
Integrated or Independent Actions of Metformin in Target Tissues Underlying Its Current Use and New Possible Applications in the Endocrine and Metabolic Disorder Area

Abstract
:
1. Introduction
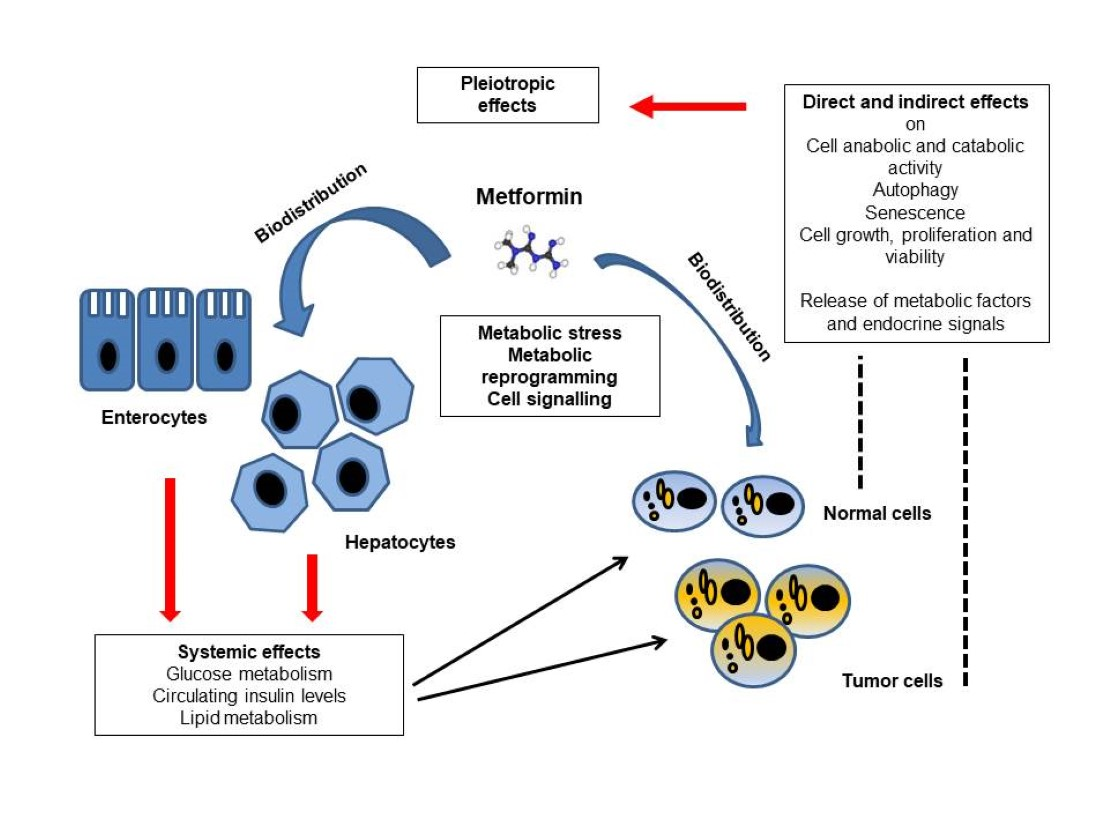
2. Metformin Absorption, Distribution and Elimination
3. Therapeutic Effects of Metformin
4. Molecular Mechanisms Underlying Metformin Actions
4.1. Direct and Indirect Molecular Targets of Metformin within Cells
4.2. Inhibition of Hepatic Gluconeogenesis
4.3. Other Actions Related to Metformin Benefits for Metabolic Health
4.4. Possible Mechanisms Underlying the Effects of Metformin in Patients with PCOS
4.5. Pharmacogenomic Studies for Metformin Action
5. The Role of Pyruvate Metabolism in Metformin Actions
5.1. The PDH Complex Activity: Regulatory Mechanisms and Consequences of Congenital or Acquired Deficiency
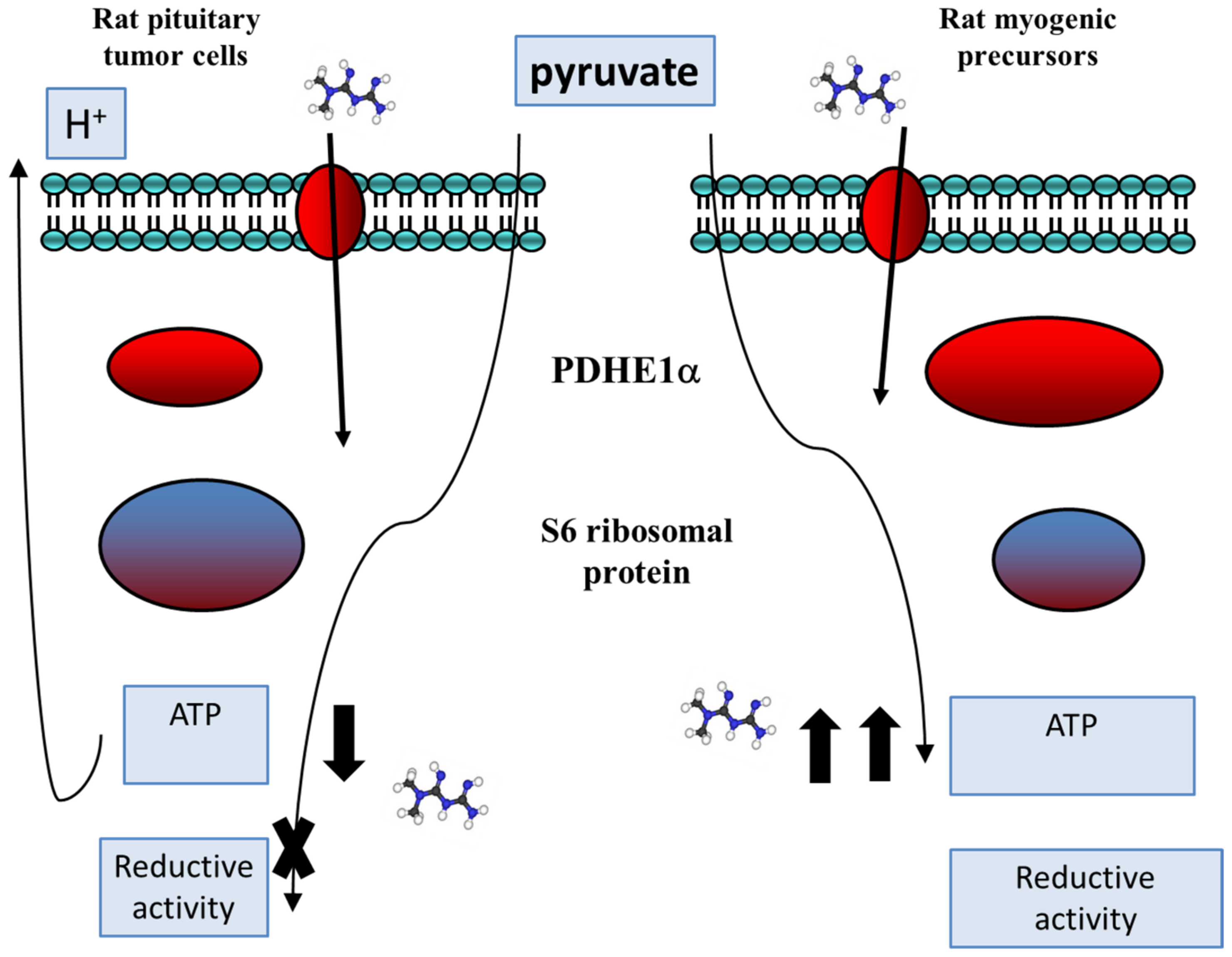
5.2. Metformin Impact on Pyruvate Metabolism
6. Metabolic Rearrangements That Occur upon Metformin Treatment in Normal and Tumor Cells
Metabolic Rearrangements in Pituitary Tumor Cells Compared with Normal Proliferating Cells
7. Cell Signaling and the Anticancer Activity of Metformin
8. Metformin Actions on Pituitary Tumor Cells and Gastroenteropancreatic Neuroendocrine Tumor Cells
8.1. Pituitary Tumors
8.2. Neuroendocrine Tumors
9. Conclusions and Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AC | adenylyl cyclase |
| ACC1 | acetyl-CoA carboxylase A1 |
| AIP | aryl hydrocarbon receptor-interacting protein |
| AMPK | AMP-activated protein kinase |
| AMP | adenosine monophosphate |
| ATM | ataxia-telangiectasia mutated kinase |
| ATP | adenosine triphosphate |
| CNS | central nervous system |
| CREB | cyclic AMP response element binding protein |
| CRTC2 | CREB regulated transcription factor 2 |
| ERK | extracellular signal-regulated kinases |
| ETC | electron transport chain |
| ERRα | estrogen receptor relatedα |
| FBP1 | fructose bisphosphatase-1 |
| FOXO1 | forkhead box O1 |
| FOXO3 | forkhead box O3 |
| fructose 1,6-P2 | fructose 1,6 bisphosphate |
| fructose 2,6-P2 | fructose 2,6 bisphosphate |
| GLP-1 | glucagon-like peptide-1 |
| GDF15 | growth differentiation factor-15 |
| GHRH | growth hormone-releasing hormone |
| GSK-3 | glycogen synthase kinase 3 |
| HIF1α | hypoxia inducible factor 1 subunit α |
| HNF4α | hepatocyte nuclear factor 4α |
| LDH | lactate dehydrogenase |
| MAPK | mitogen-activated protein kinase |
| MATE | multidrug and toxin extrusion proteins |
| MCT1 MCT4 | H+⁄ lactate monocarboxylate transporterH+⁄ lactate monocarboxylate transporter |
| MEK | mitogen-activated protein kinase kinase |
| mGPDH | mitochondrial glycerophosphate dehydrogenase |
| mTORC1 | mammalian target of rapamycin complex 1 |
| NADH | nicotinamide adenine dinucleotide |
| NET | neuroendocrine tumor |
| NRF-1 | respiratory factor 1 |
| NRF-2 | nuclear factor erythroid 2-related factor |
| OCT1 | organic cation transporter subtype 1 |
| PC | pyruvate carboxylase |
| PCOS | polycystic ovary syndrome |
| PDH | pyruvate dehydrogenase |
| PDHE1α | pyruvate dehydrogenase E1αprotein |
| PDK | pyruvate dehydrogenase kinase |
| PDP | pyruvate dehydrogenase phosphatase |
| PEP | 2-phosphoenolpyruvate |
| PEPCK | phosphoenolpyruvate carboxykinase |
| PFK1 | phosphofructokinase-1 |
| PFS | progression free survival |
| PI3K | phosphoinositide 3-kinase |
| PGC1-α | peroxisome proliferator-activated receptor gamma coactivator 1-alpha |
| PMAT | membrane monoamine transporter |
| PPARα/δ | peroxisome proliferator-activated receptor α/δ |
| p70S6K | p70 S6 protein kinase |
| ROS | reactive oxygen species |
| Shh | Sonic Hedgehog |
| STAT3 | signal transducer and activator of transcription 3 |
| SIRT | sirtuin |
| SREBP1 | sterol regulatory element-binding protein 1 |
| TCA cycle | tricarboxylic acid cycle |
| TSC | tuberous sclerosis complex |
References
- Sanchez-Rangel, E.; Inzucchi, S.E. Metformin: Clinical use in type 2 diabetes. Diabetologia 2017, 60, 1586–1593. [Google Scholar] [CrossRef]
- Dabrowski, M. Diabetes, Antidiabetic Medications and Cancer Risk in Type 2 Diabetes: Focus on SGLT-2 Inhibitors. Int. J. Mol. Sci. 2021, 22, 1680. [Google Scholar] [CrossRef]
- Bost, F.; Rena, G.; Viollet, B. Editorial: Metformin: Beyond diabetes. Front. Endocrinol. 2019, 10, 851. [Google Scholar] [CrossRef]
- Barzilai, N.; Crandall, J.P.; Kritchevsky, S.B.; Espeland, M.A. Metformin as a Tool to Target Aging. Cell Metab. 2016, 23, 1060–1065. [Google Scholar] [CrossRef] [Green Version]
- Day, E.A.; Ford, R.J.; Steinberg, G.R. AMPK as a Therapeutic Target for Treating Metabolic Diseases. Trends Endocrinol. Metab. 2017, 28, 545–560. [Google Scholar] [CrossRef]
- Rena, G.; Lang, C.C. Reporpusing metformin for cardiovascular disease. Circulation 2018, 137, 422–424. [Google Scholar] [CrossRef] [Green Version]
- Vancura, A.; Bu, P.; Bhagwat, M.; Zeng, J.; Vancurova, I. Metformin as an anticancer agent. Trends Pharmacol. Sci. 2018, 39, 867–878. [Google Scholar] [CrossRef]
- Yerevanian, A.; Soukas, A.A. Metformin: Mechanism in human obesity and weight loss. Curr. Obes. Rep. 2019, 8, 156–164. [Google Scholar] [CrossRef]
- Onken, B.; Driscoll, M. Metformin induces a dietary restriction-like state and the oxidative stress response to extend C. elegans healthspan via AMPK, LKB1, and SKN-1. PLoS ONE 2010, 5, e8758. [Google Scholar] [CrossRef]
- Johnson, S.C.; Rabinovitch, P.S.; Kaeberlein, M. mTOR is a key modulator of ageing and age-related disease. Nature 2013, 493, 338–345. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Zhou, B.; Oshiro-Rapley, N.; Li, M.; Paulo, J.A.; Webster, C.M.; Mou, F.; Kacergis, M.C.; Talkowski, M.E.; Carr, C.E.; et al. An ancient, unified mechanism for metformin growth inhibition in C. elegans and cancer. Cell 2016, 167, 1705–1718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrzejewski, S.; Siegel, P.M.; St-Pierre, J. Metabolic profiles associated with metformin efficacy in cancer. Front. Endocrinol. 2018, 9, 372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thakur, S.; Daley, B.; Klubo-Gwiezdzinska, J. The role of an antidiabetic drug—Metformin—In the treatment of endocrine tumors. J. Mol. Endocrinol. 2019, 63, R17–R35. [Google Scholar] [CrossRef]
- Günther, T.; Tulipano, G.; Dournaud, P.; Bousquet, C.; Csaba, Z.; Kreienkamp, H.J.; Lupp, A.; Korbonitz, M.; Castano, J.P.; Wester, H.-J.; et al. International Union of Basic and Clinical Pharmacology. CV. Somatostatin Receptors: Structure, Function, Ligands, and New Nomenclature. Pharmacol. Rev. 2018, 70, 763–835. [Google Scholar] [CrossRef] [Green Version]
- Tulipano, G.; Giustina, A. Autophagy in normal pituitary and pituitary tumor cells and its potential role in the actions of somatostatin receptor ligands in acromegaly. Rev. Endocr. Metab. Disord. 2021, 22, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Kim, W.G.; Zhao, L.; Enomoto, K.; Willingam, M.; Cheng, S.-Y. Metformin blocks obesity associated progression of obesity-activated thyroid cancer in a mouse model. Oncotarget 2016, 7, 23. [Google Scholar]
- Pusceddu, S.; Vernieri, C.; Prinzi, N.; Torchio, M.; Coppa, J.; Antista, M.; Niger, M.; Milione, M.; Giacomelli, L.; Corti, F.; et al. The potential role of metformin in the treatment of patients with pancreatic neuroendocrine tumors: A review of preclinical to clinical evidence. Ther. Adv. Gastroenterol. 2020, 13, 1756284820927271. [Google Scholar] [CrossRef]
- Thakur, S.; Daley, B.; Gaskins, K.; Vasko, V.V.; Boufraqech, M.; Patel, D.; Sourbier, C.; Reece, J.; Cheng, S.Y.; Kebebew, E.; et al. Metformin targets Mitochondrial Glycerophosphate Dehydrogenase (mGPDH) to control Rate of Oxidative Phosphorylation and growth of thyroid cancer in vitro and in vivo. Clin. Cancer Res. 2018, 24, 4030–4043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tulipano, G. How treatments with endocrine and metabolic drugs influence pituitary cell function. Endocr. Connect. 2020, 9, R14–R27. [Google Scholar] [CrossRef] [Green Version]
- Tulipano, G.; Paghera, S.; Missale, M.C.; Giustina, A. Differential activity of metformin on reductive activity and energy production in pituitary tumor cells compared to myogenic precursors. Endocrine 2020, 69, 604–614. [Google Scholar] [CrossRef] [PubMed]
- Costa, D.; Ceccato, F.; Lauretta, R.; Mercuri, V.; D’Amico, T.; De Vito, C.; Scaroni, C.; Appetecchia, M.; Gargiulo, P. The prevalance of secondary neoplasms in acromegalic patients: Possible preventive and/or protective role of metformin. Int. J. Clin. Oncol. 2021, 26, 1015–1021. [Google Scholar] [CrossRef] [PubMed]
- Portari, L.H.C.; Correa-Silva, S.R.; Abucham, J. Prolactin response to metformin in cabergoline-resistant prolactinomas: A pilot study. Neuroendocrinology 2021. Epub ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Kaneto, H.; Kimura, T.; Obata, A.; Shimoda, M.; Kaku, K. Multifaceted mechanisms of action of metformin which have been unraveled one after another in the long history. Int. J. Mol. Sci. 2021, 22, 2596. [Google Scholar] [CrossRef] [PubMed]
