Iron Therapy in Chronic Kidney Disease: Days of Future Past


Abstract
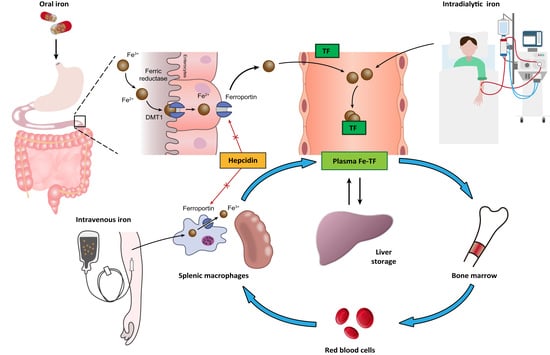
:
1. Introduction
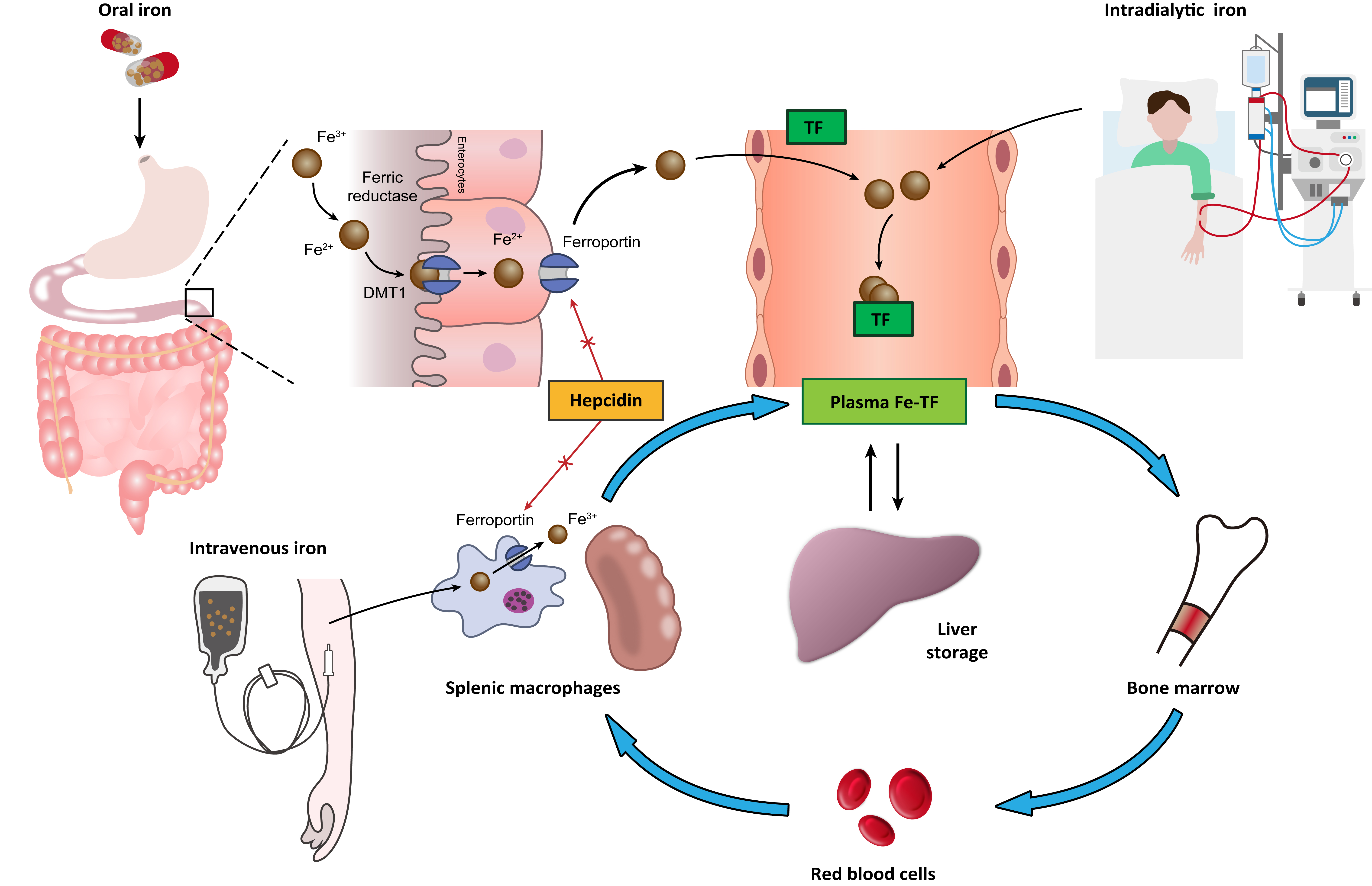
2. Normal Iron Metabolism
3. Pathophysiology of Renal Anemia
3.1. Relative Erythropoietin Deficiency
3.2. Shortened RBC Lifespan and Increased Blood Loss
3.3. Chronic Inflammation and Iron Deficiency
3.4. Copper Deficiency
3.5. Vitamin B12 and Folate Deficiency
3.6. Aluminum Overload
4. Strategies for Iron Management in Patients with Chronic Kidney Disease and End-Stage Renal Disease
4.1. Absolute versus Relative (Functional) Iron Deficiency
4.2. Optimal Target of Iron Supplementation
4.3. Iron Chelation Therapy for Iron Overload
5. Current Advances in Oral Iron Supplementation
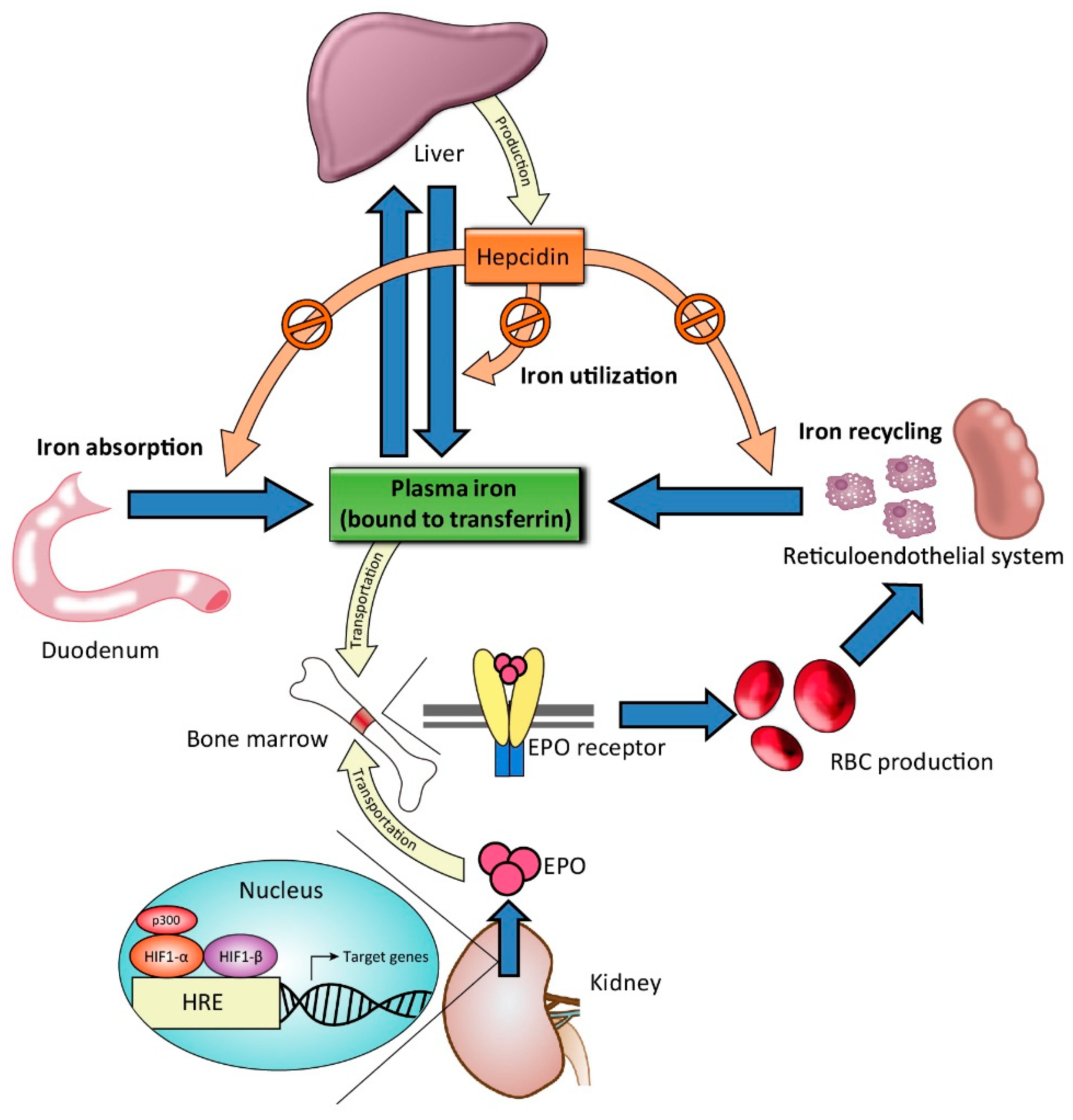
5.1. Ferric Citrate
5.1.1. Effects of Ferric Citrate on Phosphate Control
5.1.2. Effects of Ferric Citrate on Iron Status and Hemoglobin Level
5.1.3. Effects of Ferric Citrate on Fibroblast Growth Factor 23
5.2. Ferric Maltol
5.3. Heme Iron Polypeptide
5.4. Sucrosomial Iron
6. Current Advances in Intravenous Iron Supplementation
7. The Rising Star: Hypoxia-Inducible Factor Prolyl Hydroxylase Inhibitors
7.1. Mechanism of Action of Hypoxia-Inducible Factor Prolyl Hydroxylase Inhibitors
7.2. Clinical Trials of Hypoxia-Inducible Factor Prolyl Hydroxylase Inhibitors
7.3. Safety Concerns Regarding Hypoxia-Inducible Factor Prolyl Hydroxylase Inhibitors
7.3.1. Tumorigenesis
7.3.2. Angiogenesis
7.3.3. Adverse Cardiovascular Events
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McLean, E.; Cogswell, M.; Egli, I.; Wojdyla, D.; de Benoist, B. Worldwide prevalence of anaemia, WHO Vitamin and Mineral Nutrition Information System, 1993–2005. Public Health Nutr. 2009, 12, 444–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atkinson, M.A.; Warady, B.A. Anemia in chronic kidney disease. Pediatr. Nephrol. 2018, 33, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Babitt, J.L.; Lin, H.Y. Mechanisms of anemia in CKD. J. Am. Soc. Nephrol. 2012, 23, 1631–1634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drüeke, T.B.; Locatelli, F.; Clyne, N.; Eckardt, K.U.; Macdougall, I.C.; Tsakiris, D.; Burger, H.U.; Scherhag, A. Normalization of hemoglobin level in patients with chronic kidney disease and anemia. N. Engl. J. Med. 2006, 355, 2071–2084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.K.; Szczech, L.; Tang, K.L.; Barnhart, H.; Sapp, S.; Wolfson, M.; Reddan, D. Correction of anemia with epoetin alfa in chronic kidney disease. N. Engl. J. Med. 2006, 355, 2085–2098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drueke, T.B.; Parfrey, P.S. Summary of the KDIGO guideline on anemia and comment: Reading between the (guide)line(s). Kidney Int. 2012, 82, 952–960. [Google Scholar] [CrossRef] [Green Version]
- Gafter-Gvili, A.; Schechter, A.; Rozen-Zvi, B. Iron Deficiency Anemia in Chronic Kidney Disease. Acta Haematol. 2019, 142, 44–50. [Google Scholar] [CrossRef]
- Fleming, R.E.; Bacon, B.R. Orchestration of Iron Homeostasis. N. Engl. J. Med. 2005, 352, 1741–1744. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, M.I.; Solak, Y.; Covic, A.; Goldsmith, D.; Kanbay, M. Renal anemia of inflammation: The name is self-explanatory. Blood Purif. 2011, 32, 220–225. [Google Scholar] [CrossRef]
- Panwar, B.; Gutiérrez, O.M. Disorders of Iron Metabolism and Anemia in Chronic Kidney Disease. Semin. Nephrol. 2016, 36, 252–261. [Google Scholar] [CrossRef]
- Batchelor, E.K.; Kapitsinou, P.; Pergola, P.E.; Kovesdy, C.P.; Jalal, D.I. Iron Deficiency in Chronic Kidney Disease: Updates on Pathophysiology, Diagnosis, and Treatment. J. Am. Soc. Nephrol. 2020, 31, 456–468. [Google Scholar] [CrossRef]
- Gluba-Brzozka, A.; Franczyk, B.; Olszewski, R.; Rysz, J. The Influence of Inflammation on Anemia in CKD Patients. Int. J. Mol. Sci. 2020, 21, 725. [Google Scholar] [CrossRef] [Green Version]
- Zaritsky, J.; Young, B.; Wang, H.J.; Westerman, M.; Olbina, G.; Nemeth, E.; Ganz, T.; Rivera, S.; Nissenson, A.R.; Salusky, I.B. Hepcidin--A potential novel biomarker for iron status in chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2009, 4, 1051–1056. [Google Scholar] [CrossRef] [PubMed]
- Santos-Silva, A.; Ribeiro, S.; Reis, F.; Belo, L. Hepcidin in chronic kidney disease anemia. Vitam. Horm. 2019, 110, 243–264. [Google Scholar] [PubMed]
- Suzuki, N.; Gradin, K.; Poellinger, L.; Yamamoto, M. Regulation of hypoxia-inducible gene expression after HIF activation. Exp. Cell Res. 2017, 356, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wei, Q.; Guo, C.; Dong, G.; Liu, Y.; Tang, C.; Dong, Z. Hypoxia, HIF, and Associated Signaling Networks in Chronic Kidney Disease. Int. J. Mol. Sci. 2017, 18, 950. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.E.; Ho, V.; Arany, Z.; Krainc, D.; Galson, D.; Tendler, D.; Livingston, D.M.; Bunn, H.F. Erythropoietin gene regulation depends on heme-dependent oxygen sensing and assembly of interacting transcription factors. Kidney Int. 1997, 51, 548–552. [Google Scholar] [CrossRef] [Green Version]
