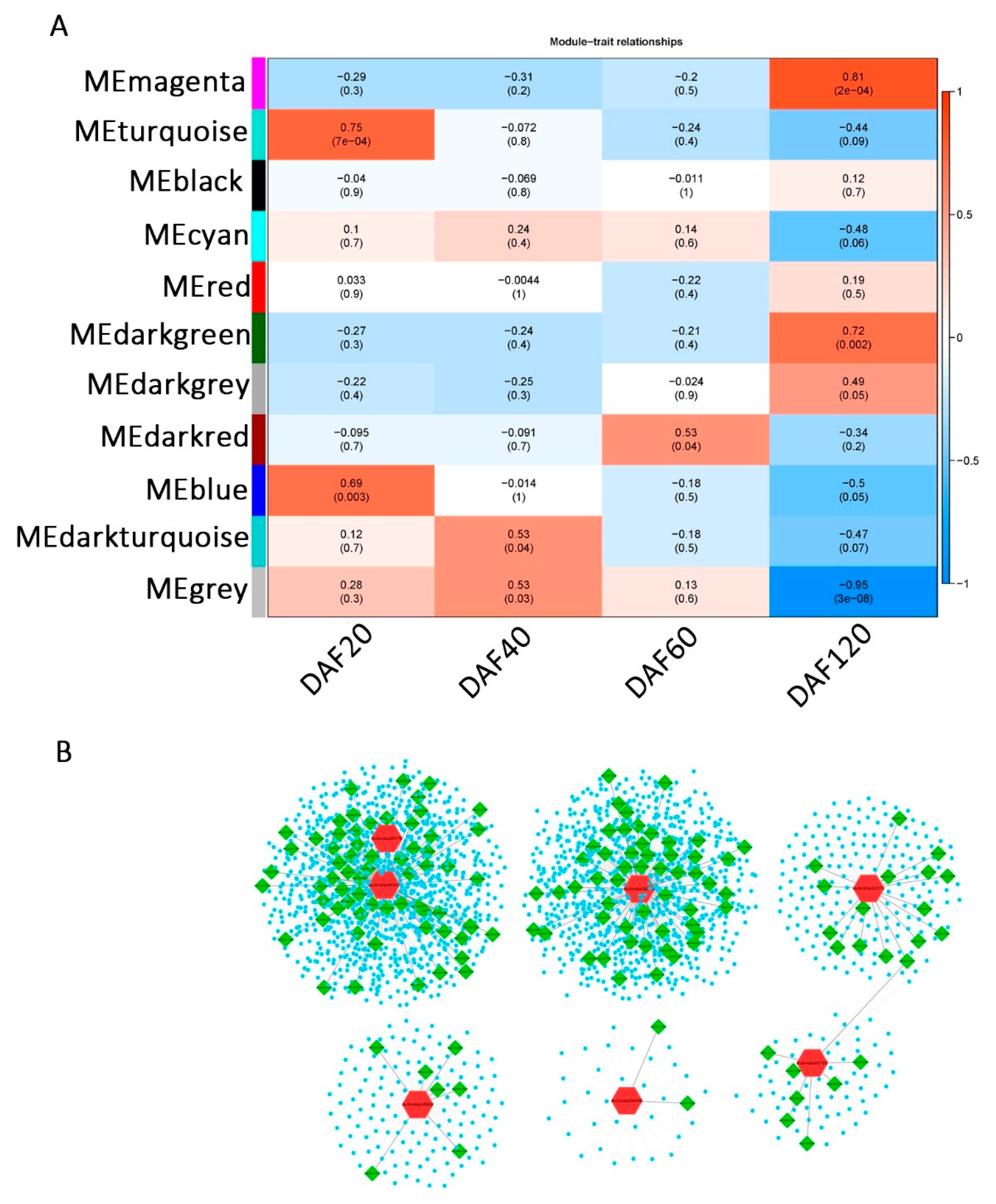
2.4. Weighted Gene Co-Expression Network
2.5. Overexpression of AcGGP3 (Actinidia32270) Increases AsA Accumulation in Kiwifruit
2.6. Differential Expression between Wild-Type and GGP3-Overexpressing Transgenic Lines of Kiwifruit
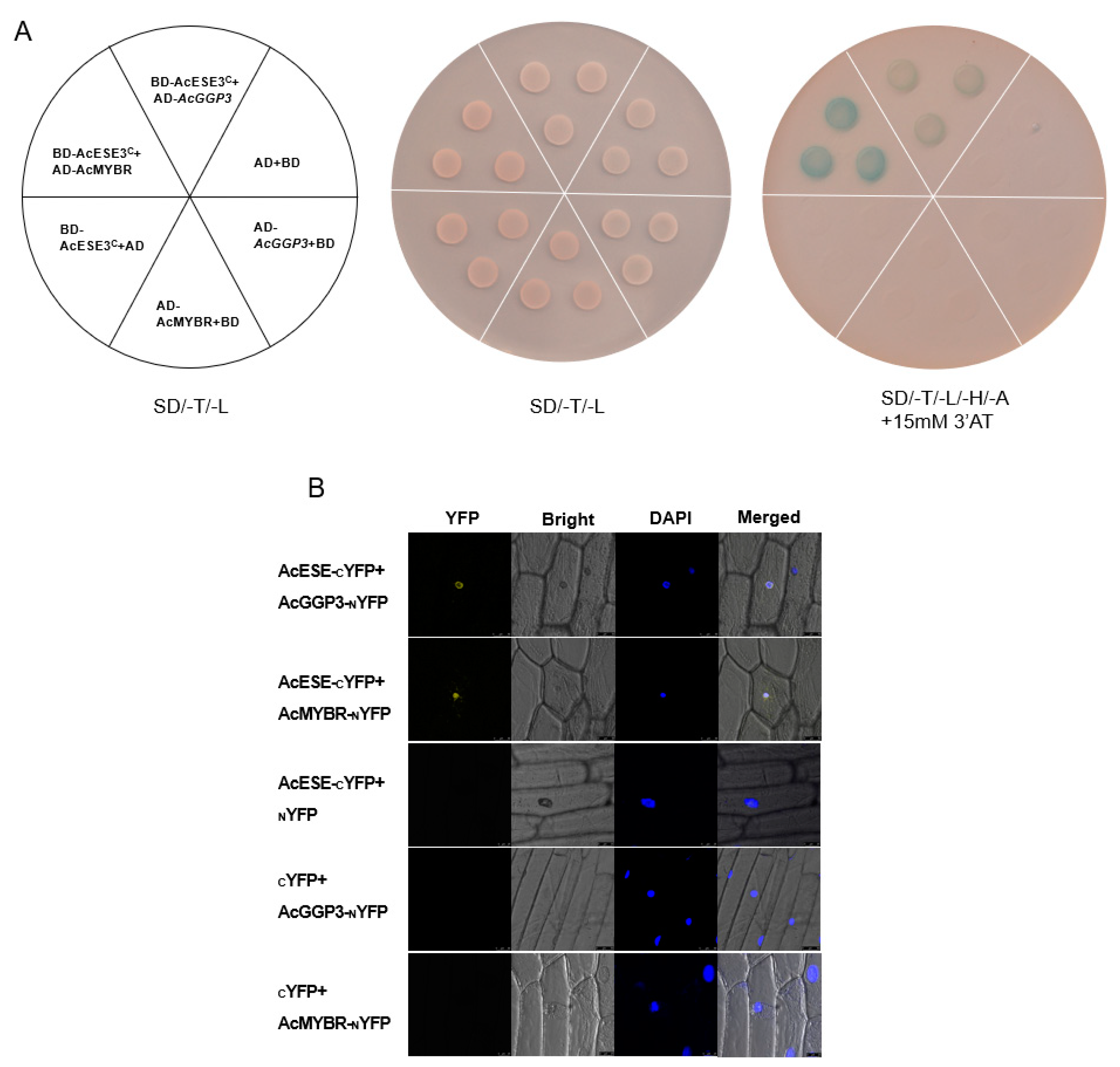
2.7. The Transcription Tactor AcESE3 Interacts with AcGGP3 and AcMYBR
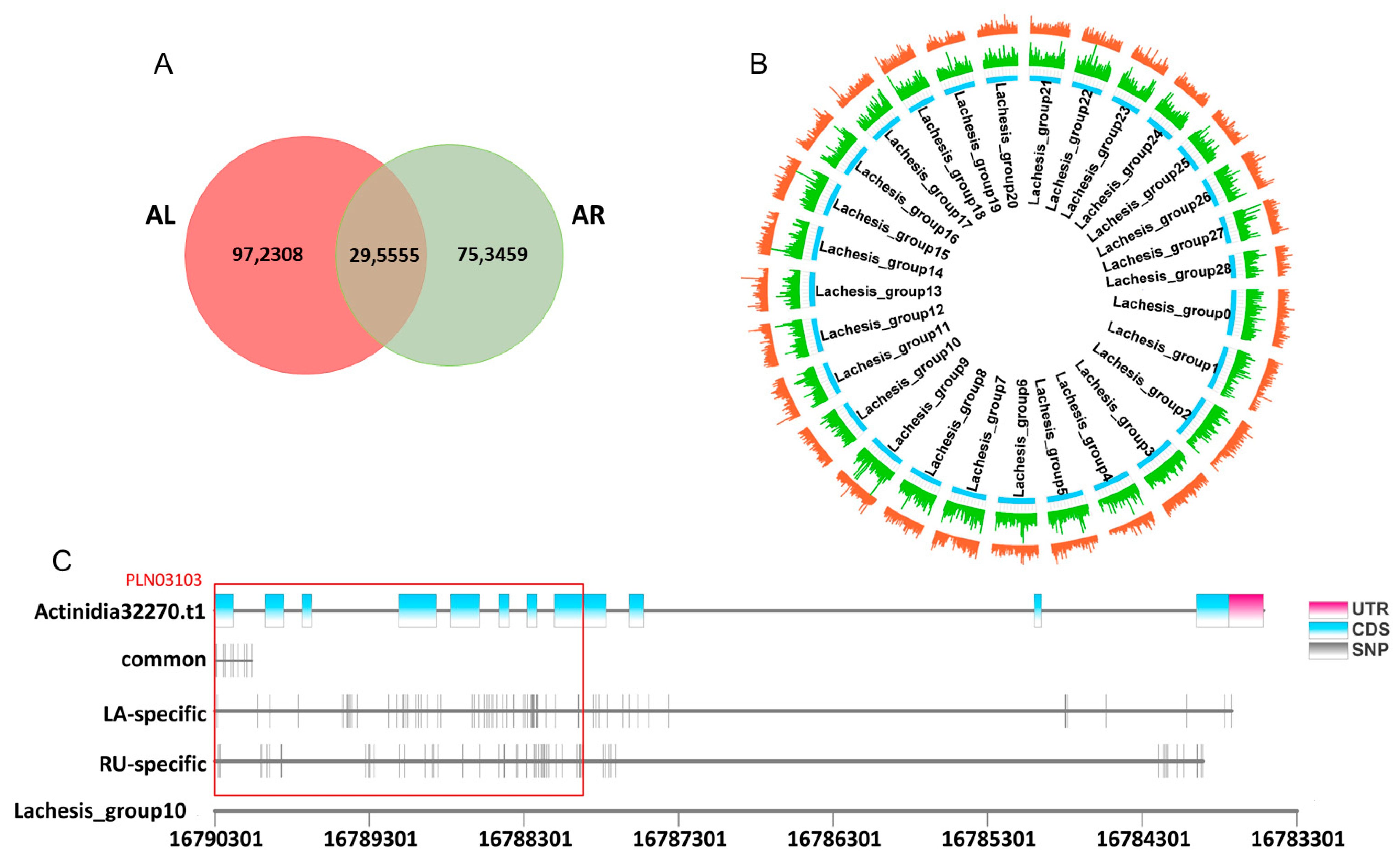
2.8. Species-Specific SNPs Might Promote the Differentiation of AsA between A. latifolia and A. rufa
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Measurement of AsA Contents by HPLC
4.3. RNA Isolation and Illumina RNA-seq
4.4. Transcriptome Analysis
4.5. Weighted Gene Co-Expression Network Construction
4.6. GO Enrichment Analysis
4.7. Quantitative Real-Time PCR Analysis
4.8. Gene Clone and Plasmid Constructs
4.9. Genetic Transformation in Kiwifruit
4.10. Yeast Two-Hybrid (Y2H) Assay
4.11. Bimolecular Fluorescence Complementation Assay (BiFC)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nishikimi, M.; Fukuyama, R.; Minoshima, S.; Shimizu, N.; Yagi, K. Cloning and chromosomal mapping of the human nonfunctional gene for L-gulono-gamma-lactone oxidase, the enzyme for L-ascorbic acid biosynthesis missing in man. J. Biol. Chem. 1994, 269, 13685–13688. [Google Scholar] [CrossRef]
- Foyer, C.H. Ascorbic acid. In Antioxidants in Higher Plants; CRC Press: Boca Raton, FL, USA, 2017; pp. 31–58. [Google Scholar]
- Iqbal, K.; Khan, A.; Khattak, M. Biological significance of ascorbic acid (vitamin C) in human health-a review. Pak. J. Nutr. 2004, 3, 5–13. [Google Scholar] [CrossRef] [Green Version]
- Mellidou, I.; Kanellis, A.K. Genetic control of ascorbic acid biosynthesis and recycling in horticultural crops. J. Mol. Struct. 2017, 5, 50. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, G.L.; Jones, M.A.; Smirnoff, N. The biosynthetic pathway of vitamin C in higher plants. Nature 1998, 393, 365–369. [Google Scholar] [CrossRef]
