
A Bioinformatic Workflow for InDel Analysis in the Wheat Multi-Copy α-Gliadin Gene Family Engineered with CRISPR/Cas9



Abstract
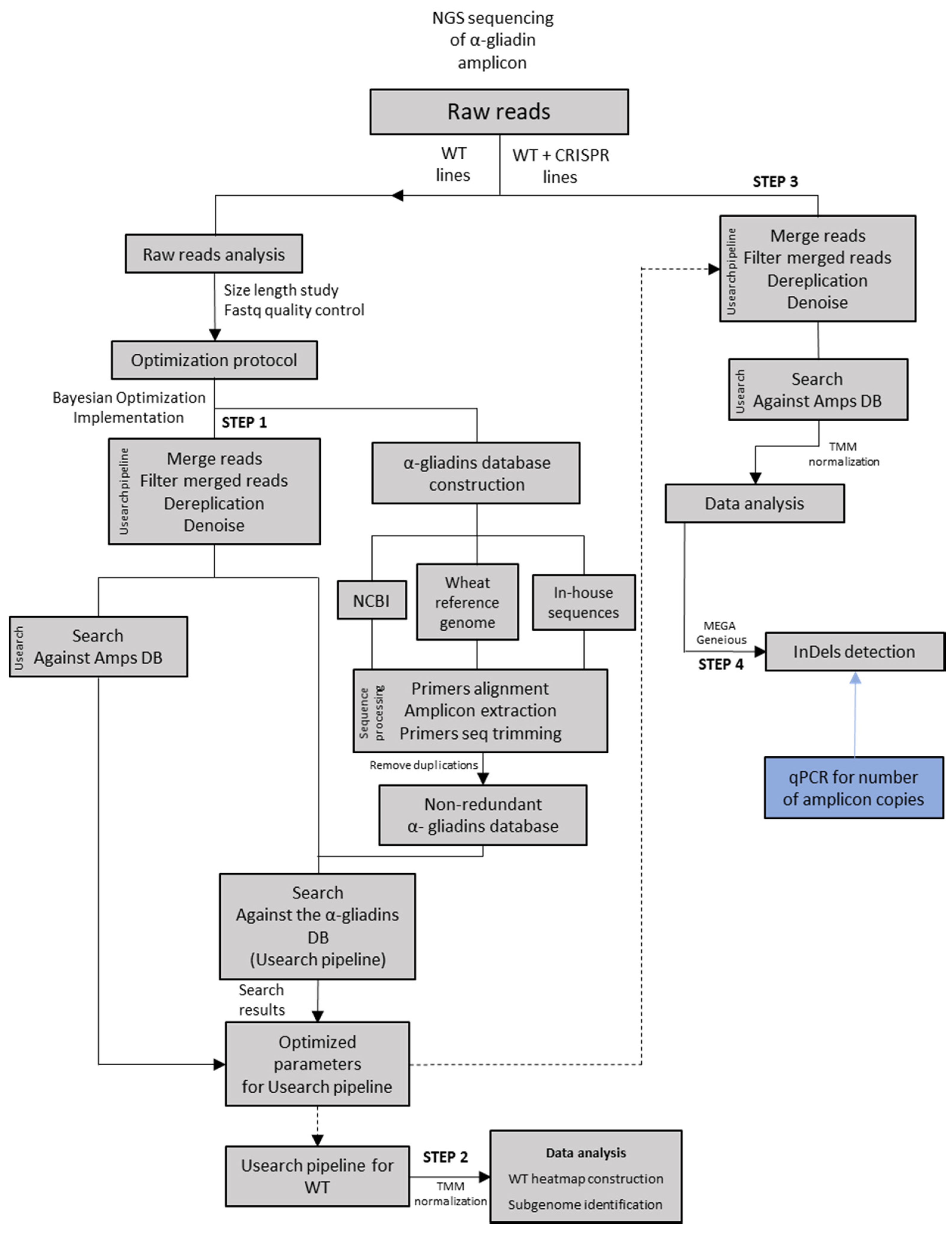
:1. Introduction
2. Results
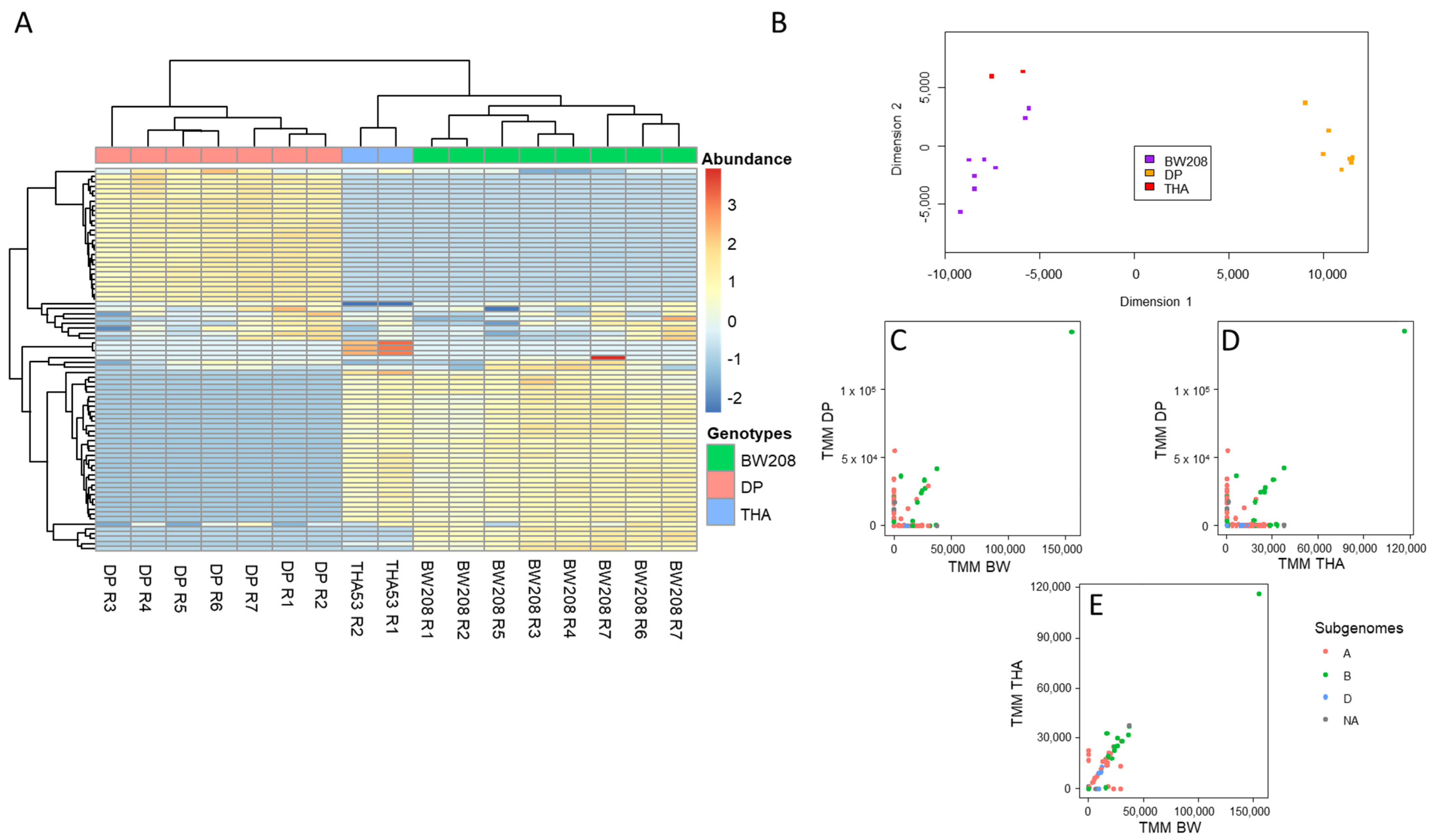
2.1. Bayesian Optimization of Bioinformatic Pipeline Parameters in WT Lines
2.2. Implementation of Optimized Bioinformatic Pipeline in WT and CRISPR Lines
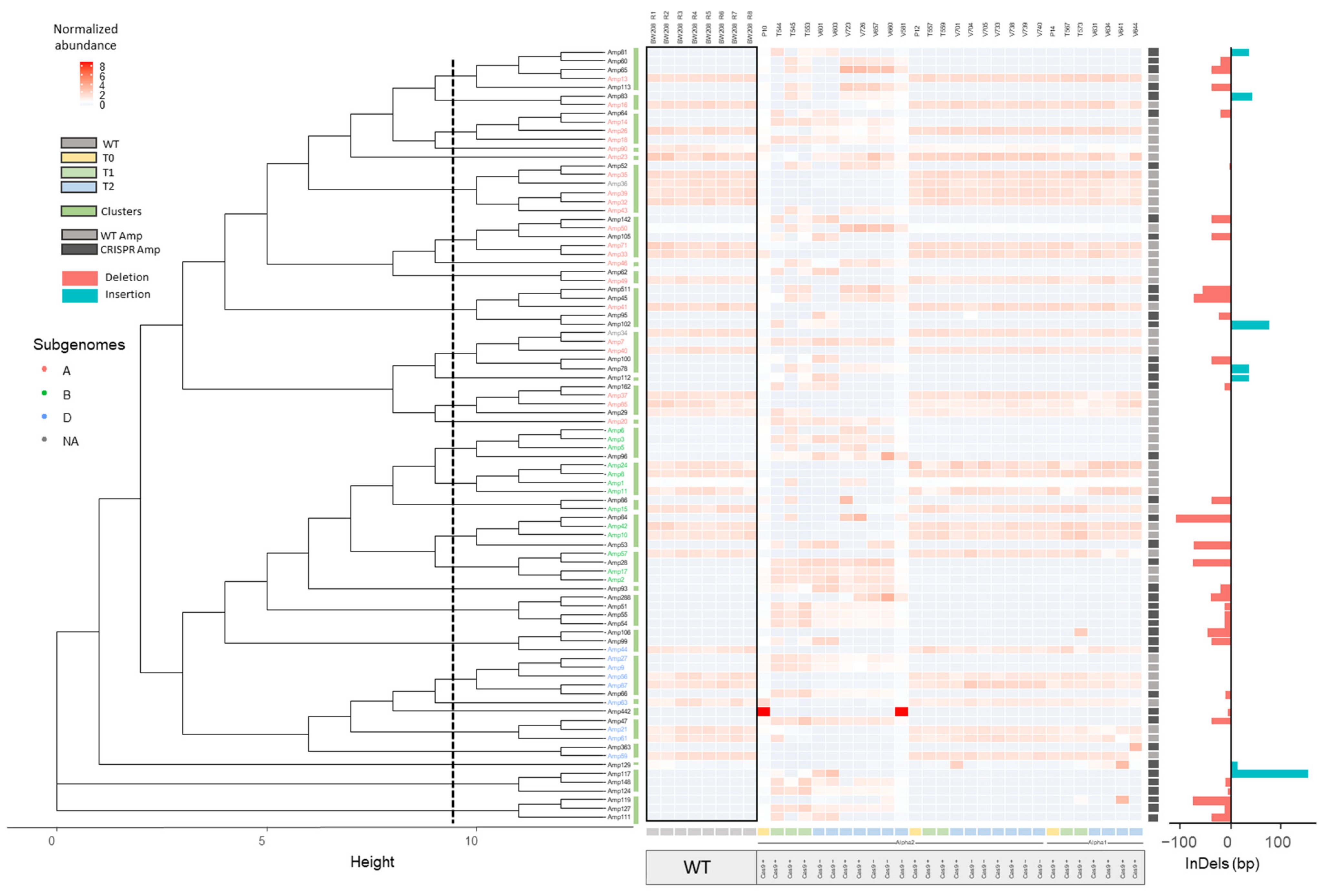
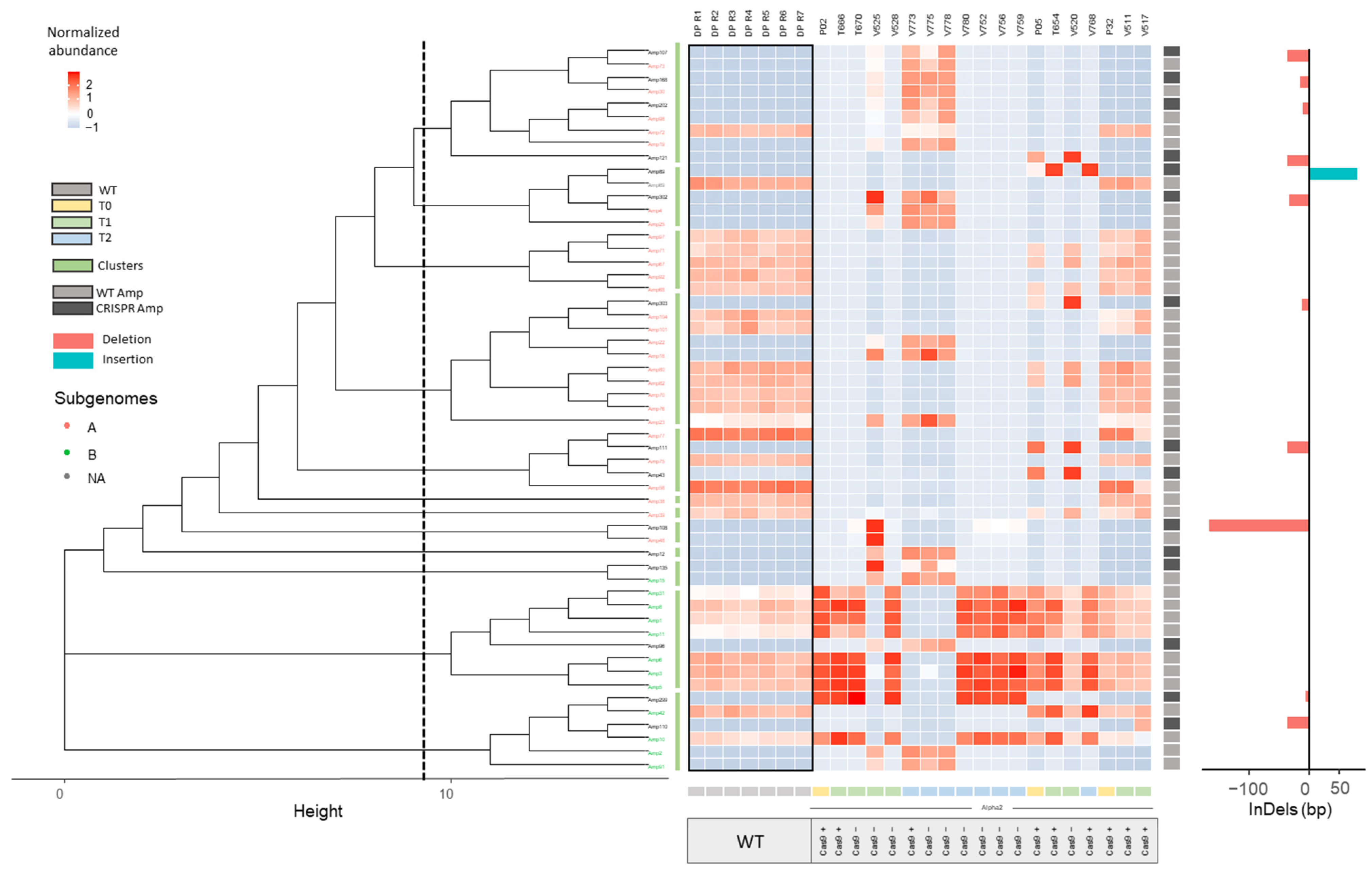
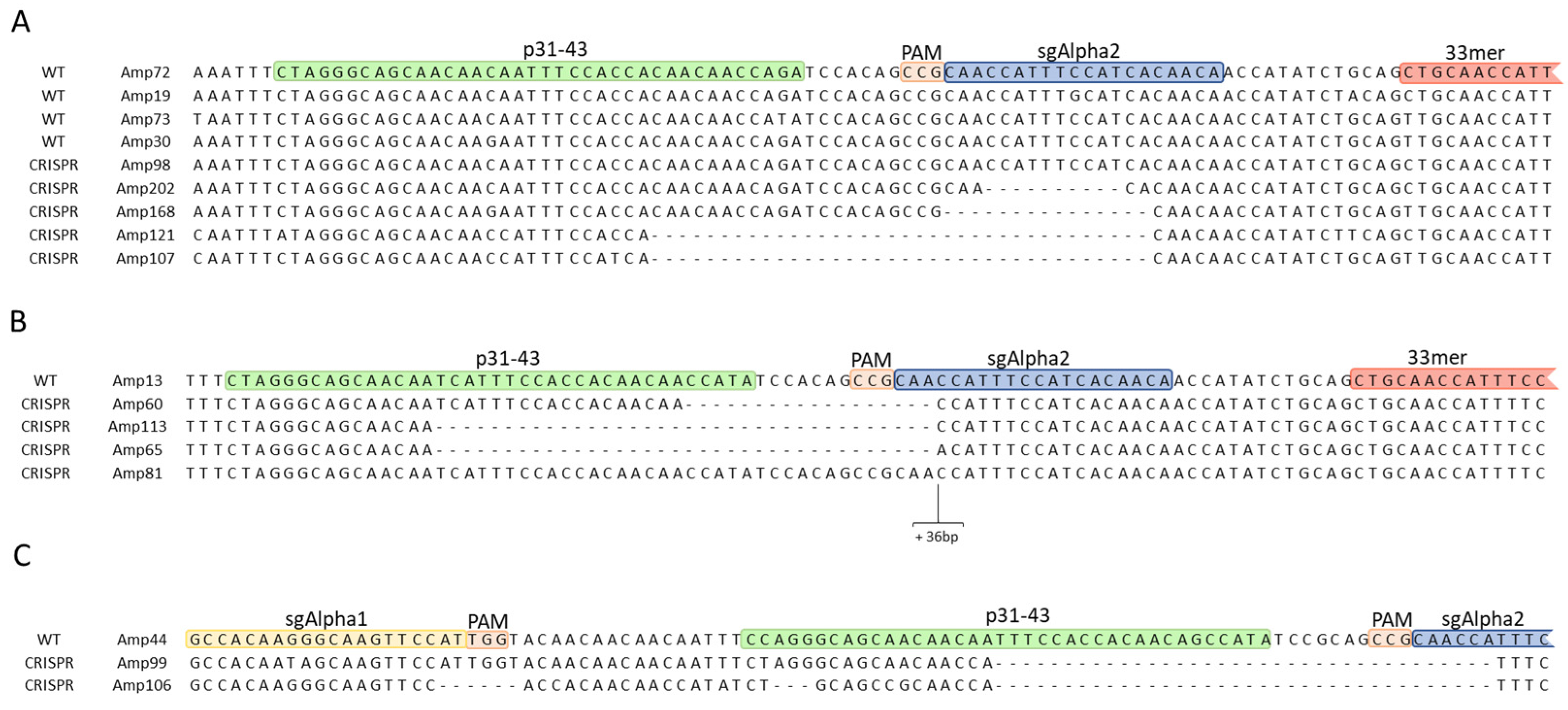
2.2.1. InDel Identification and Characterization in CRISPR/Cas9 Lines
2.2.2. Offspring Analysis in CRISPR/Cas9 Lines
2.3. qPCR Amplicon Copy Number in CRISPR/Cas9 Lines
3. Discussion
4. Materials and Methods
4.1. Plant Material
4.2. Next Generation Sequencing Data
4.3. Construction of a Non-Redundant α-Gliadin Amplicon Database
4.4. Bayesian Optimization
4.5. Optimized Protocol for CRISPR/Cas9 Edited Lines
4.6. Dendrogram Clusters and InDels Analysis
4.7. qPCR Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Doudna, J.A.; Charpentier, E. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346. [Google Scholar] [CrossRef]
- Wiles, M.V.; Qin, W.; Cheng, A.W.; Wang, H. CRISPR–Cas9-mediated genome editing and guide RNA design. Mamm. Genome 2015, 26, 501–510. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Li, C.; Gao, C. Applications of CRISPR–Cas in agriculture and plant biotechnology. Nat. Rev. Mol. Cell Biol. 2020, 21, 661–677. [Google Scholar] [CrossRef]
- Shewry, P.R. Wheat. J. Exp. Bot. 2009, 60, 1537–1553. [Google Scholar] [CrossRef]
- García-Molina, M.D.; Giménez, M.J.; Sánchez-León, S.; Barro, F. Gluten Free Wheat: Are We There? Nutrients 2019, 11, 487. [Google Scholar] [CrossRef] [Green Version]
- Shewry, P.R.; Halford, N.G. Cereal seed storage proteins: Structures, properties and role in grain utilization. J. Exp. Bot. 2002, 53, 947–958. [Google Scholar] [CrossRef] [Green Version]
- Juhász, A.; Belova, T.; Florides, C.G.; Maulis, C.; Fischer, I.; Gell, G.; Birinyi, Z.; Ong, J.; Keeble-Gagnère, G.; Maharajan, A. Genome mapping of seed-borne allergens and immunoresponsive proteins in wheat. Sci. Adv. 2018, 4, eaar8602. [Google Scholar] [CrossRef] [Green Version]
- Larre, C.; Lupi, R.; Gombaud, G.; Brossard, C.; Branlard, G.; Moneret-Vautrin, D.A.; Rogniaux, H.; Denery-Papini, S. Assessment of allergenicity of diploid and hexaploid wheat genotypes: Identification of allergens in the albumin/globulin fraction. J. Proteom. 2011, 74, 1279–1289. [Google Scholar] [CrossRef] [PubMed]
