
Therapeutic Potential of TAAR1 Agonists in Schizophrenia: Evidence from Preclinical Models and Clinical Studies


 and
and
Abstract
:1. Introduction
1.1. Schizophrenia: A Severe Psychiatric Disease with Significant Unmet Needs
1.2. Current Pharmacologic Treatments
2. TAAR1 As a Novel Therapeutic Target for the Treatment of Schizophrenia
2.1. Trace Amines and TAAR1
2.2. Development of Synthetic TAAR1 Ligands

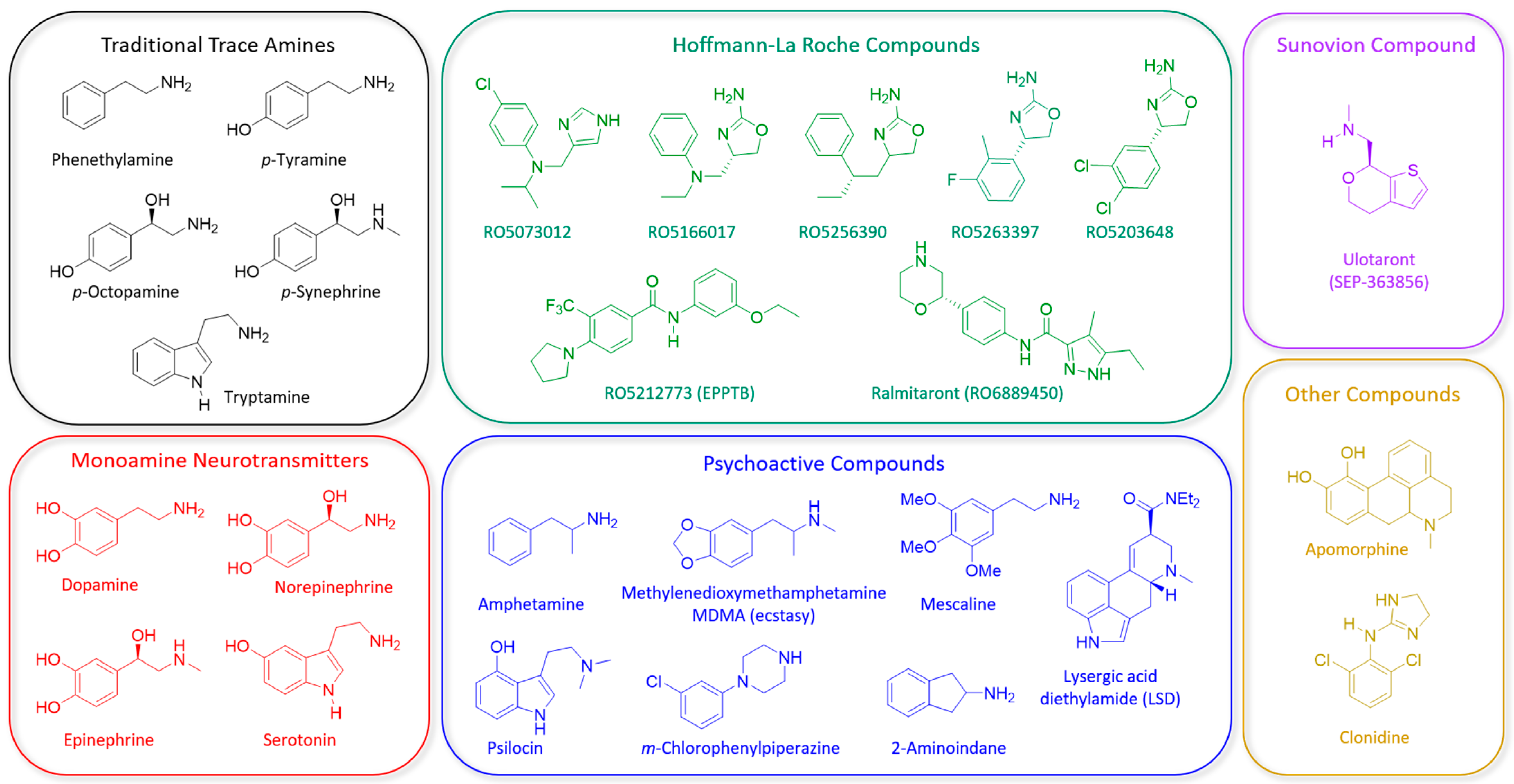{kind=link}
{kind=link}
| Compound (Company) | Human Receptor Profile | Behavioral Effects in Preclinical Models &Assays Relevant to Positive, Negative and Cognitive Symptoms of Schizophrenia | Clinical Trials in Schizophrenia Patients |
|---|---|---|---|
| RO5166017 F. Hoffmann-La Roche | TAAR1 Full Agonist [86] | N/A | |
| RO5256390 F. Hoffmann-La Roche | TAAR1 Full Agonist [74] |
| N/A |
| RO5203648 F. Hoffmann-La Roche | TAAR1 Partial Agonist [111] |
| N/A |
| RO5263397 F. Hoffmann-La Roche | TAAR1 Partial Agonist [74] |
| N/A |
| RO5073012 F. Hoffmann-La Roche | TAAR1 Partial Agonist [121] |
| N/A |
| Ralmitaront (RO6889450) F. Hoffmann-La Roche | TAAR1 Partial Agonist | N/A | |
| Ulotaront (SEP-363856) Sunovion Pharmaceuticals | TAAR1 Full Agonist and 5-HT1A Partial Agonist [106] |
|
|
3. Preclinical Evidence for TAAR1 as a Therapeutic Target for Schizophrenia
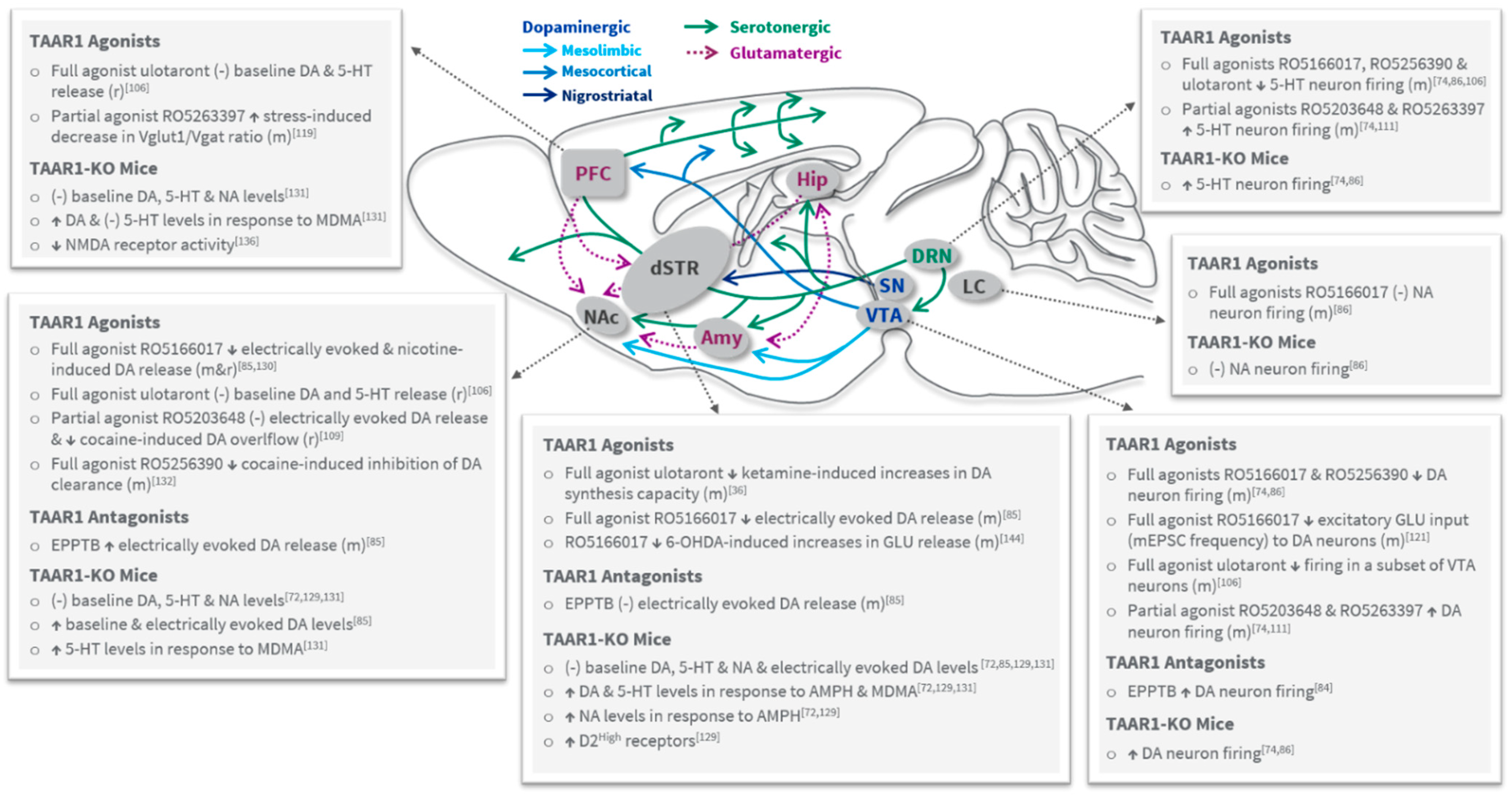
3.1. Role of TAAR1 in Psychosis and Dopaminergic Tone
3.2. Role of TAAR1 in Cognition, Negative Symptoms, Mood and Anxiety
3.3. TAAR1 Effects on Serotonergic, Noradrenergic and Glutamatergic Systems
4. Additional Considerations for TAAR1 Agonists as Therapeutic Agents for Schizophrenia
5. Clinical Evidence for TAAR1 Agonists for the Treatment of Schizophrenia
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Disease, D.G.B.; Injury, I.; Prevalence, C. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1789–1858. [Google Scholar]
- Keeley, J.W.; Gaebel, W. Symptom rating scales for schizophrenia and other primary psychotic disorders in ICD-11. Epidemiology Psychiatr. Sci. 2018, 27, 219–224. [Google Scholar] [CrossRef] [Green Version]
- Biedermann, F.; Fleischhacker, W.W. Psychotic disorders in DSM-5 and ICD-11. CNS Spectr. 2016, 21, 349–354. [Google Scholar] [CrossRef]
- Owen, J.M.; Sawa, A.; Mortensen, P.B. Schizophrenia. Lancet 2016, 388, 86–97. [Google Scholar] [CrossRef] [Green Version]
- Tsai, J.; Rosenheck, R.A. Psychiatric comorbidity among adults with schizophrenia: A latent class analysis. Psychiatry Res. 2013, 210, 16–20. [Google Scholar] [CrossRef] [Green Version]
- Kooyman, I.; Dean, K.; Harvey, S.; Walsh, E. Outcomes of public concern in schizophrenia. Br. J. Psychiatry 2007, 191, s29–s36. [Google Scholar] [CrossRef] [Green Version]
- Marwaha, S.; Johnson, S. Schizophrenia and employment—A review. Soc. Psychiatry Psychiatr. Epidemiol. 2004, 39, 337–349. [Google Scholar] [CrossRef]
- Sher, L.; Kahn, R.S. Suicide in Schizophrenia: An Educational Overview. Medicina 2019, 55, 361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chesney, E.; Goodwin, G.M.; Fazel, S. Risks of all-cause and suicide mortality in mental disorders: A meta-review. World Psychiatry 2014, 13, 153–160. [Google Scholar] [CrossRef] [Green Version]
- Laursen, T.M.; Plana-Ripoll, O.; Andersen, P.K.; McGrath, J.J.; Toender, A.; Nordentoft, M.; Canudas-Romo, V.; Erlangsen, A. Cause-specific life years lost among persons diagnosed with schizophrenia: Is it getting better or worse? Schizophr. Res. 2019, 206, 284–290. [Google Scholar] [CrossRef]
- Correll, C.U.; Solmi, M.; Veronese, N.; Bortolato, B.; Rosson, S.; Santonastaso, P.; Thapa-Chhetri, N.; Fornaro, M.; Gallicchio, D.; Collantoni, E.; et al. Prevalence, incidence and mortality from cardiovascular disease in patients with pooled and specific severe mental illness: A large-scale meta-analysis of 3,211,768 patients and 113,383,368 controls. World Psychiatry 2017, 16, 163–180. [Google Scholar] [CrossRef] [Green Version]
- Vancampfort, D.; Stubbs, B.; Mitchell, A.J.; De Hert, M.; Wampers, M.; Ward, P.B.; Rosenbaum, S.; Correll, C.U. Risk of metabolic syndrome and its components in people with schizophrenia and related psychotic disorders, bipolar disorder and major depressive disorder: A systematic review and meta-analysis. World Psychiatry 2015, 14, 339–347. [Google Scholar] [CrossRef]
- Alnæs, D.; Kaufmann, T.; van der Meer, D.; Córdova-Palomera, A.; Rokicki, J.; Moberget, T.; Bettella, F.; Agartz, I.; Barch, D.M.; Bertolino, A.; et al. Brain Heterogeneity in Schizophrenia and Its Association with Polygenic Risk. JAMA Psychiatry 2019, 76, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Ripke, S.; Neale, B.M.; Corvin, A.; Walters, J.T.; Farh, K.H.; Holmans, P.A.; Milanova, V. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014, 511, 421–427. [Google Scholar]
- Fatemi, S.H.; Folsom, T.D. The Neurodevelopmental Hypothesis of Schizophrenia, Revisited. Schizophr. Bull. 2009, 35, 528–548. [Google Scholar] [CrossRef]
- Kirkpatrick, B.; Miller, B.J. Inflammation and Schizophrenia. Schizophr. Bull. 2013, 39, 1174–1179. [Google Scholar] [CrossRef] [Green Version]
- Eaton, W.W.; Byrne, M.; Ewald, H.; Mors, O.; Chen, C.-Y.; Agerbo, E.; Mortensen, P.B. Association of Schizophrenia and Autoimmune Diseases: Linkage of Danish National Registers. Am. J. Psychiatry 2006, 163, 521–528. [Google Scholar] [CrossRef]
- Horvath, S.; Mirnics, K. Immune System Disturbances in Schizophrenia. Biol. Psychiatry 2014, 75, 316–323. [Google Scholar] [CrossRef] [Green Version]