- Linster, C.L.; Adler, L.N.; Webb, K.; Christensen, K.C.; Brenner, C.; Clarke, S.G. A second GDP-L-galactose phosphorylase in Arabidopsis en route to vitamin C. Covalent intermediate and substrate requirements for the conserved reaction. J. Biol. Chem. 2008, 283, 18483–18492. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, G.; Ishikawa, T.; Pornsaksit, V.; Smirnoff, N. Evolution of alternative biosynthetic pathways for vitamin C following plastid acquisition in photosynthetic eukaryotes. eLife 2015, 4, e06369. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Ma, F.; Liu, J.; Li, J. Shading the whole vines during young fruit development decreases ascorbate accumulation in kiwi. Physiol. Plant. 2010, 140, 225–237. [Google Scholar] [CrossRef]
- Laing, W.A.; Bulley, S.; Wright, M.; Cooney, J.; Jensen, D.; Barraclough, D.; MacRae, E. A highly specific L-galactose-1-phosphate phosphatase on the path to ascorbate biosynthesis. Proc. Natl. Acad. Sci. USA 2004, 101, 16976–16981. [Google Scholar] [CrossRef] [Green Version]
- Bulley, S.M.; Laing, W. Ascorbic acid-related genes. In The Kiwifruit Genome, 1st ed.; Testolin, R., Huang, H., Ferguson, A.R., Eds.; Springer: Berlin/Heidelberg, Germany, 2016; pp. 163–177. [Google Scholar]
- Iqbal, Y.; Ihsanullah, I.; Shaheen, N.; Hussain, I. Significance of Vitamin C in Plants. J. Chem. Soc. Pak. 2009, 31, 169–170. [Google Scholar]
- Matić, D. The function and metabolism of ascorbic acid in plants. Bachelor’s Thesis, University of Zagreb, Zagreb, Croatia, 2014. [Google Scholar]
- Horemans, N.; Foyer, C.H.; Asard, H. Transport and action of ascorbate at the plant plasma membrane. Trends Plant Sci. 2000, 5, 263–267. [Google Scholar] [CrossRef]
- Horemans, N.; Foyer, C.H.; Potters, G.; Asard, H. Ascorbate function and associated transport systems in plants. Plant Physiol. Biochem. 2000, 38, 531–540. [Google Scholar] [CrossRef]
