
Dystroglycanopathy: From Elucidation of Molecular and Pathological Mechanisms to Development of Treatment Methods

Abstract
:1. Introduction
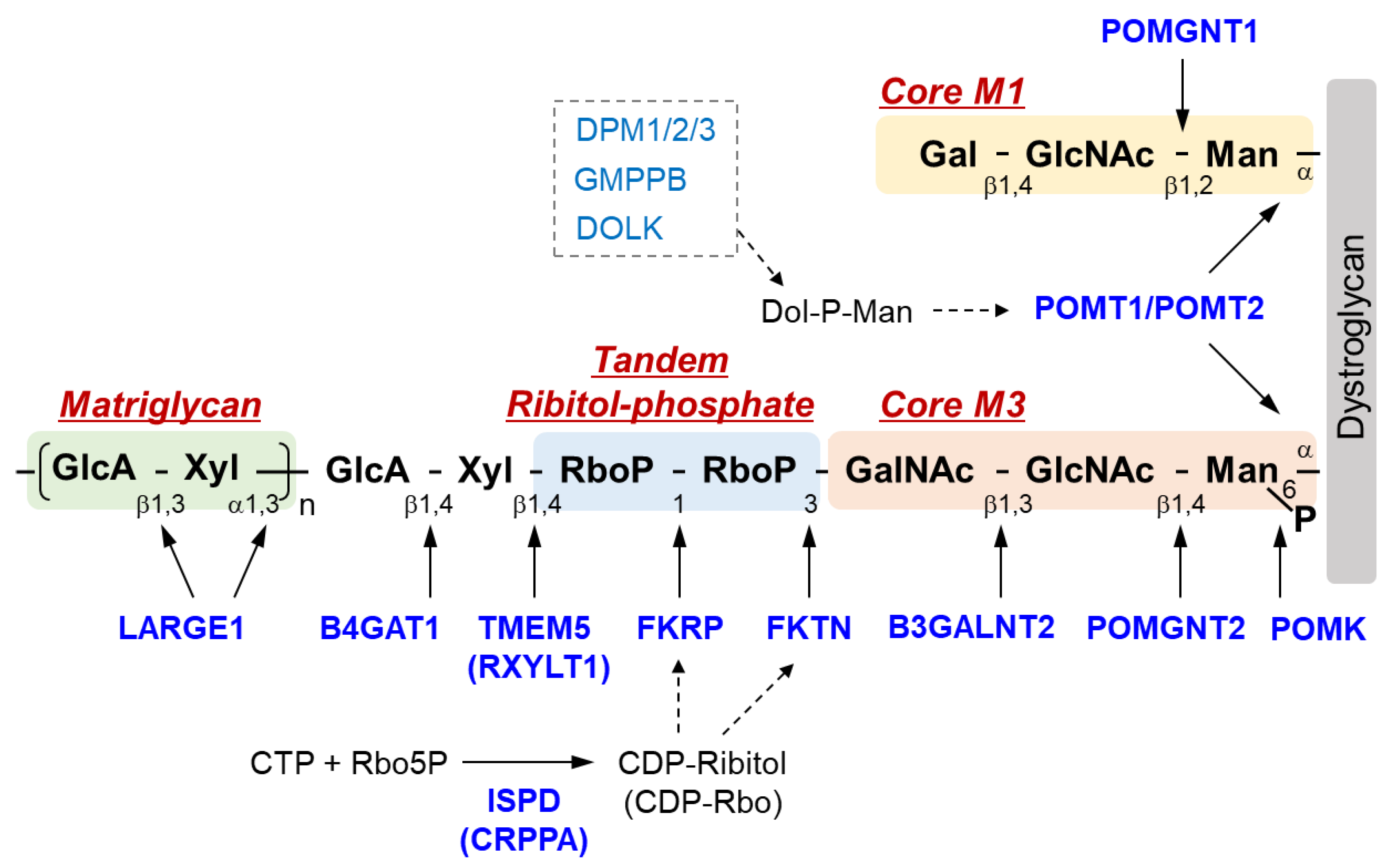
2. Sugar Chain Structure of DG and Functions of Causative Genes of DGpathy
3. DGpathy Model Mice
4. Pathological Mechanism of DGpathy
4.1. Muscular Dystrophy
4.2. Central Nervous System Abnormalities
4.3. Cardiomyopathy
5. Treatment Methods
5.1. Gene Therapy
5.2. Pharmacotherapy
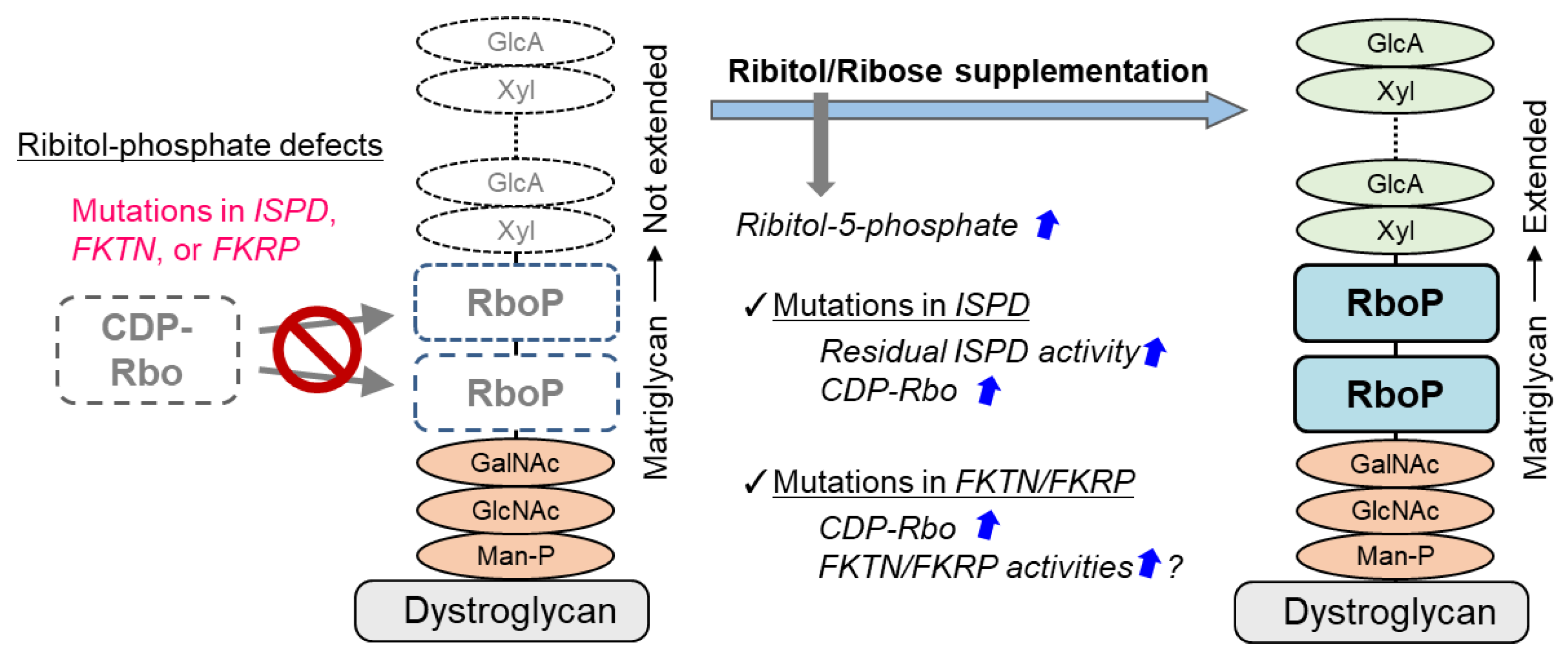
5.3. Ribitol Supplementation Therapy
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Ibraghimov-Beskrovnaya, O.; Ervasti, J.M.; Leveille, C.J.; Slaughter, C.A.; Sernett, S.W.; Campbell, K.P. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature 1992, 355, 696–702. [Google Scholar] [CrossRef]
- Kanagawa, M.; Toda, T. Ribitol-phosphate-a newly identified posttranslational glycosylation unit in mammals: Structure, modification enzymes and relationship to human diseases. J. Biochem. 2018, 163, 359–369. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, Y.K.; Ogawa, M.; Tagawa, K.; Noguchi, S.; Ishihara, T.; Nonaka, I.; Arahata, K. Selective deficiency of alpha-dystroglycan in Fukuyama-type congenital muscular dystrophy. Neurology 2001, 57, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Michele, D.E.; Barresi, R.; Kanagawa, M.; Saito, F.; Cohn, R.D.; Satz, J.S.; Dollar, J.; Nishino, I.; Kelley, R.I.; Somer, H.; et al. Post-translational disruption of dystroglycan-ligand interactions in congenital muscular dystrophies. Nature 2002, 418, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Brockington, M.; Blake, D.J.; Prandini, P.; Brown, S.C.; Torelli, S.; Benson, M.A.; Ponting, C.P.; Estournet, B.; Romero, N.B.; Mercuri, E.; et al. Mutations in the fukutin-related protein gene (FKRP) cause a form of congenital muscular dystrophy with secondary laminin alpha2 deficiency and abnormal glycosylation of alpha-dystroglycan. Am. J. Hum. Genet. 2001, 69, 1198–1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brockington, M.; Yuva, Y.; Prandini, P.; Brown, S.C.; Torelli, S.; Benson, M.A.; Herrmann, R.; Anderson, L.V.; Bashir, R.; Burgunder, J.M.; et al. Mutations in the fukutin-related protein gene (FKRP) identify limb girdle muscular dystrophy 2I as a milder allelic variant of congenital muscular dystrophy MDC1C. Hum. Mol. Genet. 2001, 10, 2851–2859. [Google Scholar] [CrossRef]
- Han, R.; Kanagawa, M.; Yoshida-Moriguchi, T.; Rader, E.P.; Ng, R.A.; Michele, D.E.; Muirhead, D.E.; Kunz, S.; Moore, S.A.; Iannaccone, S.T.; et al. Basal lamina strengthens cell membrane integrity via the laminin G domain-binding motif of alpha-dystroglycan. Proc. Natl. Acad. Sci. USA 2009, 106, 12573–12579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanagawa, M.; Toda, T. Muscular dystrophy with ribitol-phosphate deficiency: A novel post-translational mechanism in dystroglycanopathy. J. Neuromuscul. Dis. 2017, 4, 259–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godfrey, C.; Foley, A.R.; Clement, E.; Muntoni, F. Dystroglycanopathies: Coming into focus. Curr. Opin. Genet. Dev. 2011, 21, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, C.; Clement, E.; Mein, R.; Brockington, M.; Smith, J.; Talim, B.; Straub, V.; Robb, S.; Quinlivan, R.; Feng, L.; et al. Refining genotype phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan. Brain 2007, 130, 2725–2735. [Google Scholar] [CrossRef] [Green Version]
