
Discovery of Novel Trace Amine-Associated Receptor 5 (TAAR5) Antagonists Using a Deep Convolutional Neural Network

 ,
, 

Abstract
:1. Introduction
2. Results
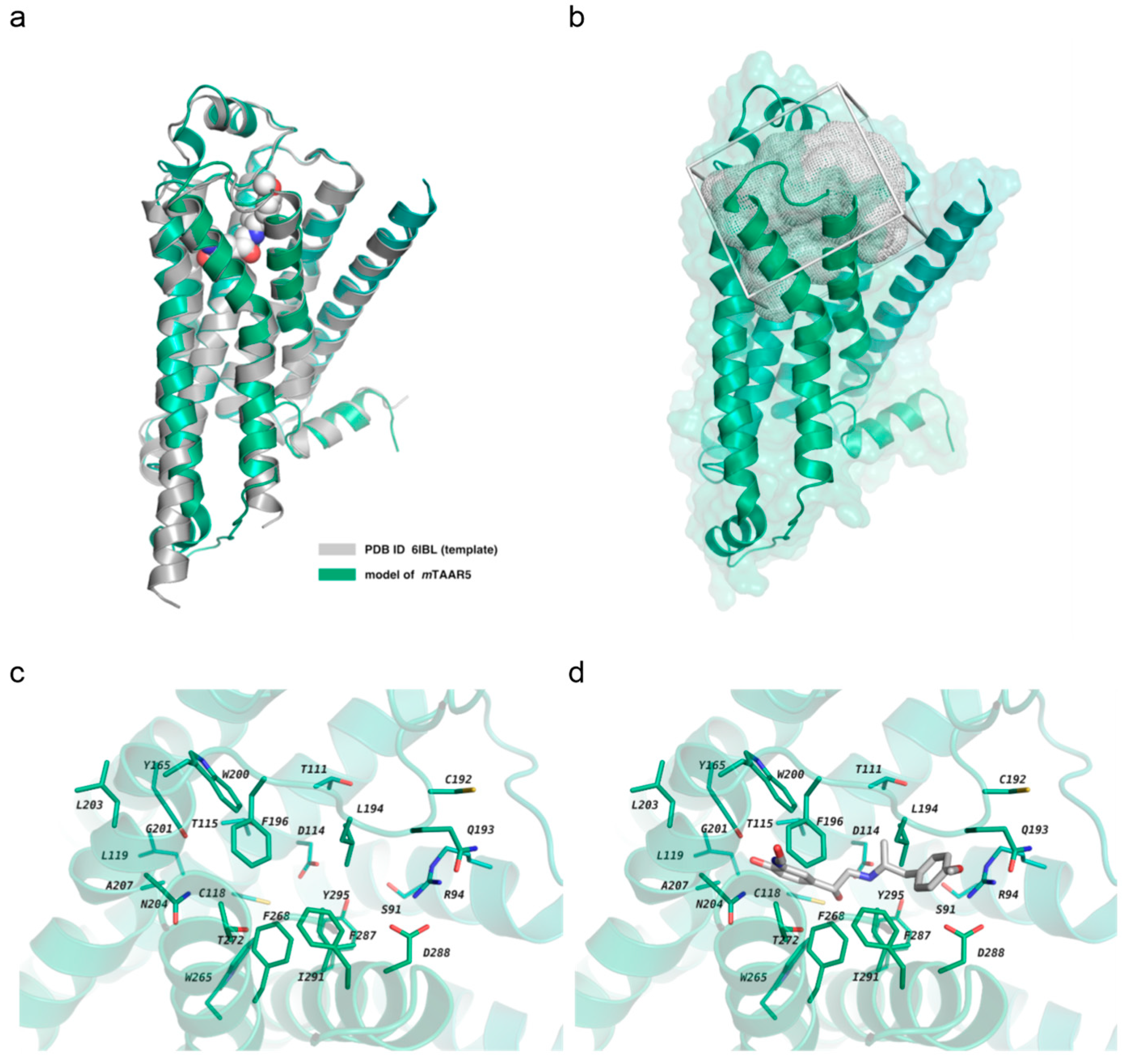
2.1. Homology Modeling and In Silico Screening
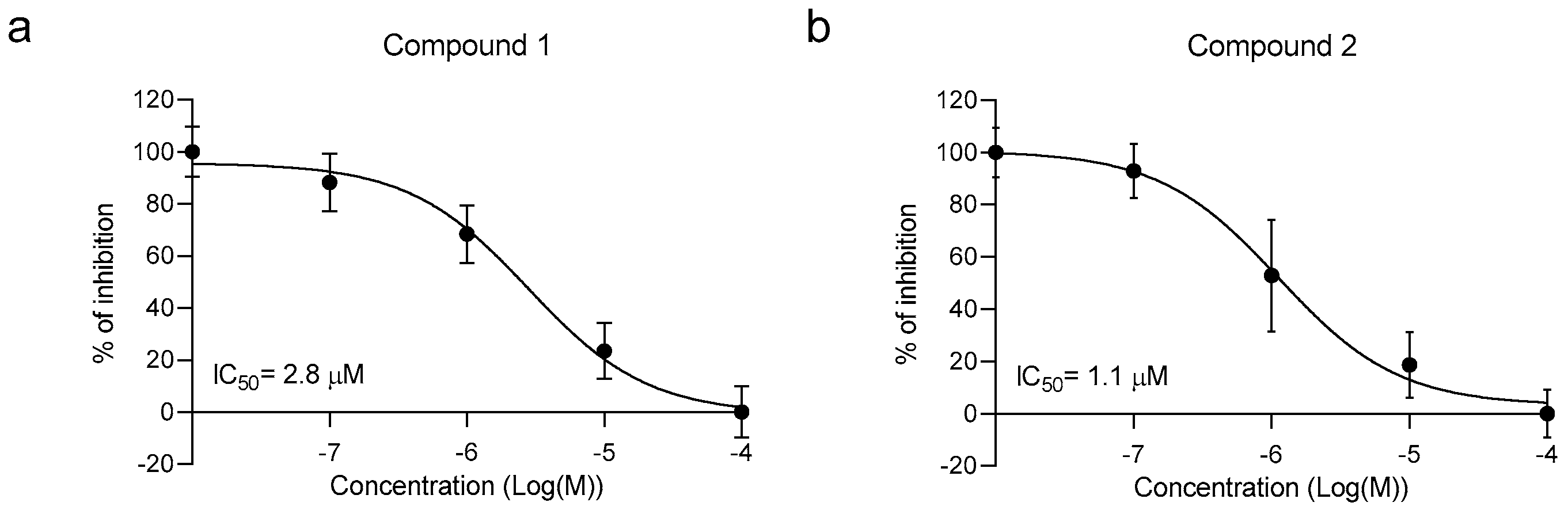
2.2. Library In Vitro Screening
2.3. TAAR5 Antagonists Inhibit ERK and CREB Phosphorylation
2.4. In Silico Prediction of ADME Properties
3. Discussion
4. Materials and Methods
4.1. Homology Modeling
4.2. Virtual Screening Method
4.3. Cell Culture and Transfection
4.4. BRET Assay
4.5. Phospho-ERK and Phospho-CREB Assays
4.6. Western Blot Analysis and Antibodies
4.7. In Silico ADME Properties Prediction
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Gainetdinov, R.R.; Hoener, M.C.; Berry, M.D. Trace Amines and Their Receptors. Pharmacol. Rev. 2018, 70, 549–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry, M.D. Mammalian central nervous system trace amines. Pharmacologic amphetamines, physiologic neuromodulators. J. Neurochem. 2004, 90, 257–271. [Google Scholar] [CrossRef] [PubMed]
- Bunzow, J.R.; Sonders, M.S.; Arttamangkul, S.; Harrison, L.M.; Zhang, G.; Quigley, D.I.; Darland, T.; Suchland, K.L.; Pasumamula, S.; Kennedy, J.L.; et al. Amphetamine, 3,4-methylenedioxymethamphetamine, lysergic acid diethylamide, and metabolites of the catecholamine neurotransmitters are agonists of a rat trace amine receptor. Mol. Pharmacol. 2001, 60, 1181–1188. [Google Scholar] [CrossRef] [PubMed]
- Borowsky, B.; Adham, N.; Jones, K.A.; Raddatz, R.; Artymyshyn, R.; Ogozalek, K.L.; Durkin, M.M.; Lakhlani, P.P.; Bonini, J.A.; Pathirana, S.; et al. Trace amines: Identification of a family of mammalian G protein-coupled receptors. Proc. Natl. Acad. Sci. USA 2001, 98, 8966–8971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liberles, S.D.; Buck, L.B. A second class of chemosensory receptors in the olfactory epithelium. Nature 2006, 442, 645–650. [Google Scholar] [CrossRef]
- Espinoza, S.; Ghisi, V.; Emanuele, M.; Leo, D.; Sukhanov, I.; Sotnikova, T.D.; Chieregatti, E.; Gainetdinov, R.R. Postsynaptic D2 dopamine receptor supersensitivity in the striatum of mice lacking TAAR1. Neuropharmacology 2015, 93, 308–313. [Google Scholar] [CrossRef]
