
Ru(III) Complexes with Lonidamine-Modified Ligands

 ,
,  ,
,  , , , ,
, , , , 

Abstract
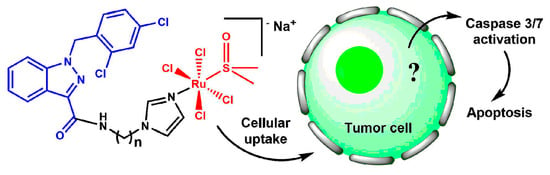
:
1. Introduction
2. Results and Discussion
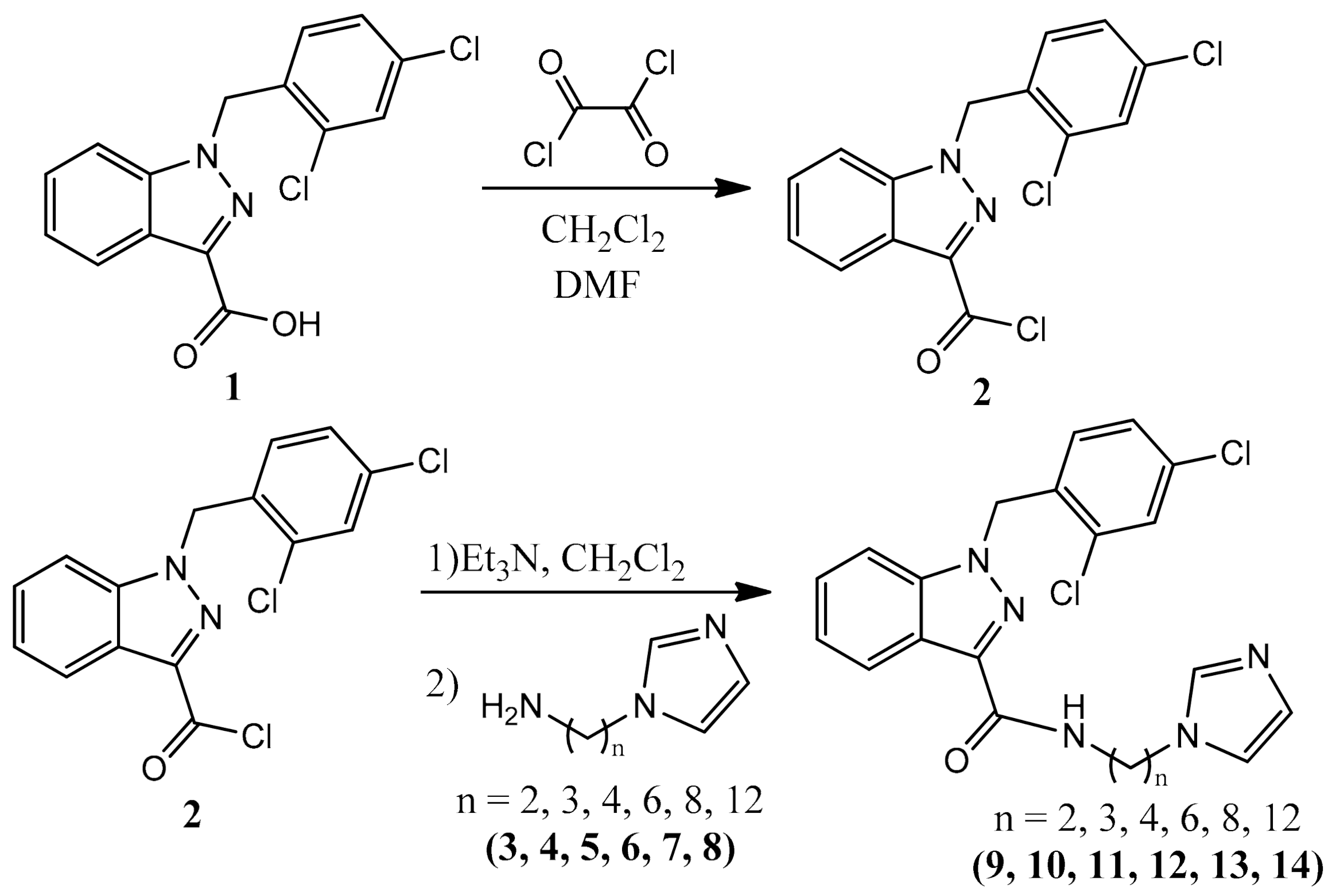
2.1. Synthesis
2.2. Electrochemical Studies of Ru(III) Complexes
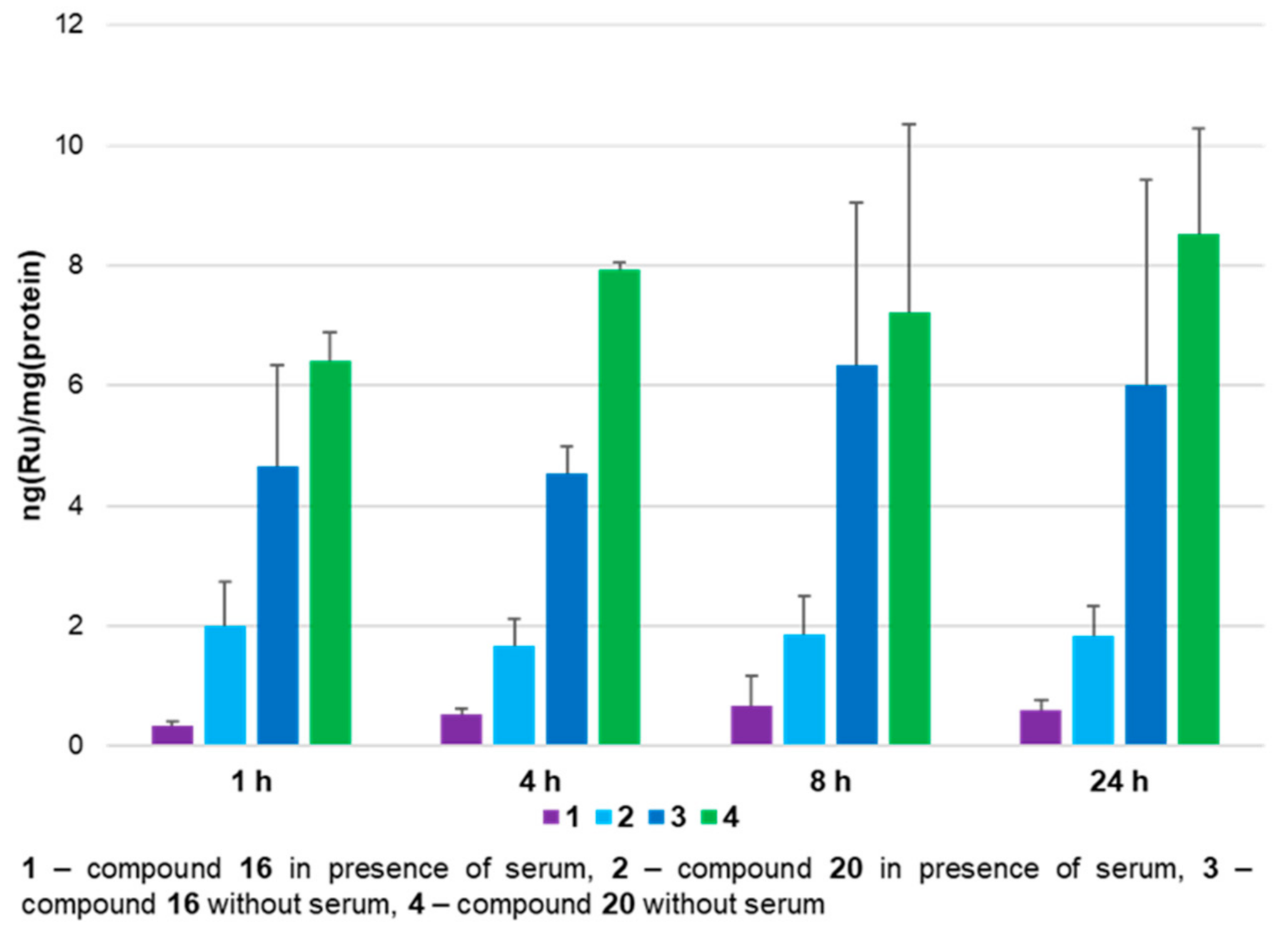
2.3. Cellular Ruthenium Accumulation
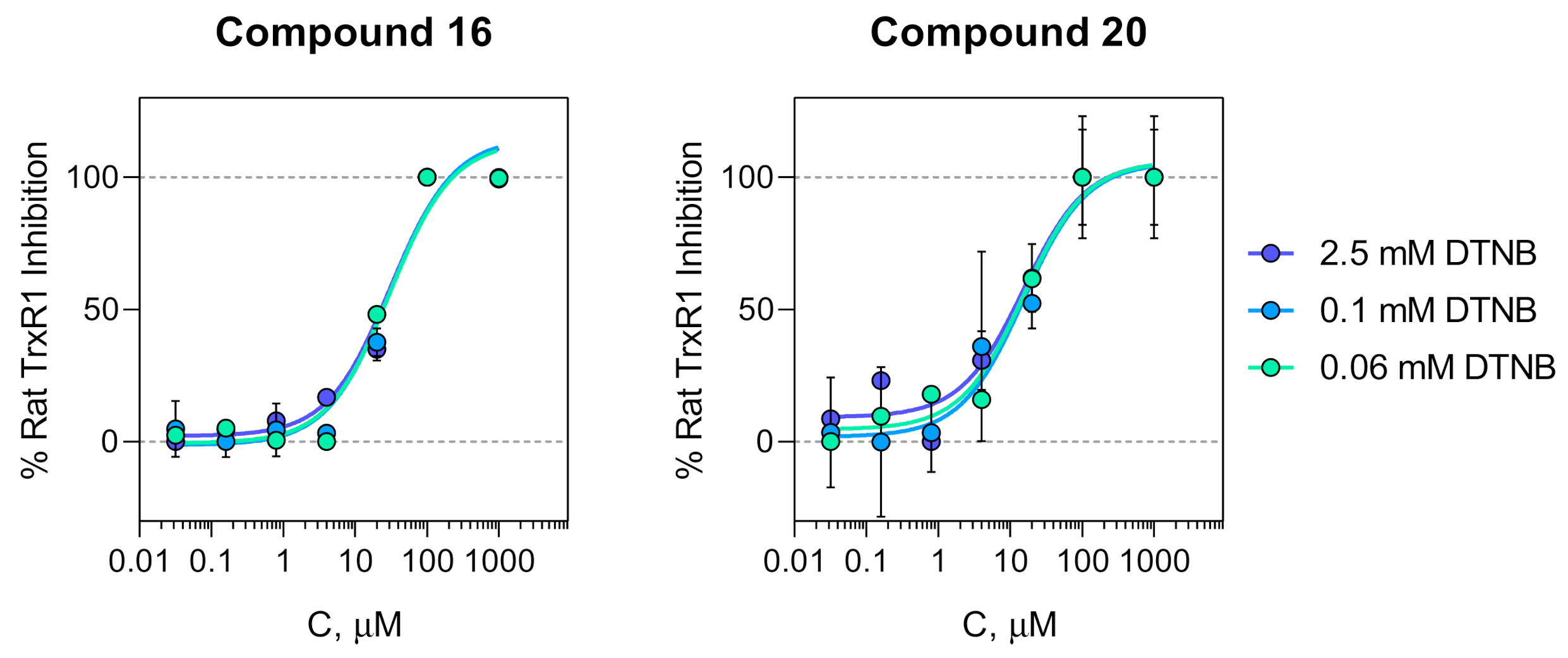
2.4. Inhibition of TrxR
2.5. Cell Death Studies
2.6. Tolerance of 16 In Vivo
3. Materials and Methods
3.1. N-(2-(1H-Imidazol-1-yl)ethyl)-1-(2,4-dichlorobenzyl)-1H-indazole-3-carboxamide (9)

3.2. N-(3-(1H-Imidazol-1-yl)propyl)-1-(2,4-dichlorobenzyl)-1H-indazole-3-carboxamide (10)

3.3. N-(4-(1H-Imidazol-1-yl)butyl)-1-(2,4-dichlorobenzyl)-1H-indazole-3-carboxamide (11)

3.4. N-(6-(1H-Imidazol-1-yl)hexyl)-1-(2,4-dichlorobenzyl)-1H-indazole-3-carboxamide (12)

3.5. N-(8-(1H-Imidazol-1-yl)octyl)-1-(2,4-dichlorobenzyl)-1H-indazole-3-carboxamide (13)

3.6. N-(12-(1H-Imidazol-1-yl)dodecyl)-1-(2,4-dichlorobenzyl)-1H-indazole-3-carboxamide (14)

3.7. Na[trans-Ru(DMSO)(C20H17Cl2N5O)Cl4] (15)
3.8. Na[trans-Ru(DMSO)(C21H19Cl2N5O)Cl4] (16)
3.9. Na[trans-Ru(DMSO)(C22H21Cl2N5O)Cl4] (17)
3.10. Na[trans-Ru(DMSO)(C24H25Cl2N5O)Cl4] (18)
3.11. Na[trans-Ru(DMSO)(C26H29Cl2N5O)Cl4] (19)
3.12. Na[trans-Ru(DMSO)(C30H37Cl2N5O)Cl4] (20)
3.13. Electrochemical Activity
3.14. Stability
3.15. Lipophilicity
3.16. Cellular Accumulation of Ruthenium Determined by Atomic Absorption Spectrometry
3.17. Inhibition of TrxR
3.17.1. Inhibition of Purified Protein
3.17.2. Inhibition of Intracellular TrxR1
3.18. Cell Death Studies
3.19. In Vivo Acute Toxicity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Rademaker-Lakhai, J.M.; van den Bongard, D.; Pluim, D.; Beijnen, J.H.; Schellens, J.H. A Phase I and pharmacological study with imidazolium-trans-DMSO-imidazole-tetrachlororuthenate, a novel ruthenium anticancer agent. Clin. Cancer Res. 2004, 10, 3717–3727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakupec, M.A.; Arion, V.B.; Kapitza, S.; Reisner, E.; Eichinger, A.; Pongratz, M.; Marian, B.; Graf von Keyserlingk, N.; Keppler, B.K. KP1019 (FFC14A) from bench to bedside: Preclinical and early clinical development—An overview. Int. J. Clin. Pharmacol. Ther. 2005, 43, 595–596. [Google Scholar] [CrossRef] [PubMed]
- Hartinger, C.G.; Jakupec, M.A.; Zorbas-Seifried, S.; Groessl, M.; Egger, A.; Berger, W.; Zorbas, H.; Dyson, P.J.; Keppler, B.K. KP1019, A New Redox-Active Anticancer Agent—Preclinical Development and Results of a Clinical Phase I Study in Tumor Patients. Chem. Biodivers. 2008, 5, 2140–2155. [Google Scholar] [CrossRef] [PubMed]
