
Enmein Decreases Synaptic Glutamate Release and Protects against Kainic Acid-Induced Brain Injury in Rats

 , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. The Influence of Enmein on Glutamate Release in Rat Cerebrocortical Nerve Terminals
2.2. The Influence of Enmein on Cytosolic Free Ca2+ Concentration ([Ca2+]C) and Synaptosomal Plasma Membrane Potential
2.3. The Reduction in N- and P/Q-type Ca2+ Channel Activities Is Involved in the Enmein-Induced Inhibition of Glutamate Release
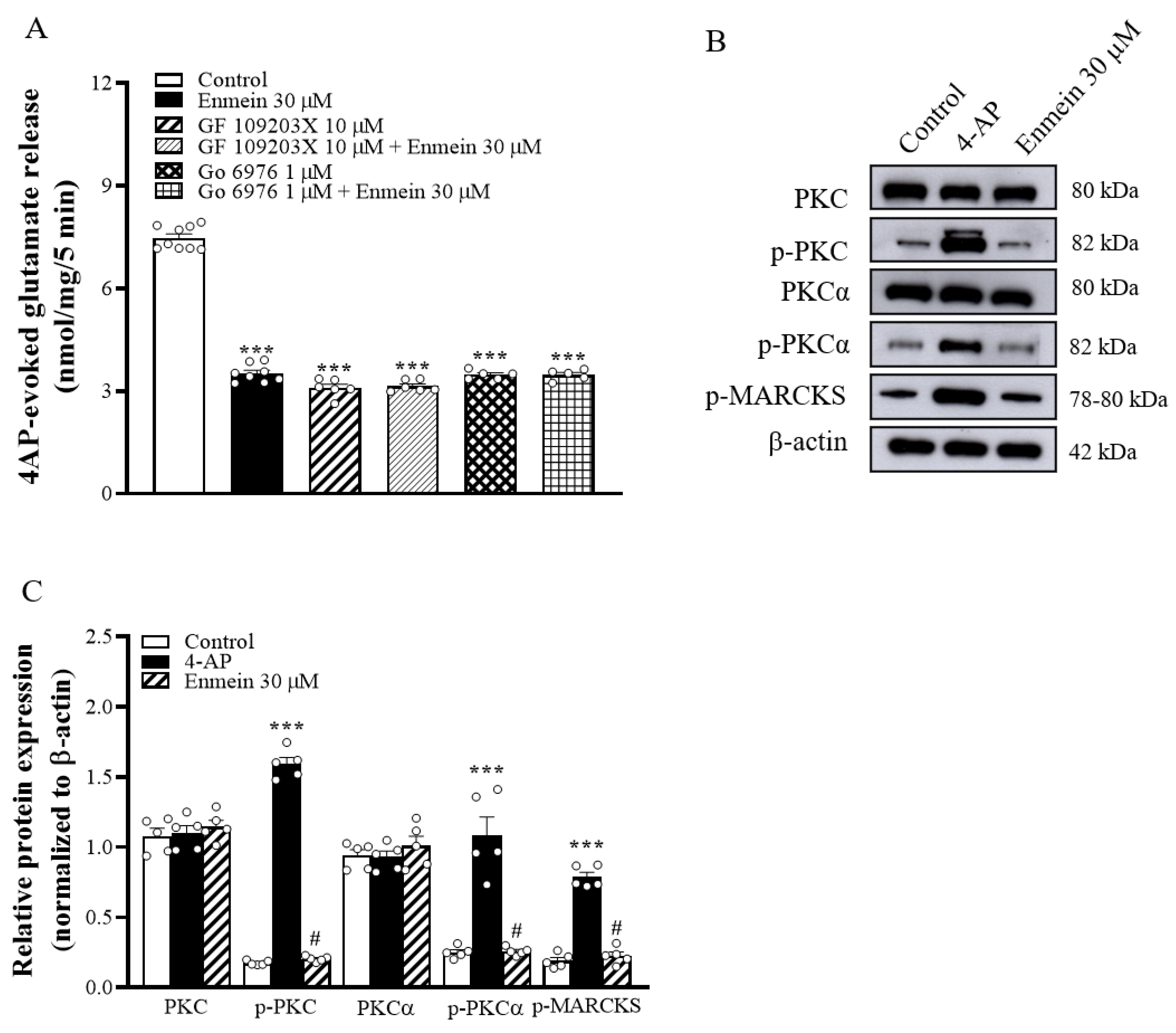
2.4. The Suppression of PKC/MARCKS Pathways Is Linked to Enmein-Induced Inhibition of Glutamate Release
2.5. The Influence of Enmein on Neuronal Death in the Hippocampi of KA Rats
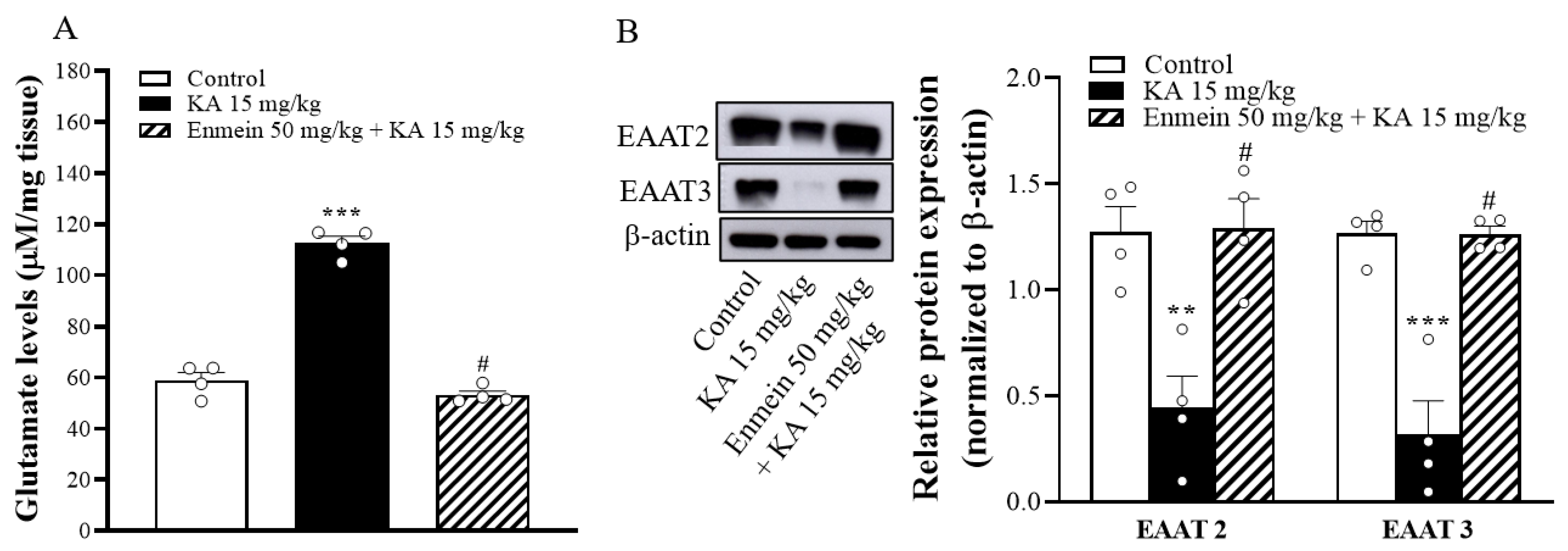
2.6. The Influence of Enmein on Glutamate Elevation and the Expression of EAAT2 and EAAT3 in the Hippocampi of KA Rats
2.7. The Influence of Enmein on the Expression of Synaptic Marker Proteins (Synaptophysin and PSD95) in the Hippocampi of KA Rats
2.8. The Influence of Enmein on the Activation of Glial Cells in the Hippocampi of KA Rats
3. Discussion
3.1. Inhibition of Presynaptic Glutamate Release by Enmein
3.2. Prevention of Glutamate Excitotoxicity by Enmein
4. Materials and Methods
4.1. Animals
4.2. Materials
4.3. Preparation of Synaptosomes
4.4. Determination of Glutamate Release
4.5. Determination of Exocytosis
4.6. Determination of Cytosolic Free Ca2+ Concentration ([Ca2+]C)
4.7. Determination of Synaptosomal Membrane Potential
4.8. Western Blot
4.9. Immunohistochemistry
4.10. Glutamate Levels in Brain Tissue from High-Performance Liquid Chromatography (HPLC)
4.11. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Headley, P.M.; Grillner, S. Excitatory amino acids and synaptic transmission: The evidence for a physiological function. Trends Pharmacol. Sci. 1990, 11, 205–211. [Google Scholar] [CrossRef]
- Javitt, D.C. Glutamate as a therapeutic target in psychiatric disorders. Mol. Psychiatry 2004, 9, 984–997, 979. [Google Scholar] [CrossRef] [Green Version]
- Meldrum, B.; Garthwaite, J. Excitatory amino acid neurotoxicity and neurodegenerative disease. Trends Pharmacol. Sci. 1990, 11, 379–387. [Google Scholar] [CrossRef]
- Dong, X.X.; Wang, Y.; Qin, Z.H. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 2009, 30, 379–387. [Google Scholar] [CrossRef] [Green Version]