- Clement, E.; Mercuri, E.; Godfrey, C.; Smith, J.; Robb, S.; Kinali, M.; Straub, V.; Bushby, K.; Manzur, A.; Talim, B.; et al. Brain involvement in muscular dystrophies with defective dystroglycan glycosylation. Ann. Neurol. 2008, 64, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Manya, H.; Chiba, A.; Yoshida, A.; Wang, X.; Chiba, Y.; Jigami, Y.; Margolis, R.U.; Endo, T. Demonstration of mammalian protein O-mannosyltransferase activity: Coexpression of POMT1 and POMT2 required for enzymatic activity. Proc. Natl. Acad. Sci. USA 2004, 101, 500–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, A.; Kobayashi, K.; Manya, H.; Taniguchi, K.; Kano, H.; Mizuno, M.; Inazu, T.; Mitsuhashi, H.; Takahashi, S.; Takeuchi, M.; et al. Muscular dystrophy and neuronal migration disorder caused by mutations in a glycosyltransferase, POMGnT1. Dev. Cell 2001, 1, 717–724. [Google Scholar] [CrossRef] [Green Version]
- Yoshida-Moriguchi, T.; Willer, T.; Anderson, M.E.; Venzke, D.; Whyte, T.; Muntoni, F.; Lee, H.; Nelson, S.F.; Yu, L.; Campbell, K.P. SGK196 is a glycosylation-specific O-mannose kinase required for dystroglycan function. Science 2013, 341, 896–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanagawa, M.; Kobayashi, K.; Tajiri, M.; Manya, H.; Kuga, A.; Yamaguchi, Y.; Akasaka-Manya, K.; Furukawa, J.I.; Mizuno, M.; Kawakami, H.; et al. Identification of a post-translational modification with ribitol-phosphate and its defect in muscular dystrophy. Cell Rep. 2016, 14, 2209–2223. [Google Scholar] [CrossRef] [Green Version]
- Riemersma, M.; Froese, D.S.; van Tol, W.; Engelke, U.F.; Kopec, J.; van Scherpenzeel, M.; Ashikov, A.; Krojer, T.; von Delft, F.; Tessari, M.; et al. Human ISPD is a cytidyltransferase required for dystroglycan O-mannosylation. Chem. Biol. 2015, 22, 1643–1652. [Google Scholar] [CrossRef]
- Gerin, I.; Ury, B.; Breloy, I.; Bouchet-Seraphin, C.; Bolsée, J.; Halbout, M.; Graff, J.; Vertommen, D.; Muccioli, G.G.; Seta, N.; et al. ISPD produces CDP-ribitol used by FKTN and FKRP to transfer ribitol phosphate onto alpha-dystroglycan. Nat. Commun. 2016, 7, 11534. [Google Scholar] [CrossRef]
- Manya, H.; Yamaguchi, Y.; Kanagawa, M.; Kobayashi, K.; Tajiri, M.; Akasaka-Manya, K.; Kawakami, H.; Mizuno, M.; Wada, Y.; Toda, T.; et al. The muscular dystrophy gene TMEM5 encodes a ribitol beta1,4-xylosyltransferase required for the functional glycosylation of dystroglycan. J. Biol. Chem. 2016, 291, 24618–24627. [Google Scholar] [CrossRef] [Green Version]
- Willer, T.; Inamori, K.; Venzke, D.; Harvey, C.; Morgensen, G.; Hara, Y.; Beltrán Valero de Bernabé, D.; Yu, L.; Wright, K.M.; Campbell, K.P. The glucuronyltransferase B4GAT1 is required for initiation of LARGE-mediated alpha-dystroglycan functional glycosylation. eLife 2014, 3, e03941. [Google Scholar] [CrossRef]
- Inamori, K.; Yoshida-Moriguchi, T.; Hara, Y.; Anderson, M.E.; Yu, L.; Campbell, K.P. Dystroglycan function requires xylosyl- and glucuronyltransferase activities of LARGE. Science 2012, 335, 93–96. [Google Scholar] [CrossRef] [Green Version]
- Goddeeris, M.M.; Wu, B.; Venzke, D.; Yoshida-Moriguchi, T.; Saito, F.; Matsumura, K.; Moore, S.A.; Campbell, K.P. LARGE glycans on dystroglycan function as a tunable matrix scaffold to prevent dystrophy. Nature 2013, 503, 136–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briggs, D.C.; Yoshida-Moriguchi, T.; Zheng, T.; Venzke, D.; Anderson, M.E.; Strazzulli, A.; Moracci, M.; Yu, L.; Hohenester, E.; Campbell, K.P. Structural basis of laminin binding to the LARGE glycans on dystroglycan. Nat. Chem. Biol. 2016, 12, 810–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuwabara, N.; Manya, H.; Yamada, T.; Tateno, H.; Kanagawa, M.; Kobayashi, K.; Akasaka-Manya, K.; Hirose, Y.; Mizuno, M.; Ikeguchi, M.; et al. Carbohydrate-binding domain of the POMGnT1 stem region modulates O-mannosylation sites of α-dystroglycan. Proc. Natl. Acad. Sci. USA 2016, 113, 9280–9285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walimbe, A.S.; Okuma, H.; Joseph, S.; Yang, T.; Yonekawa, T.; Hord, J.M.; Venzke, D.; Anderson, M.E.; Torelli, S.; Manzur, A.; et al. POMK regulates dystroglycan function via LARGE1-mediated elongation of matriglycan. eLife 2020, 9, e61388. [Google Scholar] [CrossRef] [PubMed]
