Regulatory Connections between Iron and Glucose Metabolism



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Iron and Energy Metabolism
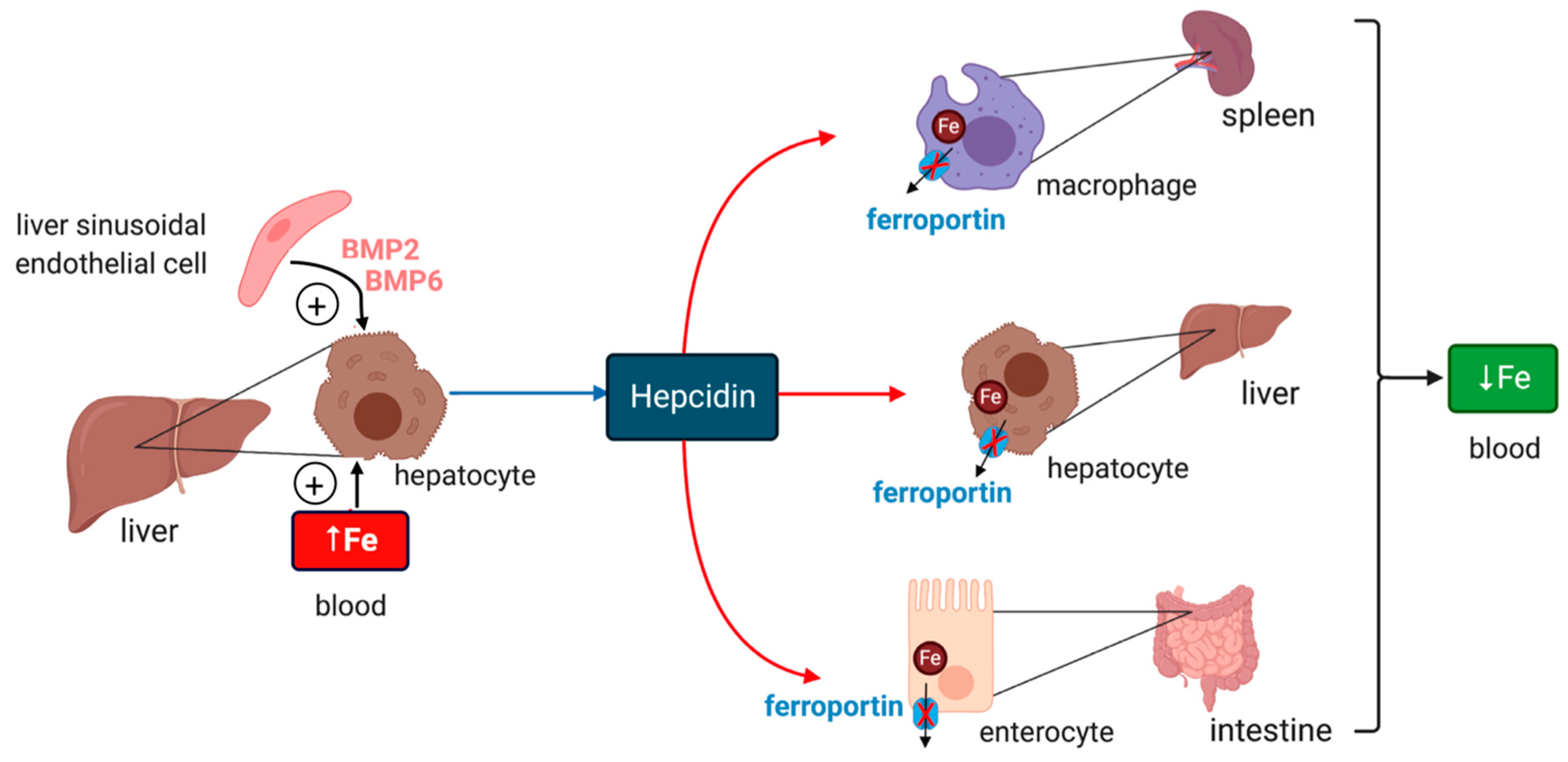
2. Systemic Iron Metabolism
3. Cellular Iron Metabolism
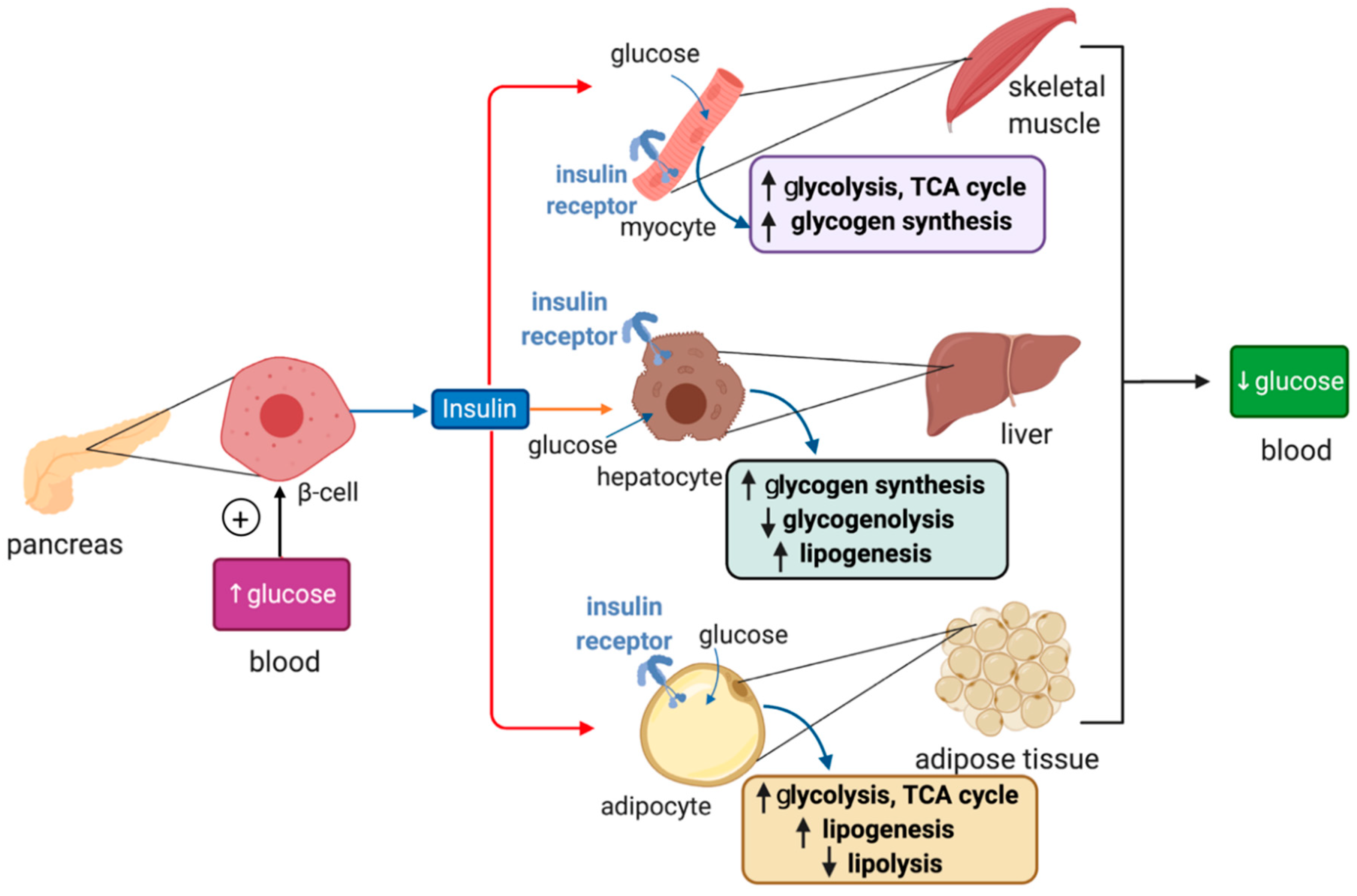
4. Overview of Glucose Metabolism
5. Insights on Iron and Glucose Control from Metabolomics Data
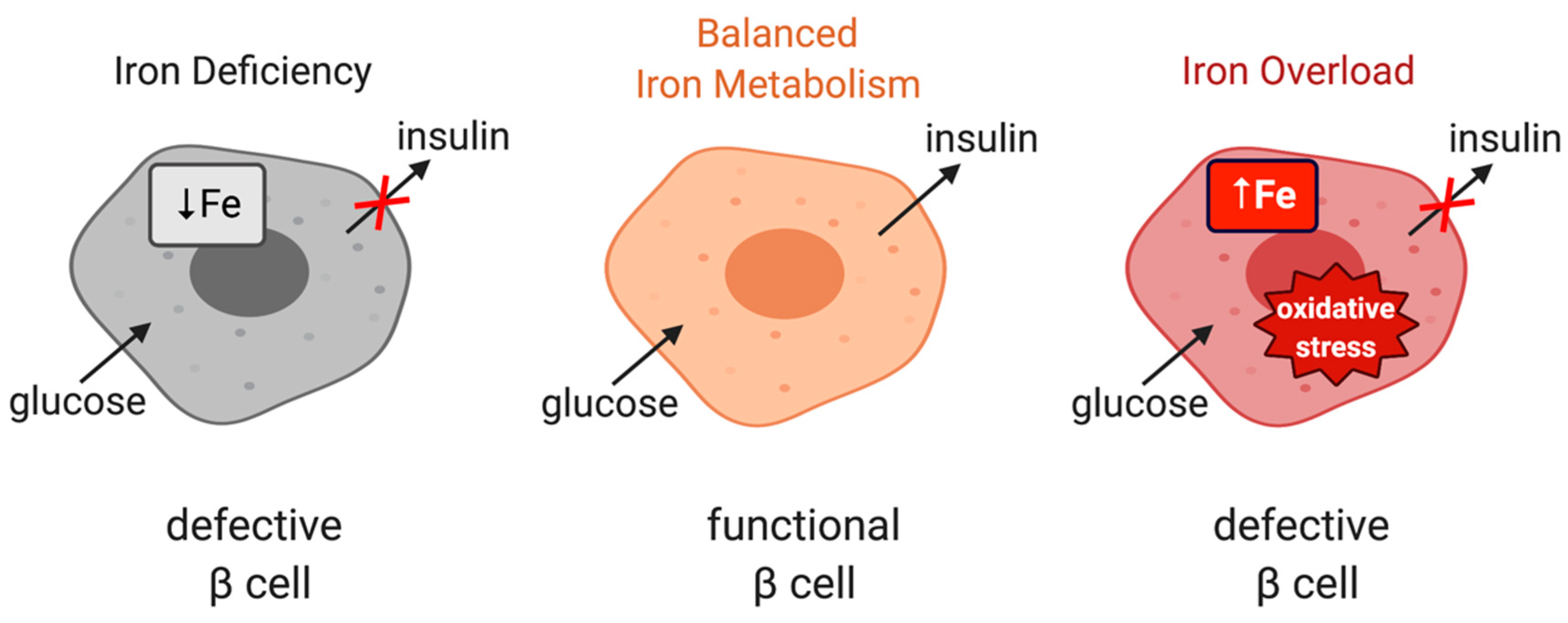
6. Iron and Glucose Metabolism in Human Disease
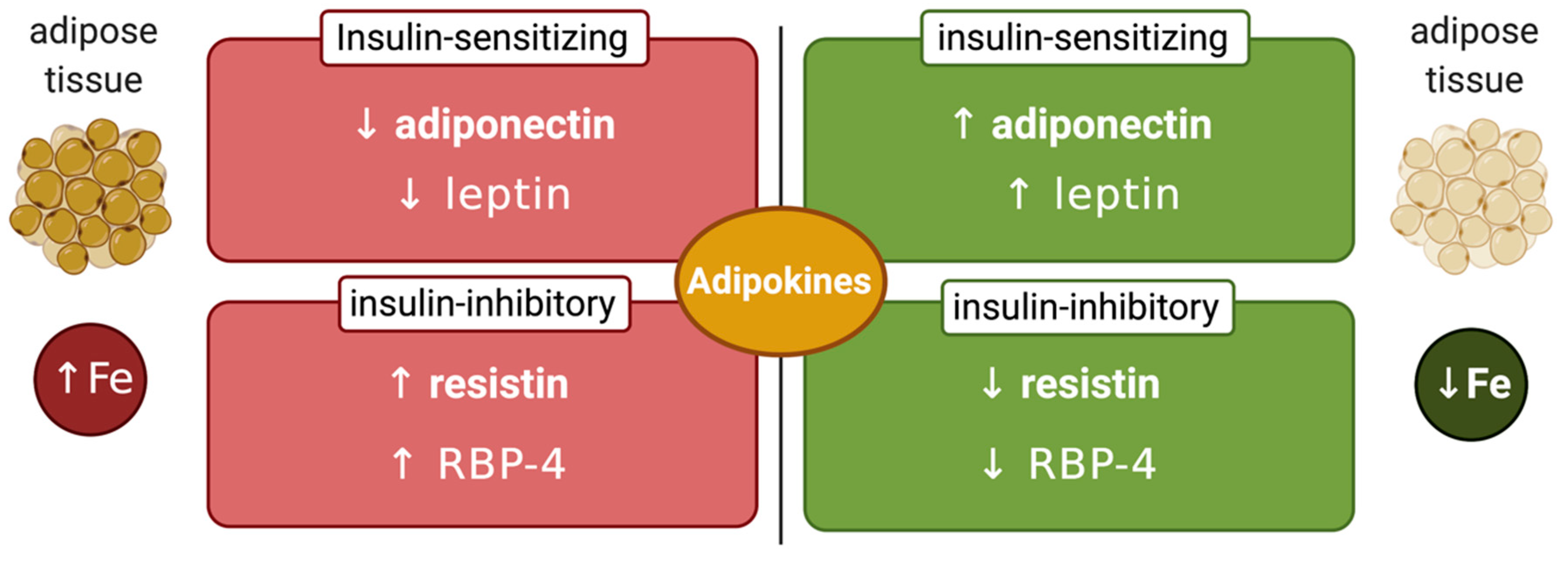
7. Links Between Systemic Iron and Glucose Metabolism
8. Links between Cellular Iron and Glucose Metabolism
9. IRP1 and Glucose Metabolism
10. IRP2 and Glucose Metabolism
11. Conclusions
Funding
Conflicts of Interest
Abbreviations
| TCA | Tricarboxylic acid |
| HO-1 | Heme oxygenase 1 |
| DMT1 | Divalent metal transporter 1 |
| DCYTB | Duodenal cytochrome b |
| HIFα | Hypoxia inducible factor α |
| NTBI | Non-transferrin-bound iron |
| BMP | Bone morphogenetic protein 6 |
| HJV | Hemojuvelin |
| TfR | Transferrin receptor |
| IRP | Iron regulatory protein |
| IRE | Iron responsive element |
| ALAS2 | Aminolevulinic acid synthase 2 |
| FBXL5 | F-box/LRR-repeat protein 5 |
| SGLT1 | Sodium–glucose co-transporter 1 |
| GSK3 | Glycogen synthase kinase 3 |
| HSL | Hormone-sensitive lipase |
| RBP-4 | Retinol-binding protein 4 |
| MetS | Metabolic syndrome |
| DIOS | Dysmetabolic iron overload syndrome |
| NAFLD | Non-alcoholic fatty liver disease |
| NASH | Non-alcoholic steatohepatitis |
| HCC | Hepatocellular carcinoma |
| SCFA | Short chain fatty acids |
| BCAA | Branched chain amino acids |
| LPS | Lipopolysaccharide |
| FOXO-1 | Forkhead box protein O1 |
| AMPK | AMP-dependent kinase |
| ER | Endoplasmic reticulum |
| mTORC1 | Mammalian target of rapamycin complex 1 |
| ALR | Autophagic-lysosome regeneration |
| AGBE | 1,4-alpha-glucan branching enzyme |
References
- Frausto da Silva, J.J.R.; Williams, R.J.P. The Biological Chemistry of the Elements. The Inorganic Chemistry of Life; Clarendon Press: Oxford, UK, 1991; pp. 319–369. [Google Scholar]
- Oexle, H.; Gnaiger, E.; Weiss, G. Iron-dependent changes in cellular energy metabolism: Influence on citric acid cycle and oxidative phosphorylation. Biochim. Biophys. Acta 1999, 1413, 99–107. [Google Scholar] [CrossRef] [Green Version]
- Volani, C.; Doerrier, C.; Demetz, E.; Haschka, D.; Paglia, G.; Lavdas, A.A.; Gnaiger, E.; Weiss, G. Dietary iron loading negatively affects liver mitochondrial function. Metallomics 2017, 9, 1634–1644. [Google Scholar] [CrossRef]
- Galaris, D.; Barbouti, A.; Pantopoulos, K. Iron homeostasis and oxidative stress: An intimate relationship. Biochim. Biophys. Acta Mol. Cell. Res. 2019, 1866, 118535. [Google Scholar] [CrossRef]
- Katsarou, A.; Pantopoulos, K. Basics and principles of cellular and systemic iron homeostasis. Mol. Asp. Med. 2020. [Google Scholar] [CrossRef]
- Gkouvatsos, K.; Papanikolaou, G.; Pantopoulos, K. Regulation of iron transport and the role of transferrin. Biochim. Biophys. Acta Gen. Subj. 2012, 1820, 188–202. [Google Scholar] [CrossRef]
- Shah, Y.M.; Xie, L. Hypoxia-Inducible Factors Link Iron Homeostasis and Erythropoiesis. Gastroenterology 2014, 146, 630–642. [Google Scholar] [CrossRef] [Green Version]
- Ganz, T. Systemic Iron Homeostasis. Physiol. Rev. 2013, 93, 1721–1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bessman, N.J.; Mathieu, J.R.R.; Renassia, C.; Zhou, L.; Fung, T.C.; Fernandez, K.C.; Austin, C.; Moeller, J.B.; Zumerle, S.; Louis, S.; et al. Dendritic cell–derived hepcidin sequesters iron from the microbiota to promote mucosal healing. Science 2020, 368, 186–189. [Google Scholar] [CrossRef] [PubMed]
- Malerba, M.; Louis, S.; Cuvellier, S.; Shambat, S.M.; Hua, C.; Gomart, C.; Fouet, A.; Ortonne, N.; Decousser, J.-W.; Zinkernagel, A.S.; et al. Epidermal hepcidin is required for neutrophil response to bacterial infection. J. Clin. Investig. 2020, 130, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Lakhal-Littleton, S.; Wolna, M.; Chung, Y.J.; Christian, H.C.; Heather, L.C.; Brescia, M.; Ball, V.; Diaz, R.; Santos, A.; Biggs, D.; et al. An essential cell-autonomous role for hepcidin in cardiac iron homeostasis. eLife 2016, 5, 2171. [Google Scholar] [CrossRef]
