
Neurofibromin Deficiency Causes Epidermal Growth Factor Receptor Upregulation through the Activation of Ras/ERK/SP1 Signaling Pathway in Neurofibromatosis Type 1-Associated Malignant Peripheral Nerve Sheet Tumor

 , , and
, , and 
Abstract
:1. Introduction
2. Results
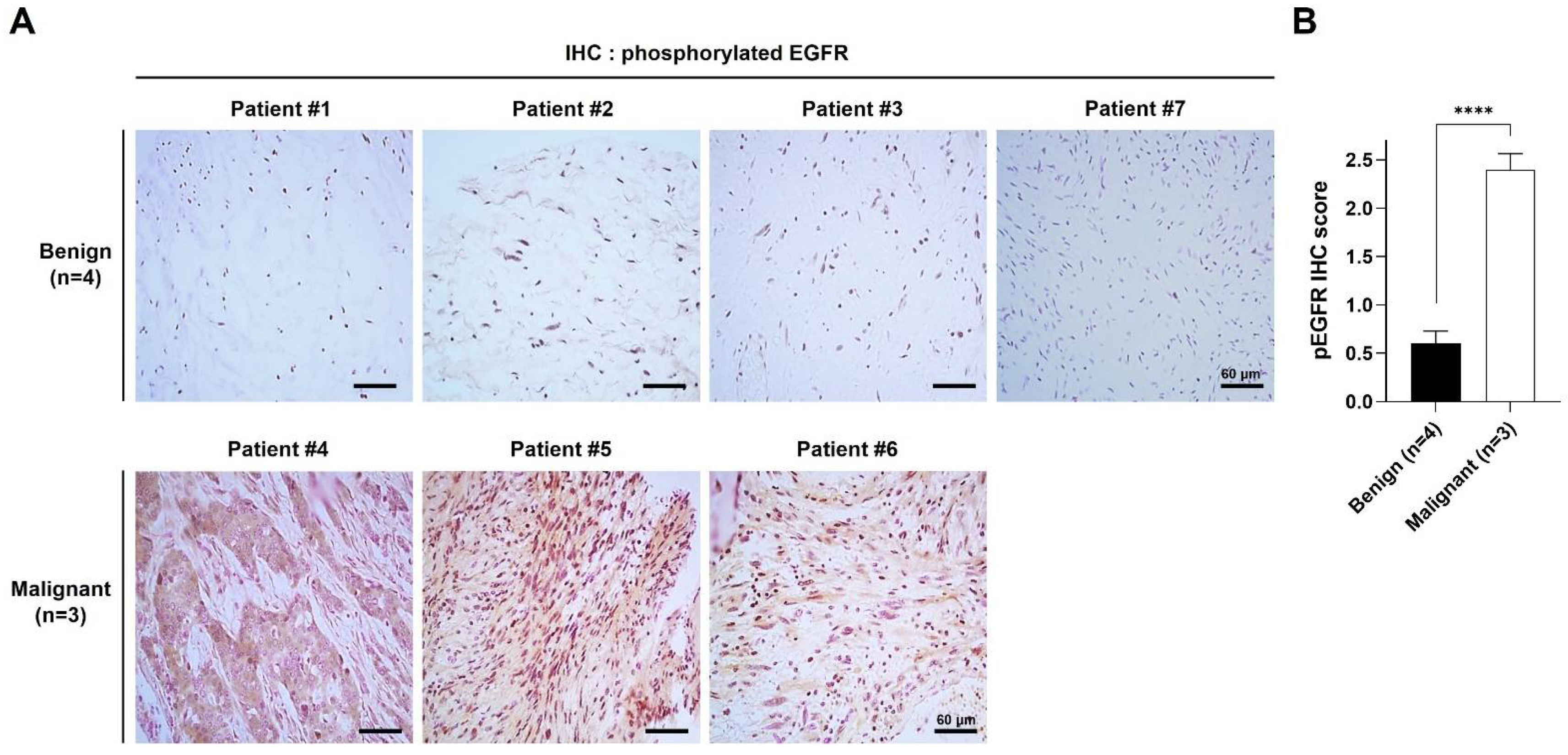
2.1. pEGFR Was Upregulated in the MPNST Tissues in Patients with NF1
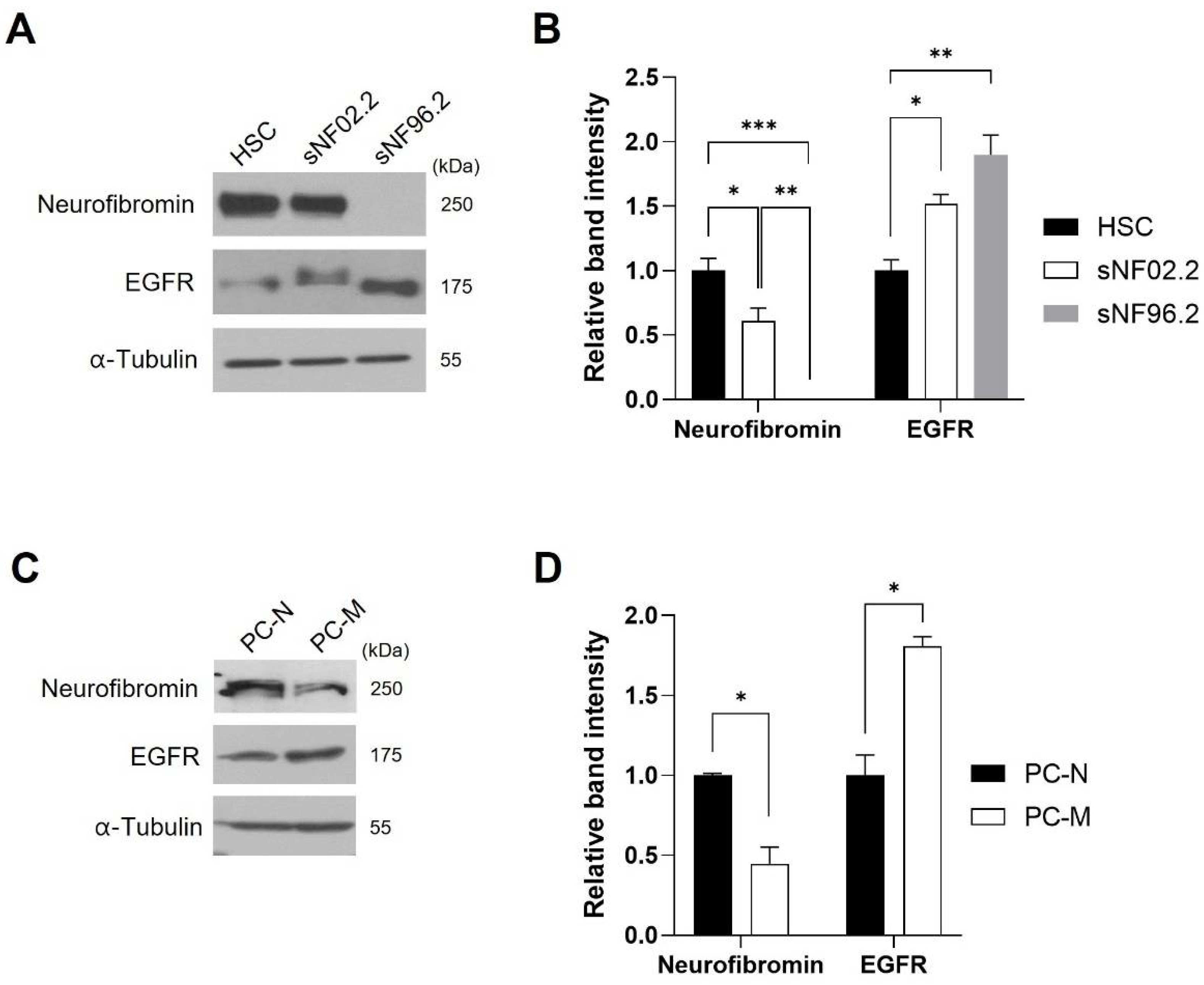
2.2. EGFR Expression Level Was Inversely Corelated with Neurofibromin Expression Level in the MPNST Cells
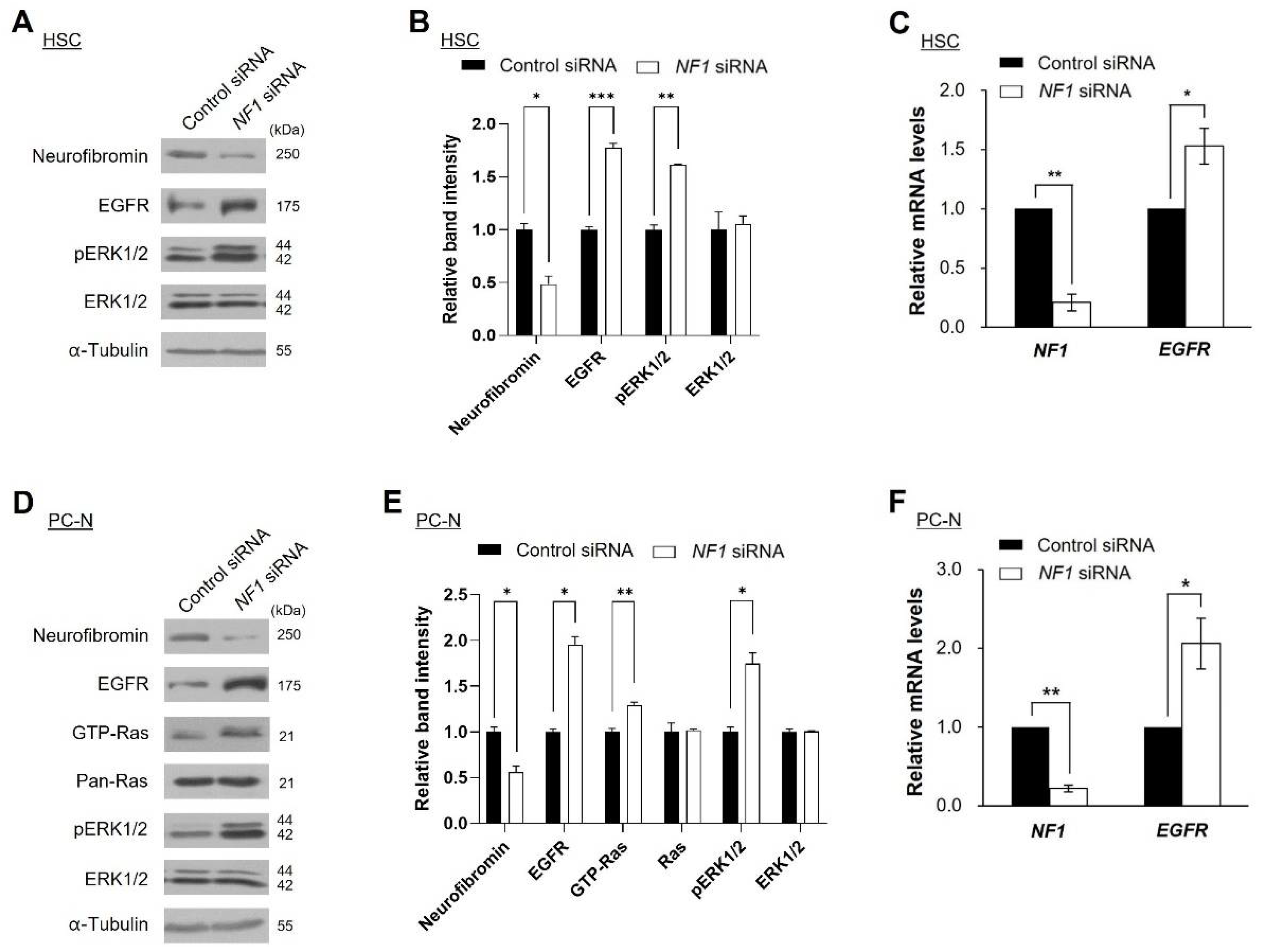
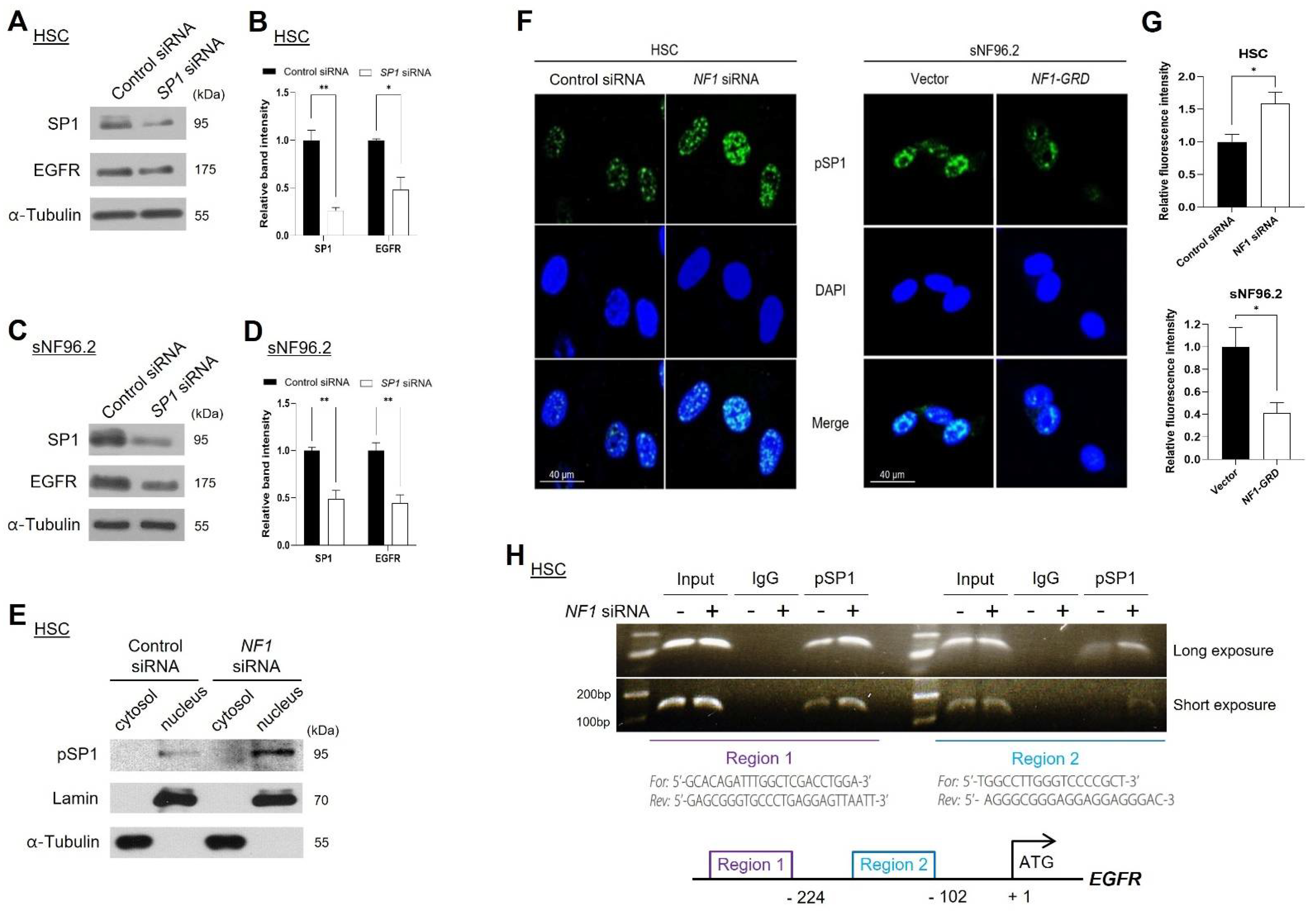
2.3. Knockdown of NF1 Promotes EGFR Expression
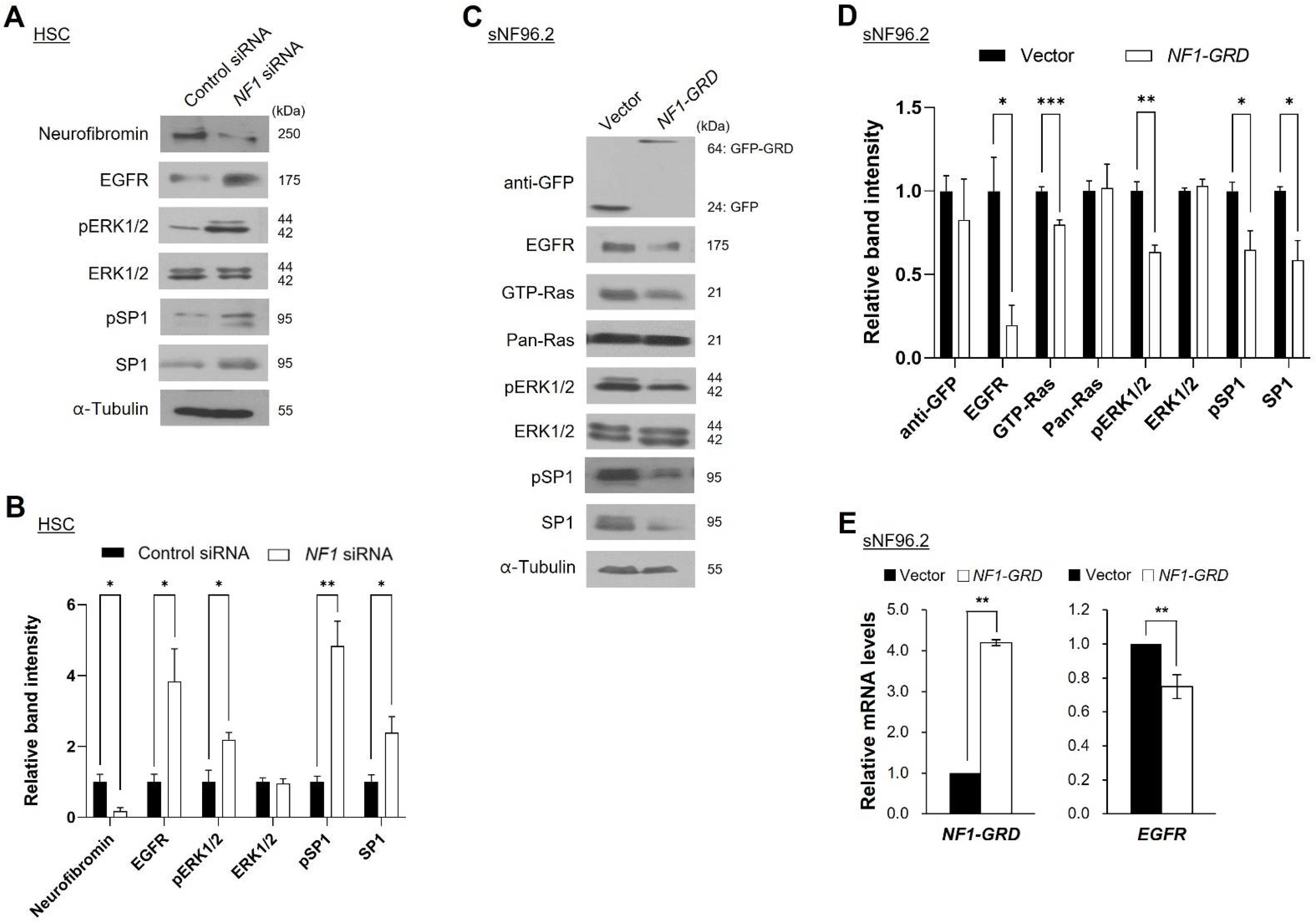
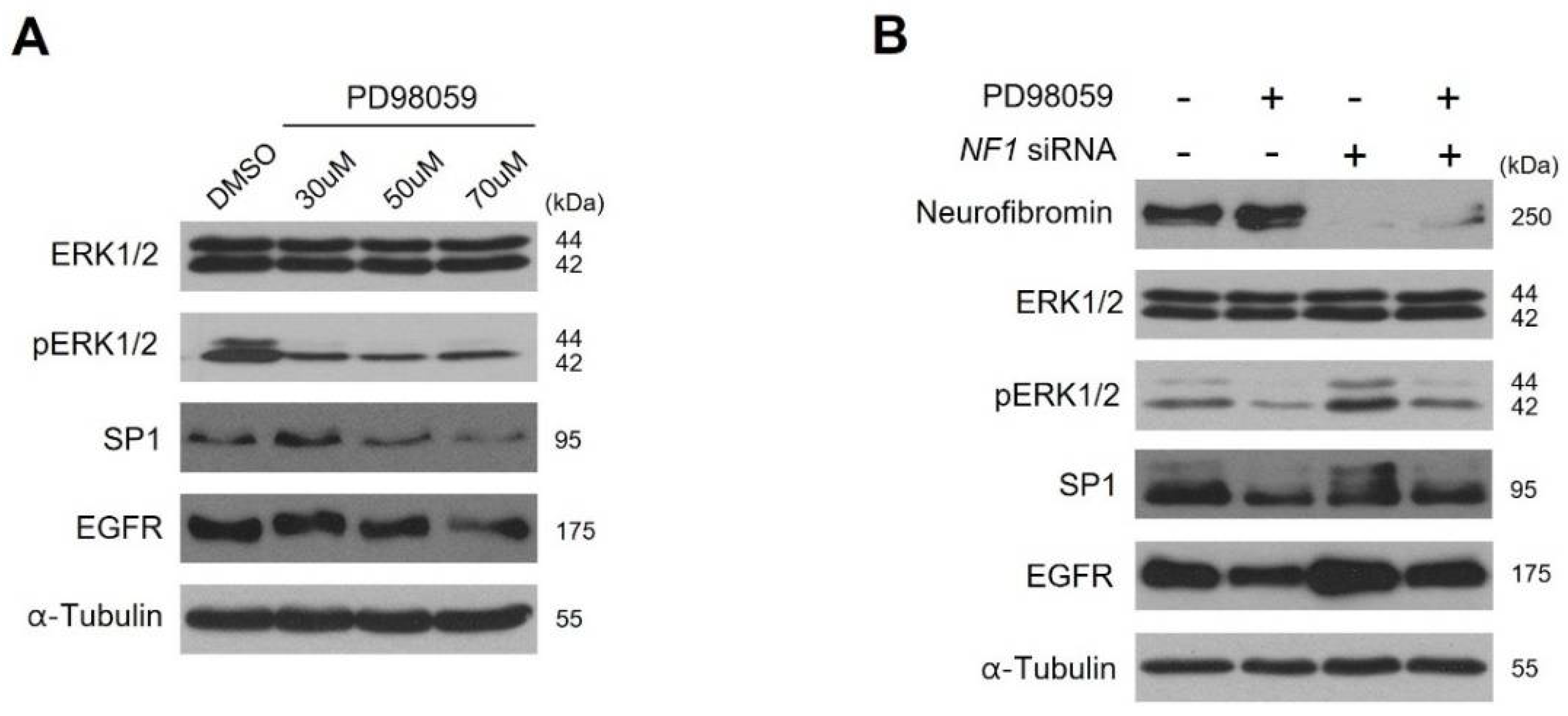
2.4. Neurofibromin Modulates the Ras/ERK/SP1 Dependent EGFR Expression
2.5. Inhibition of ERK1/2 Caused Downregulation of SP1 and There by Augmentation of EGFR Expression
2.6. Neurofibromin Regulated EGFR Expression through Controlling the Nuclear Localization and Binding Directly of SP1 to EGFR Promoter Regions
2.7. Combined EGFR Inhibitor and Doxorubicin Treatment Showed Synergistically Enhanced Antiproliferative Effect in NF1-Deficient MPNST Cells
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Patient Samples
4.3. Cell Proliferation Assays
4.4. Subcellular Fractionations and Western Blot
4.5. Chromatin Immunoprecipitation (ChIP) Assay
4.6. Immunocytochemistry
4.7. Immunohistochemistry (IHC)
4.8. Plasmids, Short Interfering RNAs (siRNAs), and Transfection
4.9. Quantitative Real-Time Reverse Transcription-Polymerase Chain Reaction (qRT-PCR)
4.10. Ras Activation Assay
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Barker, D.; Wright, E.; Nguyen, K.; Cannon, L.; Fain, P.; Goldgar, D.; Bishop, D.; Carey, J.; Baty, B.; Kivlin, J. Gene for von Recklinghausen neurofibromatosis is in the pericentromeric region of chromosome 17. Science 1987, 236, 1100–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legius, E.; Marchuk, D.A.; Collins, F.S.; Glover, T.W. Somatic deletion of the neurofibromatosis type 1 gene in a neurofibrosarcoma supports a tumour suppressor gene hypothesis. Nat. Genet. 