- Espinoza, S.; Leo, D.; Sotnikova, T.D.; Shahid, M.; Kaariainen, T.M.; Gainetdinov, R.R. Biochemical and Functional Characterization of the Trace Amine-Associated Receptor 1 (TAAR1) Agonist RO5263397. Front. Pharmacol. 2018, 9, 645. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, S.; Salahpour, A.; Masri, B.; Sotnikova, T.D.; Messa, M.; Barak, L.S.; Caron, M.G.; Gainetdinov, R.R. Functional interaction between trace amine-associated receptor 1 and dopamine D2 receptor. Mol. Pharmacol. 2011, 80, 416–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harmeier, A.; Obermueller, S.; Meyer, C.A.; Revel, F.G.; Buchy, D.; Chaboz, S.; Dernick, G.; Wettstein, J.G.; Iglesias, A.; Rolink, A.; et al. Trace amine-associated receptor 1 activation silences GSK3beta signaling of TAAR1 and D2R heteromers. Eur. Neuropsychopharmacol. 2015, 25, 2049–2061. [Google Scholar] [CrossRef] [PubMed]
- Leo, D.; Mus, L.; Espinoza, S.; Hoener, M.C.; Sotnikova, T.D.; Gainetdinov, R.R. Taar1-mediated modulation of presynaptic dopaminergic neurotransmission: Role of D2 dopamine autoreceptors. Neuropharmacology 2014, 81, 283–291. [Google Scholar] [CrossRef]
- Revel, F.G.; Moreau, J.L.; Gainetdinov, R.R.; Bradaia, A.; Sotnikova, T.D.; Mory, R.; Durkin, S.; Zbinden, K.G.; Norcross, R.; Meyer, C.A.; et al. TAAR1 activation modulates monoaminergic neurotransmission, preventing hyperdopaminergic and hypoglutamatergic activity. Proc. Natl. Acad. Sci. USA 2011, 108, 8485–8490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolinsky, T.D.; Swanson, C.J.; Smith, K.E.; Zhong, H.; Borowsky, B.; Seeman, P.; Branchek, T.; Gerald, C.P. The Trace Amine 1 receptor knockout mouse: An animal model with relevance to schizophrenia. Genes Brain Behav. 2007, 6, 628–639. [Google Scholar] [CrossRef] [PubMed]
- Lindemann, L.; Meyer, C.A.; Jeanneau, K.; Bradaia, A.; Ozmen, L.; Bluethmann, H.; Bettler, B.; Wettstein, J.G.; Borroni, E.; Moreau, J.L.; et al. Trace amine-associated receptor 1 modulates dopaminergic activity. J. Pharmacol. Exp. Ther. 2008, 324, 948–956. [Google Scholar] [CrossRef] [Green Version]
- Sukhanov, I.; Caffino, L.; Efimova, E.V.; Espinoza, S.; Sotnikova, T.D.; Cervo, L.; Fumagalli, F.; Gainetdinov, R.R. Increased context-dependent conditioning to amphetamine in mice lacking TAAR1. Pharmacol. Res. 2016, 103, 206–214. [Google Scholar] [CrossRef]
- Koblan, K.S.; Kent, J.; Hopkins, S.C.; Krystal, J.H.; Cheng, H.; Goldman, R.; Loebel, A. A Non-D2-Receptor-Binding Drug for the Treatment of Schizophrenia. N. Engl. J. Med. 2020, 382, 1497–1506. [Google Scholar] [CrossRef]