- Lentz, F.; Drescher, A.; Lindauer, A.; Henke, M.; Hilger, R.A.; Hartinger, C.G.; Scheulen, M.E.; Dittrich, C.; Keppler, B.K.; Jaehde, U. Pharmacokinetics of a novel anticancer ruthenium complex (KP1019, FFC14A) in a phase I dose-escalation study. Anticancer Drugs 2009, 20, 97–103. [Google Scholar] [CrossRef]
- Alessio, E. Thirty Years of the Drug Candidate NAMI-A and the Myths in the Field of Ruthenium Anticancer Compounds: A Personal Perspective. Eur. J. Inorg. Chem. 2017, 2017, 1549–1560. [Google Scholar] [CrossRef]
- Alessio, E.; Messori, L. NAMI-A and KP1019/1339, Two Iconic Ruthenium Anticancer Drug Candidates Face-to-Face: A Case Story in Medicinal Inorganic Chemistry. Molecules 2019, 24, 1995. [Google Scholar] [CrossRef] [Green Version]
- Coverdale, J.P.C.; Laroiya-McCarron, T.; Romero-Canelón, I. Designing Ruthenium Anticancer Drugs: What Have We Learnt from the Key Drug Candidates? Inorganics 2019, 7, 31. [Google Scholar] [CrossRef] [Green Version]
- Sava, G.; Gagliardi, R.; Bergamo, A.; Alessio, E.; Mestroni, G. Treatment of metastases of solid mouse tumours by NAMI-A: Comparison with cisplatin, cyclophosphamide and dacarbazine. Anticancer Res. 1999, 19, 969–972. [Google Scholar]
- Vadori, M.; Florio, C.; Groppo, B.; Cocchietto, M.; Pacor, S.; Zorzet, S.; Candussio, L.; Sava, G. The antimetastatic drug NAMI-A potentiates the phenylephrine-induced contraction of aortic smooth muscle cells and induces a transient increase in systolic blood pressure. J. Biol. Inorg. Chem. 2015, 20, 831–840. [Google Scholar] [CrossRef]
- Brescacin, L.; Masi, A.; Sava, G.; Bergamo, A. Effects of the ruthenium-based drug NAMI-A on the roles played by TGF-β1 in the metastatic process. J. Biol. Inorg. Chem. 2015, 20, 1163–1173. [Google Scholar] [CrossRef]
- Bergamo, A.; Dyson, P.J.; Sava, G. The mechanism of tumour cell death by metal-based anticancer drugs is not only a matter of DNA interactions. Coord. Chem. Rev. 2018, 360, 17–33. [Google Scholar] [CrossRef]
- Hartinger, C.G.; Zorbas-Seifried, S.; Jakupec, M.A.; Kynast, B.; Zorbas, H.; Keppler, B.K. From bench to bedside—Preclinical and early clinical development of the anticancer agent indazolium trans-[tetrachlorobis(1H-indazole)ruthenate(III)] (KP1019 or FFC14A). J. Inorg. Biochem. 2006, 100, 891–904. [Google Scholar] [CrossRef]
- Burris, H.A.; Bakewell, S.; Bendell, J.C.; Infante, J.; Jones, S.F.; Spigel, D.R.; Weiss, G.J.; Ramanathan, R.K.; Ogden, A.; Von Hoff, D. Safety and activity of IT-139, a ruthenium-based compound, in patients with advanced solid tumours: A first-in-human, open-label, dose-escalation phase I study with expansion cohort. ESMO Open 2016, 1, e000154. [Google Scholar] [CrossRef] [Green Version]