- Pantopoulos, K. Inherited Disorders of Iron Overload. Front. Nutr. 2018, 5, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, G.; Ganz, T.; Goodnough, L.T. Anemia of inflammation. Blood 2019, 133, 40–50. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.-Y.; Babitt, J.L. Liver iron sensing and body iron homeostasis. Blood 2019, 133, 18–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, M.J.; Lebrón, J.A.; Bjorkman, P.J. Crystal structure of the hereditary haemochromatosis protein HFE complexed with transferrin receptor. Nat. Cell Biol. 2000, 403, 46–53. [Google Scholar] [CrossRef]
- Schmidt, P.J. Regulation of Iron Metabolism by Hepcidin under Conditions of Inflammation. J. Biol. Chem. 2015, 290, 18975–18983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinbicker, A.U.; Sachidanandan, C.; Vonner, A.J.; Yusuf, R.Z.; Deng, D.Y.; Lai, C.S.; Rauwerdink, K.M.; Winn, J.C.; Saez, B.; Cook, C.M.; et al. Inhibition of bone morphogenetic protein signaling attenuates anemia associated with inflammation. Blood 2011, 117, 4915–4923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asshoff, M.; Petzer, V.; Warr, M.R.; Haschka, D.; Tymoszuk, P.; Demetz, E.; Seifert, M.; Posch, W.; Nairz, M.; Maciejewski, P.; et al. Momelotinib inhibits ACVR1/ALK2, decreases hepcidin production, and ameliorates anemia of chronic disease in rodents. Blood 2017, 129, 1823–1830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.-H.; Li, C.; Xu, X.; Zheng, Y.; Xiao, C.; Zerfas, P.; Cooperman, S.; Eckhaus, M.; Rouault, T.; Mishra, L.; et al. A role of SMAD4 in iron metabolism through the positive regulation of hepcidin expression. Cell Metab. 2005, 2, 399–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayeur, C.; Lohmeyer, L.K.; Leyton, P.; Kao, S.M.; Pappas, A.E.; Kolodziej, S.A.; Spagnolli, E.; Yu, B.; Galdos, R.L.; Yu, P.B.; et al. The type I BMP receptor Alk3 is required for the induction of hepatic hepcidin gene expression by interleukin-6. Blood 2014, 123, 2261–2268. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Pantopoulos, K. Regulation of cellular iron metabolism. Biochem. J. 2011, 434, 365–381. [Google Scholar] [CrossRef] [Green Version]
- Walden, W.E.; Selezneva, A.I.; Dupuy, J.; Volbeda, A.; Fontecilla-Camps, J.C.; Theil, E.C.; Volz, K.; Özcan, U.; Cao, Q.; Yilmaz, E.; et al. Structure of Dual Function Iron Regulatory Protein 1 Complexed with Ferritin IRE-RNA. Science 2006, 314, 1903–1908. [Google Scholar] [CrossRef] [PubMed]
- Salahudeen, A.A.; Thompson, J.W.; Ruiz, J.C.; Ma, H.-W.; Kinch, L.N.; Li, Q.; Grishin, N.V.; Bruick, R.K. An E3 Ligase Possessing an Iron-Responsive Hemerythrin Domain Is a Regulator of Iron Homeostasis. Science 2009, 326, 722–726. [Google Scholar] [CrossRef] [Green Version]
- Vashisht, A.A.; Zumbrennen, K.B.; Huang, X.; Powers, D.N.; Durazo, A.; Sun, D.; Bhaskaran, N.; Persson, A.; Uhlen, M.; Sangfelt, O.; et al. Control of Iron Homeostasis by an Iron-Regulated Ubiquitin Ligase. Science 2009, 326, 718–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, A.; Hornby, P.J. Intestinal SGLT1 in metabolic health and disease. Am. J. Physiol. Liver Physiol. 2016, 310, G887–G898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kellett, G.L.; Brot-Laroche, E. Apical GLUT2: A Major Pathway of Intestinal Sugar Absorption. Diabetes 2005, 54, 3056–3062. [Google Scholar] [CrossRef] [Green Version]
- Navale, A.M.; Paranjape, A.N. Glucose transporters: Physiological and pathological roles. Biophys. Rev. 2016, 8, 5–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCulloch, L.J.; Van De Bunt, M.; Braun, M.; Frayn, K.N.; Clark, A.; Gloyn, A.L. GLUT2 (SLC2A2) is not the principal glucose transporter in human pancreatic beta cells: Implications for understanding genetic association signals at this locus. Mol. Genet. Metab. 2011, 104, 648–653. [Google Scholar] [CrossRef]
- Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef] [Green Version]
- Volani, C.; Paglia, G.; Smarason, S.V.; Pramstaller, P.P.; Demetz, E.; Pfeifhofer-Obermair, C.; Weiss, G. Metabolic Signature of Dietary Iron Overload in a Mouse Model. Cells 2018, 7, 264. [Google Scholar] [CrossRef] [Green Version]
- Stechemesser, L.; Eder, S.K.; Wagner, A.; Patsch, W.; Feldman, A.; Strasser, M.; Auer, S.; Niederseer, D.; Huber-Schönauer, U.; Paulweber, B.; et al. Metabolomic profiling identifies potential pathways involved in the interaction of iron homeostasis with glucose metabolism. Mol. Metab. 2017, 6, 38–47. [Google Scholar] [CrossRef]