- González, J.C.; Egea, J.; Del Carmen Godino, M.; Fernandez-Gomez, F.J.; Sánchez-Prieto, J.; Gandía, L.; García, A.G.; Jordán, J.; Hernández-Guijo, J.M. Neuroprotectant minocycline depresses glutamatergic neurotransmission and Ca2+ signalling in hippocampal neurons. Eur. J. Neurosci. 2007, 26, 2481–2495. [Google Scholar] [CrossRef]
- Lazarevic, V.; Yang, Y.; Ivanova, D.; Fejtova, A.; Svenningsson, P. Riluzole attenuates the efficacy of glutamatergic transmission by interfering with the size of the readily releasable neurotransmitter pool. Neuropharmacology 2018, 143, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zhao, B.; She, Y.; Song, X. Dexmedetomidine ameliorates lidocaine-induced spinal neurotoxicity via inhibiting glutamate release and the PKC pathway. Neurotoxicology 2018, 69, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, T.; Reker, D.; Schneider, P.; Schneider, G. Counting on natural products for drug design. Nat. Chem. 2016, 8, 531–541. [Google Scholar] [CrossRef]
- Chang, Y.; Lu, C.W.; Lin, T.Y.; Huang, S.K.; Wang, S.J. Baicalein, a Constituent of Scutellaria baicalensis, Reduces Glutamate Release and Protects Neuronal Cell Against Kainic Acid-Induced Excitotoxicity in Rats. Am. J. Chin. Med. 2016, 44, 943–962. [Google Scholar] [CrossRef]
- Lu, C.W.; Hsieh, H.L.; Lin, T.Y.; Hsieh, T.Y.; Huang, S.K.; Wang, S.J. Echinacoside, an Active Constituent of Cistanche Herba, Exerts a Neuroprotective Effect in a Kainic Acid Rat Model by Inhibiting Inflammatory Processes and Activating the Akt/GSK3β Pathway. Biol. Pharm. Bull. 2018, 41, 1685–1693. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.W.; Lin, T.Y.; Wang, S.J. 11-Keto-β-Boswellic Acid Attenuates Glutamate Release and Kainic Acid-Induced Excitotoxicity in the Rat Hippocampus. Planta Med. 2020, 86, 434–441. [Google Scholar] [CrossRef]
- Lu, C.W.; Lin, T.Y.; Pan, T.L.; Wang, P.W.; Chiu, K.M.; Lee, M.Y.; Wang, S.J. Asiatic Acid Prevents Cognitive Deficits by Inhibiting Calpain Activation and Preserving Synaptic and Mitochondrial Function in Rats with Kainic Acid-Induced Seizure. Biomedicines 2021, 9, 284. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, S.; Kan, J.; Zhang, J.; Zhou, L.; Huang, Y.; Zhang, Y. Chinese Herbal Medicine Interventions in Neurological Disorder Therapeutics by Regulating Glutamate Signaling. Curr. Neuropharmacol. 2020, 18, 260–276. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Tian, X. Enmein type diterpenoids from Isodon japonica. Phytochemistry 2001, 58, 543–546. [Google Scholar] [CrossRef]