- Espinoza, S.; Sukhanov, I.; Efimova, E.V.; Kozlova, A.; Antonova, K.A.; Illiano, P.; Leo, D.; Merkulyeva, N.; Kalinina, D.; Musienko, P.; et al. Trace Amine-Associated Receptor 5 Provides Olfactory Input Into Limbic Brain Areas and Modulates Emotional Behaviors and Serotonin Transmission. Front. Mol. Neurosci. 2020, 13, 18. [Google Scholar] [CrossRef]
- Dinter, J.; Muhlhaus, J.; Wienchol, C.L.; Yi, C.X.; Nurnberg, D.; Morin, S.; Gruters, A.; Kohrle, J.; Schoneberg, T.; Tschop, M.; et al. Inverse agonistic action of 3-iodothyronamine at the human trace amine-associated receptor 5. PLoS ONE 2015, 10, e0117774. [Google Scholar] [CrossRef] [Green Version]
- Vaganova, A.N.; Murtazina, R.Z.; Shemyakova, T.S.; Prjibelski, A.D.; Katolikova, N.V.; Gainetdinov, R.R. Pattern of TAAR5 Expression in the Human Brain Based on Transcriptome Datasets Analysis. Int. J. Mol. Sci. 2021, 22, 8802. [Google Scholar] [CrossRef]
- Gaudel, F.; Guiraudie-Capraz, G.; Feron, F. Limbic Expression of mRNA Coding for Chemoreceptors in Human Brain-Lessons from Brain Atlases. Int. J. Mol. Sci. 2021, 22, 6858. [Google Scholar] [CrossRef]
- Efimova, E.V.; Kozlova, A.A.; Razenkova, V.; Katolikova, N.V.; Antonova, K.A.; Sotnikova, T.D.; Merkulyeva, N.S.; Veshchitskii, A.S.; Kalinina, D.S.; Korzhevskii, D.E.; et al. Increased dopamine transmission and adult neurogenesis in trace amine-associated receptor 5 (TAAR5) knockout mice. Neuropharmacology 2021, 182, 108373. [Google Scholar] [CrossRef]
- Kalinina, D.S.; Ptukha, M.A.; Goriainova, A.V.; Merkulyeva, N.S.; Kozlova, A.A.; Murtazina, R.Z.; Shemiakova, T.S.; Kuvarzin, S.R.; Vaganova, A.N.; Volnova, A.B.; et al. Role of the trace amine associated receptor 5 (TAAR5) in the sensorimotor functions. Sci. Rep. 2021, 11, 23092. [Google Scholar] [CrossRef]
- Maggi, S.; Bon, C.; Gustincich, S.; Tucci, V.; Gainetdinov, R.R.; Espinoza, S. Improved Cognitive Performances in Trace Amine-Associated Receptor 5 (TAAR5) Knock-out Mice. Preprints 2022. [Google Scholar] [CrossRef]
- Gisladottir, R.S.; Ivarsdottir, E.V.; Helgason, A.; Jonsson, L.; Hannesdottir, N.K.; Rutsdottir, G.; Arnadottir, G.A.; Skuladottir, A.; Jonsson, B.A.; Norddahl, G.L.; et al. Sequence Variants in TAAR5 and Other Loci Affect Human Odor Perception and Naming. Curr. Biol. 2020, 30, 4643–4653.e3. [Google Scholar] [CrossRef]
- Li, Q.; Korzan, W.J.; Ferrero, D.M.; Chang, R.B.; Roy, D.S.; Buchi, M.; Lemon, J.K.; Kaur, A.W.; Stowers, L.; Fendt, M.; et al. Synchronous evolution of an odor biosynthesis pathway and behavioral response. Curr. Biol. 2013, 23, 11–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aleksandrov, A.A.; Dmitrieva, E.S.; Volnova, A.B.; Knyazeva, V.M.; Gerasimov, A.S.; Gainetdinov, R.R. TAAR5 receptor agonist affects sensory gating in rats. Neurosci. Lett. 2018, 666, 144–147. [Google Scholar] [CrossRef]
