Multi-Omics Analysis of Small RNA, Transcriptome, and Degradome in T. turgidum—Regulatory Networks of Grain Development and Abiotic Stress Response




Abstract
:1. Introduction
2. Results
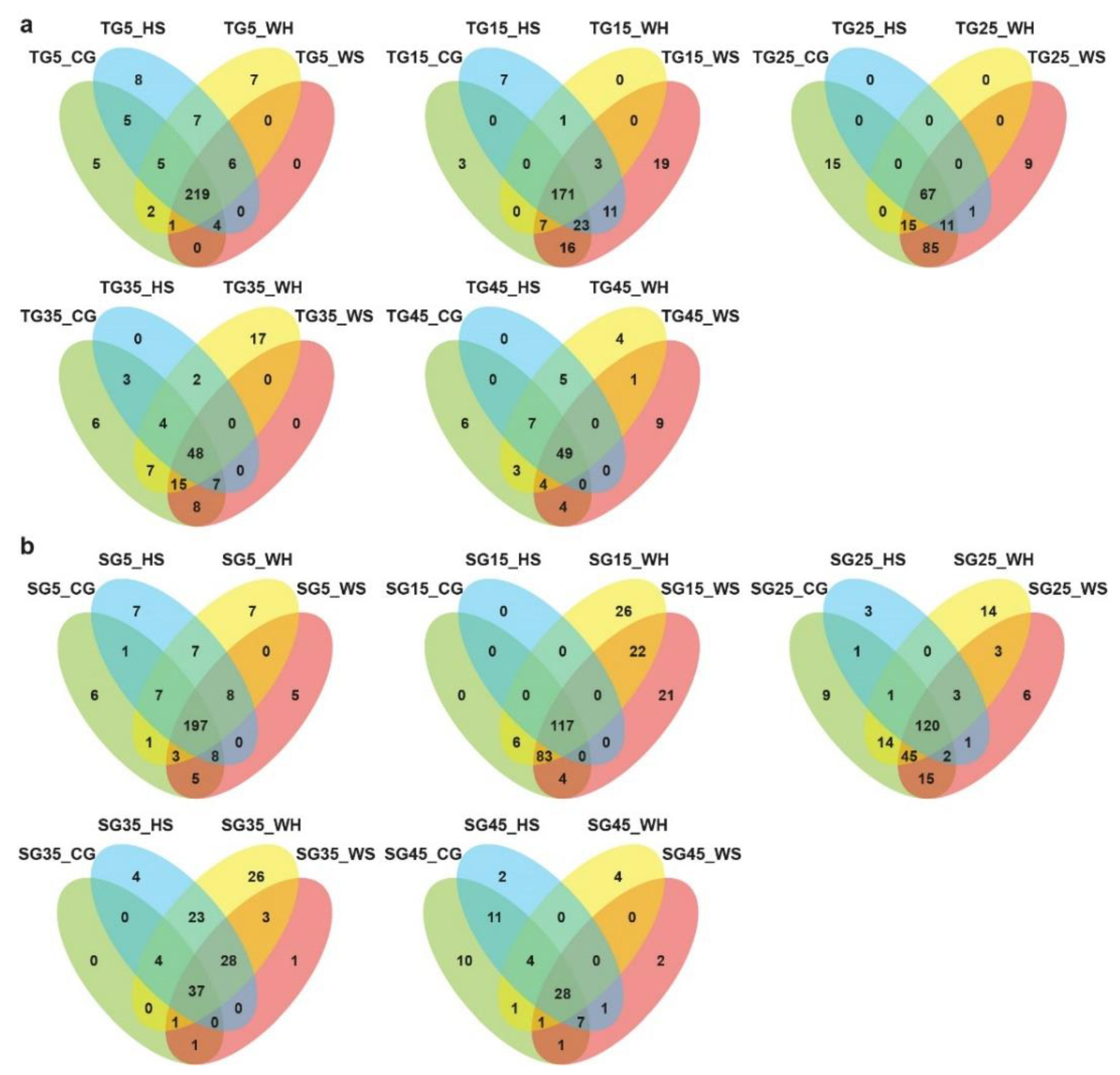
2.1. Conserved and Novel miRNAs Revealed by sRNA Sequencing
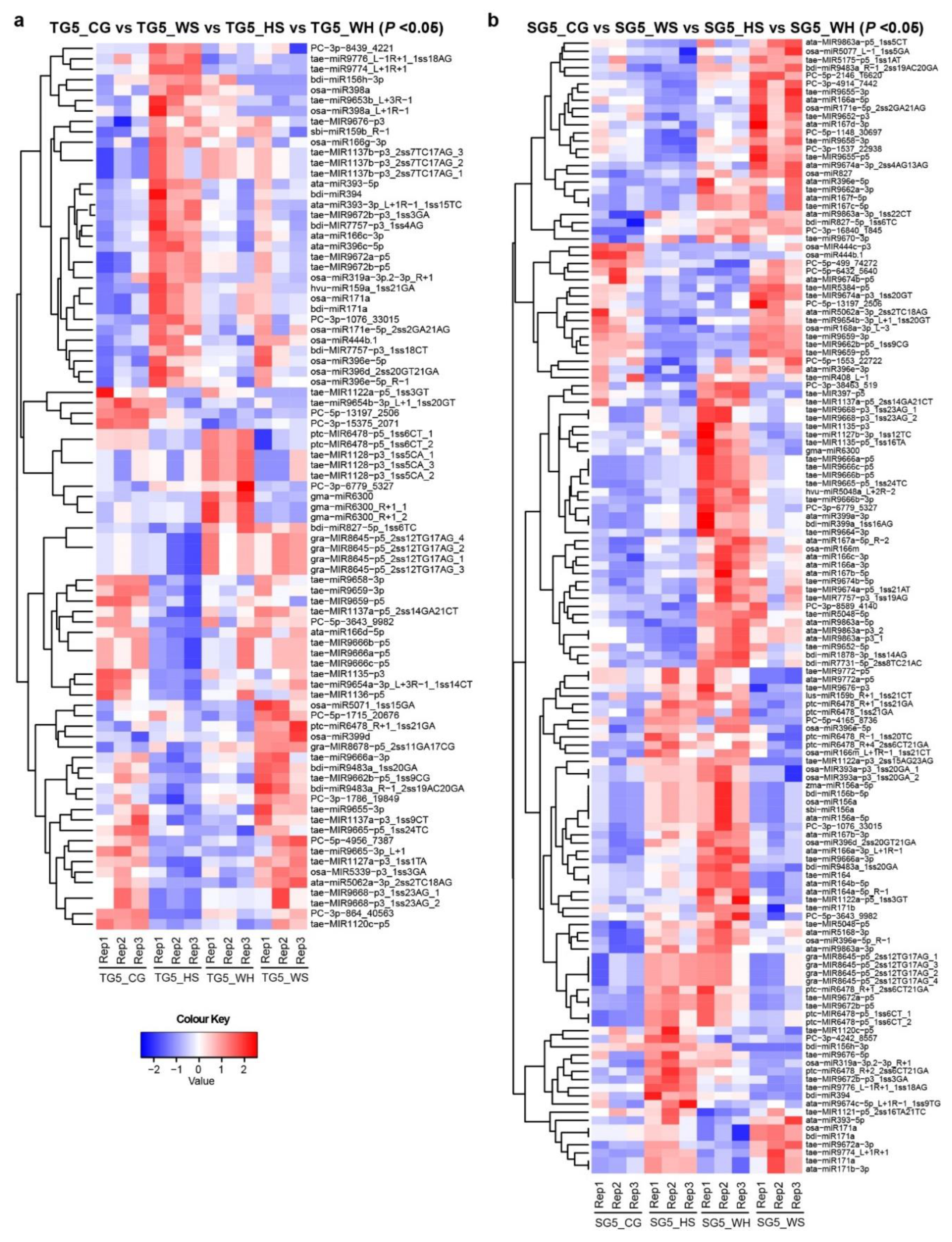
2.2. DEM (Differentially Expressed miRNAs) Dependent on Stress, Genotype, and Time-Point
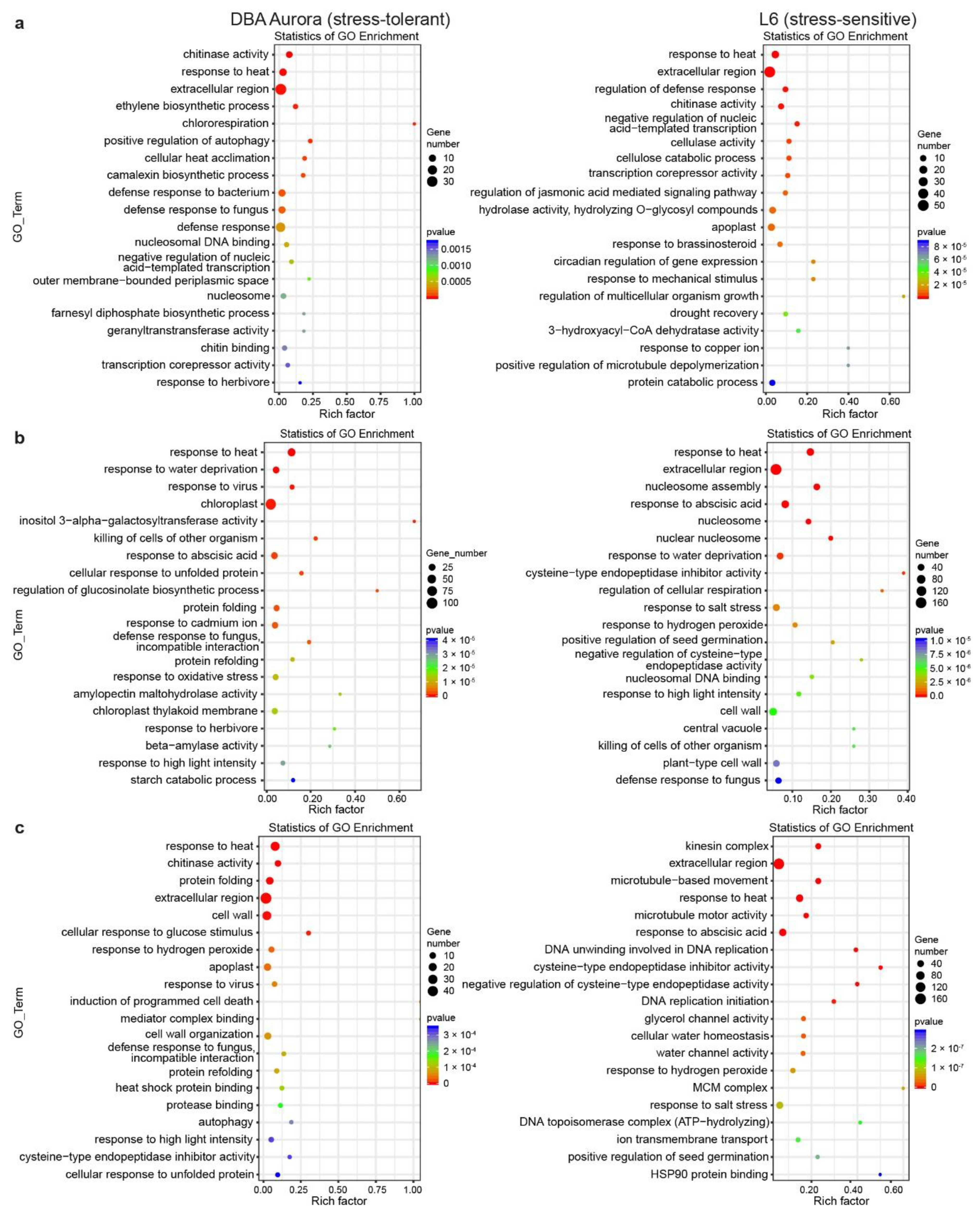
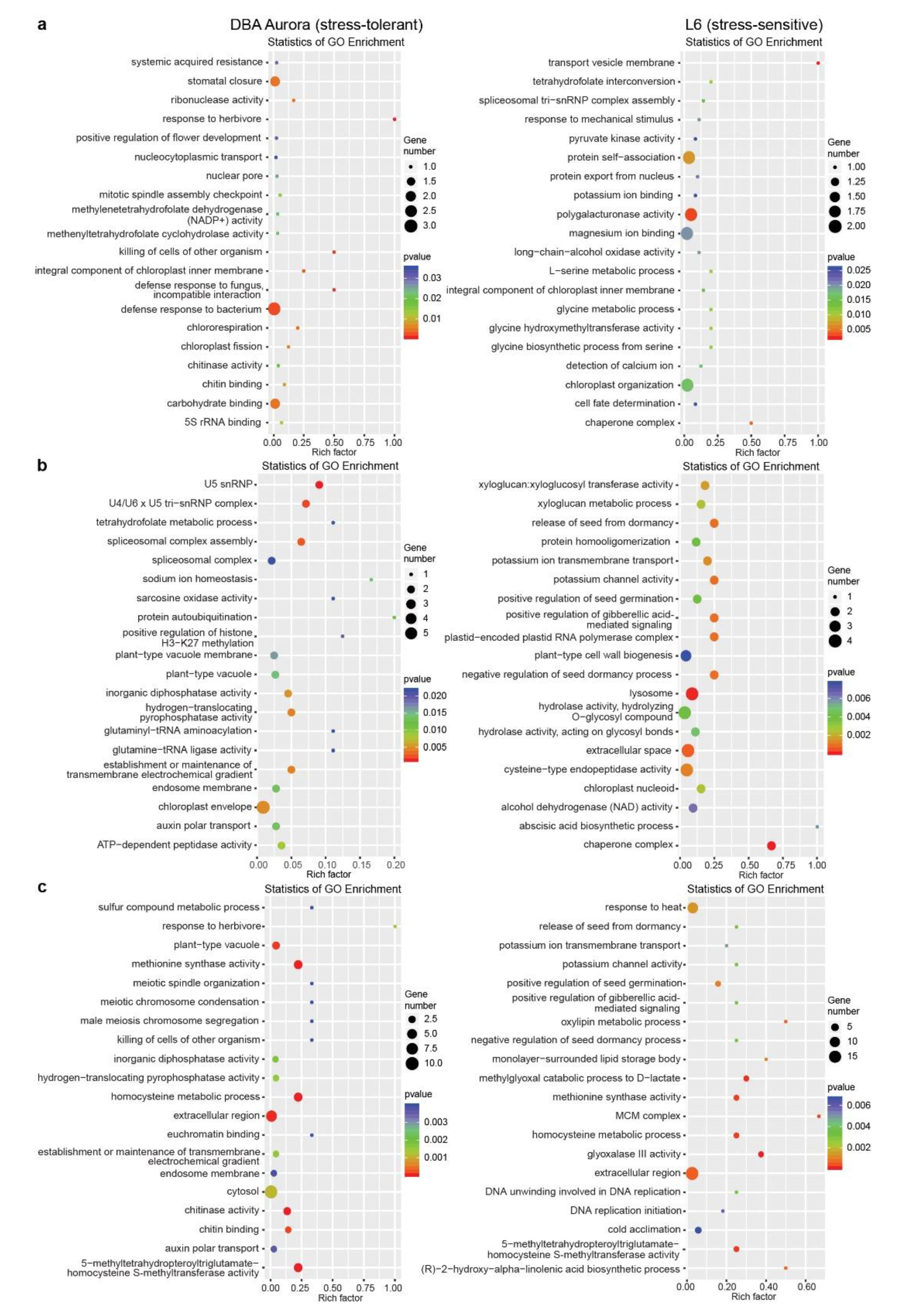
2.3. Transcriptome Sequencing Identified DEGs (Differentially Expressed Genes) in Response to Different Types of Stress
2.4. Genotype-Specific Changes in DEG Expression
2.5. SNP (Single-Nucleotide Polymorphisms) and AS (Alternative Splicing) Analysis
2.6. Degradome Sequencing and miRNA-Regulated mRNAs with Stress-Responsive Profiles
2.7. Multi-Omics Analysis: Stress-Responsive and Genotype-Dependent miRNA–mRNA Modules
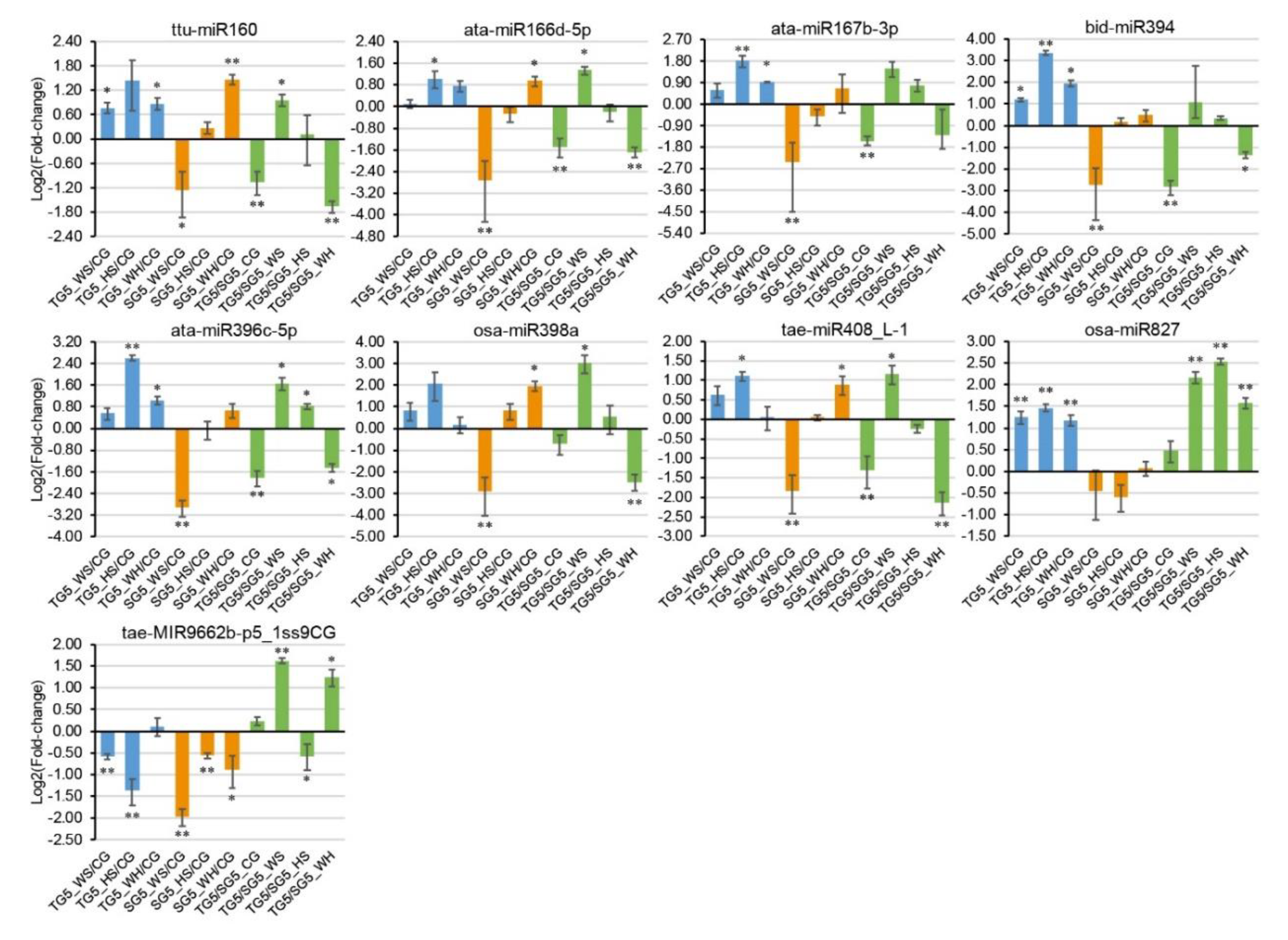
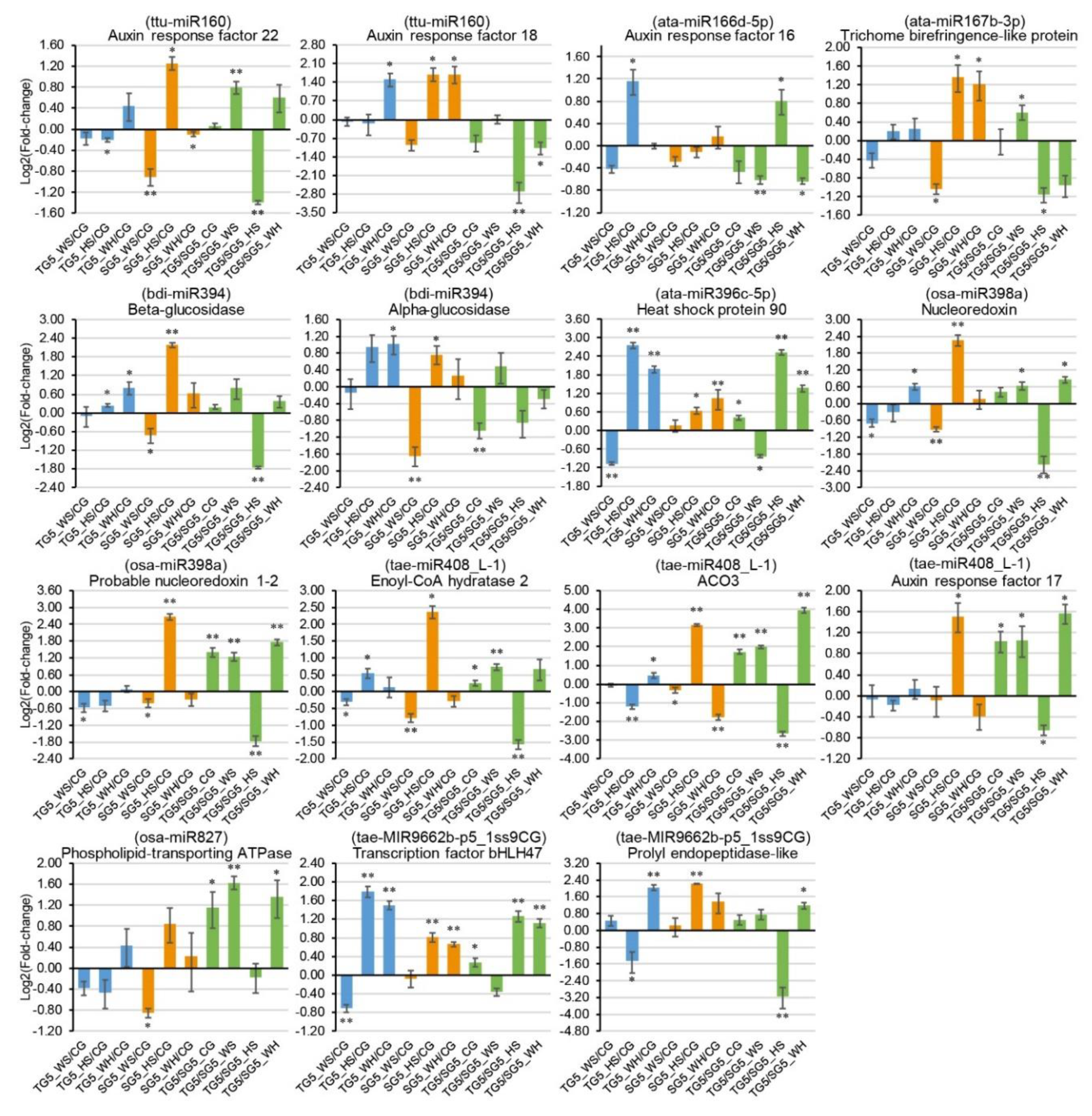
2.8. qPCR Analysis of DEMs and DEGs
3. Discussion
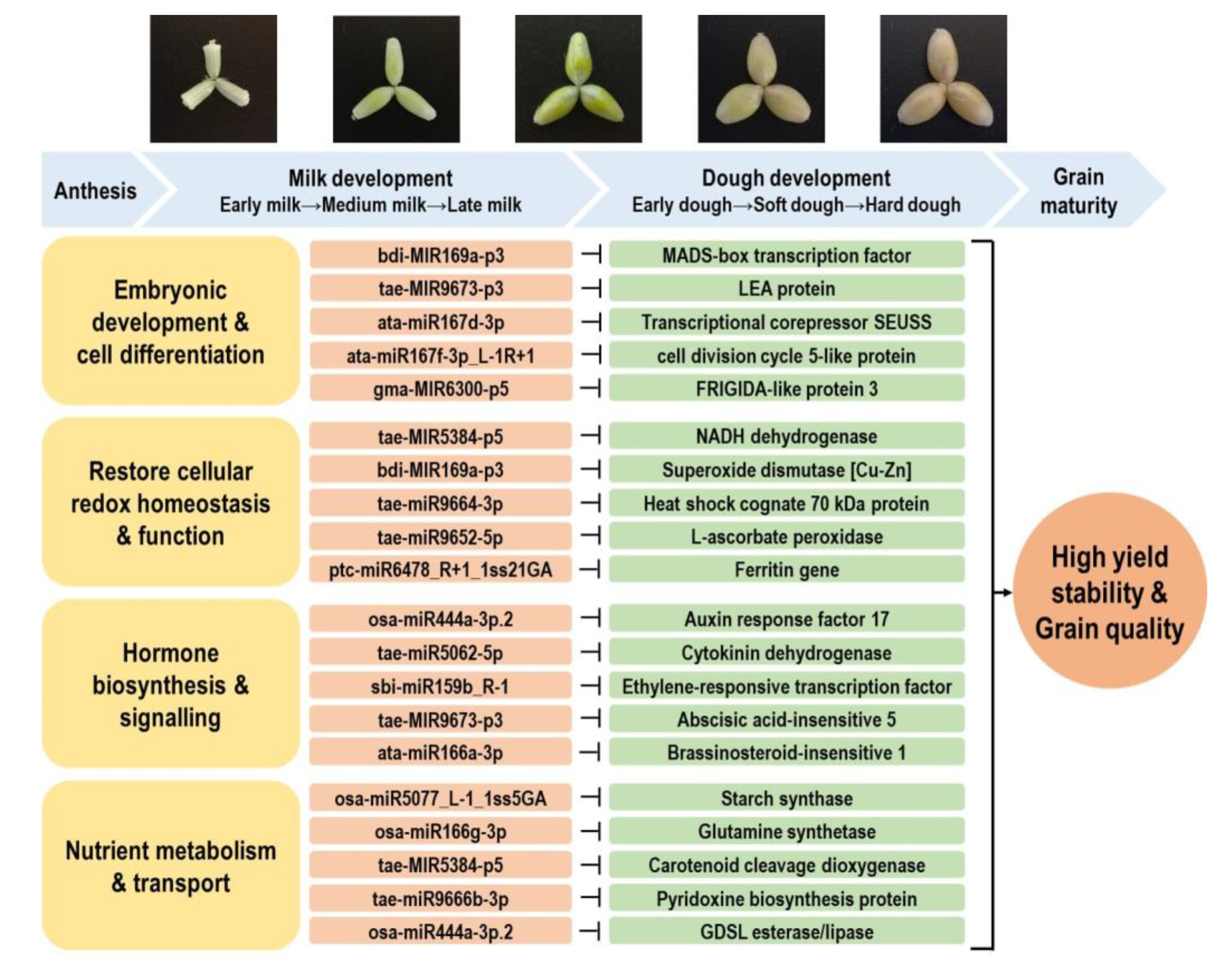
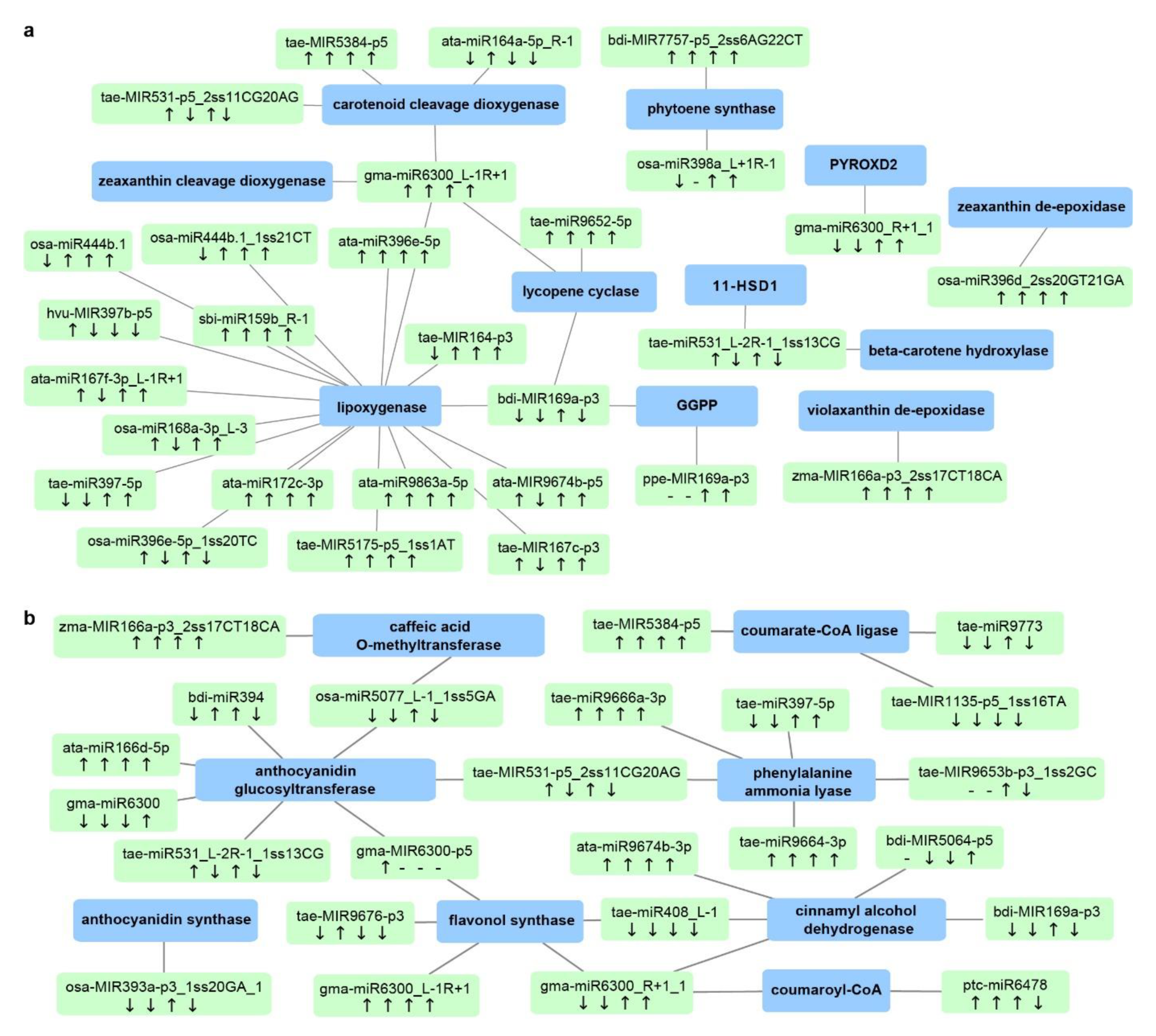
3.1. Functional Roles of T. turgidum miRNA–mRNA Modules at Different Stages of Grain Development
3.2. Stress-Responsive miRNA–mRNA Modules Contribute to Stable Grain Development and Grain Quality Characteristic under Different Stresses
4. Materials and Methods
4.1. Collection of Plant Materials and the Application of Stress
4.2. Small RNA Sequencing and Data Analysis
4.3. Sequencing and Bioinformatics Analysis of Transcriptome Libraries
4.4. Sequencing and Bioinformatics Analysis of Degradome Libraries
4.5. Multi-Omics Anlysis and Enrichment Analysis of GO Terms and KEGG Pathways