1993, 3, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Ratner, N.; Miller, S.J. A RASopathy gene commonly mutated in cancer: The neurofibromatosis type 1 tumour suppressor. Nat. Rev. Cancer 2015, 15, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Anastasaki, C.; Gutmann, D.H. Neuronal NF1/RAS regulation of cyclic AMP requires atypical PKC activation. Hum. Mol. Genet 2014, 23, 6712–6721. [Google Scholar] [CrossRef] [Green Version]
- Crawford, A.H. Neurofibromatosis in children. Acta Orthop. Scand. 1986, 57, 8–60. [Google Scholar] [CrossRef]
- Ferner, R.E.; Gutmann, D.H. Neurofibromatosis type 1 (NF1): Diagnosis and management. Handb. Clin. Neurol. 2013, 115, 939–955. [Google Scholar]
- Cichowski, K.; Jacks, T. NF1 tumor suppressor gene function: Narrowing the GAP. Cell 2001, 104, 593–604. [Google Scholar] [CrossRef] [Green Version]
- Kluwe, L.; Friedrich, R.; Mautner, V.F. Loss of NF1 allele in Schwann cells but not in fibroblasts derived from an NF1-associated neurofibroma. Genes Chromosomes Cancer 1999, 24, 283–285. [Google Scholar] [CrossRef]
- Huson, S.; Harper, P.; Compston, D. Von Recklinghausen neurofibromatosis: A clinical and population study in south-east Wales. Brain 1988, 111, 1355–1381. [Google Scholar] [CrossRef]
- Evans, D.G.R.; Baser, M.E.; McGaughran, J.; Sharif, S.; Howard, E.; Moran, A. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J. Med. Genet. 2002, 39, 311–314. [Google Scholar] [CrossRef] [Green Version]
- Brennan, M.F.; Antonescu, C.R.; Moraco, N.; Singer, S. Lessons learned from the study of 10,000 patients with soft tissue sarcoma. Ann. Surg. 2014, 260, 416–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hruban, R.H.; Shiu, M.H.; Senie, R.T.; Woodruff, J.M. Malignant peripheral nerve sheath tumors of the buttock and lower extremity. A study of 43 cases. Cancer 1990, 66, 1253–1265. [Google Scholar] [CrossRef]