- Pugh, C.W. Modulation of the Hypoxic Response. Adv. Exp. Med. Biol. 2016, 903, 259–271. [Google Scholar]
- Wong, M.M.Y.; Tu, C.; Li, Y.; Perlman, R.L.; Pecoits-Filho, R.; Lopes, A.A.; Narita, I.; Reichel, H.; Port, F.K.; Sukul, N.; et al. Anemia and iron deficiency among chronic kidney disease Stages 3-5ND patients in the Chronic Kidney Disease Outcomes and Practice Patterns Study: Often unmeasured, variably treated. Clin. Kidney J. 2020, 13, 613–624. [Google Scholar] [CrossRef]
- Souma, T.; Yamazaki, S.; Moriguchi, T.; Suzuki, N.; Hirano, I.; Pan, X.; Minegishi, N.; Abe, M.; Kiyomoto, H.; Ito, S.; et al. Plasticity of renal erythropoietin-producing cells governs fibrosis. J. Am. Soc. Nephrol. 2013, 24, 1599–1616. [Google Scholar] [CrossRef] [Green Version]
- Bernhardt, W.M.; Wiesener, M.S.; Scigalla, P.; Chou, J.; Schmieder, R.E.; Günzler, V.; Eckardt, K.U. Inhibition of prolyl hydroxylases increases erythropoietin production in ESRD. J. Am. Soc. Nephrol. 2010, 21, 2151–2156. [Google Scholar] [CrossRef] [PubMed]
- Ly, J.; Marticorena, R.; Donnelly, S. Red blood cell survival in chronic renal failure. Am. J. Kidney Dis. 2004, 44, 715–719. [Google Scholar] [CrossRef]
- Wu, S.G.; Jeng, F.R.; Wei, S.Y.; Su, C.Z.; Chung, T.C.; Chang, W.J.; Chang, H.W. Red blood cell osmotic fragility in chronically hemodialyzed patients. Nephron 1998, 78, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Sethi, S.K.; Bansal, S.B.; Wadhwani, N.; Makasana, M.; Nandwani, A.; Kher, V.; Raina, R. Myelofibrosis-Induced Erythropoietin-Resistant Anemia Due to Severe Refractory Hyperparathyroidism. Kidney Int. Rep. 2018, 3, 1010–1014. [Google Scholar] [CrossRef] [PubMed]
- Watts, D.; Gaete, D.; Rodriguez, D.; Hoogewijs, D.; Rauner, M.; Sormendi, S.; Wielockx, B. Hypoxia Pathway Proteins are Master Regulators of Erythropoiesis. Int. J. Mol. Sci. 2020, 21, 8131. [Google Scholar] [CrossRef]
- Balla, S.; Ismail, A. Impact of Hemodialysis on Serum Zinc and Copper Level in CKD Patients. J. App. Pharm. Sci. 2016, 6, 165–168. [Google Scholar] [CrossRef] [Green Version]
- Myint, Z.W.; Oo, T.H.; Thein, K.Z.; Tun, A.M.; Saeed, H. Copper deficiency anemia: Review article. Ann. Hematol. 2018, 97, 1527–1534. [Google Scholar] [CrossRef]
- Fishman, S.M.; Christian, P.; West, K.P. The role of vitamins in the prevention and control of anaemia. Public Health Nutr. 2000, 3, 125–150. [Google Scholar] [CrossRef] [Green Version]
- Lazarus, B.; Grams, M.E. Proton Pump Inhibitors in Kidney Disease. Clin. J. Am. Soc. Nephrol. 2018, 13, 1458–1459. [Google Scholar] [CrossRef] [Green Version]
- Chandna, S.M.; Tattersall, J.E.; Nevett, G.; Tew, C.J.; O’Sullivan, J.; Greenwood, R.N.; Farrington, K. Low Serum Vitamin B12 Levels in Chronic High-Flux Haemodialysis Patients. Nephron 1997, 75, 259–263. [Google Scholar] [CrossRef]
- Herbert, V.; Zalusky, R. Interrelations of vitamin B12 and folic acid metabolism: Folic acid clearance studies. J. Clin. Investig. 1962, 41, 1263–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teschner, M.; Kosch, M.; Schaefer, R.M. Folate metabolism in renal failure. Nephrol. Dial. Transplant. 2002, 17 (Suppl. 5), 24–27. [Google Scholar]
- Fernández Martín, J.L.; Cannata, J.B. Evolution of the aluminum concentration in the final dialysis solution: Multicenter study in Spanish dialysis centers. Nefrologia 2000, 20, 342–347. [Google Scholar] [PubMed]
- Pérez, G.; Pregi, N.; Vittori, D.; Di Risio, C.; Garbossa, G.; Nesse, A. Aluminum exposure affects transferrin-dependent and -independent iron uptake by K562 cells. Biochim. Biophys. Acta 2005, 1745, 124–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vittori, D.; Pregi, N.; Pérez, G.; Garbossa, G.; Nesse, A. The distinct erythropoietin functions that promote cell survival and proliferation are affected by aluminum exposure through mechanisms involving erythropoietin receptor. Biochim. Biophys. Acta 2005, 1743, 29–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bazzoni, G.B.; Bollini, A.N.; Hernández, G.N.; Contini Mdel, C.; Chiarotto, M.M.; Rasia, M.L. In vivo effect of aluminium upon the physical properties of the erythrocyte membrane. J. Inorg. Biochem. 2005, 99, 822–827. [Google Scholar] [CrossRef]
- de la Serna, F.J.; Praga, M.; Gilsanz, F.; Rodicio, J.L.; Ruilope, L.M.; Alcazar, J.M. Improvement in the erythropoiesis of chronic haemodialysis patients with desferrioxamine. Lancet 1988, 1, 1009–1011. [Google Scholar] [CrossRef]
- Ueda, N.; Takasawa, K. Impact of Inflammation on Ferritin, Hepcidin and the Management of Iron Deficiency Anemia in Chronic Kidney Disease. Nutrients 2018, 10, 1173. [Google Scholar] [CrossRef] [Green Version]
- Wish, J.B. Assessing iron status: Beyond serum ferritin and transferrin saturation. Clin. J. Am. Soc. Nephrol. 2006, 1 (Suppl. 1), S4-8. [Google Scholar] [CrossRef]
- Tarng, D.C.; Huang, T.P. A parallel, comparative study of intravenous iron versus intravenous ascorbic acid for erythropoietin-hyporesponsive anaemia in haemodialysis patients with iron overload. Nephrol. Dial. Transplant. 1998, 13, 2867–2872. [Google Scholar] [CrossRef]