4.6. qPCR Quantification of miRNAs and Target Genes with Stress-Responsive Patterns
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sissons, M. Role of durum wheat composition on the quality of pasta and bread. Food 2008, 2, 75–90. [Google Scholar]
- Troccoli, A.; Borrelli, G.; De Vita, P.; Fares, C.; Di Fonzo, N. Mini review: Durum wheat quality: A multidisciplinary concept. J. Cereal Sci. 2000, 32, 99–113. [Google Scholar] [CrossRef]
- Barnabas, B.; Jaeger, K.; Feher, A. The effect of drought and heat stress on reproductive processes in cereals. Plant Cell Environ. 2008, 31, 11–38. [Google Scholar] [CrossRef] [PubMed]
- Dong, B.; Yang, H.; Liu, H.; Qiao, Y.; Zhang, M.; Wang, Y.; Xie, Z.; Liu, M. Effects of shading stress on grain number, yield, and photosynthesis during early reproductive growth in wheat. Crop Sci. 2019, 59, 363–378. [Google Scholar] [CrossRef]
- Liu, H.; Able, A.J.; Able, J.A. Genotypic performance of Australian durum under single and combined water-deficit and heat stress during reproduction. Sci. Rep. 2019, 9, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Searle, I.R.; Mather, D.E.; Able, A.J.; Able, J.A. Morphological, physiological and yield responses of durum wheat to pre-anthesis water-deficit stress are genotype-dependent. Crop Pasture Sci. 2015, 66, 1024–1038. [Google Scholar] [CrossRef]
- Liu, H.; Bruce, D.R.; Sissons, M.; Able, A.J.; Able, J.A. Genotype-dependent changes in the phenolic content of durum under water-deficit stress. Cereal Chem. 2018, 95, 59–78. [Google Scholar] [CrossRef]