- Cheng, S.-F.; Chen, Y.-I.; Chang, C.-Y.; Peng, Y.; Liao, S.-L. Malignant peripheral nerve sheath tumor of the orbit: Malignant transformation from neurofibroma without neurofibromatosis. Ophthalmic Plast. Reconstr. Surg. 2008, 24, 413–415. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Liu, X.; Liu, Z.; Yang, S.; Liao, H.-I.; Jiang, L.; Wei, F. Malignant peripheral nerve sheath tumours of the spine: Clinical manifestations, classification, treatment, and prognostic factors. Eur. Spine J. 2012, 21, 897–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carroll, S.L. The challenge of cancer genomics in rare nervous system neoplasms: Malignant peripheral nerve sheath tumors as a paradigm for cross-species comparative oncogenomics. Am. J. Pathol. 2016, 186, 464–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olayioye, M.A.; Neve, R.M.; Lane, H.A.; Hynes, N.E. The ErbB signaling network: Receptor heterodimerization in development and cancer. EMBO Rep. 2000, 19, 3159–3167. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Gureasko, J.; Shen, K.; Cole, P.A.; Kuriyan, J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 2006, 125, 1137–1149. [Google Scholar] [CrossRef] [Green Version]
- Seshacharyulu, P.; Ponnusamy, M.P.; Haridas, D.; Jain, M.; Ganti, A.K.; Batra, S.K. Targeting the EGFR signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 15–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabone-Eglinger, S.; Bahleda, R.; Côté, J.-F.; Terrier, P.; Vidaud, D.; Cayre, A.; Beauchet, A.; Théou-Anton, N.; Terrier-Lacombe, M.-J.; Lemoine, A. Frequent EGFR positivity and overexpression in high-grade areas of human MPNSTs. Sarcoma 2008, 2008, 849156. [Google Scholar] [CrossRef] [Green Version]
- Tawbi, H.; Thomas, D.; Lucas, D.R.; Biermann, J.S.; Schuetze, S.M.; Hart, A.L.; Chugh, R.; Baker, L.H. Epidermal growth factor receptor expression and mutational analysis in synovial sarcomas and malignant peripheral nerve sheath tumors. Oncologist 2008, 13, 459–466. [Google Scholar] [CrossRef] [Green Version]