- Jensen, J.B.; Sundelin, E.I.; Jakobsen, S.; Gormsen, L.C.; Munk, O.L.; Frokiaer, J.; Jessen, N. [11C]-labeled metformin distribution in the liver and small intestine using dynamic positron emission tomography in mice demonstrates tissue-specific transporter dependency. Diabetes 2016, 65, 1724–1730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, J.; Lips, K.S.; Metzner, L.; Neubert, R.H.; Koepsell, H.; Brandsch, M. Drug specificity and intestinal membrane localization of human organic cation transporters (OCT). Biochem. Pharmacol. 2005, 70, 1851–1860. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Xia, L.; Wang, J. Metformin transport by a newly cloned proton stimulated organic cation transporter (plasma membrane monoamine transporter) expressed in human intestine. Drug Metab. Dispos. 2007, 35, 1956–1962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, Y.; Brown, C.; Castro, R.A.; Shi, R.J.; Lin, E.T.; Owen, R.P.; Sheardown, S.A.; Yue, L.; Burchard, E.G.; Brett, C.M.; et al. Effect of genetic variation in the organic cation transporter 1, OCT1, on metformin pharmacokinetics. Clin. Pharmacol. Ther. 2008, 83, 273–280. [Google Scholar] [CrossRef] [Green Version]
- Lee, N.; Duan, H.; Hebert, M.F.; Liang, C.J.; Rice, K.M.; Wang, J. Taste of a pill: Organic cation transporter-3 (OCT3) mediates metformin accumulation and secretion in salivary glands. J. Biol. Chem. 2014, 289, 27055–27064. [Google Scholar] [CrossRef] [Green Version]
- Gormsen, L.C.; Sundelin, E.I.; Jensen, J.B.; Vendelbo, M.H.; Jakobsen, S.; Munk, O.L.; Hougaard Christensen, M.M.; Brøsen, K.; Frøkiær, J.; Jessen, N. In Vivo Imaging of Human 11C-Metformin in Peripheral Organs: Dosimetry, Biodistribution, and Kinetic Analyses. J. Nucl. Med. 2016, 57, 1920–1926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanke, N.; Turk, D.; Selzer, D.; Ishiguro, N.; Ebner, T.; Wiebe, S.; Muller, F.; Stopfer, P.; Nock, V.; Lehr, T. A comprehensive whole-body physiologically based pharmacokinetic drug-drug-gene interaction model of metformin and cimetidine in healthy adults and renally impaired individuals. Clin. Pharmacokinet. 2020, 59, 1419–1431. [Google Scholar] [CrossRef]
- Tucker, G.T.; Casey, C.; Phillips, P.J.; Connor, H.; Ward, J.D.; Woods, H.F. Metformin kinetics in healthy subjects and in patients with diabetes mellitus. Br. J. Clin. Pharmacol. 1981, 12, 235–246. [Google Scholar] [CrossRef]
- Wilcock, C.; Bailey, C.J. Accumulation of metformin by tissues of the normal and diabetic mouse. Xenobiotica 1994, 24, 49–57. [Google Scholar] [CrossRef]
- Sundelin, E.; Jensen, J.B.; Jakobsen, S.; Gormsen, L.C.; Jessen, N. Metformin Biodistribution: A Key to Mechanisms of Action? J. Clin. Endocrinol. Metab. 2020, 105, dgaa332. [Google Scholar] [CrossRef] [PubMed]
- LaMoia, T.E.; Schulman, G.I. Cellular and molecular mechanism of metformin action. Endocr. Rev. 2021, 42, 77–96. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Borrego, M.C.; Fuentes-Fayos, A.C.; Herrera Martinez, A.D.; L-Lopez, F.L.; Ibanez-Costa, A.; Moreno-Moreno, P.; Alhambra-Expósito, M.R.; Barrera-Martín, A.; Blanco-Acevedo, C.; Dios, E.; et al. Biguanides exert antitumoral actions in pituitary tumor cells through AMPK-dependent and –independent mechanisms. J. Clin. Endocrinol. Metabol. 2019, 104, 3501–3513. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.J. Metformin: Historical overview. Diabetologia 2017, 60, 1566–1576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pernicova, I.; Korbonits, M. Metformin-mode of action and clinical implications for diabetes and cancer. Nat. Rev. Endocrinol. 2014, 10, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef] [Green Version]
- Agius, L.; Ford, B.E.; Cachra, S.S. The metformin mechanism on gluconeogenesis and AMPK activation: The metabolic perspective. Int. J. Mol. Sci. 2020, 21, 3240. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Buse, J.B.; Kim, T.; Burns, C.; Skare, S.; Baron, A.; Fineman, M. Once-daily delayed-release metformin lowers plasma glucose and enhances fasting and postprandial GLP-1 and PYY: Results from two randomised trials. Diabetologia 2016, 59, 1645–1654. [Google Scholar] [CrossRef] [Green Version]
- McCreight, L.J.; Bailey, C.J.; Pearson, E.R. Metformin and the gastrointestinal tract. Diabetologia 2016, 59, 426–435. [Google Scholar] [CrossRef] [Green Version]
- Lord, J.M.; Flight, I.H.K.; Norman, R.J. Metformin in polycystic ovary syndrome: Systematic review and metanalysis. BMJ 2003, 327, 951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diamanti-Kandarakis, E.; Dunaif, A. Insulin resistance and the polycystic ovary syndrome revisited: An update on mechanisms and Implications. Endocr. Rev. 2012, 33, 981–1030. [Google Scholar] [CrossRef] [PubMed]
- Naderpoor, N.; Shorakae, S.; de Courten, B.; Misso, M.L.; Moran, L.J.; Teede, H.J. Metformin and lifestyle modification in polycystic ovary syndrome: Systematic review and meta-analysis. Hum. Reprod. Update 2015, 21, 560–574. [Google Scholar] [CrossRef]
- Dadachanji, R.; Shaikh, N.; Mukherjee, S. Genetic Variants Associated with Hyperandrogenemia in PCOS Pathophysiology. Genet. Res. Int. 2018, 2018, 7624932. [Google Scholar] [CrossRef]
- Zhang, S.; Tu, H.; Yao, J.; Le, J.; Jiang, Z.; Tang, Q.; Zhang, R.; Huo, P.; Lei, X. Combined use of Diane-35 and metformin improves the ovulation in the PCOS rat model possibly via regulating glycolysis pathway. Reprod. Biol. Endocrinol. 2020, 18, 58. [Google Scholar] [CrossRef] [PubMed]
- Shpakov, A.O. Improvement Effect of Metformin on Female and Male Reproduction in Endocrine Pathologies and Its Mechanisms. Pharmaceuticals 2021, 14, 42. [Google Scholar] [CrossRef] [PubMed]
- Diamanti-Kandarakis, E.; Christakou, C.D.; Kandaraki, E.; Economou, F.N. Metformin: An old medication of new fashion: Evolving new molecular mechanisms and clinical implications in polycystic ovary syndrome. Eur. J. Endocrinol. 2010, 162, 193–212. [Google Scholar] [CrossRef] [PubMed]
- Viollet, B.; Guigas, B.; Sanz Garcia, N.; Leclerc, J.; Foretz, M.; Andreelli, F. Cellular and molecular mechanisms of metformin: An overview. Clin. Sci. 2012, 122, 253–270. [Google Scholar] [CrossRef] [Green Version]
- Andrae, F.; Abbott, D.; Stridsklev, S.; Schmedes, A.V.; Odsaeter, I.H.; Vanky, E.; Salvesen, O. Sustained maternal hyperandrogenism during PCOS pregnancy reduced by metformin in non-obese women carrying a male fetus. J. Clin. Endocrinol. Metab. 2020, 105, 3762–3770. [Google Scholar] [CrossRef]
- De Medeiros, S.F.; Rodgers, R.J.; Norman, R.J. Adipocyte and steroidogenic cell cross-talk in polycystic ovary syndrome. Hum. Reprod. Update 2021, 27, 771–796. [Google Scholar] [CrossRef] [PubMed]
- León-González, A.J.; Jiménez-Vacas, J.M.; Fuentes-Fayos, A.C.; Sarmento-Cabral, A.; Herrera-Martínez, A.D.; Gahete, M.D.; Luque, R.M. Role of metformin and other metabolic drugs in the prevention and therapy of endocrine-related cancers. Curr. Opin. Pharmacol. 2021, 60, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Demarè, S.; Kothari, A.; Calcutt, N.A.; Ferryhough, P. Metformin as a potential therapeutic for neurological disease: Mobilizing AMPK to repair the nervous system. Expert Rev. Neurother. 2021, 21, 45–63. [Google Scholar] [CrossRef] [PubMed]
- Zu, T.; Guo, S.; Bardhi, O.; Ryskamp, D.A.; Li, J.; Khoramian Tusi, S.; Engelbrecht, A.; Klippel, K.; Chakrabarty, P.; Nguyen, L.; et al. Metformin inhibits RAN translation through PKR pathway and mitigates disease in C9orf72 ALS/FTD mice. Proc. Natl. Acad. Sci. USA 2020, 117, 18591–18599. [Google Scholar] [CrossRef]
- Hervas, D.; Fornes-Ferrer, V.; Gomez-Escribano, A.P.; Sequedo, M.D.; Peiró, C.; Millán, J.M.; Vázquez-Manrique, R.P. Metformin intake associates with better cognitive function in patients with Huntington’s disease. PLoS ONE 2017, 12, e0179283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, Y.; Tong, Y.; Guo, Y.; Lang, X.; Huang, X.; Xie, X.; Guan, Y.; Li, Z. Metformin Attenuates the Metabolic Disturbance and Depression-like Behaviors Induced by Corticosterone and Mediates the Glucose Metabolism Pathway. Pharmacopsychiatry 2021, 54, 131–141. [Google Scholar] [CrossRef] [PubMed]
- UK Prospective Study (UKPDS) Group. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). Lancet 1998, 352, 854–865. [Google Scholar] [CrossRef]
- Rena, G.; Mordi, I.R.; Lang, C.C. Metformin: Still the sweet spot for CV protection in diabetes? Curr. Opin. Pharmacol. 2020, 54, 202–208. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Wondisford, F.E. Metformin action: Concentrations matter. Cell Metab. 2015, 21, 159–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontaine, E. Metformin induced mitochondrial complex I inhibition: Facts, uncertainties, and consequences. Front. Endocrinol. 2018, 9, 753. [Google Scholar] [CrossRef]
- Song, R. Mechanism of Metformin: A Tale of Two Sites. Diabetes Care 2016, 39, 187–189. [Google Scholar] [CrossRef] [Green Version]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000, 348, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Andrzejewski, S.; Gravel, S.P.; Pollak, M.; St-Pierre, J. Metformin directly acts on mitochondria to alter cellular bioenergetics. Cancer Metab. 2014, 2, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bridges, H.R.; Jones, A.J.; Pollak, M.N.; Hirst, J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem. J. 2014, 462, 475–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife 2014, 3, e02242. [Google Scholar] [CrossRef]