- Aprile, A.; Havlickova, L.; Panna, R.; Marè, C.; Borrelli, G.M.; Marone, D.; Perrotta, C.; Rampino, P.; De Bellis, L.; Curn, V. Different stress responsive strategies to drought and heat in two durum wheat cultivars with contrasting water use efficiency. BMC Genom. 2013, 14, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tardieu, F.; Tuberosa, R. Dissection and modelling of abiotic stress tolerance in plants. Curr. Opin. Plant Biol. 2010, 13, 206–212. [Google Scholar] [CrossRef]
- Ramu, V.S.; Paramanantham, A.; Ramegowda, V.; Mohan-Raju, B.; Udayakumar, M.; Senthil-Kumar, M. Transcriptome analysis of sunflower genotypes with contrasting oxidative stress tolerance reveals individual-and combined-biotic and abiotic stress tolerance mechanisms. PLoS ONE 2016, 11, e0157522. [Google Scholar] [CrossRef] [Green Version]
- Maccaferri, M.; Harris, N.S.; Twardziok, S.O.; Pasam, R.K.; Gundlach, H.; Spannagl, M.; Ormanbekova, D.; Lux, T.; Prade, V.M.; Milner, S.G. Durum wheat genome highlights past domestication signatures and future improvement targets. Nat. Genet. 2019, 51, 885–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krasileva, K.V.; Buffalo, V.; Bailey, P.; Pearce, S.; Ayling, S.; Tabbita, F.; Soria, M.; Wang, S.; Akhunov, E.; Uauy, C.; et al. Separating homeologs by phasing in the tetraploid wheat transcriptome. Genome Biol. 2013, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallory, A.C.; Vaucheret, H. Functions of microRNAs and related small RNAs in plants. Nat. Genet. 2006, 38, S31–S36. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wang, Q. MicroRNA-based biotechnology for plant improvement. J. Cell. Physiol. 2015, 230, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Able, A.J.; Able, J.A. SMARTER de-stressed cereal breeding. Trends Plant Sci. 2016, 21, 909–925. [Google Scholar] [CrossRef]