- Drogan, D.; Dunn, W.B.; Lin, W.; Buijsse, B.; Boeing, H.; Langenberg, C.; Brown, M.; Floegel, A.; Dietrich, S.; Rolandsson, O.; et al. Untargeted Metabolic Profiling Identifies Altered Serum Metabolites of Type 2 Diabetes Mellitus in a Prospective, Nested Case Control Study. Clin. Chem. 2015, 61, 487–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Floegel, A.; Stefan, N.; Yu, Z.; Mühlenbruch, K.; Drogan, D.; Joost, H.-G.; Fritsche, A.; Häring, H.-U.; De Angelis, M.H.; Peters, A.; et al. Identification of Serum Metabolites Associated With Risk of Type 2 Diabetes Using a Targeted Metabolomic Approach. Diabetes 2012, 62, 639–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang-Sattler, R.; Yu, Z.; Herder, C.; Messias, A.C.; Floegel, A.; He, Y.; Heim, K.; Campillos, M.; Holzapfel, C.; Thorand, B.; et al. Novel biomarkers for pre-diabetes identified by metabolomics. Mol. Syst. Biol. 2012, 8, 615. [Google Scholar] [CrossRef] [PubMed]
- Rhee, E.P.; Cheng, S.; Larson, M.G.; Walford, G.A.; Lewis, G.D.; McCabe, E.; Yang, E.; Farrell, L.; Fox, C.S.; O’Donnell, C.J.; et al. Lipid profiling identifies a triacylglycerol signature of insulin resistance and improves diabetes prediction in humans. J. Clin. Investig. 2011, 121, 1402–1411. [Google Scholar] [CrossRef] [Green Version]
- Simcox, J.A.; McClain, D.A. Iron and Diabetes Risk. Cell Metab. 2013, 17, 329–341. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Real, J.M.; Manco, M. Effects of iron overload on chronic metabolic diseases. Lancet Diabetes Endocrinol. 2014, 2, 513–526. [Google Scholar] [CrossRef]
- Utzschneider, K.M.; Kowdley, K.V. Hereditary hemochromatosis and diabetes mellitus: Implications for clinical practice. Nat. Rev. Endocrinol. 2010, 6, 26–33. [Google Scholar] [CrossRef]
- De Sanctis, V.; Soliman, A.T.; Elsedfy, H.; Pepe, A.; Kattamis, C.; El Kholy, M.; Yassin, M. Diabetes and Glucose Metabolism in Thalassemia Major: An Update. Expert Rev. Hematol. 2016, 9, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Bozzini, C.; Girelli, D.; Olivieri, O.; Martinelli, N.; Bassi, A.; De Matteis, G.; Tenuti, I.; Lotto, V.; Friso, S.; Pizzolo, F.; et al. Prevalence of Body Iron Excess in the Metabolic Syndrome. Diabetes Care 2005, 28, 2061–2063. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Real, J.M.; Ricart, W.; Arroyo, E.; Balançá, R.; Casamitjana-Abella, R.; Cabrero, D.; Fernández-Castañer, M.; Soler, J. Serum ferritin as a component of the insulin resistance syndrome. Diabetes Care 1998, 21, 62–68. [Google Scholar] [CrossRef]
- Lee, D.H.; Liu, D.Y.; Jacobs, D.R., Jr.; Shin, H.R.; Song, K.; Lee, I.K.; Kim, B.; Hider, R.C. Common presence of non-transferrin-bound iron among patients with type 2 diabetes. Diabetes Care 2006, 29, 1090–1095. [Google Scholar] [CrossRef] [PubMed]
- Mendler, M.-H.; Turlin, B.; Moirand, R.; Jouanolle, A.-M.; Sapey, T.; Guyader, D.; Le Gall, J.-Y.; Brissot, P.; David, V.; Deugnier, Y. Insulin resistance–associated hepatic iron overload. Gastroenterology 1999, 117, 1155–1163. [Google Scholar] [CrossRef]
- Deugnier, Y.; Bardou-Jacquet, E.; Lainé, F. Dysmetabolic iron overload syndrome (DIOS). Presse Méd. 2017, 46, e306–e311. [Google Scholar] [CrossRef] [PubMed]
- Aigner, E.; Feldman, A.; Datz, C. Obesity as an Emerging Risk Factor for Iron Deficiency. Nutrients 2014, 6, 3587–3600. [Google Scholar] [CrossRef] [PubMed]
- Bellentani, S. The epidemiology of non-alcoholic fatty liver disease. Liver Int. 2017, 37, 81–84. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef]
- Corradini, E.; Pietrangelo, A. Iron and steatohepatitis. J. Gastroenterol. Hepatol. 2012, 27, 42–46. [Google Scholar] [CrossRef] [Green Version]
- Sorrentino, P.; D’Angelo, S.; Ferbo, U.; Micheli, P.; Bracigliano, A.; Vecchione, R. Liver iron excess in patients with hepatocellular carcinoma developed on non-alcoholic steato-hepatitis. J. Hepatol. 2009, 50, 351–357. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Fracanzani, A.L.; Fargion, S.; Valenti, L. Iron in fatty liver and in the metabolic syndrome: A promising therapeutic target. J. Hepatol. 2011, 55, 920–932. [Google Scholar] [CrossRef]
- Valenti, L.; Fracanzani, A.L.; Dongiovanni, P.; Bugianesi, E.; Marchesini, G.; Manzini, P.; Vanni, E.; Fargion, S. Iron Depletion by Phlebotomy Improves Insulin Resistance in Patients With Nonalcoholic Fatty Liver Disease and Hyperferritinemia: Evidence from a Case-Control Study. Am. J. Gastroenterol. 2007, 102, 1251–1258. [Google Scholar] [CrossRef]