- Sekar, A.; Adolfsson, R.; Bialas, A.R.; De Rivera, H.; Davis, A.; Hammond, T.R.; Kamitaki, N.; Tooley, K.; Presumey, J.; Baum, M.; et al. Schizophrenia risk from complex variation of complement component 4. Nature 2016, 530, 177–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varese, F.; Smeets, F.; Drukker, M.; Lieverse, R.; Lataster, T.; Viechtbauer, W.; Read, J.; van Os, J.; Bentall, R.P. Childhood Adversities Increase the Risk of Psychosis: A Meta-analysis of Patient-Control, Prospective- and Cross-sectional Cohort Studies. Schizophr. Bull. 2012, 38, 661–671. [Google Scholar] [CrossRef]
- Krabbendam, L.; van Os, J. Schizophrenia and urbanicity: A major environmental influence--conditional on genetic risk. Schizophr. Bull. 2005, 31, 795–799. [Google Scholar] [CrossRef]
- Werner, S.; Malaspina, D.; Rabinowitz, J. Socioeconomic Status at Birth Is Associated With Risk of Schizophrenia: Population-Based Multilevel Study. Schizophr. Bull. 2006, 33, 1373–1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaar, S.J.; Natesan, S.; McCutcheon, R.; Howes, O.D. Antipsychotics: Mechanisms underlying clinical response and side-effects and novel treatment approaches based on pathophysiology. Neuropharmacology 2020, 172, 107704. [Google Scholar] [CrossRef]
- Janowsky, D.S.; Risch, C. Amphetamine psychosis and psychotic symptoms. Psychopharmacology 1979, 65, 73–77. [Google Scholar] [CrossRef]
- Howes, O.D.; Kambeitz, J.; Kim, E.; Stahl, D.; Slifstein, M.; Abi-Dargham, A.; Kapur, S. The Nature of Dopamine Dysfunction in Schizophrenia and What This Means for Treatment. Arch. Gen. Psychiatry 2012, 69, 776–786. [Google Scholar] [CrossRef] [Green Version]
- Laruelle, M.; Abi-Dargham, A.; Gil, R.; Kegeles, L.; Innis, R. Increased dopamine transmission in schizophrenia: Relationship to illness phases. Biol. Psychiatry 1999, 46, 56–72. [Google Scholar] [CrossRef]
- Jauhar, S.; Nour, M.; Veronese, M.; Rogdaki, M.; Bonoldi, I.; Azis, M.; Turkheimer, F.; McGuire, P.; Young, A.; Howes, O.D. A Test of the Transdiagnostic Dopamine Hypothesis of Psychosis Using Positron Emission Tomographic Imaging in Bipolar Affective Disorder and Schizophrenia. JAMA Psychiatry 2017, 74, 1206–1213. [Google Scholar] [CrossRef] [Green Version]
- Abi-Dargham, A.; Rodenhiser, J.; Printz, D.; Zea-Ponce, Y.; Gil, R.; Kegeles, L.S.; Weiss, R.; Cooper, T.B.; Mann, J.J.; Van Heertum, R.L.; et al. Increased baseline occupancy of D2 receptors by dopamine in schizophrenia. Proc. Natl. Acad. Sci. USA 2000, 97, 8104–8109. [Google Scholar] [CrossRef] [Green Version]
- Selvaraj, S.; Arnone, D.; Cappai, A.; Howes, O. Alterations in the serotonin system in schizophrenia: A systematic review and meta-analysis of postmortem and molecular imaging studies. Neurosci. Biobehav. Rev. 2014, 45, 233–245. [Google Scholar] [CrossRef] [PubMed]
- McCutcheon, R.; Krystal, J.H.; Howes, O.D. Dopamine and glutamate in schizophrenia: Biology, symptoms and treatment. World Psychiatry 2020, 19, 15–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coyle, J.T. Glutamate and Schizophrenia: Beyond the Dopamine Hypothesis. Cell. Mol. Neurobiol. 2006, 26, 363–382. [Google Scholar] [CrossRef]
- Javitt, D.C.; Zukin, S. Recent advances in the phencyclidine model of schizophrenia. Am. J. Psychiatry 1991, 148, 1301–1308. [Google Scholar] [CrossRef]
- Moghaddam, B.; Krystal, J.H. Capturing the Angel in “Angel Dust”: Twenty Years of Translational Neuroscience Studies of NMDA Receptor Antagonists in Animals and Humans. Schizophr. Bull. 2012, 38, 942–949. [Google Scholar] [CrossRef] [Green Version]
- Eggers, A.E. A serotonin hypothesis of schizophrenia. Med. Hypotheses 2013, 80, 791–794. [Google Scholar] [CrossRef]
- Shah, U.H.; Gaitonde, S.A.; Moreno, J.L.; Glennon, R.A.; Dukat, M.; González-Maeso, J. Revised Pharmacophore Model for 5-HT2A Receptor Antagonists Derived from the Atypical Antipsychotic Agent Risperidone. ACS Chem. Neurosci. 2019, 10, 2318–2331. [Google Scholar] [CrossRef]
- Kokkinou, M.; Irvine, E.E.; Bonsall, D.R.; Natesan, S.; Wells, L.A.; Smith, M.; Glegola, J.; Paul, E.J.; Tossell, K.; Veronese, M.; et al. Reproducing the dopamine pathophysiology of schizophrenia and approaches to ameliorate it: A translational imaging study with ketamine. Mol. Psychiatry 2021, 26, 2562–2576. [Google Scholar] [CrossRef]
- Keepers, G.A.; Fochtmann, L.J.; Anzia, J.M.; Benjamin, S.; Lyness, J.M.; Mojtabai, R.; Servis, M.; Walaszek, A.; Buckley, P.; Lenzenweger, M.F.; et al. The American Psychiatric Association Practice Guideline for the Treatment of Patients with Schizophrenia. Am. J. Psychiatry 2020, 177, 868–872. [Google Scholar] [CrossRef]
- Leucht, S.; Chaimani, A.; Leucht, C.; Huhn, M.; Mavridis, D.; Helfer, B.; Samara, M.; Cipriani, A.; Geddes, J.R.; Salanti, G.; et al. 60 years of placebo-controlled antipsychotic drug trials in acute schizophrenia: Meta-regression of predictors of placebo response. Schizophr. Res. 2018, 201, 315–323. [Google Scholar] [CrossRef] [Green Version]
- Gaebel, W.; Stricker, J.; Riesbeck, M. The long-term antipsychotic treatment of schizophrenia: A selective review of clinical guidelines and clinical case examples. Schizophr. Res. 2020, 225, 4–14. [Google Scholar] [CrossRef]
- Howes, O.; Egerton, A.; Allan, V.; McGuire, P.; Stokes, P.; Kapur, S. Mechanisms Underlying Psychosis and Antipsychotic Treatment Response in Schizophrenia: Insights from PET and SPECT Imaging. Curr. Pharm. Des. 2009, 15, 2550–2559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siafis, S.; Tzachanis, D.; Samara, M.; Papazisis, G. Antipsychotic Drugs: From Receptor-binding Profiles to Metabolic Side Effects. Curr. Neuropharmacol. 2018, 16, 1210–1223. [Google Scholar] [CrossRef] [PubMed]