- Cameron, A.R.; Logie, L.; Patel, K.; Erhardt, S.; Bacon, S.; Middleton, P.; Harthill, J.; Forteath, C.; Coats, J.T.; Kerra, C.; et al. Metformin selectively targets redox control of complex I energy transduction. Redox Biol. 2018, 14, 187–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zong, Y.; Zhang, C.S.; Li, M.; Wang, W.; Wang, Z.; Hawley, S.A.; Ma, T.; Feng, J.W.; Tian, X.; Qi, Q.; et al. Hierarchical activation of compartmentalized pools of AMPK depends on severity of nutrient or energy stress. Cell Res. 2019, 29, 460–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.S.; Hawley, S.A.; Zong, Y.; Li, M.; Wang, Z.; Gray, A.; Ma, T.; Cui, J.; Feng, J.W.; Zhu, M.; et al. Fructose-1, 6-bisphosphate and aldolase mediate glucose sensing by AMPK. Nature 2017, 548, 112–116. [Google Scholar] [CrossRef]
- Alshawi, A.; Agius, L. Low metformin causes a more oxidized mitochondrial NADH/NAD redox state in hepatocytes and inhibits gluconeogenesis by a redox-independent mechanism. J. Biol. Chem. 2019, 294, 2839–2853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vytla, V.S.; Ochs, R.S. Metformin increases mitochondrial energy formation in L6 muscle cell cultures. J. Biol. Chem. 2013, 288, 20369–20377. [Google Scholar] [CrossRef] [Green Version]
- Piwkowska, A.; Rogack, D.; Jankowski, M.; Dominiczak, M.H.; Stepinski, J.K.; Angielski, S. Metformin induces suppression of NAD(P)H oxidase activity in podocytes. Biochem. Biophys. Res. Commun. 2010, 393, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Rogacka, D.; Piwkowska, A. Beneficial effects of metformin on glomerular podocytes in diabetes. Biochem. Pharmacol. 2021, 192, 114687. [Google Scholar] [CrossRef]
- Madiraju, A.K.; Erion, D.M.; Rahimi, Y.; Zhang, X.M.; Braddock, D.T.; Albright, R.A.; Prigaro, B.J.; Wood, J.L.; Bhanot, S.; MacDonald, M.J.; et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature 2014, 510, 542–546. [Google Scholar] [CrossRef] [Green Version]
- Madiraju, A.K.; Qiu, Y.; Perry, R.J.; Rahimi, Y.; Zhang, X.M.; Zhang, D.; Camporez, J.G.; Cline, G.W.; Butrico, G.M.; Kemp, B.E.; et al. Metformin inhibits gluconeogenesis via a redox-dependent mechanism in vivo. Nat. Med. 2018, 24, 1384–1394. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Meng, S.; Chang, E.; Beckwith-Fickas, K.; Xiong, L.; Cole, R.N.; Radovick, S.; Wondisford, F.E.; He, L. Low concentrations of metformin suppress glucose production in hepatocytes through AMP-activated protein kinase (AMPK). J. Biol. Chem. 2014, 289, 20435–20446. [Google Scholar] [CrossRef] [Green Version]
- Woods, A.; Williams, J.R.; Muckett, P.J.; Mayer, F.V.; Liljevald, M.; Bohlooly-Y, M.; Carling, D. Liver-Specific Activation of AMPK Prevents Steatosis on a High-Fructose Diet. Cell Rep. 2017, 18, 3043–3051. [Google Scholar] [CrossRef] [Green Version]
- Rada, P.; Mosquera, A.; Muntané, J.; Ferrandiz, F.; Rodriguez-Mañas, L.; de Pablo, F.; González-Canudas, J.; Valverde, Á.M. Differential effects of metformin glycinate and hydrochloride in glucose production, AMPK phosphorylation and insulin sensitivity in hepatocytes from non-diabetic and diabetic mice. Food Chem. Toxicol. 2019, 123, 470–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, R.A.; Chu, Q.; Xie, J.; Foretz, M.; Viollet, B.; Birnbaum, M.J. Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature 2013, 494, 256–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johanns, M.; Lai, Y.C.; Hsu, M.F.; Jacobs, R.; Vertommen, D.; Van Sande, J.; Dumont, J.E.; Woods, A.; Carling, D.; Hue, L.; et al. AMPK antagonizes hepatic glucagon-stimulated cyclic AMP signalling via phosphorylation-induced activation of cyclic nucleotide phosphodiesterase 4B. Nat. Commun. 2016, 7, 10856. [Google Scholar] [CrossRef]
- He, L.; Sabet, A.; Djedjos, S.; Miller, R.; Sun, X.; Hussain, M.A.; Radovick, S.; Wondisford, F.E. Metformin and insulin suppress hepatic gluconeogenesis through phosphorylation of CREB binding protein. Cell 2009, 137, 635–646. [Google Scholar] [CrossRef] [Green Version]
- Foretz, M.; Hébrard, S.; Leclerc, J.; Zarrinpashneh, E.; Soty, M.; Mithieux, G.; Sakamoto, K.; Andreelli, F.; Viollet, B. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/ AMPK pathway via a decrease in hepatic energy state. J. Clin. Investig. 2010, 120, 2355–2369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konopka, A.R.; Esponda, R.R.; Robinson, M.M.; Johnson, M.L.; Carter, R.E.; Schiavon, M.; Cobelli, C.; Wondisford, F.E.; Lanza, I.R.; Nair, K.S.; et al. Hyperglucagonemia mitigates the effect of metformin on glucose production in prediabetes. Cell Rep. 2016, 15, 1394–1400. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.S.; Lee, J.; Lee, H.S.; Song, J.E.; Kim, D.H.; Song, H.T. Offset of apparent hyperpolarized 13 C lactate flux by the use of adjuvant metformin in ionizing radiation therapy in vivo. NMR Biomed. 2021, 3, e4561. [Google Scholar] [CrossRef] [PubMed]
- Williamson, D.H.; Lund, P.; Krebs, H.A. The redox state of free nicotinamide-adenine dinucleotide in the cytoplasm and mitochondria of rat liver. Biochem. J. 1967, 103, 514–527. [Google Scholar] [CrossRef] [PubMed]
- El-Mir, M.Y.; Nogueira, V.; Fontaine, E.; Avéret, N.; Rigoulet, M.; Leverve, X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J. Biol. Chem. 2000, 275, 223–228. [Google Scholar] [CrossRef] [Green Version]
- MacDonald, M.J.; Ansari, I.H.; Longacre, M.J.; Stoker, S.W. Metformin’s Therapeutic Efficacy in the Treatment of Diabetes Does Not Involve Inhibition of Mitochondrial Glycerol Phosphate Dehydrogenase. Diabetes 2021, 70, 1575–1580. [Google Scholar] [CrossRef]
- Hue, L.; Bartrons, R. Role of fructose 2, 6-bisphosphate in the control by glucagon of gluconeogenesis from various precursors in isolated rat hepatocytes. Biochem. J. 1984, 218, 165–170. [Google Scholar] [CrossRef] [Green Version]
- Calza, G.; Nyberg, E.; Mäkinen, M.; Soliymani, R.; Cascone, A.; Lindholm, D.; Barborini, E.; Baumann, M.; Lalowski, M.; Eriksson, O. Lactate-induced glucose output is unchanged by metformin at a therapeutic concentration—A mass spectrometry imaging study of the perfused rat liver. Front. Pharmacol. 2018, 9, 141. [Google Scholar] [CrossRef] [Green Version]
- Hunter, R.W.; Hughey, C.C.; Lantier, L.; Sundelin, E.I.; Peggie, M.; Zeqiraj, E.; Sicheri, F.; Jessen, N.; Wasserman, D.H.; Sakamoto, K. Metformin reduces liver glucose production by inhibition of fructose-1-6-bisphosphatase. Nat. Med. 2018, 24, 1395–1406. [Google Scholar] [CrossRef] [Green Version]
- Moonira, T.; Chachra, S.S.; Ford, X.B.E.; Marin, X.S.; Alshawi, A.; Adam-Primus, N.S.; Arden, C.; Al-Oanzi, Z.H.; Foretz, X.M.; Viollet, B.; et al. Metformin lowers glucose 6-phosphate in hepatocytes by activation of glycolysis downstream of glucose phosphorylation. J. Biol. Chem. 2020, 295, 3330–3346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Oanzi, Z.H.; Fountana, S.; Moonira, T.; Tudhope, S.J.; Petrie, J.L.; Alshawi, A.; Patman, G.; Arden, C.; Reeves, H.L.; Agius, L. Opposite effects of a glucokinase activator and metformin on glucose regulated gene expression in hepatocytes. Diabetes Obes. Metab. 2017, 19, 1078–1087. [Google Scholar] [CrossRef] [PubMed]
- Salvatore, T.; Pafundi, P.C.; Morgillo, F.; Di Liello, R.; Galiero, R.; Nevola, R.; Marfella, R.; Monaco, L.; Rinaldi, L.; Adinolfi, L.E. Metformin: An old drug against old age and associated morbidities. Diabetes Res. Clin. Pract. 2020, 160, 108025. [Google Scholar] [CrossRef] [PubMed]
- Cokorinos, E.C.; Delmore, J.; Reyes, A.R.; Albuquerque, B.; Kjøbsted, R.; Jørgensen, N.O.; Tran, J.L.; Jatkar, A.; Cialdea, K.; Esquejo, R.M.; et al. Activation of Skeletal Muscle AMPK Promotes Glucose Disposal and Glucose Lowering in Non-human Primates and Mice. Cell Metab. 2017, 25, 1147–1159. [Google Scholar] [CrossRef] [PubMed]
- Rivera, M.E.; Lyon, E.S.; Vaughan, R.A. Effect of metformin on myotube BCAA catabolism. J. Cell Biochem. 2020, 121, 816–827. [Google Scholar] [CrossRef]
- He, L. Metformin and Systemic Metabolism. Trends Pharmacol. Sci. 2020, 41, 868–881. [Google Scholar] [CrossRef] [PubMed]
- Ito, J.; Nogami, M.; Morita, Y.; Sakaguchi, K.; Komada, H.; Hirota, Y.; Sugawara, K.; Tamori, Y.; Zeng, F.; Murakami, T.; et al. Dose-dependent accumulation of glucose in the intestinal wall and lumen induced by metformin as revealed by [18F]-labelled fluorodeoxyglucose positron emission tomography-MRI. Diabetes Obes. Metab. 2021, 23, 692–699. [Google Scholar] [CrossRef] [PubMed]
- Buse, J.B.; DeFronzo, R.A.; Rosenstock, J.; Kim, T.; Burns, C.; Skare, S.; Baron, A.; Fineman, M. The Primary Glucose-Lowering Effect of Metformin Resides in the Gut, Not the Circulation: Results from Short-term Pharmacokinetic and 12-Week Dose-Ranging Studies. Diabetes Care 2016, 39, 198–205. [Google Scholar] [CrossRef] [Green Version]
- Gontier, E.; Fourme, E.; Wartski, M.; Blondet, C.; Bonardel, G.; Le Stanc, E.; Mantzarides, M.; Foehrenbach, H.; Pecking, A.-P.; Alberini, J.-L. High and typical 18F-FDG bowel uptake in patients treated with metformin. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.J.; Wilcock, C.; Day, C. Effect of metformin on glucose metabolism in the splanchnic bed. Br. J. Pharmacol. 1992, 105, 1009–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, D.J.; Preston, T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes 2016, 7, 189–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Zhao, Y.; Xu, J.; Xue, Z.; Zhang, M.; Pang, X.; Zhang, X.; Zhao, L. Modulation of gut microbiota by berberine and metformin during the treatment of high-fat diet-induced obesity in rats. Sci. Rep. 2015, 5, 14405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Xu, J.H.; Yu, T.; Chen, Q.-K. Effects of berberine and metformin on intestinal inflammation and gut microbiome composition in db/db mice. Biomed. Pharmacother. 2019, 118, 109131. [Google Scholar] [CrossRef]