- Catassi, C.; Gatti, S.; Fasano, A. The new epidemiology of celiac disease. J. Pediatr. Gastroenterol. Nutr. 2014, 59, S7–S9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mustalahti, K.; Catassi, C.; Reunanen, A.; Fabiani, E.; Heier, M.; McMillan, S.; Murray, L.; Metzger, M.-H.; Gasparin, M.; Bravi, E. The prevalence of celiac disease in Europe: Results of a centralized, international mass screening project. Ann. Med. 2010, 42, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Ludvigsson, J.F.; Bai, J.C.; Biagi, F.; Card, T.R.; Ciacci, C.; Ciclitira, P.J.; Green, P.H.R.; Hadjivassiliou, M.; Holdoway, A.; Van Heel, D.A. Diagnosis and management of adult coeliac disease: Guidelines from the British Society of Gastroenterology. Gut 2014, 63, 1210–1228. [Google Scholar] [CrossRef] [PubMed]
- Sollid, L.M.; Tye-Din, J.A.; Qiao, S.-W.; Anderson, R.P.; Gianfrani, C.; Koning, F. Update 2020: Nomenclature and listing of celiac disease–relevant gluten epitopes recognized by CD4+ T cells. Immunogenetics 2020, 72, 85–88. [Google Scholar] [CrossRef]
- Tye-Din, J.A.; Stewart, J.A.; Dromey, J.A.; Beissbarth, T.; van Heel, D.A.; Tatham, A.; Henderson, K.; Mannering, S.I.; Gianfrani, C.; Jewell, D.P. Comprehensive, quantitative mapping of T cell epitopes in gluten in celiac disease. Sci. Transl. Med. 2010, 2, 41ra51. [Google Scholar] [CrossRef] [PubMed]
- Comino, I.; Fernández-Bañares, F.; Esteve, M.; Ortigosa, L.; Castillejo, G.; Fambuena, B.; Ribes-Koninckx, C.; Sierra, C.; Rodríguez-Herrera, A.; Salazar, J.C.; et al. Fecal Gluten Peptides Reveal Limitations of Serological Tests and Food Questionnaires for Monitoring Gluten-Free Diet in Celiac Disease Patients. Am. J. Gastroenterol. 2016, 111, 1456. [Google Scholar] [CrossRef] [Green Version]
- Rai, S.; Kaur, A.; Chopra, C.S. Gluten-Free Products for Celiac Susceptible People. Front. Nutr. 2018, 5, 116. [Google Scholar] [CrossRef] [PubMed]
- Gil-Humanes, J.; Pistón, F.; Tollefsen, S.; Sollid, L.M.; Barro, F. Effective shutdown in the expression of celiac disease-related wheat gliadin T-cell epitopes by RNA interference. Proc. Natl. Acad. Sci. USA 2010, 107, 17023–17028. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-León, S.; Gil-Humanes, J.; Ozuna, C.V.; Giménez, M.J.; Sousa, C.; Voytas, D.F.; Barro, F. Low-gluten, nontransgenic wheat engineered with CRISPR/Cas9. Plant Biotechnol. J. 2018, 16, 902–910. [Google Scholar] [CrossRef]
- Turnbull, C.; Lillemo, M.; Hvoslef-Eide, T.A.K. Global Regulation of Genetically Modified Crops Amid the Gene Edited Crop Boom—A Review. Front. Plant Sci. 2021, 12, 258. [Google Scholar] [CrossRef]