- Pillinger, T.; McCutcheon, R.; Vano, L.; Mizuno, Y.; Arumuham, A.; Hindley, G.; Beck, K.; Natesan, S.; Efthimiou, O.; Cipriani, A.; et al. Comparative effects of 18 antipsychotics on metabolic function in patients with schizophrenia, predictors of metabolic dysregulation, and association with psychopathology: A systematic review and network meta-analysis. Lancet Psychiatry 2020, 7, 64–77. [Google Scholar] [CrossRef]
- Meyer, J.M. Antipsychotics and Metabolics in the Post-CATIE Era. Neurobiol. Child. 2010, 4, 23–42. [Google Scholar] [CrossRef]
- Henderson, D.C.; Vincenzi, B.; Andrea, N.V.; Ulloa, M.; Copeland, P.M. Pathophysiological mechanisms of increased cardiometabolic risk in people with schizophrenia and other severe mental illnesses. Lancet Psychiatry 2015, 2, 452–464. [Google Scholar] [CrossRef]
- Roerig, L.J.; Steffen, K.J.; Mitchell, J.E. Atypical antipsychotic-induced weight gain: Insights into mechanisms of action. CNS Drugs 2011, 25, 1035–1059. [Google Scholar] [CrossRef]
- Raben, A.T.; Marshe, V.S.; Chintoh, A.; Gorbovskaya, I.; Müller, D.J.; Hahn, M.K. The Complex Relationship between Antipsychotic-Induced Weight Gain and Therapeutic Benefits: A Systematic Review and Implications for Treatment. Front. Neurosci. 2018, 11, 741. [Google Scholar] [CrossRef]
- Fusar-Poli, P.; Papanastasiou, E.; Stahl, D.; Rocchetti, M.; Carpenter, W.; Shergill, S.; McGuire, P. Treatments of Negative Symptoms in Schizophrenia: Meta-Analysis of 168 Randomized Placebo-Controlled Trials. Schizophr. Bull. 2015, 41, 892–899. [Google Scholar] [CrossRef] [PubMed]
- Correll, C.U.; Schooler, N.R. Negative Symptoms in Schizophrenia: A Review and Clinical Guide for Recognition, Assessment, and Treatment. Neuropsychiatr. Dis. Treat. 2020, 16, 519–534. [Google Scholar] [CrossRef] [Green Version]
- Kahn, R.S.; Keefe, R.S. Schizophrenia is a cognitive illness: Time for a change in focus. JAMA Psychiatry 2013, 70, 1107–1112. [Google Scholar] [CrossRef]
- McCleery, A.; Nuechterlein, K.H. Cognitive impairment in psychotic illness: Prevalence, profile of impairment, developmental course, and treatment considerations. Dialog. Clin. Neurosci. 2019, 21, 239–248. [Google Scholar] [CrossRef]
- Torrisi, S.; Laudani, S.; Contarini, G.; De Luca, A.; Geraci, F.; Managò, F.; Papaleo, F.; Salomone, S.; Drago, F.; Leggio, G. Dopamine, Cognitive Impairments and Second-Generation Antipsychotics: From Mechanistic Advances to More Personalized Treatments. Pharmaceuticals 2020, 13, 365. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.B.; Barnes, T.R.; Davies, L.; Dunn, G.; Lloyd, H.; Hayhurst, K.P.; Lewis, S.W. Randomized controlled trial of the effect on Quality of Life of second- vs first-generation antipsychotic drugs in schizophrenia: Cost Utility of the Latest Antipsychotic Drugs in Schizophrenia Study (CUtLASS 1). Arch. Gen. Psychiatry 2006, 63, 1079–1087. [Google Scholar] [CrossRef] [Green Version]
- Kahn, R.S.; Fleischhacker, W.W.; Boter, H.; Davidson, M.; Vergouwe, Y.; Keet, I.P.; Gheorghe, M.D.; Rybakowski, J.K.; Galderisi, S.; Libiger, J.; et al. Effectiveness of antipsychotic drugs in first-episode schizophrenia and schizophreniform disorder: An open randomised clinical trial. Lancet 2008, 371, 1085–1097. [Google Scholar] [CrossRef] [Green Version]
- Lieberman, J.A.; Stroup, T.S.; McEvoy, J.P.; Swartz, M.S.; Rosenheck, R.A.; Perkins, D.O.; Keefe, R.S.E.; Davis, S.M.; Davis, C.E.; Lebowitz, B.D.; et al. Effectiveness of Antipsychotic Drugs in Patients with Chronic Schizophrenia. N. Engl. J. Med. 2005, 353, 1209–1223. [Google Scholar] [CrossRef] [Green Version]
- Mizuno, Y.; McCutcheon, R.A.; Brugger, S.P.; Howes, O.D. Heterogeneity and efficacy of antipsychotic treatment for schizophrenia with or without treatment resistance: A meta-analysis. Neuropsychopharmacology 2019, 45, 622–631. [Google Scholar] [CrossRef]
- Hippius, H. The history of clozapine. Psychopharmacology 1989, 99, S3–S5. [Google Scholar] [CrossRef]
- Potkin, S.G.; Kane, J.M.; Correll, C.U.; Lindenmayer, J.-P.; Agid, O.; Marder, S.R.; Olfson, M.; Howes, O.D. The neurobiology of treatment-resistant schizophrenia: Paths to antipsychotic resistance and a roadmap for future research. NPJ Schizophr. 2020, 6, 1–10. [Google Scholar] [CrossRef]
- Brown, E.; Bedi, G.; McGorry, P.; O’Donoghue, B. Rates and Predictors of Relapse in First-Episode Psychosis: An Australian Cohort Study. Schizophr. Bull. Open 2020, 1. [Google Scholar] [CrossRef] [Green Version]
- Girgis, R.R.; Zoghbi, A.W.; Javitt, D.C.; Lieberman, J.A. The past and future of novel, non-dopamine-2 receptor therapeutics for schizophrenia: A critical and comprehensive review. J. Psychiatr. Res. 2019, 108, 57–83. [Google Scholar] [CrossRef]
- Berry, M.D. Mammalian central nervous system trace amines. Pharmacologic amphetamines, physiologic neuromodulators. J. Neurochem. 2004, 90, 257–271. [Google Scholar] [CrossRef]
- Durden, D.A.; Philips, S.R. Kinetic measurements of the turnover rates of phenylethylamine and tryptamine in vivo in the rat brain. J. Neurochem. 1980, 34, 1725–1732. [Google Scholar]
- Borowsky, B.; Adham, N.; Jones, K.A.; Raddatz, R.; Artymyshyn, R.; Ogozalek, K.L.; Durkin, M.M.; Lakhlani, P.P.; Bonini, J.A.; Pathirana, S.; et al. Trace amines: Identification of a family of mammalian G protein-coupled receptors. Proc. Natl. Acad. Sci. USA 2001, 98, 8966–8971. [Google Scholar] [CrossRef] [Green Version]