- Bakewell, S.; Conde, I.; Fallah, Y.; McCoy, M.; Jin, L.; Shajahan-Haq, A.N. Inhibition of DNA Repair Pathways and Induction of ROS Are Potential Mechanisms of Action of the Small Molecule Inhibitor BOLD-100 in Breast Cancer. Cancers 2020, 12, 2647. [Google Scholar] [CrossRef]
- Neuditschko, B.; Legin, A.A.; Baier, D.; Schintlmeister, A.; Reipert, S.; Wagner, M.; Keppler, B.K.; Berger, W.; Meier-Menches, S.M.; Gerner, C. Interaction with Ribosomal Proteins Accompanies Stress Induction of the Anticancer Metallodrug BOLD-100/KP1339 in the Endoplasmic Reticulum. Angew. Chem. Int. Ed. 2021, 60, 5063–5068. [Google Scholar] [CrossRef]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Pelicano, H.; Martin, D.S.; Xu, R.H.; Huang, P. Glycolysis inhibition for anticancer treatment. Oncogene 2006, 25, 4633–4646. [Google Scholar] [CrossRef] [Green Version]
- Lis, P.; Dyląg, M.; Niedźwiecka, K.; Ko, Y.H.; Pedersen, P.L.; Goffeau, A.; Ułaszewski, S. The HK2 Dependent “Warburg Effect” and Mitochondrial Oxidative Phosphorylation in Cancer: Targets for Effective Therapy with 3-Bromopyruvate. Molecules 2016, 21, 1730. [Google Scholar] [CrossRef] [Green Version]
- Floridi, A.; Paggi, M.G.; D’Atri, S.; De Martino, C.; Marcante, M.L.; Silvestrini, B.; Caputo, A. Effect of Lonidamine on the Energy Metabolism of Ehrlich Ascites Tumor Cells. Cancer Res. 1981, 41, 4661–4666. [Google Scholar]
- Nath, K.; Guo, L.; Nancolas, B.; Nelson, D.S.; Shestov, A.A.; Lee, S.-C.; Roman, J.; Zhou, R.; Leeper, D.B.; Halestrap, A.P.; et al. Mechanism of antineoplastic activity of lonidamine. Biochim. Biophys. Acta BBA Rev. Cancer 2016, 1866, 151–162. [Google Scholar] [CrossRef] [Green Version]
- Berruti, A.; Bitossi, R.; Gorzegno, G.; Bottini, A.; Alquati, P.; De Matteis, A.; Nuzzo, F.; Giardina, G.; Danese, S.; De Lena, M.; et al. Time to progression in metastatic breast cancer patients treated with epirubicin is not improved by the addition of either cisplatin or lonidamine: Final results of a phase III study with a factorial design. J. Clin. Oncol. 2002, 20, 4150–4159. [Google Scholar] [CrossRef]
- Armarego, W.L.F.; Chai, C. Purification of Laboratory Chemicals, 5th ed.; Butterworth-Heinemann: Oxford, UK, 2003; p. 608. [Google Scholar]
- Cervantes-Madrid, D.; Romero, Y.; Dueñas-González, A. Reviving Lonidamine and 6-Diazo-5-oxo-L-norleucine to Be Used in Combination for Metabolic Cancer Therapy. BioMed Res. Int. 2015, 2015, 690492. [Google Scholar] [CrossRef] [Green Version]