- Wang, Y.; Li, K.; Chen, L.; Zou, Y.; Liu, H.; Tian, Y.; Li, D.; Wang, R.; Zhao, F.; Ferguson, B.J.; et al. microRNA167-directed regulation of the auxin response factors, GmARF8a and GmARF8b, is required for soybean nodulation and lateral root development. Plant Physiol. 2015, 168, 984–999. [Google Scholar] [CrossRef] [Green Version]
- Alptekin, B.; Langridge, P.; Budak, H. Abiotic stress miRNomes in the Triticeae. Funct. Integr. Genom. 2016, 17, 145–170. [Google Scholar] [CrossRef] [Green Version]
- Budak, H.; Akpinar, B.A. Plant miRNAs: Biogenesis, organization and origins. Funct. Integr. Genom. 2015, 15, 523–531. [Google Scholar] [CrossRef]
- Akpinar, B.A.; Kantar, M.; Budak, H. Root precursors of microRNAs in wild emmer and modern wheats show major differences in response to drought stress. Funct. Integr. Genom. 2015, 15, 587–598. [Google Scholar] [CrossRef]
- Zuluaga, D.L.; Liuzzi, V.; Curci, P.L.; Sonnante, G. MicroRNAs in durum wheat seedlings under chronic and short-term nitrogen stress. Funct. Integr. Genom. 2018, 18, 645–657. [Google Scholar] [CrossRef]
- Zuluaga, D.L.; De Paola, D.; Janni, M.; Curci, P.L.; Sonnante, G. Durum wheat miRNAs in response to nitrogen starvation at the grain filling stage. PLoS ONE 2017, 12, e0183253. [Google Scholar] [CrossRef]
- Giusti, L.; Mica, E.; Bertolini, E.; De Leonardis, A.M.; Faccioli, P.; Cattivelli, L.; Crosatti, C. microRNAs differentially modulated in response to heat and drought stress in durum wheat cultivars with contrasting water use efficiency. Funct. Integr. Genom. 2017, 17, 293–309. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Searle, I.R.; Watson-Haigh, N.S.; Baumann, U.; Mather, D.E.; Able, A.J.; Able, J.A. Genome-wide identification of microRNAs in leaves and the developing head of four durum genotypes during water deficit stress. PLoS ONE 2015, 10, e0142799. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Ma, L.; Geng, Y.; Hao, C.; Chen, X.; Zhang, X. Small RNA and degradome sequencing reveal complex roles of miRNAs and their targets in developing wheat grains. PLoS ONE 2015, 10, e0139658. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xu, Z.; Mo, Q.; Zou, C.; Li, W.; Xu, Y.; Xie, C. Combined small RNA and degradome sequencing reveals novel miRNAs and their targets in response to low nitrate availability in maize. Ann. Bot. 2013, 112, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Gu, L.; Li, P.; Song, X.; Wei, L.; Chen, Z.; Cao, X. Degradome sequencing reveals endogenous small RNA targets in rice (Oryza sativa L. ssp. indica). Front. Biol. 2010, 5, 67–90. [Google Scholar] [CrossRef]
- Ozhuner, E.; Eldem, V.; Ipek, A.; Okay, S.; Sakcali, S.; Zhang, B.; Boke, H.; Unver, T. Boron stress responsive microRNAs and their targets in barley. PLoS ONE 2013, 8, e59543. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Able, A.J.; Able, J.A. Integrated analysis of small RNA, transcriptome and degradome sequencing reveals the water-deficit and heat stress response network in durum wheat. Int. J. Mol. Sci. 2020, 21, 6017. [Google Scholar] [CrossRef]
- Zhong, M.; Huang, F.; Luo, R.; Lv, Y.; Ali, U.; Sheng, Z.; Tang, S.; Wei, X.; Hu, P. The effect of cadmium on the microRNAome, degradome and transcriptome of rice seedlings. Plant Growth Regul. 2020, 90, 15–27. [Google Scholar] [CrossRef]