- Kuwabara, N.; Imae, R.; Manya, H.; Tanaka, T.; Mizuno, M.; Tsumoto, H.; Kanagawa, M.; Kobayashi, K.; Toda, T.; Senda, T.; et al. Crystal structures of fukutin-related protein (FKRP), a ribitol-phosphate transferase related to muscular dystrophy. Nat. Commun. 2020, 11, 303. [Google Scholar] [CrossRef]
- Van Tol, W.; Ashikov, A.; Korsch, E.; Abu Bakar, N.; Willemsen, M.A.; Thiel, C.; Lefeber, D.J. A mutation in mannose-phosphate-dolichol utilization defect 1 reveals clinical symptoms of congenital disorders of glycosylation type I and dystroglycanopathy. JIMD Rep. 2019, 50, 31–39. [Google Scholar] [CrossRef]
- Larson, A.A.; Baker, P.R., 2nd; Milev, M.P.; Press, C.A.; Sokol, R.J.; Cox, M.O.; Lekostaj, J.K.; Stence, A.A.; Bossler, A.D.; Mueller, J.M.; et al. TRAPPC11 and GOSR2 mutations associate with hypoglycosylation of alpha-dystroglycan and muscular dystrophy. Skelet. Muscle 2018, 8, 17. [Google Scholar] [CrossRef]
- Grewal, P.K.; Holzfeind, P.J.; Bittner, R.E.; Hewitt, J.E. Mutant glycosyltransferase and altered glycosylation of alpha-dystroglycan in the myodystrophy mouse. Nat. Genet. 2001, 28, 151–154. [Google Scholar] [CrossRef]
- Moore, S.A.; Saito, F.; Chen, J.; Michele, D.E.; Henry, M.D.; Messing, A.; Cohn, R.D.; Ross-Barta, S.E.; Westra, S.; Williamson, R.A.; et al. Deletion of brain dystroglycan recapitulates aspects of congenital muscular dystrophy. Nature 2002, 418, 422–425. [Google Scholar] [CrossRef]
- Levedakou, E.N.; Chen, X.J.; Soliven, B.; Popko, B. Disruption of the mouse Large gene in the enr and myd mutants results in nerve, muscle, and neuromuscular junction defects. Mol. Cell Neurosci. 2005, 28, 757–769. [Google Scholar] [CrossRef]
- Lee, Y.; Kameya, S.; Cox, G.A.; Hsu, J.; Hicks, W.; Maddatu, T.P.; Smith, R.S.; Naggert, J.K.; Peachey, N.S.; Nishina, P.M. Ocular abnormalities in Large(myd) and Large(vls) mice, spontaneous models for muscle, eye, and brain diseases. Mol. Cell Neurosci. 2005, 30, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Ball, S.L.; Yang, Y.; Mei, P.; Zhang, L.; Shi, H.; Kaminski, H.J.; Lemmon, V.P.; Hu, H. A genetic model for muscle-eye-brain disease in mice lacking protein O-mannose 1,2-N-acetylglucosaminyltransferase (POMGnT1). Mech. Dev. 2006, 123, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Miyagoe-Suzuki, Y.; Masubuchi, N.; Miyamoto, K.; Wada, M.R.; Yuasa, S.; Saito, F.; Matsumura, K.; Kanesaki, H.; Kudo, A.; Manya, H.; et al. Reduced proliferative activity of primary POMGnT1-null myoblasts in vitro. Mech. Dev. 2009, 126, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Yagi, H.; Nakagawa, N.; Saito, T.; Kiyonari, H.; Abe, T.; Toda, T.; Wu, S.W.; Khoo, K.H.; Oka, S.; Kato, K. AGO61-dependent GlcNAc modification primes the formation of functional glycans on alpha-dystroglycan. Sci. Rep. 2013, 3, 3288. [Google Scholar] [CrossRef] [Green Version]
- Willer, T.; Prados, B.; Falcón-Pérez, J.M.; Renner-Müller, I.; Przemeck, G.K.; Lommel, M.; Coloma, A.; Valero, M.C.; de Angelis, M.H.; Tanner, W.; et al. Targeted disruption of the Walker-Warburg syndrome gene Pomt1 in mouse results in embryonic lethality. Proc. Natl. Acad. Sci. USA 2004, 101, 14126–14131. [Google Scholar] [CrossRef] [Green Version]
- Kurahashi, H.; Taniguchi, M.; Meno, C.; Taniguchi, Y.; Takeda, S.; Horie, M.; Otani, H.; Toda, T. Basement membrane fragility underlies embryonic lethality in fukutin-null mice. Neurobiol. Dis. 2005, 19, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Henry, M.D.; Campbell, K.P. A role for dystroglycan in basement membrane assembly. Cell 1998, 95, 859–870. [Google Scholar] [CrossRef] [Green Version]