- Carroll, S.L.; Ratner, N. How does the Schwann cell lineage form tumors in NF1? Glia 2008, 56, 1590–1605. [Google Scholar] [CrossRef] [Green Version]
- DeClue, J.E.; Heffelfinger, S.; Benvenuto, G.; Ling, B.; Li, S.; Rui, W.; Vass, W.C.; Viskochil, D.; Ratner, N. Epidermal growth factor receptor expression in neurofibromatosis type 1-related tumors and NF1 animal models. J. Clin. Investig. 2000, 105, 1233–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, D.; Tainsky, M.A.; Haddad, R. Oncogene Mutation Survey in MPNST Cell Lines Enhances the Dominant Role of Hyperactive Ras in NF1 Associated Pro-Survival and Malignancy. Transl. Oncogenomics 2012, 5, 1–7. [Google Scholar] [CrossRef]
- Kahen, E.J.; Brohl, A.; Yu, D.; Welch, D.; Cubitt, C.L.; Lee, J.K.; Chen, Y.; Yoder, S.J.; Teer, J.K.; Zhang, Y.O. Neurofibromin level directs RAS pathway signaling and mediates sensitivity to targeted agents in malignant peripheral nerve sheath tumors. Oncotarget 2018, 9, 22571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.-J.; Park, H.-J.; Kim, Y.-H.; Kim, B.-Y.; Jin, H.-S.; Kim, H.J.; Han, J.-H.; Yim, H.; Jeong, S.-Y. Inhibition of Bcl-xL by ABT-737 enhances chemotherapy sensitivity in neurofibromatosis type 1-associated malignant peripheral nerve sheath tumor cells. J. Mol. Med. 2012, 30, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Kageyama, R.; Merlino, G.T.; Pastan, I. A transcription factor active on the epidermal growth factor receptor gene. Proc. Natl. Acad. Sci. USA 1988, 85, 5016–5020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merchant, J.L.; Du, M.; Todisco, A. Sp1 phosphorylation by Erk 2 stimulates DNA binding. Biochem. Biophys. Res. Commun. 1999, 254, 454–461. [Google Scholar] [CrossRef]
- Ito, T.; Kitamura, H.; Uwatoko, C.; Azumano, M.; Itoh, K.; Kuwahara, J. Interaction of Sp1 zinc finger with transport factor in the nuclear localization of transcription factor Sp1. Biochem. Biophys. Res. Commun. 