- Dujic, T.; Causevic, A.; Bego, T.; Malenica, M.; Velija-Asimi, Z.; Pearson, E.R.; Semiz, S. Organic cation transporter 1 variants and gastrointestinal side effects of metformin in patients with Type 2 diabetes. Diabet. Med. 2016, 33, 511–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morita, Y.; Nogami, M.; Sakaguchi, K.; Okada, Y.; Hirota, Y.; Sugawara, K.; Tamori, Y.; Zeng, F.; Murakami, T.; Ogawa, W. Enhanced Release of Glucose into the Intraluminal Space of the Intestine Associated with Metformin Treatment as Revealed by [18F] Fluorodeoxyglucose PET-MRI. Diabetes Care 2020, 43, 1796–1802. [Google Scholar] [CrossRef] [PubMed]
- Chau-Van, C.; Gamba, M.; Salvi, R.; Gaillard, R.C.; Pralong, F.P. Metformin inhibits adenosine 5′-monophosphate-activated kinase activation and prevents increases in neuropeptide Y expression in cultured hypothalamic neurons. Endocrinology 2007, 148, 507–511. [Google Scholar] [CrossRef]
- Lopez, M. Hypothalamic AMPK and energy balance. Eur. J. Clin. Investig. 2018, 48, e12996. [Google Scholar] [CrossRef] [Green Version]
- Lien, F.; Berthier, A.; Bouchaert, E.; Gheeraert, C.; Alexandre, J.; Porez, G.; Prawitt, J.; Dehondt, H.; Ploton, M.; Colin, S.; et al. Metformin interferes with bile acid homeostasis through AMPK-FXR crosstalk. J. Clin. Investig. 2014, 124, 1037–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhre, R.E.; Wewer Albrechtsen, N.J.; Larsen, O.; Jepsen, S.L.; Balk-Møller, E.; Andersen, D.B.; Deacon, C.F.; Schoonjans, K.; Reimann, F.; Gribble, F.M.; et al. Bile acids are important direct and indirect regulators of the secretion of appetite- and metabolism-regulating hormones from the gut and pancreas. Mol. Metab. 2018, 11, 84–95. [Google Scholar] [CrossRef]
- Coll, A.P.; Chen, M.; Taskar, P.; Rimmington, D.; Patel, S.; Tadross, J.A.; Cimino, I.; Yang, M.; Welsh, P.; Virtue, S.; et al. GDF15 mediates the effects of metformin on body weight and energy balance. Nature 2020, 578, 444–448. [Google Scholar] [CrossRef]
- Day, E.A.; Ford, R.J.; Smith, B.K.; Mohammadi-Shemirani, P.; Morrow, M.R.; Gutgesell, R.M.; Lu, R.; Raphenya, A.R.; Kabiri, M.; McArthur, A.G.; et al. Metformin-induced increases in GDF15 are important for suppressing appetite and promoting weight loss. Nat. Metab. 2019, 1, 1202–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natali, A.; Nesti, L.; Venturi, E.; Shore, A.C.; Khan, F.; Gooding, K.; Gates, P.E.; Looker, H.C.; Dove, F.; Goncalves, I.; et al. Metformin is the key factor in elevated plasma growth differentiation factor-15 levels in type 2 diabetes: A nested, case–control study. Diabetes Obes. Metab. 2019, 21, 412–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napolitano, A.; Miller, S.; Nicholls, A.W.; Baker, D.; Van Horn, S.; Thomas, E.; Rajpal, D.; Spivak, A.; Brown, J.R.; Nunez, D.J. Novel gut-based pharmacology of metformin in patients with type 2 diabetes mellitus. PLoS ONE 2014, 9, e100778. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Darwish, T.; Larraufie, P.; Rimmington, D.; Cimino, I.; Goldspink, D.A.; Jenkins, B.; Koulman, A.; Brighton, C.A.; Ma, M.; et al. Inhibition of mitochondrial function by metformin increases glucose uptake, glycolysis and GDF-15 release from intestinal cells. Sci. Rep. 2021, 11, 2529. [Google Scholar] [CrossRef] [PubMed]
- Cimino, I.; Kimb, H.; Tunga, Y.C.L.; Pedersen, K.; Rimmington, D.; Tadross, J.A.; Kohnke, S.N.; Neves-Costa, A.; Barros, A.; Joaquim, S.; et al. Activation of the hypothalamic–pituitary–adrenal axis by exogenous and endogenous GDF15. Proc. Natl. Acad. Sci. USA 2021, 118, e2106868118. [Google Scholar] [CrossRef]
- Salvatore, T.; Pafundi, P.C.; Galiero, R.; Rinaldi, L.; Caturano, A.; Vetrano, E.; Aprea, C.; Albanese, G.; Di Martino, A.; Ricozzi, C.; et al. Can Metformin Exert as an Active Drug on Endothelial Dysfunction in Diabetic Subjects? Biomedicines 2021, 9, 3. [Google Scholar] [CrossRef] [PubMed]
- Ohara, M.; Yoshida-Komiya, H.; Ono-Okutsu, M.; Yamaguchi-Ito, A.; Takahashi, T.; Fujimori, K. Metformin reduces androgen receptor and upregulates homeobox A10 expression in uterine endometrium in women with polycystic ovary syndrome. Reprod. Biol. Endocrinol. 2021, 19, 77. [Google Scholar] [CrossRef] [PubMed]
- Floretz, J.C. The pharmacogenetics of metformin. Diabetologia 2017, 60, 1648–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, Y.; Sheardown, S.A.; Brown, C.; Owen, R.P.; Zhang, S.; Castro, R.A.; Ianculescu, A.G.; Yue, L.; Lo, J.C.; Burchard, E.G.; et al. Effect of genetic variation in the organic cation transporter 1 (OCT1) on metformin action. J. Clin. Investig. 2007, 117, 1422–1431. [Google Scholar] [CrossRef] [Green Version]
- Zhou, K.; Bellenguez, C.; Spencer, C.C.; Bennett, A.J.; Coleman, R.L.; Tavendale, R.; Hawley, S.A.; Donnelly, L.A.; Schofield, C.; Groves, C.J.; et al. Common variants near ATM are associated with glycemic response to metformin in type 2 diabetes. Nat. Genet. 2011, 43, 117–120. [Google Scholar]
- Zhou, K.; Yee, S.W.; Seiser, E.L.; van Leeuwen, N.; Tavendale, R.; Bennett, A.J.; Groves, C.J.; Coleman, R.L.; van der Heijden, A.A.; Beulens, J.W.; et al. Variation in the glucose transporter gene SLC2A2 is associated with glycemic response to metformin. Nat. Genet. 2016, 48, 1055–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luizon, M.R.; Eckalbar, W.L.; Wang, Y.; Jones, S.C.; Smith, R.P.; Laurance, M.; Lin, L.; Gallins, P.J.; Etheridge, A.S.; Wright, F.; et al. Genomic characterization of metformin hepatic response. PLoS Genet. 2016, 12, e1006449. [Google Scholar] [CrossRef]
- García-Calzón, S.; Perfilyev, A.; Martinell, M.; Ustinova, M.; Kalamajski, S.; Franks, P.W.; Bacos, K.; Elbere, I.; Pihlajamäki, J.; Volkov, P.; et al. Epigenetic markers associated with metformin response and intolerance in drug-naïve patients with type 2 diabetes. Sci. Transl. Med. 2020, 12, eaaz1803. [Google Scholar] [CrossRef]
- Elbere, I.; Silamikelis, I.; Ustinova, M.; Kalnina, I.; Zaharenko, L.; Peculis, R.; Konrade, I.; Ciuculete, D.M.; Zhukovsky, C.; Gudra, D.; et al. Significantly altered peripheral blood cell DNA methylation profile as a result of immediate effect of metformin use in healthy individuals. Clin. Epigenetics 2018, 10, 156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rardin, M.J.; Wiley, S.E.; Naviaux, R.K.; Murphy, A.N.; Dixon, J.E. Monitoring phosphorylation of the pyruvate dehydrogenase complex. Anal. Biochem. 2009, 389, 157–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeoung, N.H. Pyruvate dehydrogenase kinases: Therapeutic targets for diabetes and cancers. Diabetes Metab. J. 2015, 39, 188–197. [Google Scholar] [CrossRef]
- Bender, T.; Martinou, J.C. The mitochondrial pyruvate carrier in health and disease: To carry or not to carry? Biochim. Biophys. Acta 2016, 1863, 2436–2442. [Google Scholar] [CrossRef]
- Linher-Melville, K.; Singh, G. The complex roles of STAT3 and STAT5 in maintaining redox balance: Lessons from STATmediated xCT expression in cancer cells. Mol. Cell. Endocrinol. 2017, 451, 40–52. [Google Scholar] [CrossRef]
- Stacpoole, P.W. Therapeutic Targeting of the Pyruvate Dehydrogenase Complex/Pyruvate Dehydrogenase Kinase (PDC/PDK) Axis in Cancer. JNCI J. Natl. Cancer Inst. 2017, 109, djx071. [Google Scholar] [CrossRef] [Green Version]
- Stacpoole, P.W. The pyruvate dehydrogenase complex as a therapeutic target for age-related diseases. Aging Cell 2012, 11, 371–377. [Google Scholar] [CrossRef]
- Patel, K.P.; O’Brien, T.W.; Subramony, S.H.; Shuster, J.; Stacpoole, P.W. The spectrum of pyruvate dehydrogenase complex deficiency: Clinical, biochemical and genetic features in 371 patients. Mol. Genet. Metab. 2011, 105, 34–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, N.E.; Han, Z.; Berendzen, K.M.; Sweeney, C.A.; Oca-Cossio, J.A.; Constantinidis, I.; Stacpoole, P.W. Magnetic resonance spectroscopic investigation of mitochondrial fuel metabolism and energetics in cultured human fibroblasts: Effects of pyruvate dehydrogenase complex deficiency and dichloroacetate. Mol. Genet. Metab. 2006, 89, 97–105. [Google Scholar] [CrossRef]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [Green Version]
- Gordan, J.D.; Thompson, C.B.; Simon, M.C. HIF and c-Myc: Sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell 2007, 12, 108–113. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Hypoxia-inducible factor 1: Regulator of mitochondrial metabolism and mediator of ischemic preconditioning. Biochim. Biophys. Acta 2011, 1813, 1263–1268. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Forbes, R.A.; Verma, A. Hypoxia-inducible factor 1 activation by aerobic glycolysis implicates the Warburg effect in carcinogenesis. J. Biol. Chem. 2002, 277, 23111–23115. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Dalgard, C.L.; Mohyeldin, A.; McFate, T.; Tait, A.S.; Verma, A. Reversible inactivation of HIF-1 prolyl hydroxylases allows cell metabolism to control basal HIF-1. J. Biol. Chem. 2005, 280, 41928–41939. [Google Scholar] [CrossRef] [Green Version]