- Valenti, L.; Fracanzani, A.L.; Dongiovanni, P.; Rovida, S.; Rametta, R.; Fatta, E.; Pulixi, E.A.; Maggioni, M.; Fargion, S. A randomized trial of iron depletion in patients with nonalcoholic fatty liver disease and hyperferritinemia. World J. Gastroenterol. 2014, 20, 3002–3010. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Valenti, L.; Fracanzani, A.L.; Gatti, S.; Cairo, G.; Fargion, S. Iron Depletion by Deferoxamine Up-Regulates Glucose Uptake and Insulin Signaling in Hepatoma Cells and in Rat Liver. Am. J. Pathol. 2008, 172, 738–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooksey, R.C.; Jones, D.; Gabrielsen, S.; Huang, J.; Simcox, J.A.; Luo, B.; Soesanto, Y.; Rienhoff, H.; Abel, E.D.; McClain, D.A. Dietary iron restriction or iron chelation protects from diabetes and loss of beta-cell function in the obese (ob/ob lep-/-) mouse. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E1236–E1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murali, A.R.; Gupta, A.; Brown, K.E. Systematic review and meta-analysis to determine the impact of iron depletion in dysmetabolic iron overload syndrome and non-alcoholic fatty liver disease. Hepatol. Res. 2017, 48, E30–E41. [Google Scholar] [CrossRef] [Green Version]
- Pietrangelo, A. Hemochromatosis: An endocrine liver disease. Hepatology 2007, 46, 1291–1301. [Google Scholar] [CrossRef]
- Wang, H.; Li, H.; Jiang, X.; Shi, W.; Shen, Z.; Li, M. Hepcidin Is Directly Regulated by Insulin and Plays an Important Role in Iron Overload in Streptozotocin-Induced Diabetic Rats. Diabetes 2014, 63, 1506–1518. [Google Scholar] [CrossRef] [Green Version]
- Aigner, E.; Felder, T.K.; Oberkofler, H.; Hahne, P.; Auer, S.; Soyal, S.; Stadlmayr, A.; Schwenoha, K.; Pirich, C.; Hengster, P.; et al. Glucose acts as a regulator of serum iron by increasing serum hepcidin concentrations. J. Nutr. Biochem. 2013, 24, 112–117. [Google Scholar] [CrossRef]
- Mao, X.; Chen, H.; Tang, J.; Wang, L.; Shu, T. Hepcidin links gluco-toxicity to pancreatic beta cell dysfunction by inhibiting Pdx-1 expression. Endocr. Connect. 2017, 6, 121–128. [Google Scholar] [CrossRef] [Green Version]
- Shu, T.; Lv, Z.; Xie, Y.; Tang, J.; Mao, X. Hepcidin as a key iron regulator mediates glucotoxicity-induced pancreatic beta-cell dysfunction. Endocr. Connect. 2019, 8, 150–161. [Google Scholar] [CrossRef]
- Tajima, S.; Ikeda, Y.; Sawada, K.; Yamano, N.; Horinouchi, Y.; Kihira, Y.; Ishizawa, K.; Izawa-Ishizawa, Y.; Kawazoe, K.; Tomita, S.; et al. Iron reduction by deferoxamine leads to amelioration of adiposity via the regulation of oxidative stress and inflammation in obese and type 2 diabetes KKAy mice. Am. J. Physiol. Metab. 2012, 302, E77–E86. [Google Scholar] [CrossRef] [Green Version]
- Vecchi, C.; Montosi, G.; Garuti, C.; Corradini, E.; Sabelli, M.; Canali, S.; Pietrangelo, A. Gluconeogenic Signals Regulate Iron Homeostasis via Hepcidin in Mice. Gastroenterology 2014, 146, 1060–1069.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, B.; Matak, P.; McKie, A.T.; Sharp, P.A. Leptin Increases the Expression of the Iron Regulatory Hormone Hepcidin in HuH7 Human Hepatoma Cells. J. Nutr. 2007, 137, 2366–2370. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Kuragano, T.; Kimura, T.; Nanami, M.; Hasuike, Y.; Nakanishi, T. Interplay of adipocyte and hepatocyte: Leptin upregulates hepcidin. Biochem. Biophys. Res. Commun. 2018, 495, 1548–1554. [Google Scholar] [CrossRef] [PubMed]
- Altamura, S.; Kopf, S.; Schmidt, J.; Müdder, K.; Da Silva, A.R.; Nawroth, P.; Muckenthaler, M.U. Uncoupled iron homeostasis in type 2 diabetes mellitus. J. Mol. Med. 2017, 95, 1387–1398. [Google Scholar] [CrossRef]
- Padda, R.S.; Gkouvatsos, K.; Guido, M.; Mui, J.; Vali, H.; Pantopoulos, K. A high-fat diet modulates iron metabolism but does not promote liver fibrosis in hemochromatotic Hjv-/- mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G251–G261. [Google Scholar] [CrossRef] [Green Version]
- Chung, J.; Kim, M.S.; Han, S.N. Diet-induced obesity leads to decreased hepatic iron storage in mice. Nutr. Res. 2011, 31, 915–921. [Google Scholar] [CrossRef]
- Sonnweber, T.; Ress, C.; Nairz, M.; Theurl, I.; Schroll, A.; Murphy, A.T.; Wroblewski, V.; Witcher, D.R.; Moser, P.; Ebenbichler, C.; et al. High-fat diet causes iron deficiency via hepcidin-independent reduction of duodenal iron absorption. J. Nutr. Biochem. 2012, 23, 1600–1608. [Google Scholar] [CrossRef]
- Meli, R.; Raso, G.M.; Irace, C.; Simeoli, R.; Di Pascale, A.; Paciello, O.; Pagano, T.B.; Calignano, A.; Colonna, A.; Santamaria, R. High Fat Diet Induces Liver Steatosis and Early Dysregulation of Iron Metabolism in Rats. PLoS ONE 2013, 8, e66570. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Lanti, C.; Gatti, S.; Rametta, R.; Recalcati, S.; Maggioni, M.; Fracanzani, A.L.; Riso, P.; Cairo, G.; Fargion, S.; et al. High Fat Diet Subverts Hepatocellular Iron Uptake Determining Dysmetabolic Iron Overload. PLoS ONE 2015, 10, e0116855. [Google Scholar] [CrossRef] [Green Version]