- Ozuna, C.V.; Iehisa, J.C.M.; Giménez, M.J.; Alvarez, J.B.; Sousa, C.; Barro, F. Diversification of the celiac disease α-gliadin complex in wheat: A 33-mer peptide with six overlapping epitopes, evolved following polyploidization. Plant J. 2015, 82, 794–805. [Google Scholar] [CrossRef] [PubMed]
- Huo, N.; Zhu, T.; Altenbach, S.; Dong, L.; Wang, Y.; Mohr, T.; Liu, Z.; Dvorak, J.; Luo, M.-C.; Gu, Y.Q. Dynamic evolution of α-gliadin prolamin gene family in homeologous genomes of hexaploid wheat. Sci. Rep. 2018, 8, 5181. [Google Scholar] [CrossRef]
- Sánchez-León, S.; Giménez, M.J.; Barro, F. The α-gliadins in Bread Wheat: Effect of Nitrogen Treatment on the Expression of the Major Ceeliac Disease Immunogenic Complex in Two RNAi Low-Gliadin Lines. Front. Plant Sci. 2021, 12, 742. [Google Scholar] [CrossRef]
- Brinkman, E.K.; Chen, T.; Amendola, M.; van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014, 42, e168. [Google Scholar] [CrossRef]
- Liu, W.; Xie, X.; Ma, X.; Li, J.; Chen, J.; Liu, Y.-G. DSDecode: A web-based tool for decoding of sequencing chromatograms for genotyping of targeted mutations. Mol. Plant 2015, 8, 1431–1433. [Google Scholar] [CrossRef]
- Brinkman, E.K.; Kousholt, A.N.; Harmsen, T.; Leemans, C.; Chen, T.; Jonkers, J.; van Steensel, B. Easy quantification of template-directed CRISPR/Cas9 editing. Nucleic Acids Res. 2018, 46, e58. [Google Scholar] [CrossRef]
- Hsiau, T.; Conant, D.; Rossi, N.; Maures, T.; Waite, K.; Yang, J.; Joshi, S.; Kelso, R.; Holden, K.; Enzmann, B.L.; et al. Inference of CRISPR Edits from Sanger Trace Data. bioRxiv 2018. [Google Scholar] [CrossRef]
- Bloh, K.; Kanchana, R.; Bialk, P.; Banas, K.; Zhang, Z.; Yoo, B.C.; Kmiec, E.B. Deconvolution of Complex DNA Repair (DECODR): Establishing a Novel Deconvolution Algorithm for Comprehensive Analysis of CRISPR-Edited Sanger Sequencing Data. Cris. J. 2021, 4, 120–131. [Google Scholar] [CrossRef]
- Liang, Z.; Chen, K.; Yan, Y.; Zhang, Y.; Gao, C. Genotyping genome-edited mutations in plants using CRISPR ribonucleoprotein complexes. Plant Biotechnol. J. 2018, 16, 2053–2062. [Google Scholar] [CrossRef] [PubMed]
- Güell, M.; Yang, L.; Church, G.M. Genome editing assessment using CRISPR Genome Analyzer (CRISPR-GA). Bioinformatics 2014, 30, 2968–2970. [Google Scholar] [PubMed] [Green Version]