- Xu, S.; Pei, L.; Li, D.; Yao, H.; Cai, H.; Yao, H.; Wu, X.; Xu, J. Synthesis and antimycobacterial evaluation of natural oridonin and its enmein-type derivatives. Fitoterapia 2014, 99, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Sun, B.; Wang, M.; Hu, X.; Gao, X.; Xu, S.; Xu, Y.; Xu, J.; Hua, H.; Li, D. Bioactive enmein-type 6,7-seco-ent-kaurane diterpenoids: Natural products, synthetic derivatives and apoptosis related mechanism. Arch. Pharm. Res. 2018, 41, 1051–1061. [Google Scholar] [CrossRef]
- Li, D.; Hu, X.; Han, T.; Liao, J.; Xiao, W.; Xu, S.; Li, Z.; Wang, Z.; Hua, H.; Xu, J. NO-Releasing Enmein-Type Diterpenoid Derivatives with Selective Antiproliferative Activity and Effects on Apoptosis-Related Proteins. Molecules 2016, 21, 1193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Yu, S.; Simonyi, A.; Sun, G.Y.; Sun, A.Y. Kainic acid-mediated excitotoxicity as a model for neurodegeneration. Mol. Neurobiol. 2005, 31, 3–16. [Google Scholar] [CrossRef]
- Nicholls, D.G. Presynaptic modulation of glutamate release. Prog. Brain Res. 1998, 116, 15–22. [Google Scholar] [CrossRef]
- Baldwin, M.L.; Rostas, J.A.; Sim, A.T. Two modes of exocytosis from synaptosomes are differentially regulated by protein phosphatase types 2A and 2B. J. Neurochem. 2003, 85, 1190–1199. [Google Scholar] [CrossRef] [Green Version]
- Vázquez, E.; Sánchez-Prieto, J. Presynaptic modulation of glutamate release targets different calcium channels in rat cerebrocortical nerve terminals. Eur. J. Neurosci. 1997, 9, 2009–2018. [Google Scholar] [CrossRef] [PubMed]
- Barrie, A.P.; Nicholls, D.G.; Sanchez-Prieto, J.; Sihra, T.S. An ion channel locus for the protein kinase C potentiation of transmitter glutamate release from guinea pig cerebrocortical synaptosomes. J. Neurochem. 1991, 57, 1398–1404. [Google Scholar] [CrossRef]
- Coffey, E.T.; Sihra, T.S.; Nicholls, D.G.; Pocock, J.M. Phosphorylation of synapsin I and MARCKS in nerve terminals is mediated by Ca2+ entry via an Aga-GI sensitive Ca2+ channel which is coupled to glutamate exocytosis. FEBS Lett. 1994, 353, 264–268. [Google Scholar] [CrossRef] [Green Version]
- Beart, P.M.; O’Shea, R.D. Transporters for L-glutamate: An update on their molecular pharmacology and pathological involvement. Br. J. Pharmacol. 2007, 150, 5–17. [Google Scholar] [CrossRef] [Green Version]