- Bunzow, J.R.; Sonders, M.S.; Arttamangkul, S.; Harrison, L.M.; Zhang, G.; Quigley, D.I.; Darland, T.; Suchland, K.L.; Pasumamula, S.; Kennedy, J.L.; et al. Amphetamine, 3,4-Methylenedioxymethamphetamine, Lysergic Acid Diethylamide, and Metabolites of the Catecholamine Neurotransmitters Are Agonists of a Rat Trace Amine Receptor. Mol. Pharmacol. 2001, 60, 1181–1188. [Google Scholar] [CrossRef]
- Berry, M.D.; Gainetdinov, R.; Hoener, M.; Shahid, M. Pharmacology of human trace amine-associated receptors: Therapeutic opportunities and challenges. Pharmacol. Ther. 2017, 180, 161–180. [Google Scholar] [CrossRef] [PubMed]
- Gainetdinov, R.R.; Hoener, M.C.; Berry, M.D. Trace Amines and Their Receptors. Pharmacol. Rev. 2018, 70, 549–620. [Google Scholar] [CrossRef] [Green Version]
- Rutigliano, G.; Zucchi, R. Molecular Variants in Human Trace Amine-Associated Receptors and Their Implications in Mental and Metabolic Disorders. Cell. Mol. Neurobiol. 2020, 40, 239–255. [Google Scholar] [CrossRef] [Green Version]
- Lindemann, L.; Hoener, M.C. A renaissance in trace amines inspired by a novel GPCR family. Trends Pharmacol. Sci. 2005, 26, 274–281. [Google Scholar] [CrossRef]
- Zucchi, R.; Chiellini, G.; Scanlan, T.S.; Grandy, D.K. Trace amine-associated receptors and their ligands. Br. J. Pharmacol. 2006, 149, 967–978. [Google Scholar] [CrossRef] [Green Version]
- Rutigliano, G.; Bräunig, J.; Del Grande, C.; Carnicelli, V.; Masci, I.; Merlino, S.; Kleinau, G.; Tessieri, L.; Pardossi, S.; Paisdzior, S.; et al. Non-Functional Trace Amine-Associated Receptor 1 Variants in Patients With Mental Disorders. Front. Pharmacol. 2019, 10, 1027. [Google Scholar] [CrossRef]
- Mühlhaus, J.; Dinter, J.; Jyrch, S.; Teumer, A.; Jacobi, S.F.; Homuth, G.; Kühnen, P.; Wiegand, S.; Grüters, A.; Völzke, H.; et al. Investigation of Naturally Occurring Single-Nucleotide Variants in Human TAAR1. Front. Pharmacol. 2017, 8, 807. [Google Scholar] [CrossRef] [Green Version]
- Pitts, M.S.; McShane, J.N.; Hoener, M.C.; Christian, S.L.; Berry, M.D. TAAR1 levels and sub-cellular distribution are cell line but not breast cancer subtype specific. Histochem. Cell Biol. 2019, 152, 155–166. [Google Scholar] [CrossRef]
- Lindemann, L.; Meyer, C.A.; Jeanneau, K.; Bradaia, A.; Ozmen, L.; Bluethmann, H.; Bettler, B.; Wettstein, J.G.; Borroni, E.; Moreau, J.-L.; et al. Trace Amine-Associated Receptor 1 Modulates Dopaminergic Activity. J. Pharmacol. Exp. Ther. 2008, 324, 948–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiellini, G.; Erba, P.; Carnicelli, V.; Manfredi, C.; Frascarelli, S.; Ghelardoni, S.; Mariani, G.; Zucchi, R. Distribution of exogenous [125I]-3-iodothyronamine in mouse in vivo: Relationship with trace amine-associated receptors. J. Endocrinol. 2012, 213, 223–230. [Google Scholar] [CrossRef]
- Revel, F.G.; Moreau, J.-L.; Pouzet, B.; Mory, R.; Bradaia, A.; Buchy, D.; Metzler, V.; Chaboz, S.; Zbinden, K.G.; Galley, G.; et al. A new perspective for schizophrenia: TAAR1 agonists reveal antipsychotic- and antidepressant-like activity, improve cognition and control body weight. Mol. Psychiatry 2012, 18, 543–556. [Google Scholar] [CrossRef] [PubMed]
- Adriaenssens, A.; Lam, B.Y.H.; Billing, L.; Skeffington, K.; Sewing, S.; Reimann, F.; Gribble, F. A Transcriptome-Led Exploration of Molecular Mechanisms Regulating Somatostatin-Producing D-Cells in the Gastric Epithelium. Endocrinology 2015, 156, 3924–3936. [Google Scholar] [CrossRef] [Green Version]
- Raab, S.; Wang, H.; Uhles, S.; Cole, N.; Alvarez-Sanchez, R.; Künnecke, B.; Ullmer, C.; Matile, H.; Bedoucha, M.; Norcross, R.D.; et al. Incretin-like effects of small molecule trace amine-associated receptor 1 agonists. Mol. Metab. 2016, 5, 47–56. [Google Scholar] [CrossRef]
- Regard, J.B.; Kataoka, H.; Cano, D.A.; Camerer, E.; Yin, L.; Zheng, Y.-W.; Scanlan, T.S.; Hebrok, M.; Coughlin, S.R. Probing cell type–specific functions of Gi in vivo identifies GPCR regulators of insulin secretion. J. Clin. Investig. 2007, 117, 4034–4043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kidd, M.; Modlin, I.M.; Gustafsson, B.I.; Drozdov, I.; Hauso, O.; Pfragner, R. Luminal regulation of normal and neoplastic human EC cell serotonin release is mediated by bile salts, amines, tastants, and olfactants. Am. J. Physiol. Liver Physiol. 2008, 295, G260–G272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, J.; Ito, M.; Nambu, H.; Fujikawa, T.; Tanaka, K.; Iwaasa, H.; Tokita, S. Anatomical and histological profiling of orphan G-protein-coupled receptor expression in gastrointestinal tract of C57BL/6J mice. Cell Tissue Res. 2009, 338, 257–269. [Google Scholar] [CrossRef]
- D’Andrea, G.; Terrazzino, S.; Fortin, D.; Farruggio, A.; Rinaldi, L.; Leon, A. HPLC electrochemical detection of trace amines in human plasma and platelets and expression of mRNA transcripts of trace amine receptors in circulating leukocytes. Neurosci. Lett. 2003, 346, 89–92. [Google Scholar] [CrossRef]
- Nelson, D.A.; Tolbert, M.D.; Singh, S.J.; Bost, K.L. Expression of Neuronal Trace Amine-associated Receptor (Taar) mRNAs in Leukocytes. J. Neuroimmunol. 2007, 192, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Wasik, A.M.; Millan, M.J.; Scanlan, T.; Barnes, N.M.; Gordon, J. Evidence for functional trace amine associated receptor-1 in normal and malignant B cells. Leuk. Res. 2012, 36, 245–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babusyte, A.; Kotthoff, M.; Fiedler, J.; Krautwurst, D. Biogenic amines activate blood leukocytes via trace amine-associated receptors TAAR1 and TAAR2. J. Leukoc. Biol. 2013, 93, 387–394. [Google Scholar] [CrossRef]