- Huang, C.; He, W.; Guo, J.; Chang, X.; Su, P.; Zhang, L. Increased sensitivity to salt stress in an ascorbate-deficient Arabidopsis mutant. J. Exp. Bot. 2005, 56, 3041–3049. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Chu, Z.; Luo, J.; Zhou, Y.; Cai, Y.; Lu, Y.; Xia, J.; Kuang, H.; Ye, Z.; Ouyang, B. The C2H2 zinc-finger protein SlZF3 regulates AsA synthesis and salt tolerance by interacting with CSN5B. Plant Biotechnol. J. 2018, 16, 1201–1213. [Google Scholar] [CrossRef] [Green Version]
- Zhong, C.; Huang, H.; Li, D.; Zhang, Q.; Li, L. Analysis of the development of the world kiwifruit industry and fresh fruit trade dynamics. China Fruits 2021, 101–108. [Google Scholar] [CrossRef]
- Huang, H.; Gong, J.; Wang, S.; He, Z.; Zhang, Z.; Li, J. Genetic diversity of kiwi plants of the genus Actinidia. Biodivers. Sci. 2000, 1–12. [Google Scholar]
- Zhang, L.; Wang, Y.; Huang, H. Correlation between the vitamin C content of kiwifruit (Actinidia Lindl.) leaves and fruits. Plant Sci. J. 2010, 28, 750–755. [Google Scholar]
- Hegedus, Z.; Zakrzewska, A.; Agoston, V.C.; Ordas, A.; Rácz, P.; Mink, M.; Spaink, H.P.; Meijer, A.H. Deep sequencing of the zebrafish transcriptome response to mycobacterium infection. Mol. Immunol. 2009, 46, 2918–2930. [Google Scholar] [CrossRef]
- Margulies, M.; Egholm, M.; Altman, W.E.; Attiya, S.; Bader, J.S.; Bemben, L.A.; Berka, J.; Braverman, M.S.; Chen, Y.J.; Chen, Z.; et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 2005, 437, 376–380. [Google Scholar] [CrossRef]
- Frishberg, A.; Peshes-Yaloz, N.; Cohn, O.; Rosentul, D.; Steuerman, Y.; Valadarsky, L.; Yankovitz, G.; Mandelboim, M.; Iraqi, F.A.; Amit, I.; et al. Cell composition analysis of bulk genomics using single-cell data. Nat. Methods 2019, 16, 327–332. [Google Scholar] [CrossRef]
- Tang, W.; Sun, X.; Yue, J.; Tang, X.; Jiao, C.; Yang, Y.; Niu, X.; Miao, M.; Zhang, D.; Huang, S.; et al. Chromosome-scale genome assembly of kiwifruit Actinidia eriantha with single-molecule sequencing and chromatin interaction mapping. GigaScience 2019, 8, giz027. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Ma, T.; Kang, M.; Ai, F.; Zhang, J.; Dong, G.; Liu, J. A high-quality Actinidia chinensis (kiwifruit) genome. Hortic. Res. 2019, 6, 117. [Google Scholar] [CrossRef] [Green Version]