- Gelemanović, A.; Vidović, T.; Stepanić, V.; Trajković, K. Identification of 37 Heterogeneous Drug Candidates for Treatment of COVID-19 via a Rational Transcriptomics-Based Drug Repurposing Approach. Pharmaceuticals 2021, 14, 87. [Google Scholar] [CrossRef]
- Gil-Moles, M.; Türck, S.; Basu, U.; Pettenuzzo, A.; Bhattacharya, S.; Rajan, A.; Ma, X.; Büssing, R.; Wölker, J.; Burmeister, H.; et al. Metallodrug Profiling against SARS-CoV-2 Target Proteins Identifies Highly Potent Inhibitors of the S/ACE2 interaction and the Papain-like Protease PLpro. Chem. Eur. J. 2021, hal-03413330. [Google Scholar] [CrossRef]
- Kenny, R.G.; Marmion, C.J. Toward Multi-Targeted Platinum and Ruthenium Drugs—A New Paradigm in Cancer Drug Treatment Regimens? Chem. Rev. 2019, 119, 1058–1137. [Google Scholar] [CrossRef]
- Kostrhunova, H.; Zajac, J.; Markova, L.; Brabec, V.; Kasparkova, J. A Multi-action PtIV Conjugate with Oleate and Cinnamate Ligands Targets Human Epithelial Growth Factor Receptor HER2 in Aggressive Breast Cancer Cells. Angew. Chem. Int. Ed. 2020, 59, 21157–21162. [Google Scholar] [CrossRef]
- Tremlett, W.D.J.; Goodman, D.M.; Steel, T.R.; Kumar, S.; Wieczorek-Błauż, A.; Walsh, F.P.; Sullivan, M.P.; Hanif, M.; Hartinger, C.G. Design concepts of half-sandwich organoruthenium anticancer agents based on bidentate bioactive ligands. Coord. Chem. Rev. 2021, 445, 213950. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, Z.; Deng, Z.; Zhu, G. Recent advances in the synthesis, stability, and activation of platinum(IV) anticancer prodrugs. Coord. Chem. Rev. 2021, 442, 213991. [Google Scholar] [CrossRef]
- Nazarov, A.A.; Gardini, D.; Baquie, M.; Juillerat-Jeanneret, L.; Serkova, T.P.; Shevtsova, E.P.; Scopelliti, R.; Dyson, P.J. Organometallic anticancer agents that interfere with cellular energy processes: A subtle approach to inducing cancer cell death. Dalton Trans. 2013, 42, 2347–2350. [Google Scholar] [CrossRef]
- Nosova, Y.N.; Foteeva, L.S.; Zenin, I.V.; Fetisov, T.I.; Kirsanov, K.I.; Yakubovskaya, M.G.; Antonenko, T.A.; Tafeenko, V.A.; Aslanov, L.A.; Lobas, A.A.; et al. Enhancing the Cytotoxic Activity of Anticancer PtIV Complexes by Introduction of Lonidamine as an Axial Ligand. Eur. J. Inorg. Chem. 2017, 2017, 1785–1791. [Google Scholar] [CrossRef]
- Okulova, Y.N.; Zenin, I.V.; Shutkov, I.A.; Kirsanov, K.I.; Kovaleva, O.N.; Lesovaya, E.A.; Fetisov, T.I.; Milaeva, E.R.; Nazarov, A.A. Antiproliferative activity of Pt(IV) complexes with lonidamine and bexarotene ligands attached via succinate-ethylenediamine linker. Inorg. Chim. Acta 2019, 495, 119010. [Google Scholar] [CrossRef]