- Pinello, L.; Canver, M.C.; Hoban, M.D.; Orkin, S.H.; Kohn, D.B.; Bauer, D.E.; Yuan, G.-C. Analyzing CRISPR genome-editing experiments with CRISPResso. Nat. Biotechnol. 2016, 34, 695–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, L.-J.; Tsai, C.-J. AGEseq: Analysis of genome editing by sequencing. Mol. Plant 2015, 8, 1428–1430. [Google Scholar] [CrossRef] [Green Version]
- Boel, A.; Steyaert, W.; De Rocker, N.; Menten, B.; Callewaert, B.; De Paepe, A.; Coucke, P.; Willaert, A. BATCH-GE: Batch analysis of Next-Generation Sequencing data for genome editing assessment. Sci. Rep. 2016, 6, 30330. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Lim, K.; Kim, J.-S.; Bae, S. Cas-analyzer: An online tool for assessing genome editing results using NGS data. Bioinformatics 2017, 33, 286–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Wang, C.; Jiao, X.; Zhang, H.; Song, L.; Li, Y.; Gao, C.; Wang, K. Hi-TOM: A platform for high-throughput tracking of mutations induced by CRISPR/Cas systems. Sci. China Life Sci. 2019, 62, 1–7. [Google Scholar] [CrossRef]
- Wang, X.; Tilford, C.; Neuhaus, I.; Mintier, G.; Guo, Q.; Feder, J.N.; Kirov, S. CRISPR-DAV: CRISPR NGS data analysis and visualization pipeline. Bioinformatics 2017, 33, 3811–3812. [Google Scholar] [CrossRef]
- Connelly, J.P.; Pruett-Miller, S.M. CRIS. py: A versatile and high-throughput analysis program for CRISPR-based genome editing. Sci. Rep. 2019, 9, 4194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Chang, H.Y.; Cho, S.W.; Ji, H.P. CRISPRpic: Fast and precise analysis for CRISPR-induced mutations via p refixed i ndex c ounting. NAR Genom. Bioinform. 2020, 2, lqaa012. [Google Scholar] [CrossRef] [Green Version]
- Reti, D.; O’brien, A.; Wetzel, P.; Tay, A.; Bauer, D.C.; Wilson, L.O.W. GOANA: A Universal High-Throughput Web Service for Assessing and Comparing the Outcome and Efficiency of Genome Editing Experiments. Cris. J. 2021, 4, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R. Usearch; Lawrence Berkeley National Lab. (LBNL): Berkeley, CA, USA, 2010. [Google Scholar]
- Appels, R.; Eversole, K.; Feuillet, C.; Keller, B.; Rogers, J.; Stein, N.; Pozniak, C.J.; Choulet, F.; Distelfeld, A.; Poland, J. Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 2018, 361, eaar7191. [Google Scholar]
- Edgar, R.C. UNOISE2: Improved error-correction for Illumina 16S and ITS amplicon sequencing. bioRxiv 2016, 81257. [Google Scholar] [CrossRef] [Green Version]
- Callahan, B.J.; McMurdie, P.J.; Holmes, S.P. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 2017, 11, 2639–2643. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [Green Version]