- Nefissi Ouertani, R.; Arasappan, D.; Abid, G.; Ben Chikha, M.; Jardak, R.; Mahmoudi, H.; Mejri, S.; Ghorbel, A.; Ruhlman, T.A.; Jansen, R.K. Transcriptomic analysis of salt-stress-responsive genes in barley roots and leaves. Int. J. Mol. Sci. 2021, 22, 8155. [Google Scholar] [CrossRef]
- Shen, C.; Wang, J.; Shi, X.; Kang, Y.; Xie, C.; Peng, L.; Dong, C.; Shen, Q.; Xu, Y. Transcriptome analysis of differentially expressed genes induced by low and high potassium levels provides insight into fruit sugar metabolism of pear. Front. Plant Sci. 2017, 8, 938. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Chen, G.; Hu, H.; Yang, X.; Zhang, Z.; Jiang, X.; Wu, F.; Shi, H.; Wang, Q.; Zhou, K.; et al. Phenotypic and genetic variation in phosphorus-deficiency-tolerance traits in Chinese wheat landraces. BMC Plant Biol. 2020, 20, 330. [Google Scholar] [CrossRef]
- Mittler, R. Oxidative stress, antioxidants and stress tolerance. Trends Plant Sci. 2002, 7, 405–410. [Google Scholar] [CrossRef]
- Gill, S.S.; Tuteja, N. Reactive oxygen species and antioxidant machinery in abiotic stress tolerance in crop plants. Plant Physiol Biochem. PPB 2010, 48, 909–930. [Google Scholar] [CrossRef]
- De Tullio, M.C. Beyond the antioxidant: The double life of vitamin C. Subcell. Biochem. 2012, 56, 49–65. [Google Scholar] [CrossRef]
- Li, J.; Li, M.; Liang, D.; Ma, F.; Lei, Y. Comparison of expression pattern, genomic structure, and promoter analysis of the gene encoding GDP-l-galactose phosphorylase from two Actinidia species. Sci. Hortic. 2014, 169, 206–213. [Google Scholar] [CrossRef]
- Li, M.; Ma, F.; Liang, D.; Li, J.; Wang, Y. Ascorbate biosynthesis during early fruit development is the main reason for its accumulation in kiwi. PLoS ONE 2010, 5, e14281. [Google Scholar] [CrossRef] [Green Version]
- Zushi, K.; Suehara, C.; Shirai, M. Effect of light intensity and wavelengths on ascorbic acid content and the antioxidant system in tomato fruit grown in vitro. Sci. Hortic. 2020, 274, 109673. [Google Scholar] [CrossRef]