- Shutkov, I.A.; Antonets, A.A.; Tyurin, V.Y.; Milaeva, E.R.; Nazarov, A.A. Ruthenium(III) Complexes of NAMI-A Type with Ligands Based on Lonidamine and Bexarotene as Antiproliferative Agents. Russ. J. Inorg. Chem. 2021, 66, 502–509. [Google Scholar] [CrossRef]
- Nosova, Y.N.; Zenin, I.V.; Maximova, V.P.; Zhidkova, E.M.; Kirsanov, K.I.; Lesovaya, E.A.; Lobas, A.A.; Gorshkov, M.V.; Kovaleva, O.N.; Milaeva, E.R.; et al. Influence of the Number of Axial Bexarotene Ligands on the Cytotoxicity of Pt(IV) Analogs of Oxaliplatin. Bioinorg. Chem. Appl. 2017, 2017, 4736321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmgren, A.; Lu, J. Thioredoxin and thioredoxin reductase: Current research with special reference to human disease. Biochem. Biophys. Res. Commun. 2010, 396, 120–124. [Google Scholar] [CrossRef] [Green Version]
- Saccoccia, F.; Angelucci, F.; Boumis, G.; Carotti, D.; Desiato, G.; Miele, A.E.; Bellelli, A. Thioredoxin reductase and its inhibitors. Curr. Protein Pept. Sci. 2014, 15, 621–646. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Zhang, J.; Peng, S.; Liu, R.; Li, X.; Hou, Y.; Han, X.; Fang, J. Thioredoxin reductase inhibitors: A patent review. Expert Opin. Ther. Pat. 2017, 27, 547–556. [Google Scholar] [CrossRef]
- Zhang, B.; Liu, Y.; Li, X.; Xu, J.; Fang, J. Small Molecules to Target the Selenoprotein Thioredoxin Reductase. Chem. Asian J. 2018, 13, 3593–3600. [Google Scholar] [CrossRef]
- Shpakovsky, D.B.; Shtil, A.A.; Kharitonashvili, E.V.; Tyurin, V.Y.; Antonenko, T.A.; Nazarov, A.A.; Osipova, V.P.; Berberova, N.T.; Foteeva, L.S.; Schmidt, C.; et al. The antioxidant 2,6-di-tert-butylphenol moiety attenuates the pro-oxidant properties of the auranofin analogue. Metallomics 2018, 10, 406–413. [Google Scholar] [CrossRef]
- Alessio, E.; Balducci, G.; Lutman, A.; Mestroni, G.; Calligaris, M.; Attia, W.M. Synthesis and characterization of two new classes of ruthenium(III)-sulfoxide complexes with nitrogen donor ligands (L): Na[trans-RuCl4(R2SO)(L)] and mer, cis-RuCl3(R2SO)(R2SO)(L). The crystal structure of Na[trans-RuCl4(DMSO)(NH3)] 2DMSO, Na[trans-RuCl4(DMSO)(Im)] H2O, Me2CO (Im = imidazole) and mer, cis-RuCl3(DMSO)(DMSO)(NH3). Inorg. Chim. Acta 1993, 203, 205–217. [Google Scholar] [CrossRef]
- Schluga, P.; Hartinger, C.G.; Egger, A.; Reisner, E.; Galanski, M.; Jakupec, M.A.; Keppler, B.K. Redox behavior of tumor-inhibiting ruthenium(III) complexes and effects of physiological reductants on their binding to GMP. Dalton Trans. 2006, 1796–1802. [Google Scholar] [CrossRef]