- Tarng, D.C. Novel aspects of vitamin C in epoetin response. J. Chin. Med. Assoc. 2007, 70, 357–360. [Google Scholar] [CrossRef] [Green Version]
- DeLoughery, T.G. Safety of Oral and Intravenous Iron. Acta Haematol. 2019, 142, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Kuo, K.L.; Hung, S.C.; Wei, Y.H.; Tarng, D.C. Intravenous iron exacerbates oxidative DNA damage in peripheral blood lymphocytes in chronic hemodialysis patients. J. Am. Soc. Nephrol. 2008, 19, 1817–1826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Franceschi, L.; Iolascon, A.; Taher, A.; Cappellini, M.D. Clinical management of iron deficiency anemia in adults: Systemic review on advances in diagnosis and treatment. Eur. J. Intern. Med. 2017, 42, 16–23. [Google Scholar] [CrossRef]
- Hung, S.C.; Kuo, K.L.; Tarng, D.C.; Hsu, C.C.; Wu, M.S.; Huang, T.P. Anaemia management in patients with chronic kidney disease: Taiwan practice guidelines. Nephrology 2014, 19, 735–739. [Google Scholar] [CrossRef]
- Tarng, D.C.; Chen, T.W.; Huang, T.P. Iron metabolism indices for early prediction of the response and resistance to erythropoietin therapy in maintenance hemodialysis patients. Am. J. Nephrol. 1995, 15, 230–237. [Google Scholar] [CrossRef]
- Tarng, D.C.; Huang, T.P.; Chen, T.W. Mathematical approach for estimating iron needs in hemodialysis patients on erythropoietin therapy. Am. J. Nephrol. 1997, 17, 158–164. [Google Scholar] [CrossRef]
- Tarng, D.C.; Huang, T.P.; Chen, T.W.; Yang, W.C. Erythropoietin hyporesponsiveness: From iron deficiency to iron overload. Kidney Int. Suppl. 1999, 69, S107–S118. [Google Scholar] [CrossRef] [Green Version]
- Fishbane, S.; Frei, G.L.; Maesaka, J. Reduction in recombinant human erythropoietin doses by the use of chronic intravenous iron supplementation. Am. J. Kidney Dis. 1995, 26, 41–46. [Google Scholar] [CrossRef]
- Besarab, A.; Amin, N.; Ahsan, M.; Vogel, S.E.; Zazuwa, G.; Frinak, S.; Zazra, J.J.; Anandan, J.V.; Gupta, A. Optimization of epoetin therapy with intravenous iron therapy in hemodialysis patients. J. Am. Soc. Nephrol. 2000, 11, 530–538. [Google Scholar]
- Coyne, D.W.; Kapoian, T.; Suki, W.; Singh, A.K.; Moran, J.E.; Dahl, N.V.; Rizkala, A.R.; Group, D.S. Ferric gluconate is highly efficacious in anemic hemodialysis patients with high serum ferritin and low transferrin saturation: Results of the Dialysis Patients’ Response to IV Iron with Elevated Ferritin (DRIVE) Study. J. Am. Soc. Nephrol. 2007, 18, 975–984. [Google Scholar] [CrossRef] [PubMed]
- Ratcliffe, L.E.; Thomas, W.; Glen, J.; Padhi, S.; Pordes, B.A.; Wonderling, D.; Connell, R.; Stephens, S.; Mikhail, A.I.; Fogarty, D.G.; et al. Diagnosis and Management of Iron Deficiency in CKD: A Summary of the NICE Guideline Recommendations and Their Rationale. Am. J. Kidney Dis. 2016, 67, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Mikhail, A.; Brown, C.; Williams, J.A.; Mathrani, V.; Shrivastava, R.; Evans, J.; Isaac, H.; Bhandari, S. Renal association clinical practice guideline on Anaemia of Chronic Kidney Disease. BMC Nephrol. 2017, 18, 345. [Google Scholar] [CrossRef] [PubMed]
- Fishbane, S.; Spinowitz, B. Update on Anemia in ESRD and Earlier Stages of CKD: Core Curriculum 2018. Am. J. Kidney Dis. 2018, 71, 423–435. [Google Scholar] [CrossRef]
- Bnaya, A.; Shavit, L.; Malyszko, J.S.; Malyszko, J.; Slotki, I. Labile plasma iron levels in chronic hemodialysis patients treated by intravenous iron supplementation. Ther. Apher. Dial. 2020, 24, 416–422. [Google Scholar] [CrossRef]
- Cappell, K.A.; Shreay, S.; Cao, Z.; Varker, H.V.; Paoli, C.J.; Gitlin, M. Red blood cell (RBC) transfusion rates among US chronic dialysis patients during changes to Medicare end-stage renal disease (ESRD) reimbursement systems and erythropoiesis stimulating agent (ESA) labels. BMC Nephrol. 2014, 15, 116. [Google Scholar] [CrossRef] [Green Version]
- Mehta, K.J.; Farnaud, S.J.; Sharp, P.A. Iron and liver fibrosis: Mechanistic and clinical aspects. World J. Gastroenterol. 2019, 25, 521–538. [Google Scholar] [CrossRef]
- Yang, W.P.; Chang, H.H.; Li, H.Y.; Lai, Y.C.; Huang, T.Y.; Tsai, K.S.; Lin, K.H.; Lin, D.T.; Jou, S.T.; Lu, M.Y.; et al. Iron Overload Associated Endocrine Dysfunction Leading to Lower Bone Mineral Density in Thalassemia Major. J. Clin. Endocrinol. Metab. 2020, 105, e1015–e1024. [Google Scholar] [CrossRef]
- Liu, Q.; Sun, L.; Tan, Y.; Wang, G.; Lin, X.; Cai, L. Role of iron deficiency and overload in the pathogenesis of diabetes and diabetic complications. Curr. Med. Chem. 2009, 16, 113–129. [Google Scholar] [CrossRef] [Green Version]
- Chacon, A.H.; Morrison, B.; Hu, S. Acquired hemochromatosis with pronounced pigment deposition of the upper eyelids. J. Clin. Aesthet. Dermatol. 2013, 6, 44–46. [Google Scholar]