- Masliah, E.; Raber, J.; Alford, M.; Mallory, M.; Mattson, M.P.; Yang, D.; Wong, D.; Mucke, L. Amyloid protein precursor stimulates excitatory amino acid transport. Implications for roles in neuroprotection and pathogenesis. J. Biol. Chem. 1998, 273, 12548–12554. [Google Scholar] [CrossRef] [Green Version]
- Sheldon, A.L.; Robinson, M.B. The role of glutamate transporters in neurodegenerative diseases and potential opportunities for intervention. Neurochem. Int. 2007, 51, 333–355. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.X.; Sun, Q.J.; Zheng, X.Y.; Lin, Y.T.; Shang, W.; Wang, A.H.; Duan, R.S.; Chi, Z.F. Abnormal expression of synaptophysin, SNAP-25, and synaptotagmin 1 in the hippocampus of kainic acid-exposed rats with behavioral deficits. Cell. Mol. Neurobiol. 2014, 34, 813–824. [Google Scholar] [CrossRef]
- Zhu, X.; Liu, J.; Huang, S.; Zhu, W.; Wang, Y.; Chen, O.; Xue, J. Neuroprotective effects of isoliquiritigenin against cognitive impairment via suppression of synaptic dysfunction, neuronal injury, and neuroinflammation in rats with kainic acid-induced seizures. Int. Immunopharmacol. 2019, 72, 358–366. [Google Scholar] [CrossRef]
- Penkowa, M.; Florit, S.; Giralt, M.; Quintana, A.; Molinero, A.; Carrasco, J.; Hidalgo, J. Metallothionein reduces central nervous system inflammation, neurodegeneration, and cell death following kainic acid-induced epileptic seizures. J. Neurosci. Res. 2005, 79, 522–534. [Google Scholar] [CrossRef] [PubMed]
- Avignone, E.; Ulmann, L.; Levavasseur, F.; Rassendren, F.; Audinat, E. Status epilepticus induces a particular microglial activation state characterized by enhanced purinergic signaling. J. Neurosci. 2008, 28, 9133–9144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, H.S.; Park, J.H.; Ahn, J.H.; Hong, S.; Cho, J.H.; Won, M.H.; Lee, C.H. The anti-inflammatory activity of duloxetine, a serotonin/norepinephrine reuptake inhibitor, prevents kainic acid-induced hippocampal neuronal death in mice. J. Neurol. Sci. 2015, 358, 390–397. [Google Scholar] [CrossRef]
- De Sousa, I.P.; Sousa Teixeira, M.V.; Jacometti Cardoso Furtado, N.A. An Overview of Biotransformation and Toxicity of Diterpenes. Molecules 2018, 23, 1387. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Liu, J.; Jia, W.; Zhao, A.; Li, T. Distinct immunosuppressive effect by Isodon serra extracts. Int. Immunopharmacol. 2005, 5, 1957–1965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.G.; Saggau, P. Presynaptic inhibition of elicited neurotransmitter release. Trends Neurosci. 1997, 20, 204–212. [Google Scholar] [CrossRef]