- Bradaia, A.; Trube, G.; Stalder, H.; Norcross, R.D.; Ozmen, L.; Wettstein, J.G.; Pinard, A.; Buchy, D.; Gassmann, M.; Hoener, M.; et al. The selective antagonist EPPTB reveals TAAR1-mediated regulatory mechanisms in dopaminergic neurons of the mesolimbic system. Proc. Natl. Acad. Sci. USA 2009, 106, 20081–20086. [Google Scholar] [CrossRef] [Green Version]
- Leo, D.; Mus, L.; Espinoza, S.; Hoener, M.; Sotnikova, T.; Gainetdinov, R. Taar1-mediated modulation of presynaptic dopaminergic neurotransmission: Role of D2 dopamine autoreceptors. Neuropharmacology 2014, 81, 283–291. [Google Scholar] [CrossRef]
- Revel, F.G.; Moreau, J.-L.; Gainetdinov, R.R.; Bradaia, A.; Sotnikova, T.D.; Mory, R.; Durkin, S.; Zbinden, K.G.; Norcross, R.; Meyer, C.A.; et al. TAAR1 activation modulates monoaminergic neurotransmission, preventing hyperdopaminergic and hypoglutamatergic activity. Proc. Natl. Acad. Sci. USA 2011, 108, 8485–8490. [Google Scholar] [CrossRef] [Green Version]
- Espinoza, S.; Ghisi, V.; Emanuele, M.; Leo, D.; Sukhanov, I.; Sotnikova, T.D.; Chieregatti, E.; Gainetdinov, R.R. Postsynaptic D2 dopamine receptor supersensitivity in the striatum of mice lacking TAAR1. Neuropharmacology 2015, 93, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Miller, G.M.; Verrico, C.D.; Jassen, A.; Konar, M.; Yang, H.; Panas, H.; Bahn, M.; Johnson, R.; Madras, B.K. Primate Trace Amine Receptor 1 Modulation by the Dopamine Transporter. J. Pharmacol. Exp. Ther. 2005, 313, 983–994. [Google Scholar] [CrossRef] [Green Version]
- Pei, Y.; Asif-Malik, A.; Canales, J.J. Trace Amines and the Trace Amine-Associated Receptor 1: Pharmacology, Neurochemistry, and Clinical Implications. Front. Neurosci. 2016, 10, 148. [Google Scholar] [CrossRef] [Green Version]
- Espinoza, S.; Salahpour, A.; Masri, B.; Sotnikova, T.D.; Messa, M.; Barak, L.S.; Caron, M.G.; Gainetdinov, R.R. Functional Interaction between Trace Amine-Associated Receptor 1 and Dopamine D2 Receptor. Mol. Pharmacol. 2011, 80, 416–425. [Google Scholar] [CrossRef] [Green Version]
- Harmeier, A.; Obermueller, S.; Meyer, C.A.; Revel, F.G.; Buchy, D.; Chaboz, S.; Dernick, G.; Wettstein, J.G.; Iglesias, A.; Rolink, A.; et al. Trace amine-associated receptor 1 activation silences GSK3β signaling of TAAR1 and D2R heteromers. Eur. Neuropsychopharmacol. 2015, 25, 2049–2061. [Google Scholar] [CrossRef]
- Panas, M.W.; Xie, Z.; Panas, H.N.; Hoener, M.C.; Vallender, E.J.; Miller, G.M. Trace Amine Associated Receptor 1 Signaling in Activated Lymphocytes. J. Neuroimmune Pharmacol. 2012, 7, 866–876. [Google Scholar] [CrossRef] [Green Version]
- Bräunig, J.; Dinter, J.; Höfig, C.S.; Paisdzior, S.; Szczepek, M.; Scheerer, P.; Rosowski, M.; Mittag, J.; Kleinau, G.; Biebermann, H. The Trace Amine-Associated Receptor 1 Agonist 3-Iodothyronamine Induces Biased Signaling at the Serotonin 1b Receptor. Front. Pharmacol. 2018, 9, 222. [Google Scholar] [CrossRef] [Green Version]
- Dinter, J.; Mühlhaus, J.; Jacobi, S.F.; Wienchol, C.L.; Cöster, M.; Meister, J.; Hoefig, C.S.; Müller, A.; Köhrle, J.; Grüters, A.; et al. 3-iodothyronamine differentially modulates α-2A-adrenergic receptor-mediated signaling. J. Mol. Endocrinol. 2015, 54, 205–216. [Google Scholar] [CrossRef] [Green Version]
- Christian, S.L.; Berry, M.D. Trace Amine-Associated Receptors as Novel Therapeutic Targets for Immunomodulatory Disorders. Front. Pharmacol. 2018, 9, 680. [Google Scholar] [CrossRef] [Green Version]
- Simmler, L.; Buchy, D.; Chaboz, S.; Hoener, M.; Liechti, M. In Vitro Characterization of Psychoactive Substances at Rat, Mouse, and Human Trace Amine-Associated Receptor 1. J. Pharmacol. Exp. Ther. 2016, 357, 134–144. [Google Scholar] [CrossRef]
- Köhrle, J.; Biebermann, H. 3-Iodothyronamine—A Thyroid Hormone Metabolite with Distinct Target Profiles and Mode of Action. Endocr. Rev. 2019, 40, 602–630. [Google Scholar] [CrossRef]
- Edelmann, M.R.; Hartung, T.; Trussardi, R.; Iding, H.; Galley, G.; Pflieger, P.; Norcross, R.D. Synthesis of enantiomerically pure [14C]-labelled morpholine derivatives for a class of trace amine-associate receptor 1 agonists. J. Label. Compd. Radiopharm. 2016, 59, 635–639. [Google Scholar] [CrossRef] [PubMed]
- Galley, G.; Stalder, H.; Goergler, A.; Hoener, M.C.; Norcross, R.D. Optimisation of imidazole compounds as selective TAAR1 agonists: Discovery of RO5073012. Bioorg. Med. Chem. Lett. 2012, 22, 5244–5248. [Google Scholar] [CrossRef]
- Galley, G.; Beurier, A.; Décoret, G.; Goergler, A.; Hutter, R.; Mohr, S.; Pähler, A.; Schmid, P.; Türck, D.; Unger, R.; et al. Discovery and Characterization of 2-Aminooxazolines as Highly Potent, Selective, and Orally Active TAAR1 Agonists. ACS Med. Chem. Lett. 2016, 7, 192–197. [Google Scholar] [CrossRef] [Green Version]
- Fowler, S.; Kletzl, H.; Finel, M.; Manevski, N.; Schmied, P.; Tuerck, D.; Norcross, R.D.; Hoener, M.; Spleiss, O.; Iglesias, V.A. A UGT2B10 Splicing Polymorphism Common in African Populations May Greatly Increase Drug Exposure. J. Pharmacol. Exp. Ther. 2014, 352, 358–367. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann-La Roche. A Trial of the Efficacy and the Safety of RO6889450 (Ralmitaront) vs Placebo in Patients with an Acute Exacerbation of Schizophrenia or Schizoaffective Disorder; Hoffmann-La Roche: Basel, Switzerland, 2020; NCT04512066. Available online: https://clinicaltrials.gov/ct2/show/NCT04512066 (accessed on 2 December 2021).