- Diez-Lopez, C.; Comin-Colet, J.; Gonzalez-Costello, J. Iron overload cardiomyopathy: From diagnosis to management. Curr. Opin. Cardiol. 2018, 33, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.; Chakraborty, A.; Nag, A.; Chattopadyay, A.; Dasgupta, A.K.; Bhattacharyya, M. Intracellular iron overload leading to DNA damage of lymphocytes and immune dysfunction in thalassemia major patients. Eur. J. Haematol. 2017, 99, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Borgna-Pignatti, C.; Marsella, M. Iron Chelation in Thalassemia Major. Clin. Ther. 2015, 37, 2866–2877. [Google Scholar] [CrossRef]
- Gattermann, N. Iron overload in myelodysplastic syndromes (MDS). Int. J. Hematol. 2018, 107, 55–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shander, A.; Sazama, K. Clinical consequences of iron overload from chronic red blood cell transfusions, its diagnosis, and its management by chelation therapy. Transfusion 2010, 50, 1144–1155. [Google Scholar] [CrossRef]
- Maker, G.L.; Siva, B.; Batty, K.T.; Trengove, R.D.; Ferrari, P.; Olynyk, J.K. Pharmacokinetics and safety of deferasirox in subjects with chronic kidney disease undergoing haemodialysis. Nephrology 2013, 18, 188–193. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Leaf, D.E. Iron Chelation as a Potential Therapeutic Strategy for AKI Prevention. J. Am. Soc. Nephrol. 2019, 30, 2060–2071. [Google Scholar] [CrossRef]
- Chen, C.H.; Shu, K.H.; Yang, Y. Long-term effects of an oral iron chelator, deferasirox, in hemodialysis patients with iron overload. Hematology 2015, 20, 304–310. [Google Scholar] [CrossRef]
- Kang, H.; Han, M.; Xue, J.; Baek, Y.; Chang, J.; Hu, S.; Nam, H.; Jo, M.J.; El Fakhri, G.; Hutchens, M.P.; et al. Renal clearable nanochelators for iron overload therapy. Nat. Commun. 2019, 10, 5134. [Google Scholar] [CrossRef] [Green Version]
- Santiago, P. Ferrous versus ferric oral iron formulations for the treatment of iron deficiency: A clinical overview. Sci. World J. 2012, 2012, 846824. [Google Scholar] [CrossRef]
- Pergola, P.E.; Fishbane, S.; Ganz, T. Novel Oral Iron Therapies for Iron Deficiency Anemia in Chronic Kidney Disease. Adv. Chronic Kidney Dis. 2019, 26, 272–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, J.B.; Sika, M.; Koury, M.J.; Chuang, P.; Schulman, G.; Smith, M.T.; Whittier, F.C.; Linfert, D.R.; Galphin, C.M.; Athreya, B.P.; et al. Ferric citrate controls phosphorus and delivers iron in patients on dialysis. J. Am. Soc. Nephrol. 2015, 26, 493–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umanath, K.; Jalal, D.I.; Greco, B.A.; Umeukeje, E.M.; Reisin, E.; Manley, J.; Zeig, S.; Negoi, D.G.; Hiremath, A.N.; Blumenthal, S.S.; et al. Ferric Citrate Reduces Intravenous Iron and Erythropoiesis-Stimulating Agent Use in ESRD. J. Am. Soc. Nephrol. 2015, 26, 2578–2587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Block, G.A.; Fishbane, S.; Rodriguez, M.; Smits, G.; Shemesh, S.; Pergola, P.E.; Wolf, M.; Chertow, G.M. A 12-week, double-blind, placebo-controlled trial of ferric citrate for the treatment of iron deficiency anemia and reduction of serum phosphate in patients with CKD Stages 3–5. Am. J. Kidney Dis. 2015, 65, 728–736. [Google Scholar] [CrossRef] [Green Version]
- Fishbane, S.; Block, G.A.; Loram, L.; Neylan, J.; Pergola, P.E.; Uhlig, K.; Chertow, G.M. Effects of Ferric Citrate in Patients with Nondialysis-Dependent CKD and Iron Deficiency Anemia. J. Am. Soc. Nephrol. 2017, 28, 1851–1858. [Google Scholar] [CrossRef] [Green Version]
- Block, G.A.; Pergola, P.E.; Fishbane, S.; Martins, J.G.; LeWinter, R.D.; Uhlig, K.; Neylan, J.F.; Chertow, G.M. Effect of ferric citrate on serum phosphate and fibroblast growth factor 23 among patients with nondialysis-dependent chronic kidney disease: Path analyses. Nephrol. Dial. Transplant. 2019, 34, 1115–1124. [Google Scholar] [CrossRef]
- Yokoyama, K.; Hirakata, H.; Akiba, T.; Fukagawa, M.; Nakayama, M.; Sawada, K.; Kumagai, Y.; Block, G.A. Ferric citrate hydrate for the treatment of hyperphosphatemia in nondialysis-dependent CKD. Clin. J. Am. Soc. Nephrol. 2014, 9, 543–552. [Google Scholar] [CrossRef] [Green Version]
- Yokoyama, K.; Fukagawa, M.; Akiba, T.; Nakayama, M.; Ito, K.; Hanaki, K.; Wolf, M.; Hirakata, H. Randomised clinical trial of ferric citrate hydrate on anaemia management in haemodialysis patients with hyperphosphataemia: ASTRIO study. Sci. Rep. 2019, 9, 8877. [Google Scholar] [CrossRef]
- Fishbane, S.; Shah, H.H. Ferric pyrophosphate citrate as an iron replacement agent for patients receiving hemodialysis. Hemodial. Int. 2017, 21 (Suppl. 1), S104–S109. [Google Scholar] [CrossRef] [Green Version]
- Sinsakul, M.; Sika, M.; Koury, M.; Shapiro, W.; Greene, T.; Dwyer, J.; Smith, M.; Korbet, S.; Lewis, J.; Collaborative Study, G. The safety and tolerability of ferric citrate as a phosphate binder in dialysis patients. Nephron. Clin. Pract. 2012, 121, c25–c29. [Google Scholar] [CrossRef]