- Van Herpen, T.W.J.M.; Goryunova, S.V.; Van Der Schoot, J.; Mitreva, M.; Salentijn, E.; Vorst, O.; Schenk, M.F.; Van Veelen, P.A.; Koning, F.; Van Soest, L.J.M. Alpha-gliadin genes from the A, B, and D genomes of wheat contain different sets of celiac disease epitopes. BMC Genom. 2006, 7, 1. [Google Scholar] [CrossRef]
- Jouanin, A.; Schaart, J.G.; Boyd, L.A.; Cockram, J.; Leigh, F.J.; Bates, R.; Wallington, E.J.; Visser, R.G.F.; Smulders, M.J.M. Outlook for coeliac disease patients: Towards bread wheat with hypoimmunogenic gluten by gene editing of α- And γ-gliadin gene families. BMC Plant Biol. 2019, 19, 333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jouanin, A.; Tenorio-Berrio, R.; Schaart, J.G.; Leigh, F.; Visser, R.G.F.; Smulders, M.J.M. Optimisation of droplet digital PCR for determining copy number variation of α-gliadin genes in mutant and gene-edited polyploid bread wheat. J. Cereal Sci. 2020, 92, 102903. [Google Scholar] [CrossRef]
- Jouanin, A.; Borm, T.; Boyd, L.A.; Cockram, J.; Leigh, F.; Santos, B.A.C.M.; Visser, R.G.F.; Smulders, M.J.M. Development of the GlutEnSeq capture system for sequencing gluten gene families in hexaploid bread wheat with deletions or mutations induced by γ-irradiation or CRISPR/Cas9. J. Cereal Sci. 2019, 88, 157–166. [Google Scholar] [CrossRef]
- Prodan, A.; Tremaroli, V.; Brolin, H.; Zwinderman, A.H.; Nieuwdorp, M.; Levin, E. Comparing bioinformatic pipelines for microbial 16S rRNA amplicon sequencing. PLoS ONE 2020, 15, e0227434. [Google Scholar] [CrossRef] [Green Version]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Pereira, M.B.; Wallroth, M.; Jonsson, V.; Kristiansson, E. Comparison of normalization methods for the analysis of metagenomic gene abundance data. BMC Genom. 2018, 19, 274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Pan, Q.; He, F.; Akhunova, A.; Chao, S.; Trick, H.; Akhunov, E. Transgenerational CRISPR-Cas9 Activity Facilitates Multiplex Gene Editing in Allopolyploid Wheat. Cris. J. 2018, 1, 65–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, A.; Liu, Y.; Wang, F.; Li, T.; Chen, Z.; Kong, D.; Bi, J.; Zhang, F.; Luo, X.; Wang, J.; et al. Enhanced rice salinity tolerance via CRISPR/Cas9-targeted mutagenesis of the OsRR22 gene. Mol. Breed. 2019, 39, 47. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Cheng, X.; Shan, Q.; Zhang, Y.; Liu, J.; Gao, C.; Qiu, J.L. Simultaneous editing of three homoeoalleles in hexaploid bread wheat confers heritable resistance to powdery mildew. Nat. Biotechnol. 2014, 32, 947–951. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wu, J.J.; Tang, T.; De Liu, K.; Dai, C. CRISPR/Cas9-mediated genome editing efficiently creates specific mutations at multiple loci using one sgRNA in Brassica napus. Sci. Rep. 2017, 7, 7489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, A.; Jia, S.; Yobi, A.; Ge, Z.; Sato, S.J.; Zhang, C.; Angelovici, R.; Clemente, T.E.; Holding, D.R. Editing of an alpha-kafirin gene family increases digestibility and protein quality in sorghum. Plant Physiol. 2018, 177, 1425–1438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melnikova, N.V.; Kudryavtseva, A.V.; Kudryavtsev, A.M. Catalogue of alleles of gliadin-coding loci in durum wheat (Triticum durum Desf.). Biochimie 2012, 94, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Liu, B.; Weeks, D.P.; Spalding, M.H.; Yang, B. Large chromosomal deletions and heritable small genetic changes induced by CRISPR/Cas9 in rice. Nucleic Acids Res. 2014, 42, 10903–10914. [Google Scholar] [CrossRef]
- Tang, T.; Yu, X.; Yang, H.; Gao, Q.; Ji, H.; Wang, Y.; Yan, G.; Peng, Y.; Luo, H.; Liu, K.; et al. Development and validation of an effective CRISPR/Cas9 vector for efficiently isolating positive transformants and transgene-free mutants in a wide range of plant species. Front. Plant Sci. 2018, 871, 1533. [Google Scholar] [CrossRef] [Green Version]
- McCarty, N.S.; Graham, A.E.; Studená, L.; Ledesma-Amaro, R. Multiplexed CRISPR technologies for gene editing and transcriptional regulation. Nat. Commun. 2020, 11, 1281. [Google Scholar] [CrossRef]
- Zhang, Z.; Mao, Y.; Ha, S.; Liu, W.; Botella, J.R.; Zhu, J.K. A multiplex CRISPR/Cas9 platform for fast and efficient editing of multiple genes in Arabidopsis. Plant Cell Rep. 2016, 35, 1519–1533. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Stoler, N.; Nekrutenko, A. Sequencing error profiles of Illumina sequencing instruments. NAR Genom. Bioinforma. 2021, 3, lqab019. [Google Scholar] [CrossRef]
- Glenn, T.C. Field guide to next-generation DNA sequencers. Mol. Ecol. Resour. 2011, 11, 759–769. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2013. [Google Scholar]
- Kolde, R.; Kolde, M.R. Package ‘pheatmap’. R Packag. 2015, 1, 790. [Google Scholar]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T. ggtree: An R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [Green Version]
- Van Rossum, G.; Drake, F.L., Jr. Python Reference Manual; Centrum voor Wiskunde en Informatica Amsterdam: Amsterdam, The Netherlands, 1995. [Google Scholar]