- Engin, A. The Pathogenesis of Obesity-Associated Adipose Tissue Inflammation. Adv. Exp. Med. Biol. 2017, 960, 221–245. [Google Scholar]
- Coimbra, S.; Catarino, C.; Santos-Silva, A. The role of adipocytes in the modulation of iron metabolism in obesity. Obes. Rev. 2013, 14, 771–779. [Google Scholar] [CrossRef] [PubMed]
- Folgueras, A.R.; Martin de Lara, F.; Pendas, A.M.; Garabaya, C.; Rodriguez, F.; Astudillo, A.; Bernal, T.; Cabanillas, R.; Lopez-Otin, C.; Velasco, G. The membrane-bound serine protease matriptase-2 (Tmprss6) is an essential regulator of iron homeostasis. Blood 2008, 112, 2539–2545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folgueras, A.R.; Freitas-Rodríguez, S.; Ramsay, A.J.; Garabaya, C.; Rodríguez, F.; Velasco, G.; López-Otín, C. Matriptase-2 deficiency protects from obesity by modulating iron homeostasis. Nat. Commun. 2018, 9, 1350. [Google Scholar] [CrossRef] [PubMed]
- Saad, M.J.A.; Santos, A.; Prada, P.O. Linking Gut Microbiota and Inflammation to Obesity and Insulin Resistance. Physiology 2016, 31, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Zmora, N.; Bashiardes, S.; Levy, M.; Elinav, E. The Role of the Immune System in Metabolic Health and Disease. Cell Metab. 2017, 25, 506–521. [Google Scholar] [CrossRef] [Green Version]
- Tilg, H.; Cani, P.D.; Mayer, E.A. Gut microbiome and liver diseases. Gut 2016, 65, 2035–2044. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, B.; Li, H. Gut Microbiota and Iron: The Crucial Actors in Health and Disease. Pharmaceuticals 2018, 11, 98. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.; Clavel, T.; Smirnov, K.; Schmidt, A.; Lagkouvardos, I.; Walker, A.; Lucio, M.; Michalke, B.; Schmitt-Kopplin, P.; Fedorak, R.; et al. Oral versus intravenous iron replacement therapy distinctly alters the gut microbiota and metabolome in patients with IBD. Gut 2016, 66, 863–871. [Google Scholar] [CrossRef]
- La Carpia, F.; Wojczyk, B.; Rebbaa, A.; Tang, A.; Hod, E.A. Chronic Transfusion and Iron Overload Modify the Mouse Gut Microbiome. Blood 2016, 128, 200. [Google Scholar] [CrossRef]
- Buhnik-Rosenblau, K.; Moshe-Belizowski, S.; Danin-Poleg, Y.; Meyron-Holtz, E.G. Genetic modification of iron metabolism in mice affects the gut microbiota. BioMetals 2012, 25, 883–892. [Google Scholar] [CrossRef]
- Das, N.K.; Schwartz, A.J.; Barthel, G.; Inohara, N.; Liu, Q.; Sankar, A.; Hill, D.R.; Ma, X.; Lamberg, O.; Schnizlein, M.K.; et al. Microbial Metabolite Signaling Is Required for Systemic Iron Homeostasis. Cell Metab. 2020, 31, 115–130.e6. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Ruscica, M.; Rametta, R.; Recalcati, S.; Steffani, L.; Gatti, S.; Girelli, D.; Cairo, G.; Magni, P.; Fargion, S.; et al. Dietary Iron Overload Induces Visceral Adipose Tissue Insulin Resistance. Am. J. Pathol. 2013, 182, 2254–2263. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Real, J.M.; Moreno-Navarrete, J.M.; Ricart, W. Circulating Retinol-Binding Protein-4 Concentration Might Reflect Insulin Resistance–Associated Iron Overload. Diabetes 2008, 57, 1918–1925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Li, Z.; Gabrielsen, J.S.; Simcox, J.A.; Lee, S.-H.; Jones, D.; Cooksey, B.; Stoddard, G.; Cefalu, W.T.; McClain, D.A. Adipocyte iron regulates leptin and food intake. J. Clin. Investig. 2015, 125, 3681–3691. [Google Scholar] [CrossRef] [Green Version]
- Gabrielsen, J.S.; Gao, Y.; Simcox, J.A.; Huang, J.; Thorup, D.; Jones, D.; Cooksey, R.C.; Gabrielsen, D.; Adams, T.D.; Hunt, S.C.; et al. Adipocyte iron regulates adiponectin and insulin sensitivity. J. Clin. Investig. 2012, 122, 3529–3540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Britton, L.; Jaskowski, L.-A.; Bridle, K.; Secondes, E.; Wallace, D.F.; Santrampurwala, N.; Reiling, J.; Miller, G.; Mangiafico, S.; Andrikopoulos, S.; et al. Ferroportin Expression in Adipocytes Does Not Contribute to Iron Homeostasis or Metabolic Responses to a High Calorie Diet. Cell. Mol. Gastroenterol. Hepatol. 2018, 5, 319–331. [Google Scholar] [CrossRef] [Green Version]
- Miranda, M.A.; Pierre, C.L.S.; Macias-Velasco, J.F.; Nguyen, H.A.; Schmidt, H.; Agnello, L.T.; Wayhart, J.P.; Lawson, H.A. Dietary iron interacts with genetic background to influence glucose homeostasis. Nutr. Metab. 2019, 16, 13. [Google Scholar] [CrossRef]