- Aleksandrov, A.A.; Dmitrieva, E.S.; Volnova, A.B.; Knyazeva, V.M.; Polyakova, N.V.; Ptukha, M.A.; Gainetdinov, R.R. Effect of alpha-NETA on auditory event related potentials in sensory gating study paradigm in mice. Neurosci. Lett. 2019, 712, 134470. [Google Scholar] [CrossRef]
- Cichero, E.; Espinoza, S.; Tonelli, M.; Franchini, S.; Gerasimov, A.S.; Sorbi, C.; Gainetdinov, R.R.; Brasili, L.; Fossa, P. A homology modelling-driven study leading to the discovery of the first mouse trace amine-associated receptor 5 (TAAR5) antagonists. MedChemComm 2016, 7, 353–364. [Google Scholar] [CrossRef]
- Lee, Y.; Warne, T.; Nehme, R.; Pandey, S.; Dwivedi-Agnihotri, H.; Chaturvedi, M.; Edwards, P.C.; Garcia-Nafria, J.; Leslie, A.G.W.; Shukla, A.K.; et al. Molecular basis of beta-arrestin coupling to formoterol-bound beta1-adrenoceptor. Nature 2020, 583, 862–866. [Google Scholar] [CrossRef]
- Espinoza, S.; Masri, B.; Salahpour, A.; Gainetdinov, R.R. BRET approaches to characterize dopamine and TAAR1 receptor pharmacology and signaling. Methods Mol. Biol. 2013, 964, 107–122. [Google Scholar]
- van de Waterbeemd, H.; Gifford, E. ADMET in silico modelling: Towards prediction paradise? Nat. Rev. Drug Discov. 2003, 2, 192–204. [Google Scholar] [CrossRef]
- Tonelli, M.; Espinoza, S.; Gainetdinov, R.R.; Cichero, E. Novel biguanide-based derivatives scouted as TAAR1 agonists: Synthesis, biological evaluation, ADME prediction and molecular docking studies. Eur. J. Med. Chem. 2017, 127, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Fan, P.; Rand, E.; Kyaw, H.; Su, K.; Madike, V.; Carter, K.C.; Li, Y. Cloning of a putative human neurotransmitter receptor expressed in skeletal muscle and brain. Biochem. Biophys. Res. Commun. 1998, 242, 575–578. [Google Scholar] [CrossRef]
- Aleksandrov, A.A.; Knyazeva, V.M.; Volnova, A.B.; Dmitrieva, E.S.; Korenkova, O.; Espinoza, S.; Gerasimov, A.; Gainetdinov, R.R. Identification of TAAR5 Agonist Activity of Alpha-NETA and Its Effect on Mismatch Negativity Amplitude in Awake Rats. Neurotox. Res. 2018, 34, 442–451. [Google Scholar] [CrossRef]