- Yokoyama, K.; Akiba, T.; Fukagawa, M.; Nakayama, M.; Sawada, K.; Kumagai, Y.; Chertow, G.M.; Hirakata, H. Long-term safety and efficacy of a novel iron-containing phosphate binder, JTT-751, in patients receiving hemodialysis. J. Ren. Nutr. 2014, 24, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, K.; Akiba, T.; Fukagawa, M.; Nakayama, M.; Hirakata, H. JTT-751 for treatment of patients with hyperphosphatemia on peritoneal dialysis. Nephron. Clin. Pract. 2014, 128, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.T.; Wu, I.W.; Chiang, S.S.; Peng, Y.S.; Shu, K.H.; Wu, M.J.; Wu, M.S. Effect of oral ferric citrate on serum phosphorus in hemodialysis patients: Multicenter, randomized, double-blind, placebo-controlled study. J. Nephrol. 2015, 28, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.Y.; Chen, Y.C.; Lin, C.H.; Wu, Y.C.; Tu, Y.K.; Tarng, D.C. Safety and efficacy of ferric citrate in phosphate reduction and iron supplementation in patients with chronic kidney disease. Oncotarget 2017, 8, 107283–107294. [Google Scholar] [CrossRef] [Green Version]
- Gulec, S.; Anderson, G.J.; Collins, J.F. Mechanistic and regulatory aspects of intestinal iron absorption. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, G397–G409. [Google Scholar] [CrossRef] [Green Version]
- Bergamaschi, G.; Di Sabatino, A.; Pasini, A.; Ubezio, C.; Costanzo, F.; Grataroli, D.; Masotti, M.; Alvisi, C.; Corazza, G.R. Intestinal expression of genes implicated in iron absorption and their regulation by hepcidin. Clin. Nutr. 2017, 36, 1427–1433. [Google Scholar] [CrossRef]
- Womack, R.; Berru, F.; Panwar, B.; Gutierrez, O.M. Effect of Ferric Citrate versus Ferrous Sulfate on Iron and Phosphate Parameters in Patients with Iron Deficiency and CKD: A Randomized Trial. Clin. J. Am. Soc. Nephrol. 2020, 15, 1251–1258. [Google Scholar] [CrossRef]
- Gutierrez, O.; Isakova, T.; Rhee, E.; Shah, A.; Holmes, J.; Collerone, G.; Juppner, H.; Wolf, M. Fibroblast growth factor-23 mitigates hyperphosphatemia but accentuates calcitriol deficiency in chronic kidney disease. J. Am. Soc. Nephrol. 2005, 16, 2205–2215. [Google Scholar] [CrossRef]
- Egli-Spichtig, D.; Imenez Silva, P.H.; Glaudemans, B.; Gehring, N.; Bettoni, C.; Zhang, M.Y.H.; Pastor-Arroyo, E.M.; Schonenberger, D.; Rajski, M.; Hoogewijs, D.; et al. Tumor necrosis factor stimulates fibroblast growth factor 23 levels in chronic kidney disease and non-renal inflammation. Kidney Int. 2019, 96, 890–905. [Google Scholar] [CrossRef]
- Durlacher-Betzer, K.; Hassan, A.; Levi, R.; Axelrod, J.; Silver, J.; Naveh-Many, T. Interleukin-6 contributes to the increase in fibroblast growth factor 23 expression in acute and chronic kidney disease. Kidney Int. 2018, 94, 315–325. [Google Scholar] [CrossRef]
- David, V.; Martin, A.; Isakova, T.; Spaulding, C.; Qi, L.; Ramirez, V.; Zumbrennen-Bullough, K.B.; Sun, C.C.; Lin, H.Y.; Babitt, J.L.; et al. Inflammation and functional iron deficiency regulate fibroblast growth factor 23 production. Kidney Int. 2016, 89, 135–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanudel, M.R.; Chua, K.; Rappaport, M.; Gabayan, V.; Valore, E.; Goltzman, D.; Ganz, T.; Nemeth, E.; Salusky, I.B. Effects of dietary iron intake and chronic kidney disease on fibroblast growth factor 23 metabolism in wild-type and hepcidin knockout mice. Am. J. Physiol. Ren. Physiol. 2016, 311, F1369–F1377. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, O.M.; Januzzi, J.L.; Isakova, T.; Laliberte, K.; Smith, K.; Collerone, G.; Sarwar, A.; Hoffmann, U.; Coglianese, E.; Christenson, R.; et al. Fibroblast growth factor 23 and left ventricular hypertrophy in chronic kidney disease. Circulation 2009, 119, 2545–2552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nam, K.H.; Kim, H.; An, S.Y.; Lee, M.; Cha, M.U.; Park, J.T.; Yoo, T.H.; Lee, K.B.; Kim, Y.H.; Sung, S.A.; et al. Circulating Fibroblast Growth Factor-23 Levels are Associated with an Increased Risk of Anemia Development in Patients with Nondialysis Chronic Kidney Disease. Sci. Rep. 2018, 8, 7294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, R.; Cai, X.; Hodakowski, A.; Lee, J.; Leonard, M.; Ricardo, A.; Chen, J.; Hamm, L.; Sondheimer, J.; Dobre, M.; et al. Fibroblast Growth Factor 23 and Anemia in the Chronic Renal Insufficiency Cohort Study. Clin. J. Am. Soc. Nephrol. 2017, 12, 1795–1803. [Google Scholar] [CrossRef]
- Czaya, B.; Faul, C. The Role of Fibroblast Growth Factor 23 in Inflammation and Anemia. Int. J. Mol. Sci. 2019, 20, 4195. [Google Scholar] [CrossRef] [Green Version]
- Bergmark, B.A.; Udell, J.A.; Morrow, D.A.; Cannon, C.P.; Steen, D.L.; Jarolim, P.; Budaj, A.; Hamm, C.; Guo, J.; Im, K.; et al. Association of Fibroblast Growth Factor 23 With Recurrent Cardiovascular Events in Patients After an Acute Coronary Syndrome: A Secondary Analysis of a Randomized Clinical Trial. JAMA Cardiol. 2018, 3, 473–480. [Google Scholar] [CrossRef] [Green Version]
- Block, G.A.; Chertow, G.M.; Cooper, K.; Xing, S.; Fouqueray, B.; Halperin, M.; Danese, M.D. Fibroblast growth factor 23 as a risk factor for cardiovascular events and mortality in patients in the EVOLVE trial. Hemodial. Int. 2020. [Google Scholar] [CrossRef]