2010, 403, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Schuch, R.; Agelopoulos, K.; Neumann, A.; Brandt, B.; Bürger, H.; Korsching, E. Site-specific chromatin immunoprecipitation: A selective method to individually analyze neighboring transcription factor binding sites in vivo. BMC Res. Notes 2012, 5, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koga, T.; Iwasaki, H.; Ishiguro, M.; Matsuzaki, A.; Kikuchi, M. Frequent genomic imbalances in chromosomes 17, 19, and 22q in peripheral nerve sheath tumours detected by comparative genomic hybridization analysis. J. Pathol. 2002, 197, 98–107. [Google Scholar] [CrossRef]
- Jeong, S.-Y.; Han, J.-H.; Park, Y.-Y.; Kim, H.-J. Identification of differentially expressed genes related to NF1-associated malignant transformation from a patient with neurofibromatosis type 1. Genes Genom. 2008, 30, 407–418. [Google Scholar]
- Dasgupta, B.; Gutmann, D.H. Neurofibromatosis 1: Closing the GAP between mice and men. Curr. Opin. Genet. Dev. 2003, 13, 20–27. [Google Scholar] [CrossRef]
- Watson, M.A.; Perry, A.; Tihan, T.; Prayson, R.A.; Guha, A.; Bridge, J.; Ferner, R.; Gutmann, D.H. Gene expression profiling reveals unique molecular subtypes of neurofibromatosis type I-associated and sporadic malignant peripheral nerve sheath tumors. Brain Pathol. 2004, 14, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Ling, B.C.; Wu, J.; Miller, S.J.; Monk, K.R.; Shamekh, R.; Rizvi, T.A.; DeCourten-Myers, G.; Vogel, K.S.; DeClue, J.E.; Ratner, N. Role for the epidermal growth factor receptor in neurofibromatosis-related peripheral nerve tumorigenesis. Cancer Cell 2005, 7, 65–75. [Google Scholar] [CrossRef] [Green Version]
- Williams, J.P.; Wu, J.; Johansson, G.; Rizvi, T.A.; Miller, S.C.; Geiger, H.; Malik, P.; Li, W.; Mukouyama, Y.S.; Cancelas, J.A.; et al. Nf1 mutation expands an EGFR-dependent peripheral nerve progenitor that confers neurofibroma tumorigenic potential. Cell Stem Cell 2008, 3, 658–669. [Google Scholar] [CrossRef] [Green Version]