- Beedle, A.M.; Turner, A.J.; Saito, Y.; Lueck, J.D.; Foltz, S.J.; Fortunato, M.J.; Nienaber, P.M.; Campbell, K.P. Mouse fukutin deletion impairs dystroglycan processing and recapitulates muscular dystrophy. J. Clin. Investig. 2012, 122, 3330–3342. [Google Scholar] [CrossRef] [Green Version]
- Kanagawa, M.; Yu, C.C.; Ito, C.; Fukada, S.; Hozoji-Inada, M.; Chiyo, T.; Kuga, A.; Matsuo, M.; Sato, K.; Yamaguchi, M.; et al. Impaired viability of muscle precursor cells in muscular dystrophy with glycosylation defects and amelioration of its severe phenotype by limited gene expression. Hum. Mol. Genet. 2013, 22, 3003–3015. [Google Scholar] [CrossRef] [Green Version]
- Sudo, A.; Kanagawa, M.; Kondo, M.; Ito, C.; Kobayashi, K.; Endo, M.; Minami, Y.; Aiba, A.; Toda, T. Temporal requirement of dystroglycan glycosylation during brain development and rescue of severe cortical dysplasia via gene delivery in the fetal stage. Hum. Mol. Genet. 2018, 27, 1174–1185. [Google Scholar] [CrossRef] [Green Version]
- Ujihara, Y.; Kanagawa, M.; Mohri, S.; Takatsu, S.; Kobayashi, K.; Toda, T.; Naruse, K.; Katanosaka, Y. Elimination of fukutin reveals cellular and molecular pathomechanisms in muscular dystrophy-associated heart failure. Nat. Commun. 2019, 10, 5754. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Li, J.; Gagen, C.S.; Gray, N.W.; Zhang, Z.; Qi, Y.; Zhang, P. Conditional knockout of protein O-mannosyltransferase 2 reveals tissue-specific roles of O-mannosyl glycosylation in brain development. J. Comp. Neurol. 2011, 519, 1320–1337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, K.; Nakahori, Y.; Miyake, M.; Matsumura, K.; Kondo-Iida, E.; Nomura, Y.; Segawa, M.; Yoshioka, M.; Saito, K.; Osawa, M.; et al. An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature 1998, 394, 388–392. [Google Scholar] [CrossRef] [PubMed]
- Kanagawa, M.; Nishimoto, A.; Chiyonobu, T.; Takeda, S.; Miyagoe-Suzuki, Y.; Wang, F.; Fujikake, N.; Taniguchi, M.; Lu, Z.; Tachikawa, M.; et al. Residual laminin-binding activity and enhanced dystroglycan glycosylation by LARGE in novel model mice to dystroglycanopathy. Hum. Mol. Genet. 2009, 18, 621–631. [Google Scholar] [CrossRef]
- Ackroyd, M.R.; Skordis, L.; Kaluarachchi, M.; Godwin, J.; Prior, S.; Fidanboylu, M.; Piercy, R.J.; Muntoni, F.; Brown, S.C. Reduced expression of fukutin related protein in mice results in a model for fukutin related protein associated muscular dystrophies. Brain 2009, 132, 439–451. [Google Scholar] [CrossRef]
- Chan, Y.M.; Keramaris-Vrantsis, E.; Lidov, H.G.; Norton, J.H.; Zinchenko, N.; Gruber, H.E.; Thresher, R.; Blake, D.J.; Ashar, J.; Rosenfeld, J.; et al. Fukutin-related protein is essential for mouse muscle, brain and eye development and mutation recapitulates the wide clinical spectrums of dystroglycanopathies. Hum. Mol. Genet. 2010, 19, 3995–4006. [Google Scholar] [CrossRef] [Green Version]
- Blaeser, A.; Keramaris, E.; Chan, Y.M.; Sparks, S.; Cowley, D.; Xiao, X.; Lu, Q.L. Mouse models of fukutin-related protein mutations show a wide range of disease phenotypes. Hum. Genet. 2013, 132, 923–934. [Google Scholar] [CrossRef]
- Ross, J.; Benn, A.; Jonuschies, J.; Boldrin, L.; Muntoni, F.; Hewitt, J.E.; Brown, S.C.; Morgan, J.E. Defects in glycosylation impair satellite stem cell function and niche composition in the muscles of the dystrophic Large(myd) mouse. Stem Cells 2012, 30, 2330–2341. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Hopkinson, M.; Kavishwar, M.; Fernandez-Fuente, M.; Brown, S.C. Prenatal muscle development in a mouse model for the secondary dystroglycanopathies. Skelet. Muscle 2016, 6, 3. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, M.; Kurahashi, H.; Noguchi, S.; Fukudome, T.; Okinaga, T.; Tsukahara, T.; Tajima, Y.; Ozono, K.; Nishino, I.; Nonaka, I.; et al. Aberrant neuromuscular junctions and delayed terminal muscle fiber maturation in alpha-dystroglycanopathies. Hum. Mol. Genet. 2006, 15, 1279–1289. [Google Scholar] [CrossRef] [Green Version]