- Aleksandrov, A.A.; Knyazeva, V.M.; Volnova, A.B.; Dmitrieva, E.S.; Polyakova, N.V. Putative TAAR5 agonist alpha-NETA affects event-related potentials in oddball paradigm in awake mice. Brain Res. Bull. 2020, 158, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Aleksandrov, A.A.; Knyazeva, V.M.; Volnova, A.B.; Dmitrieva, E.S.; Polyakova, N.V.; Gainetdinov, R.R. Trace Amine-Associated Receptor 1 Agonist Modulates Mismatch Negativity-Like Responses in Mice. Front. Pharmacol. 2019, 10, 470. [Google Scholar] [CrossRef] [PubMed]
- Stecula, A.; Hussain, M.S.; Viola, R.E. Discovery of Novel Inhibitors of a Critical Brain Enzyme Using a Homology Model and a Deep Convolutional Neural Network. J. Med. Chem. 2020, 63, 8867–8875. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. What does AlphaFold mean for drug discovery? Nat. Rev. Drug Discov. 2021, 20, 725–727. [Google Scholar] [CrossRef] [PubMed]
- Hekkelman, M.L.; de Vries, I.; Joosten, R.P.; Perrakis, A. AlphaFill: Enriching the AlphaFold models with ligands and co-factors. bioRxiv 2021. [Google Scholar] [CrossRef]
- Lefkowitz, R.J. A brief history of G-protein coupled receptors (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 2013, 52, 6366–6378. [Google Scholar] [CrossRef] [PubMed]
- Barak, L.S.; Salahpour, A.; Zhang, X.; Masri, B.; Sotnikova, T.D.; Ramsey, A.J.; Violin, J.D.; Lefkowitz, R.J.; Caron, M.G.; Gainetdinov, R.R. Pharmacological characterization of membrane-expressed human trace amine-associated receptor 1 (TAAR1) by a bioluminescence resonance energy transfer cAMP biosensor. Mol. Pharmacol. 2008, 74, 585–594. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Abagyan, R.; Totrov, M.; Kuznetsov, D. ICM—A new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem. 1994, 15, 488–506. [Google Scholar] [CrossRef]
- Hsieh, C.H.; Li, L.; Vanhauwaert, R.; Nguyen, K.T.; Davis, M.D.; Bu, G.; Wszolek, Z.K.; Wang, X. Miro1 Marks Parkinson’s Disease Subset and Miro1 Reducer Rescues Neuron Loss in Parkinson’s Models. Cell Metab. 2019, 30, 1131–1140.e7. [Google Scholar] [CrossRef] [PubMed]
- Wallach, I.; Dzamba, M.; Heifets, A. AtomNet: A Deep Convolutional Neural Network for Bioactivity Prediction in Structure-based Drug Discovery. arXiv 2015, arXiv:1510.02855. [Google Scholar]
- Hendlich, M.; Rippmann, F.; Barnickel, G. LIGSITE: Automatic and efficient detection of potential small molecule-binding sites in proteins. J. Mol. Graph. Model. 1997, 15, 359–363. [Google Scholar] [CrossRef]
- Kingma, D.P.; Ba, J. Adam: A Method for Stochastic Optimization. arXiv 2014, arXiv:1412.6980. [Google Scholar]
- SMARTS Patterns. Available online: https://github.com/rdkit/rdkit/blob/master/Data/SmartsLib/RLewis_smarts.txt (accessed on 4 February 2022).