- Ullah, M.S.; Davies, A.J.; Halestrap, A.P. The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. J. Biol. Chem. 2006, 281, 9030–9037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Floch, R.; Chiche, J.; Marchiq, I.; Naı¨ken, T.; Ilk, K.; Murray, C.M.; Critchlow, S.E.; Roux, D.; Simon, M.P.; Pouysségur, J. CD147 subunit of lactate ⁄ H+ symporters MCT1 and hypoxia-inducible MCT4 is critical for energetics and growth of glycolytic tumors. Proc. Natl Acad. Sci. USA 2011, 108, 16663–16668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnet, S.; Archer, S.L.; Allalunis-Turner, J.; Haromy, A.; Beaulieu, C.; Thompson, R.; Lee, C.T.; Lopaschuk, G.D.; Puttagunta, L.; Bonnet, S.; et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 2007, 11, 37–51. [Google Scholar] [CrossRef] [Green Version]
- Archer, S.L.; Gomberg-Maitland, M.; Maitland, M.L.; Rich, S.; Garcia, J.G.; Weir, E.K. Mitochondrial metabolism, redox signaling, and fusion: A mitochondria-ROS-HIF-1alpha-Kv1.5 O2-sensing pathway at the intersection of pulmonary hypertension and cancer. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H570–H578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathias, R.A.; Greco, T.M.; Oberstein, A.; Budayeva, H.G.; Chakrabarti, R.; Rowland, E.A.; Kang, Y.; Shenk, T.; Cristea, I.M. Sirtuin 4 is a lipoamidase regulating pyruvate dehydrogenase complex activity. Cell 2014, 159, 1615–1625. [Google Scholar] [CrossRef] [Green Version]
- Bhutia, Y.D.; Babu, E.; Ramachandran, S.; Ganapathy, V. Amino acid transporters in cancer and their relevance to “glutamine addiction”: Novel targets for the design of a new class of anticancer drugs. Cancer Res. 2015, 75, 1782–1788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kankotia, S.; Stacpoole, P.W. Dichloroacetate and cancer: New home for an orphan drug? Biochim. Biophys. Acta 2014, 1846, 617–629. [Google Scholar] [CrossRef]
- Verdin, E.; Hirschey, M.D.; Finley, L.W.; Haigis, M.C. Sirtuin regulation of mitochondria: Energy production, apoptosis, and signaling. Trends Biochem. Sci. 2010, 35, 669–675. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.W.; Lim, I.K. Sensitization of metformin-cytotoxicity by dichloroacetate via reprogramming glucose metabolism in cancer cells. Cancer Lett. 2014, 346, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Martin-Montalvo, A.; Mercken, E.M.; Mitchell, S.J.; Palacios, H.H.; Mote, P.L.; Scheibye-Knudsen, M.; Gomes, A.P.; Ward, T.M.; Minor, R.K.; Blouin, M.J.; et al. Metformin improves healthspan and lifespan in mice. Nat. Commun. 2013, 4, 2192. [Google Scholar] [CrossRef] [PubMed]
- Palma, F.R.; Ratti, B.A.; Paviani, V.; Coelho, D.R.; Miguel, R.; Danes, J.M.; Zaichik, S.V.; de Abreu, A.L.; Silva, S.O.; Chen, Y.; et al. AMPK-deficiency forces metformin-challenged cancer cells to switch from carbohydrate metabolism to ketogenesis to support energy metabolism. Oncogene 2021. Epub ahead of print. [Google Scholar] [CrossRef]
- Hakimi, P.; Yang, J.; Casadesus, G.; Massillon, D.; Tolentino-Silva, F.; Nye, C.K.; Cabrera, M.E.; Hagen, D.R.; Utter, C.B.; Baghdy, Y.; et al. Overexpression of the Cytosolic Form of Phosphoenolpyruvate Carboxykinase (GTP) in Skeletal Muscle Repatterns Energy Metabolism in the Mouse. J. Biol Chem. 2007, 282, 32844–32855. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, A.S.; Brutsaert, E.F.; Anghel, V.; Zhang, K.; Bloomgarden, N.; Pollak, M.; Mar, J.C.; Hawkins, M.; Crandall, J.P.; Barzilai, N. Metformin regulates metabolic and nonmetabolic pathways in skeletal muscle and subcutaneous adipose tissues of older adults. Aging Cell. 2018, 17, e12723. [Google Scholar] [CrossRef] [PubMed]
- Dohm, G.L.; Patel, V.K.; Kasperek, G.J. Regulation of muscle pyruvate metabolism during exercise. Biochem. Med. Metab. Biol. 1986, 35, 260–266. [Google Scholar] [CrossRef]
- Sahlin, K.; Katz, A.; Broberg, S. Tricarboxylic acid cycle intermediates in human muscle during prolonged exercise. Am. J. Physiol. 1990, 259, C834–C841. [Google Scholar] [CrossRef] [PubMed]
- Gibala, M.J.; MacLean, D.A.; Graham, T.E.; Saltin, B. Anaplerotic processes in human skeletal muscle during brief dynamic exercise. J. Physiol. 1997, 502, 703–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn, P.; Novak, M. Development of brown and white adipose tissue. J. Lipid Res. 1975, 16, 79–91. [Google Scholar] [CrossRef]
- McKee, E.E.; Bentley, A.T.; Smith, R.M., Jr.; Kraas, J.R.; Ciaccio, C.E. Guanine nucleotide transport by atractyloside-sensitive and -insensitive carriers in isolated heart mitochondria. Am. J. Physiol. Cell. Physiol. 2000, 279, C1870–C1879. [Google Scholar] [CrossRef]
- Subramanian, V.S.; Subramanya, S.B.; Said, H.M. Relative contribution of THTR-1 and THTR-2 in thiamin uptake by pancreatic acinar cells: Studies utilizing Slc19a2 and Slc19a3 knockout mouse models. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G572–G578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemos, C.; Faria, A.; Meireles, M.; Martel, F.; Monteiro, R.; Calhau, C. Thiamine is a substrate of organic cation transporters in Caco-2 cells. Eur. J. Pharmacol. 2012, 682, 37–42. [Google Scholar] [CrossRef]
- Chen, L.; Shu, Y.; Liang, X.; Chen, E.C.; Wah Yee, S.; Zur, A.A.; Li, S.; Xu, L.; Keshari, K.R.; Lin, M.J.; et al. OCT1 is a high-capacity thiamine transporter that regulates hepatic steatosis and is a target of metformin. Proc. Natl. Acad. Sci. USA 2014, 111, 9983–9988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, M.J.; Tuerkova, A.; Römer, S.; Wenzel, C.; Seitz, T.; Gaedcke, J.; Oswald, S.; Brockmöller, J.; Zdrazil, B.; Tzvetkov, M.V. Differences in Metformin and Thiamine Uptake between Human and Mouse Organic Cation Transporter 1: Structural Determinants and Potential Consequences for Intrahepatic Concentrations. Drug Metab. Dispos. 2020, 48, 1380–1392. [Google Scholar] [CrossRef] [PubMed]
- Vora, B.; Green, E.A.E.; Khuri, N.; Ballgren, F.; Sirota, M.; Giacomini, K.M. Drug-nutrient interactions: Discovering prescription drug inhibitors of the thiamine transporter ThTR-2 (SLC19A3). Am. J. Clin. Nutr. 2020, 111, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Fendt, S.M.; Bell, E.L.; Keibler, M.A.; Davidson, S.M.; Wirth, G.J.; Fiske, B.; Mayers, J.R.; Schwab, M.; Bellinger, G.; Csibi, A.; et al. Metformin decreases glucose oxidation and increases the dependency of prostate cancer cells on reductive glutamine metabolism. Cancer Res. 2013, 73, 4429–4438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deblois, G.; St-Pierre, J.; Giguere, V. The PGC-1/ERR signaling axis in cancer. Oncogene 2013, 32, 3483–3490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaube, B.; Malvi, P.; Singh, S.V.; Mohammad, N.; Viollet, B.; Bhat, M.K. AMPK maintains energy homeostasis and survival in cancer cells via regulating p38/PGC-1alpha-mediated mitochondrial biogenesis. Cell Death Discov. 2015, 1, 15063. [Google Scholar] [CrossRef] [Green Version]
- Klimcakova, E.; Chenard, V.; McGuirk, S.; Germain, D.; Avizonis, D.; Muller, W.J.; St-Pierre, J. PGC-1alpha promotes the growth of ErbB2/Neu-induced mammary tumors by regulating nutrient supply. Cancer Res. 2012, 72, 1538–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGuirk, S.; Gravel, S.P.; Deblois, G.; Papadopoli, D.J.; Faubert, B.; Wegner, A.; Hiller, K.; Avizonis, D.; Akavia, U.D.; Jones, R.G.; et al. PGC-1alpha supports glutamine metabolism in breast cancer. Cancer Metab. 2013, 1, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrzejewski, S.; Klimcakova, E.; Johnson, R.M.; Tabaries, S.; Annis, M.G.; McGuirk, S.; Northey, J.J.; Chénard, V.; Sriram, U.; Papadopoli, D.J.; et al. PGC-1a promotes breast cancer metastasis and confers bioenergetic flexibility against metabolic drugs. Cell Metab. 2017, 26, 778–787. [Google Scholar] [CrossRef]
- Canto, C.; Auwerx, J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 2009, 20, 98–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nillni, E.A. The metabolic sensor Sirt1 and the hypothalamus: Inteplay between peptide hormones and pro-hormone covertases. Mol. Cell. Endocrinol. 2016, 438, 77–88. [Google Scholar] [CrossRef]
- Canto, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef]
- Tulipano, G.; Faggi, L.; Losa, M.; Mortini, P.; Spinello, M.; Sibilia, V.; Pagani, F.; Cocchi, D.; Giustina, A. Effects of AMPK activation and combined treatment with AMPK activators and somatostatin on hormone secretion and cell growth in cultured GH-secreting pituitary tumor cells. Mol. Cell. Endocrinol. 2013, 365, 197–206. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Pei, X.; Zang, Z.; Zhou, Z.; Hu, J.; Zheng, X.; Zhang, Y.; He, J.; Duan, L.; Shen, R.; et al. Metformin inhibits proliferation and growth hormone secretion of GH3 pituitary adenoma cells. Oncotarget 2017, 8, 37538–37549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faggi, L.; Giustina, A.; Tulipano, G. Effects of metformin on cell growth and AMPK activity in pituitary adenoma cell cultures, focusing on the interaction with adenylyl cyclase activating signals. Mol. Cell. Endocrinol. 2018, 470, 60–74. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Liu, Y.; Han, G.; Deng, K.; Liu, X.; Bao, X.; Feng, M.; Yao, Y.; Lian, W.; Xing, B.; et al. Metformin inhibits growth and prolactin secretion of pituitary prolactinoma cells and xenografts. J. Cell. Mol. Med. 2018, 22, 6368–6379. [Google Scholar] [CrossRef]