- Nicholls, D.G.; Sihra, T.S.; Sanchez-Prieto, J. Calcium-dependent and -independent release of glutamate from synaptosomes monitored by continuous fluorometry. J. Neurochem. 1987, 49, 50–57. [Google Scholar] [CrossRef]
- Nichols, R.A.; Sihra, T.S.; Czernik, A.J.; Nairn, A.C.; Greengard, P. Calcium/calmodulin-dependent protein kinase II increases glutamate and noradrenaline release from synaptosomes. Nature 1990, 343, 647–651. [Google Scholar] [CrossRef]
- Ferkany, J.W.; Zaczek, R.; Coyle, J.T. Kainic acid stimulates excitatory amino acid neurotransmitter release at presynaptic receptors. Nature 1982, 298, 757–759. [Google Scholar] [CrossRef]
- Chittajallu, R.; Vignes, M.; Dev, K.K.; Barnes, J.M.; Collingridge, G.L.; Henley, J.M. Regulation of glutamate release by presynaptic kainate receptors in the hippocampus. Nature 1996, 379, 78–81. [Google Scholar] [CrossRef]
- Ben-Ari, Y. Limbic seizure and brain damage produced by kainic acid: Mechanisms and relevance to human temporal lobe epilepsy. Neuroscience 1985, 14, 375–403. [Google Scholar] [CrossRef]
- Smani, D.; Sarkar, S.; Raymick, J.; Kanungo, J.; Paule, M.G.; Gu, Q. Downregulation of 14-3-3 Proteins in a Kainic Acid-Induced Neurotoxicity Model. Mol. Neurobiol. 2018, 55, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Rose, C.R.; Felix, L.; Zeug, A.; Dietrich, D.; Reiner, A.; Henneberger, C. Astroglial Glutamate Signaling and Uptake in the Hippocampus. Front. Mol. Neurosci. 2017, 10, 451. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, M.K. Structure-Function Relationship of Transporters in the Glutamate-Glutamine Cycle of the Central Nervous System. Int. J. Mol. Sci. 2018, 19, 1177. [Google Scholar] [CrossRef] [Green Version]
- Rothstein, J.D.; Martin, L.; Levey, A.I.; Dykes-Hoberg, M.; Jin, L.; Wu, D.; Nash, N.; Kuncl, R.W. Localization of neuronal and glial glutamate transporters. Neuron 1994, 13, 713–725. [Google Scholar] [CrossRef]
- Takahashi, K.; Foster, J.B.; Lin, C.L. Glutamate transporter EAAT2: Regulation, function, and potential as a therapeutic target for neurological and psychiatric disease. Cell. Mol. Life Sci. 2015, 72, 3489–3506. [Google Scholar] [CrossRef]
- Pajarillo, E.; Rizor, A.; Lee, J.; Aschner, M.; Lee, E. The role of astrocytic glutamate transporters GLT-1 and GLAST in neurological disorders: Potential targets for neurotherapeutics. Neuropharmacology 2019, 161, 107559. [Google Scholar] [CrossRef] [PubMed]