- Hoffmann-La Roche. A Study to Assess the Effects of RO6889450 (Ralmitaront) in Participants with Schizophrenia or Schizoaffective Disorder and Negative Symptoms; Hoffmann-La Roche: Basel, Switzerland, 2018; NCT03669640. Available online: https://clinicaltrials.gov/ct2/show/NCT03669640 (accessed on 2 December 2021).
- Francesconi, V.; Cichero, E.; Kanov, E.V.; Laurini, E.; Pricl, S.; Gainetdinov, R.R.; Tonelli, M. Novel 1-Amidino-4-Phenylpiperazines as Potent Agonists at Human TAAR1 Receptor: Rational Design, Synthesis, Biological Evaluation and Molecular Docking Studies. Pharmaceuticals 2020, 13, 391. [Google Scholar] [CrossRef] [PubMed]
- Galley, G.H.M.; Norcross, R.; Pflieger, P. 5-ethyl-4-methyl-pyrazole-3-carboxamide Derivative Having Activity as Agonist of TAAR; F. Hoffmann-La Roche; WIPOI Bureau: Washington, DC, USA, 2017. [Google Scholar]
- Dedic, N.; Jones, P.G.; Hopkins, S.C.; Lew, R.; Shao, L.; Campbell, J.E.; Spear, K.L.; Large, T.H.; Campbell, U.C.; Hanania, T.; et al. SEP-363856, a Novel Psychotropic Agent with a Unique, Non-D2 Receptor Mechanism of Action. J. Pharmacol. Exp. Ther. 2019, 371, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.-F.; Thorn, D.; Zhang, Y.; Li, J.-X. Effects of Trace Amine-associated Receptor 1 Agonists on the Expression, Reconsolidation, and Extinction of Cocaine Reward Memory. Int. J. Neuropsychopharmacol. 2016, 19, pyw009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.-F.; Siemian, J.N.; Seaman, R.; Zhang, Y.; Li, J.-X. Role of TAAR1 within the Subregions of the Mesocorticolimbic Dopaminergic System in Cocaine-Seeking Behavior. J. Neurosci. 2017, 37, 882–892. [Google Scholar] [CrossRef] [Green Version]
- Pei, Y.; Lee, J.; Leo, D.; Gainetdinov, R.R.; Hoener, M.C.; Canales, J.J. Activation of the Trace Amine-Associated Receptor 1 Prevents Relapse to Cocaine Seeking. Neuropsychopharmacology 2014, 39, 2299–2308. [Google Scholar] [CrossRef] [Green Version]
- Ferragud, A.; Howell, A.D.; Moore, C.; Ta, T.L.; Hoener, M.C.; Sabino, V.; Cottone, P. The Trace Amine-Associated Receptor 1 Agonist RO5256390 Blocks Compulsive, Binge-like Eating in Rats. Neuropsychopharmacology 2016, 42, 1458–1470. [Google Scholar] [CrossRef] [PubMed]
- Revel, F.G.; Moreau, J.-L.; Gainetdinov, R.; Ferragud, A.; Velázquez-Sánchez, C.; Sotnikova, T.D.; Morairty, S.R.; Harmeier, A.; Zbinden, K.G.; Norcross, R.D.; et al. Trace Amine-Associated Receptor 1 Partial Agonism Reveals Novel Paradigm for Neuropsychiatric Therapeutics. Biol. Psychiatry 2012, 72, 934–942. [Google Scholar] [CrossRef]
- Cotter, R.; Pei, Y.; Mus, L.; Harmeier, A.; Gainetdinov, R.R.; Hoener, M.C.; Canales, J.J. The trace amine-associated receptor 1 modulates methamphetamine’s neurochemical and behavioral effects. Front. Neurosci. 2015, 9, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leo, D.; Sukhanov, I.; Zoratto, F.; Illiano, P.; Caffino, L.; Sanna, F.; Messa, G.; Emanuele, M.; Esposito, A.; Dorofeikova, M.; et al. Pronounced Hyperactivity, Cognitive Dysfunctions, and BDNF Dysregulation in Dopamine Transporter Knock-out Rats. J. Neurosci. 2018, 38, 1959–1972. [Google Scholar] [CrossRef] [Green Version]
- Jing, L.; Zhang, Y.; Li, J.-X. Effects of the Trace Amine Associated Receptor 1 Agonist RO5263397 on Abuse-Related Behavioral Indices of Methamphetamine in Rats. Int. J. Neuropsychopharmacol. 2014, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorn, D.; Jing, L.; Qiu, Y.; Gancarz-Kausch, A.M.; Galuska, C.M.; Dietz, D.; Zhang, Y.; Li, J.-X. Effects of the Trace Amine-Associated Receptor 1 Agonist RO5263397 on Abuse-Related Effects of Cocaine in Rats. Neuropsychopharmacology 2014, 39, 2309–2316. [Google Scholar] [CrossRef]
- Thorn, D.A.; Zhang, C.; Zhang, Y.; Li, J.-X. The trace amine associated receptor 1 agonist RO5263397 attenuates the induction of cocaine behavioral sensitization in rats. Neurosci. Lett. 2014, 566, 67–71. [Google Scholar] [CrossRef] [Green Version]
- Xue, Z.; Siemian, J.N.; Johnson, B.N.; Zhang, Y.; Li, J.-X. Methamphetamine-induced impulsivity during chronic methamphetamine treatment in rats: Effects of the TAAR 1 agonist RO5263397. Neuropharmacology 2018, 129, 36–46. [Google Scholar] [CrossRef]
- Pei, Y.; Mortas, P.; Hoener, M.C.; Canales, J.J. Selective activation of the trace amine-associated receptor 1 decreases cocaine’s reinforcing efficacy and prevents cocaine-induced changes in brain reward thresholds. Prog. Neuropsychopharmacol. Biol. Psychiatry 2015, 63, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, J.T.; Wang, H.; Niu, W.P.; Zhang, C.C.; Zhang, Y.; Su, Y.A. Role of trace amine-associated receptor 1 in the medial prefrontal cortex in chronic social stress-induced cognitive deficits in mice. Pharmacol. Res. 2021, 167, 105571. [Google Scholar] [CrossRef]
- Aleksandrov, A.A.; Knyazeva, V.M.; Volnova, A.; Dmitrieva, E.; Polyakova, N.V.; Gainetdinov, R. Trace Amine-Associated Receptor 1 Agonist Modulates Mismatch Negativity-Like Responses in Mice. Front. Pharmacol. 2019, 10, 470. [Google Scholar] [CrossRef]
- Revel, F.G.; Meyer, C.; Bradaia, A.; Jeanneau, K.; Calcagno, E.; André, C.B.; Haenggi, M. Brain-Specific Overexpression of Trace Amine-Associated Receptor 1 Alters Monoaminergic Neurotransmission and Decreases Sensitivity to Amphetamine. Neuropsychopharmacology 2012, 37, 2580–2592. [Google Scholar] [CrossRef] [Green Version]
- Begni, V.; Sanson, A.; Luoni, A.; Sensini, F.; Grayson, B.; Munni, S.; Neill, J.; Riva, M. Towards Novel Treatments for Schizophrenia: Molecular and Behavioural Signatures of the Psychotropic Agent SEP-363856. Int. J. Mol. Sci. 2021, 22, 4119. [Google Scholar] [CrossRef]