- Bekri, S.; Gual, P.; Anty, R.; Luciani, N.; Dahman, M.; Ramesh, B.; Iannelli, A.; Staccini–Myx, A.; Casanova, D.; Ben Amor, I.; et al. Increased Adipose Tissue Expression of Hepcidin in Severe Obesity Is Independent From Diabetes and NASH. Gastroenterology 2006, 131, 788–796. [Google Scholar] [CrossRef]
- Gotardo, E.M.F.; Dos Santos, A.N.; Miyashiro, R.A.; Gambero, S.; Rocha, T.; Ribeiro, M.L.; Gambero, A. Mice That Are Fed a High-Fat Diet Display Increased Hepcidin Expression in Adipose Tissue. J. Nutr. Sci. Vitaminol. 2013, 59, 454–461. [Google Scholar] [CrossRef] [Green Version]
- Orr, J.S.; Kennedy, A.; Anderson-Baucum, E.K.; Webb, C.D.; Fordahl, S.C.; Erikson, K.M.; Zhang, Y.; Etzerodt, A.; Moestrup, S.K.; Hasty, A.H. Obesity Alters Adipose Tissue Macrophage Iron Content and Tissue Iron Distribution. Diabetes 2014, 63, 421–432. [Google Scholar] [CrossRef] [Green Version]
- Hubler, M.J.; Peterson, K.R.; Hasty, A.H. Iron homeostasis: A new job for macrophages in adipose tissue? Trends Endocrinol. Metab. 2015, 26, 101–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, R.J.; Corvera, S.; Czech, M.P. Insulin stimulates cellular iron uptake and causes the redistribution of intracellular transferrin receptors to the plasma membrane. J. Biol. Chem. 1986, 261, 8708–8711. [Google Scholar] [PubMed]
- Biswas, S.; Tapryal, N.; Mukherjee, R.; Kumar, R.; Mukhopadhyay, C.K. Insulin promotes iron uptake in human hepatic cell by regulating transferrin receptor-1 transcription mediated by hypoxia inducible factor-1. Biochim. Biophys. Acta 2013, 1832, 293–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, A.; Basile, R.; Rumberger, J.M. Transferrin and iron induce insulin resistance of glucose transport in adipocytes. Metabolism 2006, 55, 1042–1045. [Google Scholar] [CrossRef]
- Varghese, J.; James, J.; Vaulont, S.; McKie, A.; Jacob, M. Increased intracellular iron in mouse primary hepatocytes in vitro causes activation of the Akt pathway but decreases its response to insulin. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 1870–1882. [Google Scholar] [CrossRef]
- Sung, H.K.; Song, E.; Jahng, J.W.S.; Pantopoulos, K.; Sweeney, G. Iron induces insulin resistance in cardiomyocytes via regulation of oxidative stress. Sci. Rep. 2019, 9, 4668. [Google Scholar] [CrossRef] [Green Version]
- Potashnik, R.; Kozlovsky, N.; Ben-Ezra, S.; Rudich, A.; Bashan, N. Regulation of glucose transport and GLUT-1 expression by iron chelators in muscle cells in culture. Am. J. Physiol. Metab. 1995, 269, E1052–E1058. [Google Scholar] [CrossRef]
- Paddock, M.L.; E Wiley, S.; Axelrod, H.L.; Cohen, A.E.; Roy, M.; Abresch, E.C.; Capraro, M.; Murphy, A.N.; Nechushtai, R.; Dixon, J.E.; et al. MitoNEET is a uniquely folded 2Fe 2S outer mitochondrial membrane protein stabilized by pioglitazone. Proc. Natl. Acad. Sci. USA 2007, 104, 14342–14347. [Google Scholar] [CrossRef] [Green Version]
- Kusminski, C.M.; Holland, W.L.; Sun, K.; Park, J.; Spurgin, S.B.; Lin, Y.; Askew, G.R.; Simcox, J.A.; McClain, D.A.; Li, C.; et al. MitoNEET-driven alterations in adipocyte mitochondrial activity reveal a crucial adaptive process that preserves insulin sensitivity in obesity. Nat. Med. 2012, 18, 1539–1549. [Google Scholar] [CrossRef] [Green Version]
- Mittler, R.; Darash-Yahana, M.; Sohn, Y.S.; Bai, F.; Song, L.; Cabantchik, I.Z.; Jennings, P.A.; Onuchic, J.N.; Nechushtai, R. NEET Proteins: A New Link Between Iron Metabolism, Reactive Oxygen Species, and Cancer. Antioxidants Redox Signal. 2019, 30, 1083–1095. [Google Scholar] [CrossRef]
- Kusminski, C.M.; Chen, S.; Ye, R.; Sun, K.; Wang, Q.A.; Spurgin, S.B.; Sanders, P.E.; Brozinick, J.T.; Geldenhuys, W.J.; Li, W.H.; et al. MitoNEET-Parkin Effects in Pancreatic alpha- and beta-Cells, Cellular Survival, and Intrainsular Cross Talk. Diabetes 2016, 65, 1534–1555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooksey, R.C.; Jouihan, H.A.; Ajioka, R.S.; Hazel, M.W.; Weiss, S.M.; Kushner, J.P.; McClain, D.A. Oxidative Stress, β-Cell Apoptosis, and Decreased Insulin Secretory Capacity in Mouse Models of Hemochromatosis. Endocrinology 2004, 145, 5305–5312. [Google Scholar] [CrossRef] [PubMed]
- Masuda, Y.; Ichii, H.; Vaziri, N.D. At pharmacologically relevant concentrations intravenous iron preparations cause pancreatic beta cell death. Am. J. Transl. Res. 2013, 6, 64–70. [Google Scholar] [PubMed]
- Hansen, J.B.; Tonnesen, M.F.; Madsen, A.N.; Hagedorn, P.H.; Friberg, J.; Grunnet, L.G.; Heller, R.S.; Nielsen, A.O.; Storling, J.; Baeyens, L.; et al. Divalent metal transporter 1 regulates iron-mediated ROS and pancreatic beta cell fate in response to cytokines. Cell Metab. 2012, 16, 449–461. [Google Scholar] [CrossRef] [Green Version]