- Sun, W.; Xu, X.H.; Wu, X.; Wang, Y.; Lu, X.; Sun, H.; Xie, X. Genome-wide identification of microRNAs and their targets in wild type and phyB mutant provides a key link between microRNAs and the phyB-mediated light signaling pathway in rice. Front. Plant Sci. 2015, 6, 372. [Google Scholar] [CrossRef] [Green Version]
- Ji, Y.; Chen, P.; Chen, J.; Pennerman, K.K.; Liang, X.; Yan, H.; Zhou, S.; Feng, G.; Wang, C.; Yin, G. Combinations of small RNA, RNA, and degradome sequencing uncovers the expression pattern of microRNA-mRNA pairs adapting to drought stress in leaf and root of Dactylis glomerata L. Int. J. Mol. Sci. 2018, 19, 3114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, M.; Luo, D.-X.; Khan, A.; Haq, S.U.; Gai, W.-X.; Zhang, H.-X.; Cheng, G.-X.; Muhammad, I.; Gong, Z.-H. Classification and genome-wide analysis of chitin-binding proteins gene family in pepper (Capsicum annuum L.) and transcriptional regulation to Phytophthora capsici, abiotic stresses and hormonal applications. Int. J. Mol. Sci. 2018, 19, 2216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, M.; Gai, W.-X.; Khattak, A.M.; Khan, A.; Haq, S.U.; Ma, X.; Wei, A.-M.; Muhammad, I.; Jan, I.; Gong, Z.-H. Knockdown of the chitin-binding protein family gene CaChiIV1 increased sensitivity to Phytophthora capsici and drought stress in pepper plants. Mol. Genet. Genom. 2019, 294, 1311–1326. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Able, A.J.; Able, J.A. Water-deficit stress responsive microRNAs and their targets in four durum wheat genotypes. Funct. Integr. Genom. 