- Jin, K.; Ruan, L.; Pu, J.; Zhong, A.; Wang, F.; Tan, S.; Huang, H.; Mu, J.; Yang, G. Metformin suppresses growth and adrenocorticotrophic hormone secretion in mouse pituitary corticotroph tumor AtT20 cells. Mol. Cell. Endocrinol. 2018, 478, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Abe, T.; Gotoh, J.; Fukuuchi, Y. Substratedependence of reduction of MTT: A tetrazolium dye differs in cultured astroglia and neurons. Neurochem. Int. 2002, 40, 441–448. [Google Scholar] [CrossRef]
- Wattanavanitchakorn, S.; Ansari, I.H.; El Azzouny, M.; Longacre, M.J.; Stoker, S.W.; MacDonald, M.J.; Jitrapakdee, S. Differential contribution of pyruvate carboxylation to anaplerosis and cataplerosis during non-gluconeogenic and gluconeogenic conditions in HepG2 cells. Arch. Biochem. Biophys. 2019, 676, 108124. [Google Scholar] [CrossRef] [PubMed]
- Urakami, K.; Zangiacomi, V.; Yamaguchi, C.; Kusuhara, M. Impact of 2-deoxy-D-glucose on the target metabolome profile of a human endometrial cancer cell line. Biomed. Res. 2013, 34, 221–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben Sahra, I.; Laurent, K.; Giuliano, S.; Larbret, F.; Ponzio, G.; Gounon, P.; Le Marchand-Brustel, Y.; Giorgetti-Peraldi, S.; Cormont, M.; Bertolotto, C.; et al. Targeting Cancer Cell Metabolism: The Combination of Metformin and 2-deoxyglucose Induces p53-Dependent Apoptosis in Prostate Cancer Cells. Cancer Res. 2010, 70, 2465–2475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elgendy, M.; Cirò, M.; Hosseini, A.; Weiszmqann, J.; Mazzarella, L.; Ferrari, E.; Cazzoli, R.; Curigliano, G.; DeCensi, A.; Bonanni, B.; et al. Combination of hypoglycemia and metformin impairs tumor metabolic plasticity and growth by modulating the PP2A-GSK3b- MCL-1 axis. Cancer Cell 2019, 35, 798–815. [Google Scholar] [CrossRef]
- Janzer, A.; German, N.J.; Gonzalez-Herrera, K.N.; Asara, J.M.; Haigis, M.C.; Struhl, K. Metformin and phenformin deplete tricarboxylic acid cycle and glycolytic intermediates during cell transformation and NTPs in cancer stemcells. Proc. Natl. Acad. Sci. USA 2014, 111, 105749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machado, I.F.; Teodoro, J.S.; Castela, A.C.; Palmeira, C.M.; Rolo, A.P. miR-378a-3p Participates in Metformin’s Mechanism of Action on C2C12 Cells under Hyperglycemia. Int. J. Mol. Sci. 2021, 22, 541. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, C.; Li, H.; Song, Y.; Zhao, Y.; Zhai, L.; Wang, H.; Zhong, R.; Tang, H.; Zhu, D. MiR-378 activates the pyruvate-PEP futile cycle and enhances lipolysis to ameliorate obesity in mice. EBioMedicine 2016, 5, 93–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guimaraes, T.A.; Farias, L.C.; Santos, E.S.; de Carvalho Fraga, C.A.; Orsini, L.A.; de Freitas Teles, L.; Feltenberger, J.D.; de Jeses, S.F.; de Souza, M.G. Metformin increases PDH and suppresses HIF-1α under hypoxic conditions and induces cell death in oral squamous cell carcinoma. Oncotarget 2016, 34, 55057–55068. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, Y.; Ly, R.C.; Frazier, C.V.; Yu, J.; Qin, S.; Fan, X.Y.; Goetz, M.P.; Boughey, J.C.; Weinshilboum, R.; Wang, L. The novel function of tumor protein D54 in regulating pyruvate dehydrogenase and metformin cytotoxicity in breast cancer. Cancer Metab. 2019, 7, 1. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin and reduced risk of cancer in diabetic patients. BMJ 2005, 330, 1304–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riedmaier, A.E.; Fisel, P.; Nies, A.T.; Schaeffeler, E.; Schwab, M. Metformin and cancer: From the old medicine cabinet to pharmacological pitfalls and prospects. Trends Pharmacol. Sci. 2013, 34, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.M.; Bellman, S.M.; Stephenson, M.D.; Lisy, K. Metformin reduces all-cause mortality and diseases of ageing independent of its effect on diabetes control: A systematic review and meta-analysis. Ageing Res. Rev. 2017, 40, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Vernieri, C.; Pusceddu, S.; de Braud, F. Impact of metformin on systemic metabolism of patients with advanced pancreatic neuroendocrine tumors. Front. Oncol. 2019, 9, 902. [Google Scholar] [CrossRef] [PubMed]
- Zahra, M.H.; Afify, S.M.; Hassan, G.; Nawara, H.M.; Kumon, K.; Seno, A.; Seno, M. Metformin suppresses self-renewal and stemness of cancer stem cell models derived from pluripotent stem cells. Cell Biochem. Funct. 2021. Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Giovannucci, E.; Harlan, D.M.; Archer, M.C.; Bergenstal, R.M.; Gapstur, S.M.; Habel, L.A.; Pollak, M.; Regensteiner, J.G.; Yee, D. Diabetes and cancer: A consensus report. Diabetes Care 2010, 33, 1674–1685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, J.; Lee, S.-Y.; Choi, J.W.; Lee, A.R.; Yoo, J.H.; Moon, S.-J.; Park, S.-H.; Cho, M.-L. Metformin ameliorates scleroderma via inhibiting Th17 cells and reducing mTOR-STAT3 signaling in skin fibroblasts. J. Transl. Med. 2021, 19, 192. [Google Scholar] [CrossRef]
- Longo, V.D.; Antebi, A.; Bartke, A.; Barzilai, N.; Brown-Borg, H.M.; Caruso, C.; Curiel, T.J.; de Cabo, R.; Franceschi, C.; Gems, D.; et al. Interventions to slow aging in humans: Are we ready? Aging Cell 2015, 14, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Drake, J.C.; Bruns, D.R.; Peelor, F.F., III; Biela, L.M.; Miller, R.A.; Miller, B.F.; Hamilton, K.L. Long-lived Snell dwarf mice display increased proteostatic mechanisms that are not dependent on decreased mTORC1 activity. Aging Cell 2015, 14, 474–482. [Google Scholar] [CrossRef] [Green Version]
- Wolff, C.A.; Reid, J.J.; Musci, R.V.; Bruns, D.R.; Linden, M.A.; Konopka, A.R.; Peelor, F.F.; Miller, B.F.; Hamilton, K.L. Differential effects of rapamycin and metformin in combination with rapamycin on mechanisms of proteostasis in cultured skeletal myotubes. J. Gerontol. A Biol. Sci. Med. Sci. 2020, 75, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Bharath, L.P.; Agrawal, M.; McCambridge, G.; Nicholas, D.A.; Hasturk, H.; Liu, J.; Jiang, K.; Liu, R.; Guo, Z.; Deeney, J.; et al. Metformin enhances autophagy and normalizes mitochondrial function to alleviate aging-associated inflammation. Cell Metab. 2020, 32, 44–55.e46. [Google Scholar] [CrossRef]
- Gou, S.; Qiu, L.; Yang, Q.; Li, P.; Zhou, X.; Sun, Y.; Zhou, X.; Zhao, W.; Zhai, W.; Li, G.; et al. Metformin leads to accumulation of reactive oxygen species by inhibiting the NFE2L1 expression in human hepatocellular carcinoma cells. Toxicol. Appl. Pharmacol. 2021, 420, 115523. [Google Scholar] [CrossRef] [PubMed]
- Warkad, M.S.; Kim, C.H.; Kang, B.G.; Park, S.H.; Jung, J.S.; Feng, J.H.; Inci, G.; Kim, S.C.; Suh, H.W.; Lim, S.S.; et al. Metformin-induced ROS upregulation as amplified by apigenin causes profound anticancer activity while sparing normal cells. Sci. Rep. 2021, 11, 14002. [Google Scholar] [CrossRef]
- Motoshima, H.; Goldstein, B.J.; Igata, M.; Araki, E. AMPK and cell proliferation—AMPK as a therapeutic target for atherosclerosis and cancer. J. Physiol. 2006, 574, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Towler, M.C.; Hardie, D.G. AMP-activated protein kinase in metabolic control and insulin signaling. Circ. Res. 2007, 100, 328–341. [Google Scholar] [CrossRef]
- Hardie, D.G. Keeping the home fires burning: AMP-activated protein kinase. J. R. Soc. Interface 2018, 15, 20170774. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell. Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Kjobsted, R.; Hingst, J.R.; Fentz, J.; Foretz, M.; Sanz, M.N.; Pehmøller, C.; Shum, M.; Marette, A.; Mounier, R.; Treebak, J.T.; et al. AMPK in skeletal muscle function and metabolism. FASEB J. 2018, 32, 1741–1777. [Google Scholar] [CrossRef] [Green Version]
- Deng, X.S.; Wang, S.; Deng, A.; Liu, B.; Edgerton, S.M.; Lind, S.E.; Wahdan-Alaswad, R.; Thor, A.D. Metformin targets Stat3 to inhibit cell growth and induce apoptosis in triple-negative breast cancers. Cell Cycle 2012, 11, 367–376. [Google Scholar] [CrossRef] [Green Version]
- Darnell, J.E., Jr. STAT3, HIF-1, glucose addiction and Warburg effect. Aging 2010, 2, 890–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cioce, M.; Pulito, C.; Strano, S.; Blandino, G.; Fazio, V.M. Metformin: Metabolic rewiring faces tumor heterogeneity. Cells 2020, 9, 2439. [Google Scholar] [CrossRef]
- Yin, W.; Liu, Y.; Liu, X.; Sun, B.; Yun, Z. Metformin inhibits epithelial-mesenchymal transition of oral squamous cell carcinoma via the mTOR/HIF-1α/PKM2/STAT3 pathway. Oncol. Lett. 2021, 21, 31. [Google Scholar]
- Wang, L.; Liang, D.; Xiong, X.; Lin, Y.; Zhu, J.; Yao, Z.; Wang, S.; Guo, Y.; Geary, K.; Pan, Y.; et al. Repurposing dextromethorphan and metformin for treating nicotine-induced cancer by directly targeting CHRNA7 to inhibit JAK2/STAT3/SOX2 signaling. Oncogene 2021, 40, 1974–1987. [Google Scholar] [CrossRef]
- Cheng, Z. The FoxO-Autophagy Axis in Health and Disease. Trends Endocrinol. Metab. 2019, 30, 658–671. [Google Scholar] [CrossRef]
- Shin, H.R.; Kim, H.; Kim, K.I.; Baek, S.H. Epigenetic and transcriptional regulation of autophagy. Autophagy 2016, 12, 2248–2249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coomans de Brachene, A.; Demoulin, J.B. FOXO transcription factors in cancer development and therapy. Cell Mol. Life Sci. 2016, 73, 1159–1172. [Google Scholar] [CrossRef] [PubMed]