- Reisner, E.; Arion, V.B.; Guedes da Silva, M.F.C.; Lichtenecker, R.; Eichinger, A.; Keppler, B.K.; Kukushkin, V.Y.; Pombeiro, A.J.L. Tuning of Redox Potentials for the Design of Ruthenium Anticancer Drugs—An Electrochemical Study of [trans-RuCl4L(DMSO)]- and [trans-RuCl4L2]- Complexes, where L = Imidazole, 1,2,4-Triazole, Indazole. Inorg. Chem. 2004, 43, 7083–7093. [Google Scholar] [CrossRef]
- Ravera, M.; Baracco, S.; Cassino, C.; Zanello, P.; Osella, D. Appraisal of the redox behaviour of the antimetastatic ruthenium(III) complex [ImH][RuCl4(DMSO)(Im)], NAMI-A. Dalton Trans. 2004, 2347–2351. [Google Scholar] [CrossRef]
- Reisner, E.; Arion, V.B.; Keppler, B.K.; Pombeiro, A.J.L. Electron-transfer activated metal-based anticancer drugs. Inorg. Chim. Acta 2008, 361, 1569–1583. [Google Scholar] [CrossRef]
- Oehninger, L.; Stefanopoulou, M.; Alborzinia, H.; Schur, J.; Ludewig, S.; Namikawa, K.; Muñoz-Castro, A.; Köster, R.W.; Baumann, K.; Wölfl, S.; et al. Evaluation of arene ruthenium(II) N-heterocyclic carbene complexes as organometallics interacting with thiol and selenol containing biomolecules. Dalton Trans. 2013, 42, 1657–1666. [Google Scholar] [CrossRef]
- Bakulina, O.; Bannykh, A.; Jovanović, M.; Domračeva, I.; Podolski-Renić, A.; Žalubovskis, R.; Pešić, M.; Dar’in, D.; Krasavin, M. Design, synthesis, and biological evaluation of novel derivatives of dithiodiglycolic acid prepared via oxidative coupling of thiols. J. Enzym. Inhib. Med. Chem. 2019, 34, 665–671. [Google Scholar] [CrossRef] [Green Version]
- Omata, Y.; Folan, M.; Shaw, M.; Messer, R.L.; Lockwood, P.E.; Hobbs, D.; Bouillaguet, S.; Sano, H.; Lewis, J.B.; Wataha, J.C. Sublethal concentrations of diverse gold compounds inhibit mammalian cytosolic thioredoxin reductase (TrxR1). Toxicol. In Vitro 2006, 20, 882–890. [Google Scholar] [CrossRef] [Green Version]
- Hashemy, S.I.; Ungerstedt, J.S.; Avval, F.Z.; Holmgren, A. Motexafin Gadolinium, a Tumor-selective Drug Targeting Thioredoxin Reductase and Ribonucleotide Reductase. J. Biol. Chem. 2006, 281, 10691–10697. [Google Scholar] [CrossRef] [Green Version]
- Citta, A.; Folda, A.; Scutari, G.; Cesaro, L.; Bindoli, A.; Rigobello, M.P. Inhibition of thioredoxin reductase by lanthanum chloride. J. Inorg. Biochem. 2012, 117, 18–24. [Google Scholar] [CrossRef]