2016, 17, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhu, S.; Liu, F.; Wang, W.; Wang, X.; Han, G.; Cheng, B. Identification of arbuscular mycorrhiza fungi responsive microRNAs and their regulatory network in maize. Int. J. Mol. Sci. 2018, 19, 3201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, T.; Sun, H.; Qiao, M.; Zhao, Y.; Du, Y.; Zhang, J.; Li, J.; Tang, G.; Zhao, Q. Differentially expressed microRNA cohorts in seed development may contribute to poor grain filling of inferior spikelets in rice. BMC Plant Biol. 2014, 14, 196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, Y.; Su, N.; Shen, Y.; Zhang, R.; Wu, F.; Cheng, Z.; Wang, J.; Zhang, X.; Guo, X.; Lei, C.; et al. Identification of novel MiRNAs and MiRNA expression profiling during grain development in indica rice. BMC Genom. 2012, 13, 264. [Google Scholar] [CrossRef] [Green Version]
- Curaba, J.; Spriggs, A.; Taylor, J.; Li, Z.; Helliwell, C. miRNA regulation in the early development of barley seed. BMC Plant Biol. 2012, 12, 120. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Liu, Z.; Gao, L.; Wang, L.; Gao, M.; Jiao, Z.; Qiao, H.; Yang, J.; Chen, M.; Yao, L. Genome-wide identification and characterization of microRNAs in developing grains of Zea mays L. PLoS ONE 2016, 11, e0153168. [Google Scholar] [CrossRef] [Green Version]
- Deng, X.; Liu, Y.; Xu, X.; Liu, D.; Zhu, G.; Yan, X.; Wang, Z.; Yan, Y. Comparative proteome analysis of wheat flag leaves and developing grains under water deficit. Front. Plant Sci. 2018, 9, 425. [Google Scholar] [CrossRef] [Green Version]
- Rangan, P.; Furtado, A.; Henry, R.J. The transcriptome of the developing grain: A resource for understanding seed development and the molecular control of the functional and nutritional properties of wheat. BMC Genom. 