- Karnoub, A.E.; Weinberg, R.A. Ras oncogenes: Split personalities. Nat. Rev. Mol. Cell Biol. 2008, 9, 517–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milanini-Mongiat, J.; Pouysségur, J.; Pagès, G. Identification of two Sp1 phosphorylation sites for p42/p44 mitogen-activated protein kinases: Their implication in vascular endothelial growth factor gene transcription. J. Biol. Chem. 2002, 277, 20631–20639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, C.; Santagata, S.; Tang, Z.; Shi, J.; Cao, J.; Kwon, H.; Bronson, R.T.; Whitesell, L.; Lindquist, S. Loss of tumor suppressor NF1 activates HSF1 to promote carcinogenesis. J. Clin. Investig. 2012, 122, 3742–3754. [Google Scholar] [CrossRef] [Green Version]
- Green, Y.S.; Sargis, T.; Reichert, E.C.; Rudasi, E.; Fuja, D.; Jonasch, E.; Koh, M.Y. Hypoxia-Associated Factor (HAF) Mediates Neurofibromin Ubiquitination and Degradation Leading to Ras–ERK Pathway Activation in Hypoxia. Mol. Cancer Res. 2019, 17, 1220–1232. [Google Scholar] [CrossRef] [Green Version]
- Errico, A.; Stocco, A.; Riccardi, V.M.; Gambalunga, A.; Bassetto, F.; Grigatti, M.; Ferlosio, A.; Tadini, G.; Garozzo, D.; Ferraresi, S. Neurofibromin Deficiency and Extracellular Matrix Cooperate to Increase Transforming Potential through FAK-Dependent Signaling. Cancers 2021, 13, 2329. [Google Scholar] [CrossRef]
- Wu, J.; Patmore, D.M.; Jousma, E.; Eaves, D.W.; Breving, K.; Patel, A.V.; Schwartz, E.B.; Fuchs, J.R.; Cripe, T.P.; Stemmer-Rachamimov, A.O. EGFR–STAT3 signaling promotes formation of malignant peripheral nerve sheath tumors. Oncogene 2014, 33, 173–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keizman, D.; Issakov, J.; Meller, I.; Meimon, N.; Ish-Shalom, M.; Sher, O.; Merimsky, O. Expression and significance of EGFR in malignant peripheral nerve sheath tumor. J. Neuro Oncol. 2009, 94, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Gottfried, O.N.; Viskochil, D.H.; Couldwell, W.T. Neurofibromatosis Type 1 and tumorigenesis: Molecular mechanisms and therapeutic implications. Neurosurg. Focus 2010, 28, E8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holtkamp, N.; Malzer, E.; Zietsch, J.; Okuducu, A.F.; Mucha, J.; Mawrin, C.; Mautner, V.