- Butina, D. Unsupervised Data Base Clustering Based on Daylight’s Fingerprint and Tanimoto Similarity: A Fast and Automated Way To Cluster Small and Large Data Sets. J. Chem. Inf. Comput. Sci. 1999, 39, 747–750. [Google Scholar] [CrossRef]
- Salahpour, A.; Espinoza, S.; Masri, B.; Lam, V.; Barak, L.S.; Gainetdinov, R.R. BRET biosensors to study GPCR biology, pharmacology, and signal transduction. Front. Endocrinol. 2012, 3, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ACD/Percepta Platform v14.0.0. Available online: www.acdlabs.com (accessed on 4 February 2022).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | cLogP a | MW b | HBA c | HBD d | nRot_bond e | TPSA f |
|---|---|---|---|---|---|---|
| 1a | 5.05 | 430 | 5 | 0 | 6 | 34.17 |
| 2a | 4.67 | 405 | 5 | 1 | 9 | 48.95 |
| 1 | 3.07 | 350 | 4 | 1 | 7 | 35.58 |
| 2 | 1.64 | 343 | 6 | 1 | 5 | 54.04 |
| Compound | HIA (%) a | LogBB b | LogPS c | Vd (l/kg) d | %PPB e | LogKa HSAf | %F (Oral) g |
|---|---|---|---|---|---|---|---|
| 1a | 100 | 0.56 | −1.1 | 6.9 | 97% | 4.50 | 89.3% |
| 2a | 100 | 0.31 | −1.3 | 4.3 | 98% | 4.24 | 99.3% |
| 1 | 100 | 0.46 | −1.8 | 4.4 | 77 | 3.43 | 99.5 |
| 2 | 100 | −0.07 | −1.9 | 2 | 71 | 3.90 | 99.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bon, C.; Chern, T.-R.; Cichero, E.; O’Brien, T.E.; Gustincich, S.; Gainetdinov, R.R.; Espinoza, S. Discovery of Novel Trace Amine-Associated Receptor 5 (TAAR5) Antagonists Using a Deep Convolutional Neural Network. Int. J. Mol. Sci. 2022, 23, 3127. https://doi.org/10.3390/ijms23063127
Bon C, Chern T-R, Cichero E, O’Brien TE, Gustincich S, Gainetdinov RR, Espinoza S. Discovery of Novel Trace Amine-Associated Receptor 5 (TAAR5) Antagonists Using a Deep Convolutional Neural Network. International Journal of Molecular Sciences. 2022; 23(6):3127. https://doi.org/10.3390/ijms23063127
Chicago/Turabian StyleBon, Carlotta, Ting-Rong Chern, Elena Cichero, Terrence E. O’Brien, Stefano Gustincich, Raul R. Gainetdinov, and Stefano Espinoza. 2022. "Discovery of Novel Trace Amine-Associated Receptor 5 (TAAR5) Antagonists Using a Deep Convolutional Neural Network" International Journal of Molecular Sciences 23, no. 6: 3127. https://doi.org/10.3390/ijms23063127
APA StyleBon, C., Chern, T. -R., Cichero, E., O’Brien, T. E., Gustincich, S., Gainetdinov, R. R., & Espinoza, S. (2022). Discovery of Novel Trace Amine-Associated Receptor 5 (TAAR5) Antagonists Using a Deep Convolutional Neural Network. International Journal of Molecular Sciences, 23(6), 3127. https://doi.org/10.3390/ijms23063127