- Stacey, J.; Isaac, P.G. Isolation of DNA from plants. In Protocols for Nucleic Acid Analysis by Nonradioactive Probes; Springer: Berlin/Heidelberg, Germany, 1994; pp. 9–15. [Google Scholar]
- Giménez, M.J.; Pistón, F.; Atienza, S.G. Identification of suitable reference genes for normalization of qPCR data in comparative transcriptomics analyses in the Triticeae. Planta 2011, 233, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Maccaferri, M.; Harris, N.S.; Twardziok, S.O.; Pasam, R.K.; Gundlach, H.; Spannagl, M.; Ormanbekova, D.; Lux, T.; Prade, V.M.; Milner, S.G. Durum wheat genome highlights past domestication signatures and future improvement targets. Nat. Genet. 2019, 51, 885–895. [Google Scholar] [CrossRef] [Green Version]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT–PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Command | Option | Interval | Optimal Value |
|---|---|---|---|---|
| Merge | fastq_mergepairs | fastq_maxdiffs | (5–82) | 58.94 |
| Merge | fastq_mergepairs | fastq_maxdiffpct | (2–30) | 24.66 |
| Filter | fastq_filter | - | (0.25–1.5) | 1.13 |
| Dereplication | fastx_uniques | - | - | - |
| Denoising | unoise2 | minampsize | (2–30) | 22.47 |
| Search | search_global/search_exact | 0.99 and 1 | 0.99 |
| % of Reads | ||
|---|---|---|
| WT Lines | All Lines | |
| Pairs (raw reads) | 100 (8,205,317 reads) | 100 (39,180,517 reads) |
| Merged | 86.32 | 85.81 |
| Filtered | 88.57 | 87.47 |
| Denoised | 68.83 | 63.45 |
| Matched to the amplicon database a | 95.08 | 85.94 |
| Assigned to Amps b | 73.41 | 70.69 |
| T0 | T1 | T2 | Cas9 +/− | Total Amps/Line a | CRISPR Amps b | Non-Targeted WT Amps c | Putative Targeted WT Amps d |
|---|---|---|---|---|---|---|---|
| WT | NA | 48 | NA | NA | NA | ||
| P10 | - | - | Cas9 + | 46 | 28 | 18 | 30 |
| T544 | - | Cas9 + | 32 | 20 | 12 | 36 | |
| V601 | Cas9 − | 34 | 23 | 11 | 37 | ||
| V603 | Cas9 − | 34 | 23 | 11 | 37 | ||
| T545 | - | Cas9 + | 34 | 19 | 15 | 33 | |
| V723 | Cas9 + | 33 | 18 | 15 | 33 | ||
| V726 | Cas9 + | 33 | 18 | 15 | 33 | ||
| T553 | - | Cas9 + | 37 | 24 | 13 | 35 | |
| V657 | Cas9 − | 32 | 19 | 13 | 35 | ||
| V660 | Cas9 − | 33 | 21 | 12 | 36 | ||
| - | V581 | Cas9 − | 39 | 24 | 15 | 33 | |
| P12 | - | - | Cas9 + | 48 | 0 | 48 | 0 |
| T557 | - | Cas9 + | 48 | 0 | 48 | 0 | |
| V701 | Cas9 + | 47 | 1 | 46 | 2 | ||
| V704 | Cas9 − | 47 | 1 | 46 | 2 | ||
| V705 | Cas9 − | 48 | 0 | 48 | 0 | ||
| T559 | - | Cas9 + | 48 | 0 | 48 | 0 | |
| V733 | Cas9 + | 48 | 0 | 48 | 0 | ||
| V738 | Cas9 + | 48 | 0 | 48 | 0 | ||
| V739 | Cas9 − | 48 | 0 | 48 | 0 | ||