-F.; Schildhaus, H.-U.; Von Deimling, A. EGFR and erbB2 in malignant peripheral nerve sheath tumors and implications for targeted therapy. Neuro Oncol. 2008, 10, 946–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albritton, K.; Rankin, C.; Coffin, C.; Ratner, N.; Budd, G.; Schuetze, S.; Randall, R.; Declue, J.; Borden, E. Phase II study of erlotinib in metastatic or unresectable malignant peripheral nerve sheath tumors (MPNST). Am. J. Clin. Oncol. 2006, 24, 9518. [Google Scholar] [CrossRef]
- Kroep, J.R.; Ouali, M.; Gelderblom, H.; Le Cesne, A.; Dekker, T.; Van Glabbeke, M.; Hogendoorn, P.; Hohenberger, P. First-line chemotherapy for malignant peripheral nerve sheath tumor (MPNST) versus other histological soft tissue sarcoma subtypes and as a prognostic factor for MPNST: An EORTC soft tissue and bone sarcoma group study. Ann. Oncol. 2011, 22, 207–214. [Google Scholar] [CrossRef] [PubMed]
- D’Adamo, D.R. Appraising the current role of chemotherapy for the treatment of sarcoma. Semin. Oncol. 2011, 38, S19–S29. [Google Scholar] [CrossRef] [PubMed]
- Takemura, G.; Fujiwara, H. Doxorubicin-induced cardiomyopathy: From the cardiotoxic mechanisms to management. Prog. Cardiovasc. Dis. 2007, 49, 330–352. [Google Scholar] [CrossRef]
- Park, H.-J.; Lee, S.-J.; Sohn, Y.B.; Jin, H.-S.; Han, J.H.; Kim, Y.-B.; Yim, H.; Jeong, S.-Y. NF1 deficiency causes Bcl-xL upregulation in Schwann cells derived from neurofibromatosis type 1-associated malignant peripheral nerve sheath tumors. Int. J. Oncol. 2013, 42, 657–666. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patients | Histological Findings | Clinical Features | Genotype | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ID | Gender | Age at Diagnosis | H&E | S100 | pEGFR | Café-au-Lait Spots | Neuro-Fibromas | Freckling | Optic Glioma | Lisch Nodule | Skeletal Dysplasia | Family History | NF1 Mutation |
| P1 | Male | 59 | Benign | + | + | Y | Y | Y | N | N | N | Y | N/A |
| P2 | Male | 42 | Benign | + | + | Y | Y | Y | N | N | N | Y | N/A |