- Gravesen, E.; Hofman-Bang, J.; Mace, M.L.; Lewin, E.; Olgaard, K. High dose intravenous iron, mineral homeostasis and intact FGF23 in normal and uremic rats. BMC Nephrol. 2013, 14, 281. [Google Scholar] [CrossRef] [Green Version]
- Prats, M.; Font, R.; Garcia, C.; Cabre, C.; Jariod, M.; Vea, A.M. Effect of ferric carboxymaltose on serum phosphate and C-terminal FGF23 levels in non-dialysis chronic kidney disease patients: Post-hoc analysis of a prospective study. BMC Nephrol. 2013, 14, 167. [Google Scholar] [CrossRef] [Green Version]
- Takeda, Y.; Komaba, H.; Goto, S.; Fujii, H.; Umezu, M.; Hasegawa, H.; Fujimori, A.; Nishioka, M.; Nishi, S.; Fukagawa, M. Effect of intravenous saccharated ferric oxide on serum FGF23 and mineral metabolism in hemodialysis patients. Am. J. Nephrol. 2011, 33, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Schouten, B.J.; Hunt, P.J.; Livesey, J.H.; Frampton, C.M.; Soule, S.G. FGF23 elevation and hypophosphatemia after intravenous iron polymaltose: A prospective study. J. Clin. Endocrinol. Metab. 2009, 94, 2332–2337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iguchi, A.; Kazama, J.J.; Yamamoto, S.; Yoshita, K.; Watanabe, Y.; Iino, N.; Narita, I. Administration of Ferric Citrate Hydrate Decreases Circulating FGF23 Levels Independently of Serum Phosphate Levels in Hemodialysis Patients with Iron Deficiency. Nephron 2015, 131, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, N.; Otsuki, T.; Yoshida, Y.; Nagura, C.; Kitai, M.; Shibahara, N.; Tomita, H.; Maruyama, T.; Abe, M. Ferric Citrate Decreases Fibroblast Growth Factor 23 and Improves Erythropoietin Responsiveness in Hemodialysis Patients. Am. J. Nephrol. 2018, 47, 406–414. [Google Scholar] [CrossRef]
- Khoury, A.; Pagan, K.A.; Farland, M.Z. Ferric Maltol: A New Oral Iron Formulation for the Treatment of Iron Deficiency in Adults. Ann. Pharmacother. 2020. [Google Scholar] [CrossRef]
- Gasche, C.; Ahmad, T.; Tulassay, Z.; Baumgart, D.C.; Bokemeyer, B.; Buning, C.; Howaldt, S.; Stallmach, A.; Group, A.S. Ferric maltol is effective in correcting iron deficiency anemia in patients with inflammatory bowel disease: Results from a phase-3 clinical trial program. Inflamm. Bowel Dis. 2015, 21, 579–588. [Google Scholar] [CrossRef] [Green Version]
- Stein, J.; Aksan, A.; Farrag, K.; Dignass, A.; Radeke, H.H. Management of inflammatory bowel disease-related anemia and iron deficiency with specific reference to the role of intravenous iron in current practice. Expert Opin. Pharmacother. 2017, 18, 1721–1737. [Google Scholar] [CrossRef]
- Kumar, A.; Brookes, M.J. Iron Therapy in Inflammatory Bowel Disease. Nutrients 2020, 12, 3478. [Google Scholar] [CrossRef]
- Dull, R.B.; Davis, E. Heme iron polypeptide for the management of anaemia of chronic kidney disease. J. Clin. Pharm. Ther. 2015, 40, 386–390. [Google Scholar] [CrossRef] [Green Version]
- Nagaraju, S.P.; Cohn, A.; Akbari, A.; Davis, J.L.; Zimmerman, D.L. Heme iron polypeptide for the treatment of iron deficiency anemia in non-dialysis chronic kidney disease patients: A randomized controlled trial. BMC Nephrol. 2013, 14, 64. [Google Scholar] [CrossRef] [Green Version]
- Nissenson, A.R.; Berns, J.S.; Sakiewicz, P.; Ghaddar, S.; Moore, G.M.; Schleicher, R.B.; Seligman, P.A. Clinical evaluation of heme iron polypeptide: Sustaining a response to rHuEPO in hemodialysis patients. Am. J. Kidney Dis. 2003, 42, 325–330. [Google Scholar] [CrossRef]
- Barraclough, K.A.; Brown, F.; Hawley, C.M.; Leary, D.; Noble, E.; Campbell, S.B.; Isbel, N.M.; Mudge, D.W.; van Eps, C.L.; Johnson, D.W. A randomized controlled trial of oral heme iron polypeptide versus oral iron supplementation for the treatment of anaemia in peritoneal dialysis patients: HEMATOCRIT trial. Nephrol. Dial. Transplant. 2012, 27, 4146–4153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabiano, A.; Brilli, E.; Fogli, S.; Beconcini, D.; Carpi, S.; Tarantino, G.; Zambito, Y. Sucrosomial(R) iron absorption studied by in vitro and ex-vivo models. Eur. J. Pharm. Sci. 2018, 111, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Ramirez, S.; Brilli, E.; Tarantino, G.; Munoz, M. Sucrosomial((R)) Iron: A New Generation Iron for Improving Oral Supplementation. Pharmaceuticals 2018, 11, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riccio, E.; Sabbatini, M.; Capuano, I.; Pellegrino, A.M.; Petruzzelli, L.A.; Pisani, A. Oral Sucrosomial(R) iron versus intravenous iron for recovering iron deficiency anaemia in ND-CKD patients: A cost- minimization analysis. BMC Nephrol. 2020, 21, 57. [Google Scholar] [CrossRef] [PubMed]
- Mafodda, A.; Giuffrida, D.; Prestifilippo, A.; Azzarello, D.; Giannicola, R.; Mare, M.; Maisano, R. Oral sucrosomial iron versus intravenous iron in anemic cancer patients without iron deficiency receiving darbepoetin alfa: A pilot study. Support. Care Cancer 2017, 25, 2779–2786. [Google Scholar] [CrossRef] [Green Version]
- Abbati, G.; Incerti, F.; Boarini, C.; Pileri, F.; Bocchi, D.; Ventura, P.; Buzzetti, E.; Pietrangelo, A. Safety and efficacy of sucrosomial iron in inflammatory bowel disease patients with iron deficiency anemia. Intern. Emerg. Med. 2019, 14, 423–431. [Google Scholar] [CrossRef]