- Menon, S.; Dibble, C.C.; Talbott, G.; Hoxhaj, G.; Valvezan, A.J.; Takahashi, H.; Cantley, L.C.; Manning, B.D. Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell 2014, 156, 771–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dowling, R.J.O.; Zakikhani, M.; Fantus, I.G.; Pollak, M.; Sonenberg, N. Metformin inhibits mammalian target of rapamycin-dependent translation in breast cancer cells. Cancer Res. 2007, 67, 10804–10812. [Google Scholar] [CrossRef] [Green Version]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell. 2008, 30, 214–226. [Google Scholar] [CrossRef] [Green Version]
- Kalender, A.; Selvaraj, A.; Kim, S.Y.; Gulati, P.; Brûlé, S.; Viollet, B.; Kemp, B.E.; Bardeesy, N.; Dennis, P.; Schlager, J.J.; et al. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab. 2010, 11, 390–401. [Google Scholar] [CrossRef] [Green Version]
- Di Magno, L.; Manni, S.; Di Pastena, F.; Coni, S.; Macone, A.; Cairoli, S.; Sambucci, M.; Infante, P.; Moretti, M.; Petroni, M.; et al. Phenformin inhibits Hedgehog-dependent tumor growth through a Complex I-independent redox/corepressor module. Cell Rep. 2020, 30, 1735–1752. [Google Scholar] [CrossRef] [Green Version]
- Gonnissen, A.; Isebaert, S.; McKee, C.M.; Muschel, R.J.; Haustermans, K. The effect of metformin and GANT61 combinations on the radiosensitivity of prostate cancer cells. Int. J. Mol. Sci. 2017, 18, 399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, C.; Wang, Y.; Liu, Z.; Sun, Y.; Wang, X.; Wei, G.; Wei, J. Metformin exerts anticancer effects through the inhibition of the Sonic hedgehog signaling pathway in breast cancer. Int. J. Mol. Med. 2015, 36, 204–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Z.; Wei, B.; Lu, C.; Huang, X.; Li, P.; Chen, L. Metformin suppresses the expression of Sonic hedgehog in gastric cancer cells. Mol. Med. Rep. 2017, 15, 1909–1915. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, N.; Ogo, A.; Yamura, M.; Yamaguchi, Y.; Nakashima, H. Metformin suppresses Sonic Hedgehog expression in pancreatic cancer cells. Anticancer Res. 2014, 34, 1765–1770. [Google Scholar]
- Adalsteinsson, J.A.; Muzumdar, S.; Waldman, R.; Wu, R.; Ratner, D.; Feng, H.; Ungar, J.; Silverberg, J.I.; Olafsdottir, G.H.; Kristjansson, A.K.; et al. Metformin is associated with decreased risk of basal cell carcinoma: A whole-population case-control study from Iceland. J. Am. Acad. Dermatol. 2021, 85, 56–61. [Google Scholar] [CrossRef]
- Della Corte, C.M.; Ciaramella, V.; Di Mauro, C.; Castellone, M.D.; Papaccio, F.; Fasano, M.; Sasso, F.C.; Martinelli, E.; Troiani, T.; De Vita, F.; et al. Metformin increases antitumor activity of MEK inhibitors through GLI1 downregulation in LKB1 positive human NSCLC cancer cells. Oncotarget 2015, 7, 4265–4278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgillo, F.; Sasso, F.C.; Della Corte, C.M.; Festino, L.; Manzo, A.; Martinelli, E.; Troiani, T.; Capuano, A.; Ciardiello, F. Metformin in lung cancer: Rationale for a combination therapy. Rev. Expert Opin. Investig. Drugs 2013, 22, 1401–1409. [Google Scholar] [CrossRef]
- Morgillo, F.; Fasano, M.; Della Corte, C.M.; Sasso, C.F.; Papaccio, F.; Viscardi, G.; Esposito, G.; Di Liello, R.; Normanno, N.; Capuano, A.; et al. Results of the safety run-in part of the METAL (METformin in Advanced Lung cancer) study: A multicentre, open-label phase I–II study of metformin with erlotinib in second-line therapy of patients with stage IV non-small-cell lung cancer. ESMO Open 2017, 2, e000132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, C.; Chen, Z.; Kim, K.T.; Sun, J.; Xue, M.; Chen, G.; Li, S.; Shen, Y.; Zhu, Z.; Wang, X.; et al. Metformin alleviates hyperglycemia-induced endothelial impairment by downregulating autophagy via the Hedgehog pathway. Autophagy 2019, 15, 843–870. [Google Scholar] [CrossRef] [PubMed]
- Galdieri, L.; Gatla, H.; Vancurova, I.; Vancura, A. Activation of AMP-activated protein kinase by metformin induces protein acetylation in prostate and ovarian cancer cells. J. Biol. Chem. 2016, 291, 25154–25166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vancura, A.; Vancurova, I. Metformin induces protein acetylation in cancer cells. Oncotarget 2017, 8, 39939–39940. [Google Scholar] [CrossRef] [PubMed]
- Cuyas, E.; Verdura, S.; Llorach-Pares, L.; Fernandez-Arroyo, S.; Luciano-Mateo, F.; Cabre, N.; Stursa, J.; Werner, L.; Martin-Castillo, B.; Viollet, B.; et al. Metformin directly targets the H3K27me3 demethylase KDM6A/UTX. Aging Cell 2018, 17, e12772. [Google Scholar] [CrossRef]
- Spada, A.; Mantovani, G.; Lania, A.G.; Treppiedi, D.; Mangili, F.; Catalano, R.; Carosi, G.; Sala, E.; Peverelli, E. Pituitary Tumors: Genetic and Molecular Factors Underlying Pathogenesis and Clinical Behavior. Neuroendocrinology 2021. Epub ahead of print. [Google Scholar] [CrossRef]
- Asa, S.L.; Casar-Borota, O.; Chanson, P.; Delgrange, E.; Earls, P.; Ezzat, S.; Grossman, A.; Ikeda, H.; Inoshita, N.; Karavitaki, N.; et al. From pituitary adenoma to pituitary neuroendocrine tumor (PitNET): An International Pituitary Pathology Club proposal. Endocr. Relat. Cancer 2017, 24, C5–C8. [Google Scholar] [CrossRef] [PubMed]
- Giustina, A. Pituitary adenoma…nomen omen? Endocrine 2021. Epub ahead of print. [Google Scholar] [CrossRef]
- Melmed, S. Acromegaly pathogenesis and treatment. J. Clin. Investig. 2009, 119, 3189–3202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitali, E.; Peverelli, E.; Giardino, E.; Locatelli, M.; Lasio, G.B.; Beck-Peccoz, P.; Spada, A.; Lania, A.G.; Mantovani, G. Cyclic adenosine 30-50-monophosphate (cAMP) exerts proliferative and anti-proliferative effects in pituitary cells of different types by activating both cAMP-dependent protein kinase A (PKA) and exchange proteins directly activated by cAMP (Epac). Mol. Cell. Endocrinol. 2014, 383, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Liu, Y.; Gao, J.; Feng, M.; Bao, X.; Deng, K.; Yao, Y.; Wang, R. Combination treatment with bromocriptine and metformin in patients with bromocriptine-resistant prolactinomas: Pilot study. World Neurosurg. 2018, 115, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Krysiak, R.; Kowalcze, K.; Okopień, B. Vitamin D status determines the impact of metformin on circulating prolactin levels in premenopausal women. J. Clin. Pharm. Ther 2021. Epub ahead of print. [Google Scholar] [CrossRef]
- Colao, A.; Grasso, L.F.S.; Giustina, A.; Melmed, S.; Chanson, P.; Pereira, A.M.; Pivonello, R. Acromegaly. Nat. Rev. Dis. 2019, 5, 20. [Google Scholar] [CrossRef] [PubMed]
- Frara, S.; Maffezzoni, F.; Mazziotti, G.; Giustina, A. Current and emerging aspects of diabetes mellitus in acromegaly. Trends Endocrinol. Metab. 2016, 27, 470–483. [Google Scholar] [CrossRef] [PubMed]
- Giustina, A.; Barkan, A.; Beckers, A.; Biermasz, N.; Biller, B.M.K.; Boguszewski, C.; Bolanowski, M.; Bonert, V.; Bronstein, M.D.; Casanueva, F.F.; et al. A Consensus on the Diagnosis and Treatment of Acromegaly Comorbidities: An Update. J. Clin. Endocrinol. Metab. 2020, 105, dgz096. [Google Scholar] [CrossRef] [PubMed]
- Albertelli, M.; Nazzari, E.; Dotto, A.; Grasso, L.F.; Sciallero, S.; Pirchio, R.; Rebora, A.; Boschetti, M.; Pivonello, R.; Ricci Bitti, S.; et al. Possible protective role of metformin therapy on colonic polyps in acromegaly: An exploratory cross-sectional study. Eur. J. Endocrinol. 2021, 184, 419–425. [Google Scholar] [CrossRef]
- Morales, D.R.; Morris, A.D. Metformin in cancer treatment and prevention. Ann. Rev. Med. 2015, 66, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, E.J.; LeRoith, D. Obesity and diabetes: The increased risk of cancer and cancer-related mortality. Physiol. Rev. 2015, 95, 727–748. [Google Scholar] [CrossRef] [Green Version]
- Onizuka, H.; Masui, K.; Amano, K.; Kawamata, T.; Yamamoto, T.; Nagashima, Y.; Shibata, N. Metabolic Reprogramming Drives Pituitary Tumor Growth through Epigenetic Regulation of TERT. Acta Histochem. Cytochem. 2021, 5, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Oronsky, B.; Ma, P.C.; Morgensztern, D.; Carter, C.A. Nothing But NET: A Review of neuroendocrine Tumors and Carcinomas. Neoplasia 2017, 19, 991–1002. [Google Scholar] [CrossRef]
- Haugvik, S.P.; Hedenstrom, P.; Korsaeth, E.; Valente, R.; Hayes, A.; Siuka, D.; Maisonneuve, P.; Gladhaug, I.P.; Lindkvist, B.; Capurso, G. Diabetes, smoking, alcohol use, and family history of cancer as risk factors for pancreatic neuroendocrine tumors: A systematic review and meta-analysis. Neuroendocrinology 2015, 101, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Pusceddu, S.; Buzzoni, R.; Vernieri, C.; Concas, L.; Marceglia, S.; Giacomelli, L.; Milione, M.; Leuzzi, L.; Femia, D.; Formisano, B.; et al. 2016 Metformin with everolimus and octreotide in pancreatic neuroendocrine tumor patients with diabetes. Future Oncol. 2016, 12, 1251–1260. [Google Scholar] [CrossRef]
- Pusceddu, S.; Vernieri, C.; Di Maio, M.; Marconcini, R.; Spada, F.; Massironi, S.; Ibrahim, T.; Brizzi, M.P.; Campana, D.; Faggiano, A.; et al. Metformin Use Is Associated with Longer Progression-Free Survival of Patients with Diabetes and Pancreatic Neuroendocrine Tumors Receiving Everolimus and/or Somatostatin Analogues. Gastroenterology 2018, 155, 479–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vernieri, C.; Pusceddu, S.; Fuca, G.; Indelicato, P.; Centonze, G.; Castagnoli, L.; Ferrari, E.; Ajazi, A.; Pupa, S.; Casola, S.; et al. Impact of systemic and tumor lipid metabolism on everolimus efficacy in advanced pancreatic neuroendocrine tumors (pNETs). Int. J. Cancer 2019, 144, 1704–1712. [Google Scholar] [CrossRef] [PubMed]