- Nematipour, S.; Vahidinia, Z.; Nejati, M.; Naderian, H.; Beyer, C.; Azami Tameh, A. Estrogen and progesterone attenuate glutamate neurotoxicity via regulation of EAAT3 and GLT-1 in a rat model of ischemic stroke. Iran. J. Basic Med. Sci. 2020, 23, 1346–1352. [Google Scholar] [CrossRef] [PubMed]
- Green, J.L.; Dos Santos, W.F.; Fontana, A.C.K. Role of glutamate excitotoxicity and glutamate transporter EAAT2 in epilepsy: Opportunities for novel therapeutics development. Biochem. Pharmacol. 2021, 193, 114786. [Google Scholar] [CrossRef] [PubMed]
- Iovino, L.; Tremblay, M.E.; Civiero, L. Glutamate-induced excitotoxicity in Parkinson’s disease: The role of glial cells. J. Pharmacol. Sci. 2020, 144, 151–164. [Google Scholar] [CrossRef]
- Martin, S.J.; Grimwood, P.D.; Morris, R.G. Synaptic plasticity and memory: An evaluation of the hypothesis. Annu. Rev. Neurosci. 2000, 23, 649–711. [Google Scholar] [CrossRef] [Green Version]
- Haroon, E.; Miller, A.H.; Sanacora, G. Inflammation, Glutamate, and Glia: A Trio of Trouble in Mood Disorders. Neuropsychopharmacology 2017, 42, 193–215. [Google Scholar] [CrossRef]
- Tilleux, S.; Hermans, E. Neuroinflammation and regulation of glial glutamate uptake in neurological disorders. J. Neurosci. Res. 2007, 85, 2059–2070. [Google Scholar] [CrossRef] [PubMed]
- Skaper, S.D.; Facci, L.; Zusso, M.; Giusti, P. An Inflammation-Centric View of Neurological Disease: Beyond the Neuron. Front. Cell. Neurosci. 2018, 12, 72. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Cho, I.H.; Kwak, K.I.; Suh, E.C.; Seo, J.; Min, H.J.; Choi, S.Y.; Kim, C.H.; Park, S.H.; Jo, E.K.; et al. Microglial Toll-like receptor 2 contributes to kainic acid-induced glial activation and hippocampal neuronal cell death. J. Biol. Chem. 2010, 285, 39447–39457. [Google Scholar] [CrossRef] [Green Version]
- Alomar, S.Y.; Barakat, M.B.; Eldosoky, M.; Atef, H.; Mohamed, A.S.; Elhawary, R.; El-Shafey, M.; Youssef, A.M.; Elkazaz, A.Y.; Gabr, A.M.; et al. Protective effect of metformin on rat diabetic retinopathy involves suppression of toll-like receptor 4/nuclear factor-k B expression and glutamate excitotoxicity. Int. Immunopharmacol. 2021, 90, 107193. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, D.G.; Sihra, T.S. Synaptosomes possess an exocytotic pool of glutamate. Nature 1986, 321, 772–773. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.W.; Lin, T.Y.; Huang, S.K.; Wang, S.J. Echinacoside Inhibits Glutamate Release by Suppressing Voltage-Dependent Ca(2+) Entry and Protein Kinase C in Rat Cerebrocortical Nerve Terminals. Int. J. Mol. Sci. 2016, 17, 1006. [Google Scholar] [CrossRef] [Green Version]