| P3 | Female | 5 | Benign | + | + | Y | Y | Y | N | N | N | Y | N/A |
| P4 | Male | 39 | Malignant | + | ++ | Y | Y | N | N | N | N | N | N/A |
| P5 | Male | 32 | Malignant | + | ++ | Y | Y | N | N | N | N | N | c.4861_4862 GT>AG |
| P6 | Female | 41 | Malignant | + | ++ | Y | Y | Y | N | N | N | N | N/A |
| P7 | Male | 12 | Benign | + | + | Y | Y | Y | N | Y | Y | N | c.4537C>T |
| 17 | Benign | + | ++ | Y | Y | Y | N | Y | Y | N | c.4537C>T | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, G.-H.; Lee, S.-J.; Lee, C.-G.; Kim, J.; Park, E.; Jeong, S.-Y. Neurofibromin Deficiency Causes Epidermal Growth Factor Receptor Upregulation through the Activation of Ras/ERK/SP1 Signaling Pathway in Neurofibromatosis Type 1-Associated Malignant Peripheral Nerve Sheet Tumor. Int. J. Mol. Sci. 2021, 22, 13308. https://doi.org/10.3390/ijms222413308
Park G-H, Lee S-J, Lee C-G, Kim J, Park E, Jeong S-Y. Neurofibromin Deficiency Causes Epidermal Growth Factor Receptor Upregulation through the Activation of Ras/ERK/SP1 Signaling Pathway in Neurofibromatosis Type 1-Associated Malignant Peripheral Nerve Sheet Tumor. International Journal of Molecular Sciences. 2021; 22(24):13308. https://doi.org/10.3390/ijms222413308
Chicago/Turabian StylePark, Gun-Hoo, Su-Jin Lee, Chang-Gun Lee, Jeonghyun Kim, Eunkuk Park, and Seon-Yong Jeong. 2021. "Neurofibromin Deficiency Causes Epidermal Growth Factor Receptor Upregulation through the Activation of Ras/ERK/SP1 Signaling Pathway in Neurofibromatosis Type 1-Associated Malignant Peripheral Nerve Sheet Tumor" International Journal of Molecular Sciences 22, no. 24: 13308. https://doi.org/10.3390/ijms222413308
APA StylePark, G. -H., Lee, S. -J., Lee, C. -G., Kim, J., Park, E., & Jeong, S. -Y. (2021). Neurofibromin Deficiency Causes Epidermal Growth Factor Receptor Upregulation through the Activation of Ras/ERK/SP1 Signaling Pathway in Neurofibromatosis Type 1-Associated Malignant Peripheral Nerve Sheet Tumor. International Journal of Molecular Sciences, 22(24), 13308. https://doi.org/10.3390/ijms222413308