- Elli, L.; Ferretti, F.; Branchi, F.; Tomba, C.; Lombardo, V.; Scricciolo, A.; Doneda, L.; Roncoroni, L. Sucrosomial Iron Supplementation in Anemic Patients with Celiac Disease Not Tolerating Oral Ferrous Sulfate: A Prospective Study. Nutrients 2018, 10, 330. [Google Scholar] [CrossRef] [Green Version]
- Pisani, A.; Riccio, E.; Sabbatini, M.; Andreucci, M.; Del Rio, A.; Visciano, B. Effect of oral liposomal iron versus intravenous iron for treatment of iron deficiency anaemia in CKD patients: A randomized trial. Nephrol. Dial. Transplant. 2015, 30, 645–652. [Google Scholar] [CrossRef]
- Auerbach, M.; Ballard, H. Clinical use of intravenous iron: Administration, efficacy, and safety. Hematol. Am. Soc. Hematol. Educ. Progr. 2010, 2010, 338–347. [Google Scholar] [CrossRef] [Green Version]
- Girelli, D.; Ugolini, S.; Busti, F.; Marchi, G.; Castagna, A. Modern iron replacement therapy: Clinical and pathophysiological insights. Int. J. Hematol. 2018, 107, 16–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, A.; Dobrovolskaia, M.A. Immunological effects of iron oxide nanoparticles and iron-based complex drug formulations: Therapeutic benefits, toxicity, mechanistic insights, and translational considerations. Nanomedicine 2018, 14, 977–990. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Kshirsagar, A.V.; Brookhart, M.A. Safety of intravenous iron in hemodialysis patients. Hemodial. Int. 2017, 21 (Suppl. 1), S93–S103. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.; Lin, V.; Guss, C.; Pratt, R.; Ikizler, T.A.; Besarab, A. Ferric pyrophosphate citrate administered via dialysate reduces erythropoiesis-stimulating agent use and maintains hemoglobin in hemodialysis patients. Kidney Int. 2015, 88, 1187–1194. [Google Scholar] [CrossRef] [Green Version]
- Pratt, R.; Handelman, G.J.; Edwards, T.E.; Gupta, A. Ferric pyrophosphate citrate: Interactions with transferrin. Biometals 2018, 31, 1081–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pratt, R.D.; Swinkels, D.W.; Ikizler, T.A.; Gupta, A. Pharmacokinetics of Ferric Pyrophosphate Citrate, a Novel Iron Salt, Administered Intravenously to Healthy Volunteers. J. Clin. Pharmacol. 2017, 57, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Shah, H.H.; Hazzan, A.D.; Fishbane, S. Ferric Pyrophosphate Citrate: A Novel Iron Replacement Agent in Patients Undergoing Hemodialysis. Semin. Nephrol. 2016, 36, 124–129. [Google Scholar] [CrossRef]
- Fishbane, S.N.; Singh, A.K.; Cournoyer, S.H.; Jindal, K.K.; Fanti, P.; Guss, C.D.; Lin, V.H.; Pratt, R.D.; Gupta, A. Ferric pyrophosphate citrate (Triferic) administration via the dialysate maintains hemoglobin and iron balance in chronic hemodialysis patients. Nephrol. Dial. Transplant. 2015, 30, 2019–2026. [Google Scholar] [CrossRef]
- Semenza, G.L. Involvement of oxygen-sensing pathways in physiologic and pathologic erythropoiesis. Blood 2009, 114, 2015–2019. [Google Scholar] [CrossRef] [Green Version]
- Gupta, N.; Wish, J.B. Hypoxia-Inducible Factor Prolyl Hydroxylase Inhibitors: A Potential New Treatment for Anemia in Patients With CKD. Am. J. Kidney Dis. 2017, 69, 815–826. [Google Scholar] [CrossRef] [Green Version]
- Souza, E.; Cho, K.H.; Harris, S.T.; Flindt, N.R.; Watt, R.K.; Pai, A.B. Hypoxia-inducible factor prolyl hydroxylase inhibitors: A paradigm shift for treatment of anemia in chronic kidney disease? Expert Opin. Investig. Drugs 2020, 29, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Nakano, D.; Zhang, A.; Kittikulsuth, W.; Morisawa, N.; Ohsaki, H.; Suzuki, N.; Yamamoto, M.; Nishiyama, A. Effects of post-renal anemia treatment with the HIF-PHD inhibitor molidustat on adenine-induced renal anemia and kidney disease in mice. J. Pharmacol. Sci. 2020, 144, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Schellinger, I.N.; Cordasic, N.; Panesar, J.; Buchholz, B.; Jacobi, J.; Hartner, A.; Klanke, B.; Jakubiczka-Smorag, J.; Burzlaff, N.; Heinze, E.; et al. Hypoxia inducible factor stabilization improves defective ischemia-induced angiogenesis in a rodent model of chronic kidney disease. Kidney Int. 2017, 91, 616–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, M.; Joharapurkar, A.; Patel, V.; Kshirsagar, S.; Sutariya, B.; Patel, M.; Patel, H.; Patel, P.R. Pharmacological inhibition of prolyl hydroxylase protects against inflammation-induced anemia via efficient erythropoiesis and hepcidin downregulation. Eur. J. Pharmacol. 2019, 843, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, J.M.; Sharma, N.; Dikdan, S. Hypoxia-Inducible Factor and Its Role in the Management of Anemia in Chronic Kidney Disease. Int. J. Mol. Sci. 2018, 19, 389. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.; Hao, C.; Peng, X.; Lin, H.; Yin, A.; Hao, L.; Tao, Y.; Liang, X.; Liu, Z.; Xing, C.; et al. Roxadustat for Anemia in Patients with Kidney Disease Not Receiving Dialysis. N. Engl. J. Med. 2019, 381, 1001–1010. [Google Scholar] [CrossRef]