- Ramey, G.; Faye, A.; Durel, B.; Viollet, B.; Vaulont, S. Iron overload in Hepc1(-/-) mice is not impairing glucose homeostasis. FEBS Lett. 2007, 581, 1053–1057. [Google Scholar] [CrossRef] [Green Version]
- Altamura, S.; Kessler, R.; Gröne, H.-J.; Gretz, N.; Hentze, M.W.; Galy, B.; Muckenthaler, M.U. Resistance of Ferroportin to Hepcidin Binding causes Exocrine Pancreatic Failure and Fatal Iron Overload. Cell Metab. 2014, 20, 359–367. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Gabrielsen, J.S.; Cooksey, R.C.; Luo, B.; Boros, L.G.; Jones, D.; Jouihan, H.A.; Soesanto, Y.; Knecht, L.; Hazel, M.W.; et al. Increased Glucose Disposal and AMP-dependent Kinase Signaling in a Mouse Model of Hemochromatosis. J. Biol. Chem. 2007, 282, 37501–37507. [Google Scholar] [CrossRef] [Green Version]
- DeFronzo, R.A.; Tripathy, D. Skeletal Muscle Insulin Resistance Is the Primary Defect in Type 2 Diabetes. Diabetes Care 2009, 32, S157–S163. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Cui, R.; Kim, D.J.; Choi, S.; Lee, W.J.; Kang, Y.; Kim, T.H.; Moon, H.U.; Jeon, J.Y.; Han, S.J.; et al. Iron Overload Plays an Important Role in ER Stress-Induced Insulin Resistance in Human Skeletal Muscle Cells. Diabetes 2018, 67, 1912. [Google Scholar] [CrossRef]
- Cui, R.; Choi, S.-E.; Kim, T.H.; Lee, H.J.; Lee, S.J.; Kang, Y.; Jeon, J.Y.; Kim, H.J.; Lee, K.-W. Iron overload by transferrin receptor protein 1 regulation plays an important role in palmitate-induced insulin resistance in human skeletal muscle cells. FASEB J. 2018, 33, 1771–1786. [Google Scholar] [CrossRef] [Green Version]
- Ahlstrom, P.; Rai, E.; Chakma, S.; Cho, H.H.; Rengasamy, P.; Sweeney, G. Adiponectin improves insulin sensitivity via activation of autophagic flux. J. Mol. Endocrinol. 2017, 59, 339–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jahng, J.W.S.; Alsaadi, R.M.; Palanivel, R.; Song, E.; Hipolito, V.E.B.; Sung, H.K.; Botelho, R.J.; Russell, R.C.; Sweeney, G. Iron overload inhibits late stage autophagic flux leading to insulin resistance. EMBO Rep. 2019, 20, e47911. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yu, L. Development of Research into Autophagic Lysosome Reformation. Mol. Cells 2018, 41, 45–49. [Google Scholar] [PubMed]
- Meyron-Holtz, E.G.; Ghosh, M.C.; Iwai, K.; LaVaute, T.; Brazzolotto, X.; Berger, U.V.; Land, W.; Ollivierre-Wilson, H.; Grinberg, A.; Love, P.; et al. Genetic ablations of iron regulatory proteins 1 and 2 reveal why iron regulatory protein 2 dominates iron homeostasis. EMBO J. 2004, 23, 386–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, M.C.; Zhang, D.-L.; Jeong, S.Y.; Kovtunovych, G.; Ollivierre-Wilson, H.; Noguchi, A.; Tu, T.; Senecal, T.; Robinson, G.; Crooks, D.R.; et al. Deletion of Iron Regulatory Protein 1 Causes Polycythemia and Pulmonary Hypertension in Mice through Translational Derepression of HIF2α. Cell Metab. 2013, 17, 271–281. [Google Scholar] [CrossRef] [Green Version]
- Anderson, S.A.; Nizzi, C.P.; Chang, Y.-I.; Deck, K.M.; Schmidt, P.J.; Galy, B.; Damnernsawad, A.; Broman, A.T.; Kendziorski, C.; Hentze, M.W.; et al. The IRP1-HIF-2α axis coordinates iron and oxygen sensing with erythropoiesis and iron absorption. Cell Metab. 2013, 17, 282–290. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, N.; Pantopoulos, K. IRP1 regulates erythropoiesis and systemic iron homeostasis by controlling HIF2α mRNA translation. Blood 2013, 122, 1658–1668. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, N.; Pantopoulos, K. Defective glucose metabolism in Irp1-/- mice. Am. J. Hematol. 2013, 88, E66. [Google Scholar]
- Taniguchi, C.M.; Finger, E.C.; Krieg, A.J.; Wu, C.; Diep, A.N.; LaGory, E.L.; Wei, K.; McGinnis, L.M.; Yuan, J.; Kuo, C.J.; et al. Cross-talk between hypoxia and insulin signaling through Phd3 regulates hepatic glucose and lipid metabolism and ameliorates diabetes. Nat. Med. 2013, 19, 1325–1330. [Google Scholar] [CrossRef] [Green Version]
- Wei, K.; Piecewicz, S.M.; McGinnis, L.M.; Taniguchi, C.M.; Wiegand, S.J.; Anderson, K.; Chan, C.W.-M.; Mulligan, K.X.; Kuo, D.; Yuan, J.; et al. A liver Hif-2α–Irs2 pathway sensitizes hepatic insulin signaling and is modulated by Vegf inhibition. Nat. Med. 2013, 19, 1331–1337. [Google Scholar] [CrossRef]