- Herbst, R.; Iskratsch, T.; Unger, E.; Bittner, R.E. Aberrant development of neuromuscular junctions in glycosylation-defective Large(myd) mice. Neuromuscul. Disord. 2009, 19, 366–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foltz, S.J.; Modi, J.N.; Melick, G.A.; Abousaud, M.I.; Luan, J.; Fortunato, M.J.; Beedle, A.M. Abnormal skeletal muscle regeneration plus mild alterations in mature fiber type specification in Fktn-deficient dystroglycanopathy muscular dystrophy mice. PLoS ONE 2016, 11, e0147049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortiz-Cordero, C.; Bincoletto, C.; Dhoke, N.R.; Selvaraj, S.; Magli, A.; Zhou, H.; Kim, D.H.; Bang, A.G.; Perlingeiro, R.C.R. Defective autophagy and increased apoptosis contribute toward the pathogenesis of FKRP-associated muscular dystrophies. Stem Cell Rep. 2021, 16, 2752–2767. [Google Scholar] [CrossRef]
- Vannoy, C.H.; Leroy, V.; Broniowska, K.; Lu, Q.L. Metabolomics analysis of skeletal muscles from FKRP-deficient mice indicates improvement after gene replacement therapy. Sci. Rep. 2019, 9, 10070. [Google Scholar] [CrossRef]
- Aksu-Menges, E.; Akkaya-Ulum, Y.Z.; Dayangac-Erden, D.; Balci-Peynircioglu, B.; Yuzbasioglu, A.; Topaloglu, H.; Talim, B.; Balci-Hayta, B. The common miRNA signatures associated with mitochondrial dysfunction in different muscular dystrophies. Am. J. Pathol. 2020, 190, 2136–2145. [Google Scholar] [CrossRef]
- Wood, A.J.; Lin, C.H.; Li, M.; Nishtala, K.; Alaei, S.; Rossello, F.; Sonntag, C.; Hersey, L.; Miles, L.B.; Krisp, C.; et al. FKRP-dependent glycosylation of fibronectin regulates muscle pathology in muscular dystrophy. Nat. Commun. 2021, 12, 2951. [Google Scholar] [CrossRef]
- Satz, J.S.; Ostendorf, A.P.; Hou, S.; Turner, A.; Kusano, H.; Lee, J.C.; Turk, R.; Nguyen, H.; Ross-Barta, S.E.; Westra, S.; et al. Distinct functions of glial and neuronal dystroglycan in the developing and adult mouse brain. J. Neurosci. 2010, 30, 14560–14572. [Google Scholar] [CrossRef] [Green Version]
- Myshrall, T.D.; Moore, S.A.; Ostendorf, A.P.; Satz, J.S.; Kowalczyk, T.; Nguyen, H.; Daza, R.A.; Lau, C.; Campbell, K.P.; Hevner, R.F. Dystroglycan on radial glia end feet is required for pial basement membrane integrity and columnar organization of the developing cerebral cortex. J. Neuropathol. Exp. Neurol. 2012, 71, 1047–1063. [Google Scholar] [CrossRef] [Green Version]
- Nakano, I.; Funahashi, M.; Takada, K.; Toda, T. Are breaches in the glia limitans the primary cause of the micropolygyria in Fukuyama-type congenital muscular dystrophy (FCMD)? Pathological study of the cerebral cortex of an FCMD fetus. Acta Neuropathol. 1996, 91, 313–321. [Google Scholar] [CrossRef]
- Hu, H.; Yang, Y.; Eade, A.; Xiong, Y.; Qi, Y. Breaches of the pial basement membrane and disappearance of the glia limitans during development underlie the cortical lamination defect in the mouse model of muscle-eye-brain disease. J. Comp. Neurol. 2007, 501, 168–183. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, P.; Yang, Y.; Xiong, Y.; Qi, Y.; Hu, H. Differentiation and developmental origin of cerebellar granule neuron ectopia in protein O-mannose UDP-N-acetylglucosaminyl transferase 1 knockout mice. Neuroscience 2008, 152, 391–406. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, N.; Yagi, H.; Kato, K.; Takematsu, H.; Oka, S. Ectopic clustering of Cajal-Retzius and subplate cells is an initial pathological feature in Pomgnt2-knockout mice, a model of dystroglycanopathy. Sci. Rep. 2015, 5, 11163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniguchi-Ikeda, M.; Koyanagi-Aoi, M.; Maruyama, T.; Takaori, T.; Hosoya, A.; Tezuka, H.; Nagase, S.; Ishihara, T.; Kadoshima, T.; Muguruma, K.; et al. Restoration of the defect in radial glial fiber migration and cortical plate organization in a brain organoid model of Fukuyama muscular dystrophy. iScience 2021, 24, 103140. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yu, M.; Feng, G.; Hu, H.; Li, X. Breaches of the pial basement membrane are associated with defective dentate gyrus development in mouse models of congenital muscular dystrophies. Neurosci. Lett. 2011, 505, 19–24. [Google Scholar] [CrossRef] [Green Version]
- Nakanishi, T.; Sakauchi, M.; Kaneda, Y.; Tomimatsu, H.; Saito, K.; Nakazawa, M.; Osawa, M. Cardiac involvement in Fukuyama-type congenital muscular dystrophy. Pediatrics 2006, 117, e1187–e1192. [Google Scholar] [CrossRef]
- Murakami, T.; Hayashi, Y.K.; Noguchi, S.; Ogawa, M.; Nonaka, I.; Tanabe, Y.; Ogino, M.; Takada, F.; Eriguchi, M.; Kotooka, N.; et al. Fukutin gene mutations cause dilated cardiomyopathy with minimal muscle weakness. Ann. Neurol. 2006, 60, 597–602. [Google Scholar] [CrossRef] [Green Version]
- Kabaeva, Z.; Meekhof, K.E.; Michele, D.E. Sarcolemma instability during mechanical activity in Largemyd cardiac myocytes with loss of dystroglycan extracellular matrix receptor function. Hum. Mol. Genet. 2011, 20, 3346–3355. [Google Scholar] [CrossRef] [Green Version]
- Blaeser, A.; Awano, H.; Wu, B.; Lu, Q.L. Progressive dystrophic pathology in diaphragm and impairment of cardiac function in FKRP P448L mutant mice. PLoS ONE 2016, 11, e0164187. [Google Scholar] [CrossRef]
- Qiao, C.; Wang, C.H.; Zhao, C.; Lu, P.; Awano, H.; Xiao, B.; Li, J.; Yuan, Z.; Dai, Y.; Martin, C.B.; et al. Muscle and heart function restoration in a limb girdle muscular dystrophy 2I (LGMD2I) mouse model by systemic FKRP gene delivery. Mol. Ther. 2014, 22, 1890–1899. [Google Scholar] [CrossRef] [Green Version]
- Gicquel, E.; Maizonnier, N.; Foltz, S.J.; Martin, W.J.; Bourg, N.; Svinartchouk, F.; Charton, K.; Beedle, A.M.; Richard, I. AAV-mediated transfer of FKRP shows therapeutic efficacy in a murine model but requires control of gene expression. Hum. Mol. Genet. 2017, 26, 1952–1965. [Google Scholar] [CrossRef]
- Barresi, R.; Michele, D.E.; Kanagawa, M.; Harper, H.A.; Dovico, S.A.; Satz, J.S.; Moore, S.A.; Zhang, W.; Schachter, H.; Dumanski, J.P.; et al. LARGE can functionally bypass alpha-dystroglycan glycosylation defects in distinct congenital muscular dystrophies. Nat. Med. 2004, 10, 696–703. [Google Scholar] [CrossRef] [PubMed]
- Dhoke, N.R.; Kim, H.; Selvaraj, S.; Azzag, K.; Zhou, H.; Oliveira, N.A.J.; Tungtur, S.; Ortiz-Cordero, C.; Kiley, J.; Lu, Q.L.; et al. A universal gene correction approach for FKRP-associated dystroglycanopathies to enable autologous cell therapy. Cell Rep. 2021, 36, 109360. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.J.; Xu, R.; Martin, P.T. B4GALNT2 (GALGT2) gene therapy reduces skeletal muscle pathology in the FKRP P448L mouse model of limb girdle muscular dystrophy 2I. Am. J. Pathol. 2016, 186, 2429–2448. [Google Scholar] [CrossRef] [Green Version]
- Vannoy, C.H.; Xu, L.; Keramaris, E.; Lu, P.; Xiao, X.; Lu, Q.L. Adeno-associated virus-mediated overexpression of LARGE rescues alpha-dystroglycan function in dystrophic mice with mutations in the fukutin-related protein. Hum. Gene Ther. Methods 2014, 25, 187–196. [Google Scholar] [CrossRef] [Green Version]
- Saito, F.; Kanagawa, M.; Ikeda, M.; Hagiwara, H.; Masaki, T.; Ohkuma, H.; Katanosaka, Y.; Shimizu, T.; Sonoo, M.; Toda, T.; et al. Overexpression of LARGE suppresses muscle regeneration via down-regulation of insulin-like growth factor 1 and aggravates muscular dystrophy in mice. Hum. Mol. Genet. 2014, 23, 4543–4558. [Google Scholar] [CrossRef] [Green Version]