- Wang, H.S.; Zhu, Z.J.; Feng, Z.; Zhang, S.G.; Yu, C. Antisense-mediated depletion of GMPase gene expression in tobacco decreases plant tolerance to temperature stresses and alters plant development. Mol. Biol. Rep. 2012, 39, 10413–10420. [Google Scholar] [CrossRef]
- Laing, W.; Norling, C.; Brewster, D.; Wright, M.; Bulley, S. Ascorbate concentration in Arabidopsis thaliana and expression of ascorbate related genes using RNAseq in response to light and the diurnal cycle. bioRxiv 2017, 138008. [Google Scholar] [CrossRef] [Green Version]
- Plumb, W.; Townsend, A.J.; Rasool, B.; Alomrani, S.; Razak, N.; Karpinska, B.; Ruban, A.V.; Foyer, C.H. Ascorbate-mediated regulation of growth, photoprotection, and photoinhibition in Arabidopsis thaliana. J. Exp. Bot. 2018, 69, 2823–2835. [Google Scholar] [CrossRef]
- Lim, B.; Smirnoff, N.; Cobbett, C.S.; Golz, J.F. Ascorbate-deficient vtc2 mutants in Arabidopsis do not exhibit decreased growth. Front. Plant Sci. 2016, 7, 1025. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Wang, J.; Yu, Y.; Wang, F.; Dong, J.; Huang, R. D27E mutation of VTC1 impairs the interaction with CSN5B and enhances ascorbic acid biosynthesis and seedling growth in Arabidopsis. Plant Mol. Biol. 2016, 92, 473–482. [Google Scholar] [CrossRef]
- Hu, T.; Ye, J.; Tao, P.; Li, H.; Zhang, J.; Zhang, Y.; Ye, Z. The tomato HD-Zip I transcription factor SlHZ24 modulates ascorbate accumulation through positive regulation of the D-mannose/L-galactose pathway. Plant J. Cell Mol. Biol. 2016, 85, 16–29. [Google Scholar] [CrossRef] [Green Version]
- Imai, T.; Ban, Y.; Yamamoto, T.; Moriguchi, T. Ectopic overexpression of peach GDP-d-mannose pyrophosphorylase and GDP-d-mannose-3′, 5′-epimerase in transgenic tobacco. Plant Cell Tissue Organ Cult. PCTOC 2012, 111, 1–13. [Google Scholar] [CrossRef]
- Wang, L.; Meng, X.; Yang, D.; Ma, N.; Wang, G.; Meng, Q. Overexpression of tomato GDP-L-galactose phosphorylase gene in tobacco improves tolerance to chilling stress. Plant Cell Rep. 2014, 33, 1441–1451. [Google Scholar] [CrossRef]