- Ramakrishnan, S.K.; Zhang, H.; Takahashi, S.; Centofanti, B.; Periyasamy, S.; Weisz, K.; Chen, Z.; Uhler, M.D.; Rui, L.; Gonzalez, F.J.; et al. HIF2 α Is an Essential Molecular Brake for Postprandial Hepatic Glucagon Response Independent of Insulin Signaling. Cell Metab. 2016, 23, 505–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, M.; Ortega, F.; Xifra, G.; Ricart, W.; Fernández-Real, J.M.; Moreno-Navarrete, J.M. Cytosolic aconitase activity sustains adipogenic capacity of adipose tissue connecting iron metabolism and adipogenesis. FASEB J. 2015, 29, 1529–1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huynh, N.; Ou, Q.; Cox, P.; Lill, R.; King-Jones, K. Glycogen branching enzyme controls cellular iron homeostasis via Iron Regulatory Protein 1 and mitoNEET. Nat. Commun. 2019, 10, 1–18. [Google Scholar] [CrossRef]
- Ferecatu, I.; Gonçalves, S.; Golinelli-Cohen, M.-P.; Clémancey, M.; Martelli, A.; Riquier, S.; Guittet, E.; Latour, J.-M.; Puccio, H.; Drapier, J.-C.; et al. The Diabetes Drug Target MitoNEET Governs a Novel Trafficking Pathway to Rebuild an Fe-S Cluster into Cytosolic Aconitase/Iron Regulatory Protein 1. J. Biol. Chem. 2014, 289, 28070–28086. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Gallardo, A.K.; Missirlis, F. Cellular iron sensing and regulation: Nuclear IRP1 extends a classic paradigm. Biochim. Biophys. Acta Mol. Cell. Res. 2020, 1867, 118705. [Google Scholar] [CrossRef]
- Cooperman, S.S.; Meyron-Holtz, E.G.; Olivierre-Wilson, H.; Ghosh, M.C.; McConnell, J.P.; Rouault, T.A. Microcytic anemia, erythropoietic protoporphyria, and neurodegeneration in mice with targeted deletion of iron-regulatory protein 2. Blood 2005, 106, 1084–1091. [Google Scholar] [CrossRef] [Green Version]
- Galy, B.; Ferring, D.; Minana, B.; Bell, O.; Janser, H.G.; Muckenthaler, M.; Schümann, K.; Hentze, M.W. Altered body iron distribution and microcytosis in mice deficient in iron regulatory protein 2 (IRP2). Blood 2005, 106, 2580–2589. [Google Scholar] [CrossRef] [PubMed]
- LaVaute, T.; Smith, S.; Cooperman, S.; Iwai, K.; Land, W.; Meyron-Holtz, E.; Drake, S.K.; Miller, G.; Abu-Asab, M.; Tsokos, M.; et al. Targeted deletion of the gene encoding iron regulatory protein-2 causes misregulation of iron metabolism and neurodegenerative disease in mice. Nat. Genet. 2001, 27, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Galy, B.; Hölter, S.M.; Klopstock, T.; Ferring, D.; Becker, L.; Kaden, S.; Wurst, W.; Gröne, H.-J.; Hentze, M.W.; Ferring-Appel, D. Iron homeostasis in the brain: Complete iron regulatory protein 2 deficiency without symptomatic neurodegeneration in the mouse. Nat. Genet. 2006, 38, 967–969. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.C.; Ollivierre-Wilson, H.; Cooperman, S.; Rouault, T.A. Reply to “Iron homeostasis in the brain: Complete iron regulatory protein 2 deficiency without symptomatic neurodegeneration in the mouse”. Nat. Genet. 2006, 38, 969–970. [Google Scholar] [CrossRef]
- Zumbrennen-Bullough, K.B.; Becker, L.; Garrett, L.; Hölter, S.M.; Calzada-Wack, J.; Mossbrugger, I.; Quintanilla-Fend, L.; Rácz, I.; Rathkolb, B.; Klopstock, T.; et al. Abnormal Brain Iron Metabolism in Irp2 Deficient Mice Is Associated with Mild Neurological and Behavioral Impairments. PLoS ONE 2014, 9, e98072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dos Santos, M.C.F.; Anderson, C.P.; Neschen, S.; Zumbrennen-Bullough, K.B.; Romney, S.J.; Kahle-Stephan, M.; Rathkolb, B.; Gailus-Durner, V.; Fuchs, H.; Wolf, E.; et al. Irp2 regulates insulin production through iron-mediated Cdkal1-catalyzed tRNA modification. Nat. Commun. 2020, 11, 296. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fillebeen, C.; Lam, N.H.; Chow, S.; Botta, A.; Sweeney, G.; Pantopoulos, K. Regulatory Connections between Iron and Glucose Metabolism. Int. J. Mol. Sci. 2020, 21, 7773. https://doi.org/10.3390/ijms21207773
Fillebeen C, Lam NH, Chow S, Botta A, Sweeney G, Pantopoulos K. Regulatory Connections between Iron and Glucose Metabolism. International Journal of Molecular Sciences. 2020; 21(20):7773. https://doi.org/10.3390/ijms21207773
Chicago/Turabian StyleFillebeen, Carine, Nhat Hung Lam, Samantha Chow, Amy Botta, Gary Sweeney, and Kostas Pantopoulos. 2020. "Regulatory Connections between Iron and Glucose Metabolism" International Journal of Molecular Sciences 21, no. 20: 7773. https://doi.org/10.3390/ijms21207773
APA StyleFillebeen, C., Lam, N. H., Chow, S., Botta, A., Sweeney, G., & Pantopoulos, K. (2020). Regulatory Connections between Iron and Glucose Metabolism. International Journal of Molecular Sciences, 21(20), 7773. https://doi.org/10.3390/ijms21207773