- Vlotides, G.; Tanyeri, A.; Spampatti, M.; Zitzmann, K.; Chourdakis, M.; Spttl, C.; Maurer, J.; Nolting, S.; Goke, B.; Auernhammer, C.J. Anticancer effects of metformin on neuroendocrine tumor cells in vitro. Hormones 2014, 13, 498–508. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Martínez, A.D.; Pedraza-Arevalo, S.; L-López, F.; Gahete, M.D.; Gálvez-Moreno, M.A.; Castaño, J.P.; Luque, R.M. Type 2 Diabetes in Neuroendocrine Tumors: Are Biguanides and Statins Part of the Solution? J. Clin. Endocrinol. Metab. 2019, 104, 57–73. [Google Scholar] [CrossRef] [PubMed]
- Vitali, E.; Boemi, I.; Tarantola, G.; Piccini, S.; Zerbi, A.; Veronesi, G.; Baldelli, R.; Mazziotti, G.; Smiroldo, V.; Lavezzi, E.; et al. Metformin and Everolimus: A Promising combination for Neuroendocrine Tumors Treatment. Cancers 2020, 12, 2143. [Google Scholar] [CrossRef] [PubMed]
- Vitali, E.; Boemi, I.; Piccini, S.; Tarantola, G.; Smiroldo, V.; Lavezzi, E.; Brambilla, T.; Zerbi, A.; Carnaghi, C.; Mantovani, G.; et al. A novel insight into the anticancer mechanism of metformin in pancreatic neuroendocrine tumor cells. Mol. Cell. Endocrinol. 2020, 509, 110803. [Google Scholar] [CrossRef] [PubMed]
- Aristizabal Prada, E.T.; Weis, C.; Orth, M.; Lauseker, M.; Spottl, G.; Maurer, J.; Grabowski, P.; Grossman, A.; Auernhammer, C.J.; Nolting, S. GSK3alpha/beta: A Novel Therapeutic Target for Neuroendocrine Tumors. Neuroendocrinology 2018, 106, 335–351. [Google Scholar] [CrossRef]
- Missiaglia, E.; Dalai, I.; Barbi, S.; Beghelli, S.; Falconi, M.; della Peruta, M.; Piemonti, L.; Capurso, G.; Di Florio, A.; delle Fave, G.; et al. Pancreatic endocrine tumors: Expression profiling evidences a role for AKT-mTOR pathway. J. Clin. Oncol. 2010, 28, 245–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, C.F.; Ji, J.; Yuan, F.; Shi, M.; Zhang, J.; Liu, B.Y.; Zhu, Z.G. mTOR activation in well differentiated pancreatic neuroendocrine tumors: A retrospective study on 34 cases. Hepatogastroenterology 2011, 58, 2140–2143. [Google Scholar] [CrossRef]
- Raymond, E.; Dahan, L.; Raoul, J.L.; Bang, Y.J.; Borbath, I.; Lombard-Bohas, C.; Valle, J.; Metrakos, P.; Smith, D.; Vinik, A.; et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N. Engl. J. Med. 2011, 364, 501–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, J.C.; Shah, M.H.; Ito, T.; Bohas, C.L.; Wolin, E.M.; Van Cutsem, E.; Hobday, T.J.; Okusaka, T.; Capdevila, J.; de Vries, E.G.; et al. Everolimus for advanced pancreatic neuroendocrine tumors. N. Engl. J. Med. 2011, 364, 514–523. [Google Scholar] [CrossRef] [Green Version]
- O’Reilly, K.E.; Rojo, F.; She, Q.B.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006, 66, 1500–1508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svejda, B.; Kidd, M.; Kazberouk, A.; Lawrence, B.; Pfragner, R.; Modlin, I.M. Limitations in small intestinal neuroendocrine tumor therapy by mTOR kinase inhibition reflect growth factor-mediated PI3K feedback loop activation via ERK1/2 and AKT. Cancer 2011, 117, 4141–4154. [Google Scholar] [CrossRef] [PubMed]
- Tulipano, G.; Faggi, L.; Cacciamali, A.; Spinello, M.; Cocchi, D.; Giustina, A. Role of AMP-activated protein kinase (AMPK) activators in antiproliferative multi-drug pituitary tumor therapies: Effects of combined treatments with compounds affecting the mTOR-p70S6 kinase axis in cultured pituitary tumour cells. J. Neuroendocrinol. 2015, 27, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Vandamme, T.; Beyens, M.; De Beeck, K.O.; Dogan, F.; Van Koetsveld, P.M.; Pauwels, P.; Mortier, G.; Vangestel, C.; De Herder, W.; Van Camp, G.; et al. Long-term acquired everolimus resistance in pancreatic neuroendocrine tumours can be overcome with novel PI3K-AKT-mTOR inhibitors. Br. J. Cancer 2016, 114, 650–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wulffele, M.G.; Kooy, A.; de Zeeuw, D.; Stehouwer, C.D.; Gansevoort, R.T. The effect of metformin on blood pressure, plasma cholesterol and triglycerides in type 2 diabetes mellitus: A systematic review. J. Intern. Med. 2004, 256, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Lauretta, R.; Lanzolla, G.; Vici, P.; Mariani, L.; Moretti, C.; Appetecchia, M. Insulin-sensitizers, polycystic ovary syndrome and gynaecological cancer risk. Int. J. Endocrinol. 2016, 8671762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Intlekofer, A.M.; Finley, L.W.S. Metabolic signatures of cancer cells and stem cells. Nat. Metab. 2019, 1, 177–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Intracellular Actions | Mechanism Mediating Gluconeogenesis Inhibition | Reports Questioning the Suggested Mechanism |
|---|---|---|
| AMPK activation | Transcriptional regulation Downregulation of G6pc and Pck1 gene expression | 34,73,81,82 |
| Inhibition of mGPDH | Inhibition of glycerophosphate shuttle Redox-dependent inhibition of gluconeogenesis | 39,69,86,88 |
| Changes in the intracellular levels of metabolites(AMP, fructose 1,6-P2, fructose 2,6-P2) | Allosteric or substrate-dependent regulation of gluconeogenic or glycolytic enzymes Redox-independent inhibition of gluconeogenesis | |
| Changes in the intracellular levels of AMP | Inhibition of adenylate cyclase Inhibition of glucagon signaling | 73,75,81,82 |
| Target | Intracellular Actions | Effects on Metabolic Health |
|---|---|---|
| Skeletal muscle | Increased basal glucose uptake | Possible impact on glucose utilization and plasma glucose homeostasis |
| Altered BCAA catabolism | ||
| Intestine | Increased glucose uptake | Impact on glucose homeostasis and food intake |
| Increased lactate production, possibly associated with a futile enterocyte-hepatocyte futile cycle (lactate-glucose) | Some side-effects associated with metformin treatment in humans | |
| Reduced bile acid absorption, with consequences on GLP-1 and peptide YY secretion | ||
| Gut microbiota | Changes in the relative abundance of bacterial strains, possibly associated with an impact on SCFA production | Impact on glucose homeostasis, appetite and body weight gain |
| Intestine and kidney | Increased expression and release of GDF15 (increased circulating levels) | Impact on energy balance and body weight gain |
| Medio-basal hypothalamus | Decreased AMPK activity | Impact on food intake |
| Congenital PDH Complex Deficiency | Aging | Cancer |
|---|---|---|
| Aerobic glycolysis Increased lactate production Decreased flux though TCA cycle Increased oxaloacetate synthesis through PC activity Overexpression and stabilization of HIF1α, leading to further downregulation of PDH activity and oxidative phosphorylation, and increased glycolysis [129,131,132,133,134] | Increased long chain fatty acid oxidation in skeletal muscle Increased acetyl-CoA and NADH levels, leading to increased PDK activity Decreased insulin-mediated tonic stimulation of PDPs The outcome is a decrease of the PDH complex activity Increased lactate production [129,137,138] | Upregulation and stabilization of HIF1α Increased glucose uptake, glycolysis, and lactate production Increased extrusion of lactate and protons Increased PDK activity, leading to inhibition of the PDH complex activity Decreased oxidative phosphorylation Increased use of glutamine as energy substrate, upregulation of SIRT4 lipoamidase activity, leading to further downregulation of PDH complex activity [83,129,137,138,139,140,141] |
| Molecular Targets within Cells | Systemic Actions |
|---|---|
| Insulin receptor or IGF-1R/PI3K/AKT/mTOR signaling pathway (inhibition) GSK-3 (phosphorylation and inhibition) AMPK (phosphorylation and activation) AIP protein (upregulation) ACC1 (AMPK-mediated inhibition) | Increased insulin sensitivity Decreased glycemia and insulinemia Impact on lipid metabolism (in type 2 diabetic patients with a metabolic syndrome profile only) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tulipano, G. Integrated or Independent Actions of Metformin in Target Tissues Underlying Its Current Use and New Possible Applications in the Endocrine and Metabolic Disorder Area. Int. J. Mol. Sci. 2021, 22, 13068. https://doi.org/10.3390/ijms222313068
Tulipano G. Integrated or Independent Actions of Metformin in Target Tissues Underlying Its Current Use and New Possible Applications in the Endocrine and Metabolic Disorder Area. International Journal of Molecular Sciences. 2021; 22(23):13068. https://doi.org/10.3390/ijms222313068
Chicago/Turabian StyleTulipano, Giovanni. 2021. "Integrated or Independent Actions of Metformin in Target Tissues Underlying Its Current Use and New Possible Applications in the Endocrine and Metabolic Disorder Area" International Journal of Molecular Sciences 22, no. 23: 13068. https://doi.org/10.3390/ijms222313068
APA StyleTulipano, G. (2021). Integrated or Independent Actions of Metformin in Target Tissues Underlying Its Current Use and New Possible Applications in the Endocrine and Metabolic Disorder Area. International Journal of Molecular Sciences, 22(23), 13068. https://doi.org/10.3390/ijms222313068