- Koblan, K.S.; Kent, J.; Hopkins, S.C.; Krystal, J.H.; Cheng, H.; Goldman, R.; Loebel, A. A Non–D2-Receptor-Binding Drug for the Treatment of Schizophrenia. N. Engl. J. Med. 2020, 382, 1497–1506. [Google Scholar] [CrossRef]
- Correll, C.U.; Koblan, K.S.; Hopkins, S.C.; Kent, J.; Cheng, H.; Goldman, R.; Loebel, A. Safety and Effectiveness of Ulotaront (SEP-363856) in Schizophrenia: Results of a 6-month, Open-label Extension Study. NPJ Schizophr. 2021, 26, 148–149. [Google Scholar]
- Sunovion Pharmaceuticals. A Clinical Trial to Study the Efficacy and Safety of an Investigational Drug in Acutely Psychotic People with Schizophrenia; Sunovion Pharmaceuticals: Marlborough, MA, USA, 2019; NCT04072354. Available online: https://clinicaltrials.gov/ct2/show/NCT04072354 (accessed on 2 December 2021).
- Sunovion Pharmaceuticals. A Clinical Trial that Will Study the Efficacy and Safety of an Investigational Drug in Acutely Psychotic People with Schizophrenia; Sunovion Pharmaceuticals: Marlborough, MA, USA, 2019; NCT04092686. Available online: https://clinicaltrials.gov/ct2/show/NCT04092686 (accessed on 2 December 2021).
- Sunovion Pharmaceuticals. A Clinical Study to Evaluate the Long-Term Safety and Tolerability of an Investigational Drug in People with Schizophrenia; Sunovion Pharmaceuticals: Marlborough, MA, USA, 2019; NCT04109950. Available online: https://clinicaltrials.gov/ct2/show/NCT04109950 (accessed on 2 December 2021).
- Sunovion Pharmaceuticals. A Study of the Long-Term Safety and Tolerability of an Investigational Drug in People with Schizophrenia (NCT04115319); Sunovion Pharmaceuticals: Marlborough, MA, USA, 2019; NCT04115319. Available online: https://clinicaltrials.gov/ct2/show/NCT04115319 (accessed on 2 December 2021).
- Wolinsky, T.D.; Swanson, C.J.; Smith, K.E.; Zhong, H.; Borowsky, B.; Seeman, P.; Branchek, T.; Gerald, C.P. The Trace Amine 1 receptor knockout mouse: An animal model with relevance to schizophrenia. Genes Brain Behav. 2007, 6, 628–639. [Google Scholar] [CrossRef]
- Liu, J.-F.; Seaman, R.; Siemian, J.N.; Bhimani, R.; Johnson, B.; Zhang, Y.; Zhu, Q.; Hoener, M.C.; Park, J.; Dietz, D.M.; et al. Role of trace amine-associated receptor 1 in nicotine’s behavioral and neurochemical effects. Neuropsychopharmacology 2018, 43, 2435–2444. [Google Scholar] [CrossRef] [PubMed]
- Di Cara, B.; Maggio, R.; Aloisi, G.; Rivet, J.-M.; Lundius, E.G.; Yoshitake, T.; Svenningsson, P.; Brocco, M.; Gobert, A.; De Groote, L.; et al. Genetic Deletion of Trace Amine 1 Receptors Reveals Their Role in Auto-Inhibiting the Actions of Ecstasy (MDMA). J. Neurosci. 2011, 31, 16928–16940. [Google Scholar] [CrossRef]
- Asif-Malik, A.; Hoener, M.C.; Canales, J.J. Interaction Between the Trace Amine-Associated Receptor 1 and the Dopamine D2 Receptor Controls Cocaine’s Neurochemical Actions. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.; Howes, O.D.; Veronese, M.; Beck, K.; Seo, S.; Park, J.W.; Lee, J.S.; Lee, Y.-S.; Kwon, J.S. Presynaptic Dopamine Capacity in Patients with Treatment-Resistant Schizophrenia Taking Clozapine: An [18F]DOPA PET Study. Neuropsychopharmacology 2017, 42, 941–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCutcheon, R.; Beck, K.; Jauhar, S.; Howes, O.D. Defining the Locus of Dopaminergic Dysfunction in Schizophrenia: A Meta-analysis and Test of the Mesolimbic Hypothesis. Schizophr. Bull. 2018, 44, 1301–1311. [Google Scholar] [CrossRef] [Green Version]
- McCutcheon, R.A.; Marques, T.R.; Howes, O.D. Schizophrenia-An Overview. JAMA Psychiatry 2020, 77, 201–210. [Google Scholar] [CrossRef]
- Espinoza, S.; Lignani, G.; Caffino, L.; Maggi, S.; Sukhanov, I.; Leo, D.; Mus, L.; Emanuele, M.; Ronzitti, G.; Harmeier, A.; et al. TAAR1 Modulates Cortical Glutamate NMDA Receptor Function. Neuropsychopharmacology 2015, 40, 2217–2227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swerdlow, N.R.; Light, G.A.; Cadenhead, K.S.; Sprock, J.; Hsieh, M.H.; Braff, D.L. Startle gating deficits in a large cohort of patients with schizophrenia: Relationship to medications, symptoms, neurocognition, and level of function. Arch. Gen. Psychiatry 2006, 63, 1325–1335. [Google Scholar] [CrossRef] [Green Version]
- Rissling, A.J.; Miyakoshi, M.; Sugar, C.A.; Braff, D.L.; Makeig, S.; Light, G.A. Cortical substrates and functional correlates of auditory deviance processing deficits in schizophrenia. NeuroImage Clin. 2014, 6, 424–437. [Google Scholar] [CrossRef] [Green Version]
- Näätänen, R.; Kujala, T.; Kreegipuu, K.; Carlson, S.; Escera, C.; Baldeweg, T.; Ponton, C. The mismatch negativity: An index of cognitive decline in neuropsychiatric and neurological diseases and in ageing. Brain 2011, 134, 3435–3453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turetsky, B.I.; Calkins, M.E.; Light, G.A.; Olincy, A.; Radant, A.D.; Swerdlow, N.R. Neurophysiological Endophenotypes of Schizophrenia: The Viability of Selected Candidate Measures. Schizophr. Bull. 2006, 33, 69–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Light, G.A.; Swerdlow, N.R. Future clinical uses of neurophysiological biomarkers to predict and monitor treatment response for schizophrenia. Ann. N. Y. Acad. Sci. 2015, 1344, 105–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umbricht, D.; Schmid, L.; Koller, R.; Vollenweider, F.X.; Hell, D.; Javitt, D.C. Ketamine-induced deficits in auditory and visual context-dependent processing in healthy volunteers: Implications for models of cognitive deficits in schizophrenia. Arch. Gen. Psychiatry 2000, 57, 1139–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Underhill, S.M.; Colt, M.S.; Amara, S.G. Amphetamine Stimulates Endocytosis of the Norepinephrine and Neuronal Glutamate Transporters in Cultured Locus Coeruleus Neurons. Neurochem. Res. 2020, 45, 1410–1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarsson, A.; Zhang, X.; Stan, T.L.; Schintu, N.; Kadkhodaei, B.; Millan, M.J.; Perlmann, T.; Svenningsson, P. Modulation by Trace Amine-Associated Receptor 1 of Experimental Parkinsonism, L-DOPA Responsivity, and Glutamatergic Neurotransmission. J. Neurosci. 2015, 35, 14057–14069. [Google Scholar] [CrossRef]