- Whitmore, C.; Fernandez-Fuente, M.; Booler, H.; Parr, C.; Kavishwar, M.; Ashraf, A.; Lacey, E.; Kim, J.; Terry, R.; Ackroyd, M.R.; et al. The transgenic expression of LARGE exacerbates the muscle phenotype of dystroglycanopathy mice. Hum. Mol. Genet. 2014, 23, 1842–1855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, F.; Li, Z.F.; Hu, W.; Wu, X. Small molecules enhance functional O-mannosylation of Alpha-dystroglycan. Bioorg. Med. Chem. 2015, 23, 7661–7670. [Google Scholar] [CrossRef]
- Kim, J.; Lana, B.; Torelli, S.; Ryan, D.; Catapano, F.; Ala, P.; Luft, C.; Stevens, E.; Konstantinidis, E.; Louzada, S.; et al. A new patient-derived iPSC model for dystroglycanopathies validates a compound that increases glycosylation of alpha-dystroglycan. EMBO Rep. 2019, 20, e47967. [Google Scholar] [CrossRef]
- Gumlaw, N.; Sevigny, L.M.; Zhao, H.; Luo, Z.; Bangari, D.S.; Masterjohn, E.; Chen, Y.; McDonald, B.; Magnay, M.; Travaline, T.; et al. biAb mediated restoration of the linkage between dystroglycan and laminin-211 as a therapeutic approach for alpha-dystroglycanopathies. Mol. Ther. 2020, 28, 664–676. [Google Scholar] [CrossRef] [Green Version]
- Foltz, S.J.; Luan, J.; Call, J.A.; Patel, A.; Peissig, K.B.; Fortunato, M.J.; Beedle, A.M. Four-week rapamycin treatment improves muscular dystrophy in a fukutin-deficient mouse model of dystroglycanopathy. Skelet. Muscle 2016, 6, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, B.; Shah, S.N.; Lu, P.; Bollinger, L.E.; Blaeser, A.; Sparks, S.; Harper, A.D.; Lu, Q.L. Long-term treatment of tamoxifen and raloxifene alleviates dystrophic phenotype and enhances muscle functions of FKRP dystroglycanopathy. Am. J. Pathol. 2018, 188, 1069–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, T.L.; Vissing, J.; Krag, T.O. Antimyostatin treatment in health and disease: The story of great expectations and limited success. Cells 2021, 10, 533. [Google Scholar] [CrossRef]
- Leung, D.G.; Bocchieri, A.E.; Ahlawat, S.; Jacobs, M.A.; Parekh, V.S.; Braverman, V.; Summerton, K.; Mansour, J.; Stinson, N.; Bibat, G.; et al. A phase Ib/IIa, open-label, multiple ascending-dose trial of domagrozumab in fukutin-related protein limb-girdle muscular dystrophy. Muscle Nerve 2021, 64, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Serafini, P.R.; Feyder, M.J.; Hightower, R.M.; Garcia-Perez, D.; Vieira, N.M.; Lek, A.; Gibbs, D.E.; Moukha-Chafiq, O.; Augelli-Szafran, C.E.; Kawahara, G.; et al. A limb-girdle muscular dystrophy 2I model of muscular dystrophy identifies corrective drug compounds for dystroglycanopathies. JCI Insight 2018, 3, e120493. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.; Shah, S.N.; Lu, P.; Richardson, S.M.; Bollinger, L.E.; Blaeser, A.; Madden, K.L.; Sun, Y.; Luckie, T.M.; Cox, M.D.; et al. Glucocorticoid steroid and alendronate treatment alleviates dystrophic phenotype with enhanced functional glycosylation of alpha-dystroglycan in mouse model of limb-girdle muscular dystrophy with FKRPP448L mutation. Am. J. Pathol. 2016, 186, 1635–1648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniguchi-Ikeda, M.; Kobayashi, K.; Kanagawa, M.; Yu, C.C.; Mori, K.; Oda, T.; Kuga, A.; Kurahashi, H.; Akman, H.O.; DiMauro, S.; et al. Pathogenic exon-trapping by SVA retrotransposon and rescue in Fukuyama muscular dystrophy. Nature 2011, 478, 127–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Tol, W.; van Scherpenzeel, M.; Alsady, M.; Riemersma, M.; Hermans, E.; Kragt, E.; Tasca, G.; Kamsteeg, E.J.; Pennings, M.; van Beusekom, E.; et al. Cytidine diphosphate-ribitol analysis for diagnostics and treatment monitoring of cytidine diphosphate-l-ribitol pyrophosphorylase A muscular dystrophy. Clin. Chem. 2019, 65, 1295–1306. [Google Scholar] [CrossRef]