| V740 | Cas9 − | 48 | 0 | 48 | 0 | ||
| P14 | - | - | Cas9 + | 48 | 0 | 48 | 0 |
| T567 | - | Cas9 + | 48 | 0 | 48 | 0 | |
| V631 | Cas9 + | 48 | 0 | 48 | 0 | ||
| V634 | Cas9 + | 48 | 0 | 48 | 0 | ||
| T573 | - | Cas9 + | 46 | 1 | 45 | 3 | |
| V641 | Cas9 + | 49 | 2 | 47 | 1 | ||
| V644 | Cas9 + | 48 | 1 | 47 | 1 | ||
| T0 | T1 | T2 | Cas9 +/− | Total Amps/Line a | CRISPR Amps b | Non-Targeted WT Amps c | Putative Targeted WT Amps d |
|---|---|---|---|---|---|---|---|
| WT | NA | 40 | NA | NA | NA | ||
| P02 | - | - | Cas9 + | 21 | 2 | 19 | 21 |
| T666 | - | Cas9 + | 20 | 2 | 18 | 22 | |
| V773 | Cas9 + | 14 | 4 | 10 | 30 | ||
| V775 | Cas9 − | 16 | 5 | 11 | 29 | ||
| V778 | Cas9 − | 15 | 5 | 10 | 30 | ||
| V780 | Cas9 − | 23 | 4 | 19 | 21 | ||
| T670 | - | Cas9 − | 16 | 2 | 14 | 26 | |
| V752 | Cas9 − | 16 | 2 | 14 | 26 | ||
| V756 | Cas9 − | 17 | 3 | 14 | 26 | ||
| V759 | Cas9 − | 16 | 2 | 14 | 26 | ||
| V525 | - | Cas9 − | 20 | 8 | 12 | 28 | |
| V528 | - | Cas9 − | 23 | 4 | 19 | 21 | |
| P05 | - | - | Cas9 + | 25 | 3 | 22 | 18 |
| T654 | - | Cas9 + | 17 | 1 | 16 | 24 | |
| V768 | Cas9 + | 17 | 1 | 16 | 24 | ||
| V520 | - | Cas9 − | 28 | 4 | 24 | 16 | |
| P32 | - | - | Cas9 + | 40 | 0 | 40 | 0 |
| V511 | - | Cas9 + | 40 | 0 | 40 | 0 | |
| V517 | - | Cas9 + | 40 | 1 | 39 | 1 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guzmán-López, M.H.; Marín-Sanz, M.; Sánchez-León, S.; Barro, F. A Bioinformatic Workflow for InDel Analysis in the Wheat Multi-Copy α-Gliadin Gene Family Engineered with CRISPR/Cas9. Int. J. Mol. Sci. 2021, 22, 13076. https://doi.org/10.3390/ijms222313076
Guzmán-López MH, Marín-Sanz M, Sánchez-León S, Barro F. A Bioinformatic Workflow for InDel Analysis in the Wheat Multi-Copy α-Gliadin Gene Family Engineered with CRISPR/Cas9. International Journal of Molecular Sciences. 2021; 22(23):13076. https://doi.org/10.3390/ijms222313076
Chicago/Turabian StyleGuzmán-López, María H., Miriam Marín-Sanz, Susana Sánchez-León, and Francisco Barro. 2021. "A Bioinformatic Workflow for InDel Analysis in the Wheat Multi-Copy α-Gliadin Gene Family Engineered with CRISPR/Cas9" International Journal of Molecular Sciences 22, no. 23: 13076. https://doi.org/10.3390/ijms222313076
APA StyleGuzmán-López, M. H., Marín-Sanz, M., Sánchez-León, S., & Barro, F. (2021). A Bioinformatic Workflow for InDel Analysis in the Wheat Multi-Copy α-Gliadin Gene Family Engineered with CRISPR/Cas9. International Journal of Molecular Sciences, 22(23), 13076. https://doi.org/10.3390/ijms222313076