- Chen, N.; Hao, C.; Liu, B.C.; Lin, H.; Wang, C.; Xing, C.; Liang, X.; Jiang, G.; Liu, Z.; Li, X.; et al. Roxadustat Treatment for Anemia in Patients Undergoing Long-Term Dialysis. N. Engl. J. Med. 2019, 381, 1011–1022. [Google Scholar] [CrossRef]
- Fishbane, S.; El-Shahawy, M.A.; Pecoits-Filho, R. OLYMPUS: A phase 3, randomized, double-blind, placebo-controlled, international study of roxadustat efficacy in patients with non-dialysis-dependent (NDD) CKD and anemia. In Proceedings of the American Society of Nephrology Kidney Week, Washington, DC, USA, 7–10 November 2019. [Google Scholar]
- Fishbane, S.; El-Shahawy, M.A.; Pecoits-Filho, R. ROCKIES: An international, phase 3, randomized, open-label, active-controlled study of roxadustat for anemia in dialysis-dependent CKD patients. In Proceedings of the American Society of Nephrology Kidney Week, Washington, DC, USA, 7–10 November 2019. [Google Scholar]
- Chertow, G.M. PRO2TECT program: Global Phase 3 Clinical Trials of Vadadustat vs. Darbepoetin Alfa for Treatment of Anemia in Patients with Non-Dialysis-Dependent CKD. In Proceedings of the American Society of Nephrology Kidney Week, 22–25 October 2020. [Google Scholar]
- Yang, M.H.; Wu, M.Z.; Chiou, S.H.; Chen, P.M.; Chang, S.Y.; Liu, C.J.; Teng, S.C.; Wu, K.J. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat. Cell. Biol. 2008, 10, 295–305. [Google Scholar] [CrossRef]
- Provenzano, R.; Fishbane, S.; Wei, L. Pooled efficacy and cardiovascular (CV) analyses of roxadustat in the treatment of anemia in CKD patients on and not on dialysis. In Proceedings of the American Society of Nephrology Kidney Week, Washington, DC, USA, 7–10 November 2019. [Google Scholar]
- Krock, B.L.; Skuli, N.; Simon, M.C. Hypoxia-induced angiogenesis: Good and evil. Genes Cancer 2011, 2, 1117–1133. [Google Scholar] [CrossRef] [Green Version]
- Akizawa, T.; Iwasaki, M.; Yamaguchi, Y.; Majikawa, Y.; Reusch, M. Phase 3, Randomized, Double-Blind, Active-Comparator (Darbepoetin Alfa) Study of Oral Roxadustat in CKD Patients with Anemia on Hemodialysis in Japan. J. Am. Soc. Nephrol. 2020, 31, 1628–1639. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factor 1 and cardiovascular disease. Annu. Rev. Physiol. 2014, 76, 39–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Provenzano, R.; Fishbane, S.; Szczech, L.; Leong, R.; Saikali, K.G.; Zhong, M.; Lee, T.T.; Houser, M.T.; Frison, L.; Houghton, J.; et al. Pooled Analysis of Roxadustat for Anemia in Patients with Kidney Failure Incident to Dialysis. Kidney Int. Rep. 2021, in press. [Google Scholar]
- Hwang, S.; Nguyen, A.D.; Jo, Y.; Engelking, L.J.; Brugarolas, J.; DeBose-Boyd, R.A. Hypoxia-inducible factor 1alpha activates insulin-induced gene 2 (Insig-2) transcription for degradation of 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase in the liver. J. Biol. Chem. 2017, 292, 9382–9393. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Drug (Dose Strength) | Common Dosage | Advantages |
|---|---|---|
| Oral iron formulations | ||
| Ferric citrate (500 mg/capsule) | 1000 mg (equivalent to 210 mg iron) three times daily with meals | Phosphate control ↓FGF-23; ↑1,25-dihydroxyvitamin D |
| Ferric maltol (30 mg of iron/capsule) | 1 capsule twice daily before meals | High bioavailability Lipid peroxidation resistance |
| Heme iron polypeptide (11 mg of iron/tablet) | 1 tablet three times daily with meals | Absorption through the intestinal heme transporter |
| Sucrosomial iron (30 mg of iron/packet) | 1 packet once daily after a meal | Unique absorption pathways Good GI tolerance |
| IV iron formulations | ||
| Ferumoxytol (510 mg of iron/vial) | 510 mg in a 15-min infusion | Common features: 1. Allows a high-dose IV infusion to quickly be obtained 2. High stability:↓free iron toxicity 3. Low immunogenicity: ↓infusion reactions |
| Iron isomaltoside 1000 (1000 mg of iron/vial) | 1000 mg in a 15-min infusion | |
| Ferric carboxymaltose (750 mg of iron/vial) | 750–1000 mg in a 15-min infusion | |
| Intradialytic iron formulations | ||
| Ferric pyrophosphate citrate (272 mg iron/packet) | 1 packet in every 25 gallons of bicarbonate concentrate | Administered through dialysate Iron is transferred to transferrin without iron sequestration |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, K.-H.; Ho, Y.; Tarng, D.-C. Iron Therapy in Chronic Kidney Disease: Days of Future Past. Int. J. Mol. Sci. 2021, 22, 1008. https://doi.org/10.3390/ijms22031008
Lee K-H, Ho Y, Tarng D-C. Iron Therapy in Chronic Kidney Disease: Days of Future Past. International Journal of Molecular Sciences. 2021; 22(3):1008. https://doi.org/10.3390/ijms22031008
Chicago/Turabian StyleLee, Kuo-Hua, Yang Ho, and Der-Cherng Tarng. 2021. "Iron Therapy in Chronic Kidney Disease: Days of Future Past" International Journal of Molecular Sciences 22, no. 3: 1008. https://doi.org/10.3390/ijms22031008
APA StyleLee, K. -H., Ho, Y., & Tarng, D. -C. (2021). Iron Therapy in Chronic Kidney Disease: Days of Future Past. International Journal of Molecular Sciences, 22(3), 1008. https://doi.org/10.3390/ijms22031008