- McCutcheon, R.A.; Brown, K.; Nour, M.M.; Smith, S.M.; Veronese, M.; Zelaya, F.; Osugo, M.; Jauhar, S.; Hallett, W.; Mehta, M.M.; et al. Dopaminergic organization of striatum is linked to cortical activity and brain expression of genes associated with psychiatric illness. Sci. Adv. 2021, 7, eabg1512. [Google Scholar] [CrossRef]
- Cisneros, I.E.; Ghorpade, A. Methamphetamine and HIV-1-induced neurotoxicity: Role of trace amine associated receptor 1 cAMP signaling in astrocytes. Neuropharmacology 2014, 85, 499–507. [Google Scholar] [CrossRef] [Green Version]
- Ding, S.; Wang, X.; Zhuge, W.; Yang, J.; Zhuge, Q. Dopamine induces glutamate accumulation in astrocytes to disrupt neuronal function leading to pathogenesis of minimal hepatic encephalopathy. Neuroscience 2017, 365, 94–113. [Google Scholar] [CrossRef] [PubMed]
- Moore, C.; Sabino, V.; Cottone, P. Trace Amine Associated Receptor 1 (TAAR1) Modulation of Food Reward. Front. Pharmacol. 2018, 9, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allison, D.B.; Mackell, J.A.; McDonnell, D.D. The Impact of Weight Gain on Quality of Life Among Persons with Schizophrenia. Psychiatr. Serv. 2003, 54, 565–567. [Google Scholar] [CrossRef] [PubMed]
- Baptista, T.; Zarate, J.; Joober, R.; Colasante, C.; Beaulieu, S.; Paez, X.; Hernandez, L. Drug induced weight gain, an impediment to successful pharmacotherapy: Focus on antipsychotics. Curr. Drug Targets 2004, 5, 279–299. [Google Scholar] [CrossRef]
- Michaelides, M.; Thanos, P.K.; Volkow, N.D.; Wang, G.-J. Dopamine-related frontostriatal abnormalities in obesity and binge-eating disorder: Emerging evidence for developmental psychopathology. Int. Rev. Psychiatry 2012, 24, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Galluppi, G.R.; Polhamus, D.G.; Fisher, J.M.; Hopkins, S.C.; Koblan, K.S. Population pharmacokinetic analysis of ulotaront in subjects with schizophrenia. CPT Pharmacomet. Syst. Pharmacol. 2021, 10, 1245–1254. [Google Scholar] [CrossRef]
- Hopkins, S.C.; Dedic, N.; Koblan, K.S. Effect of TAAR1/5-HT(1A) agonist SEP-363856 on REM sleep in humans. Transl. Psychiatry 2021, 11, 228. [Google Scholar] [CrossRef]
- Hopkins, S.C.; Cheng, O.A.; Loebel, H.; Koblan, A. Effects of SEP-363856 on negative symptoms in schizophrenia: Analysis of an acute, placebo-controlled trial of a novel psychotropic agent with no dopamine-D2/5-HT2A antagonist activity. In Proceedings of the American College of Neuropsychopharmacology Annual Meeting, Orlando, FL, USA, 8–11 December 2019. [Google Scholar]
- Dworak, H.H.S.; Koblan, K.S.; Hayes, R.; Zhu, H.; Li, Y.; Zeni, C.; Kent, J. Effects of SEP-363856, a novel TAAR1 agonist, on negative symptoms in schizophrenia: Results across an initial double-blind acute study, and a 6-month, open-label extension study. In Proceedings of the American College of Neuropsychopharmacology 59th Annual Meeting, Virtual Meeting. 6–9 December 2020. [Google Scholar]
- Hopkins, S.C.; Ogirala, A.; Loebel, A.; Koblan, K.S. Transformed PANSS Factors Intended to Reduce Pseudospecificity Among Symptom Domains and Enhance Understanding of Symptom Change in Antipsychotic-Treated Patients with Schizophrenia. Schizophr. Bull. 2017, 44, 593–602. [Google Scholar] [CrossRef]
- Krzystanek, M.; Krupka-Matuszczyk, I. An open, large, 6-month naturalistic study of outcome in schizophrenic outpatients, treated with olanzapine. Hum. Psychopharmacol. Clin. Exp. 2011, 26, 81–85. [Google Scholar] [CrossRef]
- WHO Drug Information. International Nonproprietary Names for Pharmaceutical Substances (INN). 2019, Volume 33. Available online: https://www.who.int/medicines/publications/druginformation/innlists/PL121.pdf?ua=1 (accessed on 2 December 2021).


| Positive Symptoms | Negative Symptoms | Cognitive Symptoms-Impairments in: |
|---|---|---|
|
|
|
|
|
|
|
|
|
|
| |
|
| |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dedic, N.; Dworak, H.; Zeni, C.; Rutigliano, G.; Howes, O.D. Therapeutic Potential of TAAR1 Agonists in Schizophrenia: Evidence from Preclinical Models and Clinical Studies. Int. J. Mol. Sci. 2021, 22, 13185. https://doi.org/10.3390/ijms222413185
Dedic N, Dworak H, Zeni C, Rutigliano G, Howes OD. Therapeutic Potential of TAAR1 Agonists in Schizophrenia: Evidence from Preclinical Models and Clinical Studies. International Journal of Molecular Sciences. 2021; 22(24):13185. https://doi.org/10.3390/ijms222413185
Chicago/Turabian StyleDedic, Nina, Heather Dworak, Courtney Zeni, Grazia Rutigliano, and Oliver D. Howes. 2021. "Therapeutic Potential of TAAR1 Agonists in Schizophrenia: Evidence from Preclinical Models and Clinical Studies" International Journal of Molecular Sciences 22, no. 24: 13185. https://doi.org/10.3390/ijms222413185
APA StyleDedic, N., Dworak, H., Zeni, C., Rutigliano, G., & Howes, O. D. (2021). Therapeutic Potential of TAAR1 Agonists in Schizophrenia: Evidence from Preclinical Models and Clinical Studies. International Journal of Molecular Sciences, 22(24), 13185. https://doi.org/10.3390/ijms222413185