- Cataldi, M.P.; Lu, P.; Blaeser, A.; Lu, Q.L. Ribitol restores functionally glycosylated α-dystroglycan and improves muscle function in dystrophic FKRP-mutant mice. Nat. Commun. 2018, 9, 3448. [Google Scholar] [CrossRef]
- Nickolls, A.R.; Lee, M.M.; Zukosky, K.; Mallon, B.S.; Bönnemann, C.G. Human embryoid bodies as a 3D tissue model of the extracellular matrix and α-dystroglycanopathies. Dis. Model Mech. 2020, 13, dmm042986. [Google Scholar] [CrossRef]
- Ortiz-Cordero, C.; Magli, A.; Dhoke, N.R.; Kuebler, T.; Selvaraj, S.; Oliveira, N.A.; Zhou, H.; Sham, Y.Y.; Bang, A.G.; Perlingeiro, R.C. NAD+ enhances ribitol and ribose rescue of alpha-dystroglycan functional glycosylation in human FKRP-mutant myotubes. eLife 2021, 10, e65443. [Google Scholar] [CrossRef] [PubMed]
- Bailey, E.C.; Alrowaished, S.S.; Kilroy, E.A.; Crooks, E.S.; Drinkert, D.M.; Karunasiri, C.M.; Belanger, J.J.; Khalil, A.; Kelley, J.B.; Henry, C.A. NAD+ improves neuromuscular development in a zebrafish model of FKRP-associated dystroglycanopathy. Skelet. Muscle 2019, 9, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| DGpathy Genes | Gene Functions |
|---|---|
| POMT1 | Protein O-mannosyl-transferase as a part of the POMT1/2 complex |
| POMT2 | Protein O-mannosyl-transferase as a part of the POMT1/2 complex |
| POMGNT1 | Protein O-mannose β1,2-N-acetylglucosaminyltransferase; Core M1 synthesis |
| POMGNT2 | Protein O-mannose β1,4-N-acetylglucosaminyltransferase; Core M3 synthesis |
| B3GALNT2 | β1,3-N-acetylgalactosaminyltransferase; Core M3 synthesis |
| POMK | Protein O-mannose kinase; phosphorylation of Core M3 |
| FKTN | Ribitol phosphate transferase; tandem ribitol synthesis |
| FKRP | Ribitol phosphate transferase; tandem ribitol synthesis |
| ISPD/CRPPA | CDP-ribitol pyrophosphorylase; synthesis of CDP-ribitol (donor substrate of FKTN/FKRP) |
| TMEM5/ RXYLT1 | Ribitol-5-phosphate xylosyltransferase; synthesis of linker structure between tandem ribitol and matriglycan |
| B4GAT1 | β1,4-Glucuronyltransferase; synthesis of linker structure between tandem ribitol and matriglycan |
| LARGE | Xylosyl- and glucuronyltransferase; matriglycan synthesis |
| DAG1 | Dystroglycan |
| GMPPB | GDP-mannose pyrophosphorylase required for the formation of GDP-Man; Dolichol-phosphate-mannose synthesis |
| DPM1 | Dolichol-phosphate-mannose synthase; Dolichol-phosphate-mannose synthesis |
| DPM2 | Dolichol-phosphate-mannose synthase; Dolichol-phosphate-mannose synthesis |
| DPM3 | Dolichol-phosphate-mannose synthase; Dolichol-phosphate-mannose synthesis |
| DOLK | Dolichol kinase required for the formation of dolichol-phosphate; Dolichol-phosphate-mannose synthesis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kanagawa, M. Dystroglycanopathy: From Elucidation of Molecular and Pathological Mechanisms to Development of Treatment Methods. Int. J. Mol. Sci. 2021, 22, 13162. https://doi.org/10.3390/ijms222313162
Kanagawa M. Dystroglycanopathy: From Elucidation of Molecular and Pathological Mechanisms to Development of Treatment Methods. International Journal of Molecular Sciences. 2021; 22(23):13162. https://doi.org/10.3390/ijms222313162
Chicago/Turabian StyleKanagawa, Motoi. 2021. "Dystroglycanopathy: From Elucidation of Molecular and Pathological Mechanisms to Development of Treatment Methods" International Journal of Molecular Sciences 22, no. 23: 13162. https://doi.org/10.3390/ijms222313162
APA StyleKanagawa, M. (2021). Dystroglycanopathy: From Elucidation of Molecular and Pathological Mechanisms to Development of Treatment Methods. International Journal of Molecular Sciences, 22(23), 13162. https://doi.org/10.3390/ijms222313162