- Vermes, I.; Haanen, C.; Steffens-Nakken, H.; Reutellingsperger, C. A novel assay for apoptosis Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J. Immunol. Methods 1995, 184, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Schatzschneider, U.; Niesel, J.; Ott, I.; Gust, R.; Alborzinia, H.; Wölfl, S. Cellular Uptake, Cytotoxicity, and Metabolic Profiling of Human Cancer Cells Treated with Ruthenium(II) Polypyridyl Complexes [Ru(bpy)2(N N)]Cl2 with NN = bpy, phen, dpq, dppz, and dppn. ChemMedChem 2008, 3, 1104–1109. [Google Scholar] [CrossRef]
- Appold, M.; Mari, C.; Lederle, C.; Elbert, J.; Schmidt, C.; Ott, I.; Stühn, B.; Gasser, G.; Gallei, M. Multi-stimuli responsive block copolymers as a smart release platform for a polypyridyl ruthenium complex. Polym. Chem. 2017, 8, 890–900. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, C.; Karge, B.; Misgeld, R.; Prokop, A.; Franke, R.; Brönstrup, M.; Ott, I. Gold(I) NHC Complexes: Antiproliferative Activity, Cellular Uptake, Inhibition of Mammalian and Bacterial Thioredoxin Reductases, and Gram-Positive Directed Antibacterial Effects. Chem. Eur. J. 2017, 23, 1869–1880. [Google Scholar] [CrossRef]
- Grin, A.M.; Tikhonov, I.S.; Petrova, S.A.; Pogorilyy, A.V.; Noev, N.A.; Tatarskiy, V.V.; Shpakovsky, B.D.; Milaeva, R.E.; Kalinina, V.E.; Chernov, N.N.; et al. New Derivatives of Bacteriopurpurin with Thiolated Au (I) Complexes: Dual Darkand Light Activated Antitumor Potency. Anti-Cancer Agents Med. Chem. 2020, 20, 49–58. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 15 | 16 | 17 | 18 | 19 | 20 |
| Linker, n | 2 | 3 | 4 | 6 | 8 | 12 |
| t1/2, sec | 360 ± 20 | 330 ± 20 | 360 ± 20 | 650 ± 30 | 1390 ± 70 | 2150 ± 110 |
| Log P | * | * | * | 0.6 ± 0.1 | ±0.08 | 1.3 ± 0.2 |
| Complex | Pt | GC | ||||
|---|---|---|---|---|---|---|
| Eox, V | Ered | Eox, V | Ered, V | |||
| E1ox | E2ox | E1ox | E2ox | |||
| 15 | 1.290/1.185 | 1.850/1.630 | - | 1.270/1.17 | 1.890/1.650 | - |
| 16 | 1.300/1.190 | 1.830/1.630 | −0.310/−0.090 | 1.280/1.195 | - | −0.360/−0.190 |
| 17 | 1.290/1.190 | 1.820/1.610 | −0.290/−0.080 | 1.300/1.196 | 1.870/1.640 | −0.365/−0.128 |
| 18 | 1.280/1.170 | 1.820/1.620 | −0.305/−0.095 | 1.310/1.192 | 1.860/1.600 | −0.410/−0.150 |
| 19 | 1.320/1.190 | 1.840/1.620 | −0.310/−0.105 | 1.308/1.200 | 1.950/1.620 | −0.384/−0.135 |
| 20 | - | - | - | 1.498/1.137 | 1.873/1.609 | −0.350/−0.190 |
| Complex | Pt | GC | ||||
|---|---|---|---|---|---|---|
| Eox, V | E red E1ox | Eox, V | E red | |||
| E1ox | E2ox | E2ox | E1ox | |||
| 15 | 0.410/0.350 | 1.290/1.170 | - | 1.157/1.080 | −0.290/−0.231 | |
| 16 | 0.470/0.340 | 1.280/1.190 | −0.540/−0.410 | - | −0.490/−0.380 | |
| 17 | 0.580/0.370 | - | −0.650/−0.430 | 1.150/1.430 | −0.690/−0.290 | |
| 18 | 0.570/0.340 | 1.430/1.270 | −0.420/−0.280 | 1.150/1.350 | −0.590/−0.410 | |