- Chen, W.; Hu, T.; Ye, J.; Wang, B.; Liu, G.; Wang, Y.; Yuan, L.; Li, J.; Li, F.; Ye, Z.; et al. A CCAAT-binding factor, SlNFYA10, negatively regulates ascorbate accumulation by modulating the D-mannose/L-galactose pathway in tomato. Hortic. Res. 2020, 7, 200. [Google Scholar] [CrossRef]
- Mellidou, I.; Chagné, D.; Laing, W.A.; Keulemans, J.; Davey, M.W. Allelic variation in paralogs of GDP-L-galactose phosphorylase is a major determinant of vitamin C concentrations in apple fruit. Plant Physiol. 2012, 160, 1613–1629. [Google Scholar] [CrossRef] [Green Version]
- Bulley, S.M.; Rassam, M.; Hoser, D.; Otto, W.; Schünemann, N.; Wright, M.; MacRae, E.; Gleave, A.; Laing, W. Gene expression studies in kiwifruit and gene over-expression in Arabidopsis indicates that GDP-L-galactose guanyltransferase is a major control point of vitamin C biosynthesis. J. Exp. Bot. 2009, 60, 765–778. [Google Scholar] [CrossRef] [Green Version]
- Bulley, S.; Wright, M.; Rommens, C.; Yan, H.; Rassam, M.; Lin-Wang, K.; Andre, C.; Brewster, D.; Karunairetnam, S.; Allan, A.C.; et al. Enhancing ascorbate in fruits and tubers through over-expression of the L-galactose pathway gene GDP-L-galactose phosphorylase. Plant Biotechnol. J. 2012, 10, 390–397. [Google Scholar] [CrossRef]
- Queval, G.; Noctor, G. A plate reader method for the measurement of NAD, NADP, glutathione, and ascorbate in tissue extracts: Application to redox profiling during Arabidopsis rosette development. Anal. Biochem. 2007, 363, 58–69. [Google Scholar] [CrossRef]
- Li, G.; Liu, X.; Li, J. Determination of VC content in kiwi fruit by high performance liquid chromatography. Storage Process 2016, 16, 89–93. [Google Scholar]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Duarte, J.M.; Cui, L.; Wall, P.K.; Zhang, Q.; Zhang, X.; Leebens-Mack, J.; Ma, H.; Altman, N.; dePamphilis, C.W. Expression pattern shifts following duplication indicative of subfunctionalization and neofunctionalization in regulatory genes of Arabidopsis. Mol. Biol. Evol. 2006, 23, 469–478. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinf. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef] [Green Version]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Man, Y.; Wang, Y. Light- and Temperature-induced expression of an R2R3-MYB gene regulates anthocyanin biosynthesis in red-fleshed kiwifruit. Int. J. Mol. Sci. 2019, 20, 5228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Atkinson, R.; Janssen, B. In the choice of Agrobacterium strain for transformation of kiwifruit. Acta Hortic. 2007, 753, 227–232. [Google Scholar] [CrossRef]
- Gietz, R.D.; Schiestl, R.H. High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2007, 2, 31–34. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Zhou, H.; Lin, F.; Zhao, X.; Jiang, Y.; Xu, D.; Deng, X.W. COLD-REGULATED GENE27 integrates signals from light and the circadian clock to promote hypocotyl growth in Arabidopsis. Plant Cell 2020, 32, 3155–3169. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Xie, X.; Zhong, C.; Li, D. Comparative Transcriptome Analysis Revealed the Key Genes Regulating Ascorbic Acid Synthesis in Actinidia. Int. J. Mol. Sci. 2021, 22, 12894. https://doi.org/10.3390/ijms222312894
Liu X, Xie X, Zhong C, Li D. Comparative Transcriptome Analysis Revealed the Key Genes Regulating Ascorbic Acid Synthesis in Actinidia. International Journal of Molecular Sciences. 2021; 22(23):12894. https://doi.org/10.3390/ijms222312894
Chicago/Turabian StyleLiu, Xiaoying, Xiaodong Xie, Caihong Zhong, and Dawei Li. 2021. "Comparative Transcriptome Analysis Revealed the Key Genes Regulating Ascorbic Acid Synthesis in Actinidia" International Journal of Molecular Sciences 22, no. 23: 12894. https://doi.org/10.3390/ijms222312894
APA StyleLiu, X., Xie, X., Zhong, C., & Li, D. (2021). Comparative Transcriptome Analysis Revealed the Key Genes Regulating Ascorbic Acid Synthesis in Actinidia. International Journal of Molecular Sciences, 22(23), 12894. https://doi.org/10.3390/ijms222312894