- Lin, T.-Y.; Lu, C.-W.; Wu, C.-C.; Huang, S.-K.; Wang, S.-J. Palmitoylethanolamide Inhibits Glutamate Release in Rat Cerebrocortical Nerve Terminals. Int. J. Mol. Sci. 2015, 16, 5555–5571. [Google Scholar] [CrossRef] [Green Version]
- Grynkiewicz, G.; Poenie, M.; Tsien, R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985, 260, 3440–3450. [Google Scholar] [CrossRef]
- Akerman, K.E.; Scott, I.G.; Heikkilä, J.E.; Heinonen, E. Ionic dependence of membrane potential and glutamate receptor-linked responses in synaptoneurosomes as measured with a cyanine dye, DiS-C2-(5). J. Neurochem. 1987, 48, 552–559. [Google Scholar] [CrossRef]
- Hung, Y.-C.; Kuo, Y.-H.; Hsieh, P.-W.; Hsieh, T.-Y.; Kuo, J.-R.; Wang, S.-J. Chlorogenic Acid Decreases Glutamate Release from Rat Cortical Nerve Terminals by P/Q-Type Ca2+ Channel Suppression: A Possible Neuroprotective Mechanism. Int. J. Mol. Sci. 2021, 22, 11447. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, C.-W.; Huang, Y.-C.; Chiu, K.-M.; Lee, M.-Y.; Lin, T.-Y.; Wang, S.-J. Enmein Decreases Synaptic Glutamate Release and Protects against Kainic Acid-Induced Brain Injury in Rats. Int. J. Mol. Sci. 2021, 22, 12966. https://doi.org/10.3390/ijms222312966
Lu C-W, Huang Y-C, Chiu K-M, Lee M-Y, Lin T-Y, Wang S-J. Enmein Decreases Synaptic Glutamate Release and Protects against Kainic Acid-Induced Brain Injury in Rats. International Journal of Molecular Sciences. 2021; 22(23):12966. https://doi.org/10.3390/ijms222312966
Chicago/Turabian StyleLu, Cheng-Wei, Yu-Chen Huang, Kuan-Ming Chiu, Ming-Yi Lee, Tzu-Yu Lin, and Su-Jane Wang. 2021. "Enmein Decreases Synaptic Glutamate Release and Protects against Kainic Acid-Induced Brain Injury in Rats" International Journal of Molecular Sciences 22, no. 23: 12966. https://doi.org/10.3390/ijms222312966
APA StyleLu, C. -W., Huang, Y. -C., Chiu, K. -M., Lee, M. -Y., Lin, T. -Y., & Wang, S. -J. (2021). Enmein Decreases Synaptic Glutamate Release and Protects against Kainic Acid-Induced Brain Injury in Rats. International Journal of Molecular Sciences, 22(23), 12966. https://doi.org/10.3390/ijms222312966