| 19 | 0.250/0.150 | 1.390/1.160 | −0.550/−0.420 | 1.190/1.410 | −0.600/−0.420 | |
| 20 | - | 1.410/1.290 | - | 1.470/1,111 | −0,680/−0,356 | |
| Compound | Linker, n | IC50, µM | ||||
|---|---|---|---|---|---|---|
| A549 | MCF-7 | SH-SY5Y | SW480 | HaCaT | ||
| cisplatin | N/A | 23 ± 6.5 | >30 | 9.5 ± 0.1 | 21.7 ± 0.5 | 10 ± 5 |
| lonidamine | N/A | >90 | 30 ± 10 | >30 | >90 | 3 ± 1 |
| 9 | 2 | 50 ± 16 | 35 ± 6 | ND | 48 ± 10 | 23 ± 2 |
| 10 | 3 | >90 | >90 | ND | >90 | 39 ± 4 |
| 11 | 4 | 25 ± 7 | 22 ± 6 | ND | 28 ± 8 | 45 ± 3 |
| 12 | 6 | 24 ± 10 | 17 ± 2.5 | ND | 16 ± 6 | 40 ± 4 |
| 13 | 8 | 15 ± 6 | 10 ± 2 | ND | >90 | 45 ± 2 |
| 14 | 12 | 15.0± 2.2 | 20.8± 4.7 | ND | 8.4 ± 1.6 | ND |
| 15 | 2 | >30 | 20.4 ± 0.1 | 25.8 ± 4.5 | 21.4 ± 1.8 | ND |
| 16 | 3 | >30 | 23.0 ± 0.5 | 26.1 ± 5.5 | 25 ± 7.1 | ND |
| 17 | 4 | >30 | >30 | 27.7 ± 3.3 | 21 ± 1.8 | ND |
| 18 | 6 | 12.9 ± 1.0 | 22.0 ± 0.7 | 5.1 ± 2.5 | 19.6 ± 3.9 | ND |
| 19 | 8 | 5.9 ± 2.6 | 17.06 ± 0.3 | 2.64 ± 1.5 | 9.3 ± 0.1 | ND |
| 20 | 12 | 8.1 ± 1.1 | 9.2 ± 0.1 | 6.1 ± 0.3 | 9.6 ± 1.3 | ND |
| Single Dose, mg/kg i.p. | ||||
|---|---|---|---|---|
| 70 | 80 | 90 | 100 | 110 |
| 0/6 * | 0/6 | 2/6 | 2/6 | 4/6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shutkov, I.A.; Okulova, Y.N.; Tyurin, V.Y.; Sokolova, E.V.; Babkov, D.A.; Spasov, A.A.; Gracheva, Y.A.; Schmidt, C.; Kirsanov, K.I.; Shtil, A.A.; et al. Ru(III) Complexes with Lonidamine-Modified Ligands. Int. J. Mol. Sci. 2021, 22, 13468. https://doi.org/10.3390/ijms222413468
Shutkov IA, Okulova YN, Tyurin VY, Sokolova EV, Babkov DA, Spasov AA, Gracheva YA, Schmidt C, Kirsanov KI, Shtil AA, et al. Ru(III) Complexes with Lonidamine-Modified Ligands. International Journal of Molecular Sciences. 2021; 22(24):13468. https://doi.org/10.3390/ijms222413468
Chicago/Turabian StyleShutkov, Ilya A., Yulia N. Okulova, Vladimir Yu. Tyurin, Elena V. Sokolova, Denis A. Babkov, Alexander A. Spasov, Yulia A. Gracheva, Claudia Schmidt, Kirill I. Kirsanov, Alexander A. Shtil, and et al. 2021. "Ru(III) Complexes with Lonidamine-Modified Ligands" International Journal of Molecular Sciences 22, no. 24: 13468. https://doi.org/10.3390/ijms222413468
APA StyleShutkov, I. A., Okulova, Y. N., Tyurin, V. Y., Sokolova, E. V., Babkov, D. A., Spasov, A. A., Gracheva, Y. A., Schmidt, C., Kirsanov, K. I., Shtil, A. A., Redkozubova, O. M., Shevtsova, E. F., Milaeva, E. R., Ott, I., & Nazarov, A. A. (2021). Ru(III) Complexes with Lonidamine-Modified Ligands. International Journal of Molecular Sciences, 22(24), 13468. https://doi.org/10.3390/ijms222413468