2017, 18, 766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Yang, H.; Lu, Q.; Wen, X.; Chen, F.; Peng, L.; Zhang, L.; Lu, C. PsbP-domain protein1, a nuclear-encoded thylakoid lumenal protein, is essential for photosystem I assembly in Arabidopsis. Plant Cell 2012, 24, 4992–5006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.-K.; You, Y.N.; Park, J.C.; Joung, Y.; Kim, B.-G.; Ahn, J.C.; Cho, H.S. The rice thylakoid lumenal cyclophilin OsCYP20-2 confers enhanced environmental stress tolerance in tobacco and Arabidopsis. Plant Cell Rep. 2012, 31, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Downs, C.A.; Coleman, J.S.; Heckathorn, S.A. The chloroplast 22-Ku heat-shock protein: A lumenal protein that associates with the oxygen evolving complex and protects photosystem II during heat stress. J. Plant Physiol. 1999, 155, 477–487. [Google Scholar] [CrossRef]
- Nebenführ, A.; Dixit, R. Kinesins and myosins: Molecular motors that coordinate cellular functions in plants. Ann. Rev. Plant Biol. 2018, 69, 329–361. [Google Scholar] [CrossRef]
- Li, J.; Xu, Y.; Chong, K. The novel functions of kinesin motor proteins in plants. Protoplasma 2012, 249, 95–100. [Google Scholar] [CrossRef] [Green Version]
- Galindo-Trigo, S.; Grand, T.M.; Voigt, C.A.; Smith, L.M. A malectin domain kinesin functions in pollen and seed development in Arabidopsis. J. Exp. Bot. 2020, 71, 1828–1841. [Google Scholar] [CrossRef]
- Wu, T.; Shen, Y.; Zheng, M.; Yang, C.; Chen, Y.; Feng, Z.; Liu, X.; Liu, S.; Chen, Z.; Lei, C. Gene SGL, encoding a kinesin-like protein with transactivation activity, is involved in grain length and plant height in rice. Plant Cell Rep. 2014, 33, 235–244. [Google Scholar] [CrossRef]
- Zheng, X.; Liu, H.; Ji, H.; Wang, Y.; Dong, B.; Qiao, Y.; Liu, M.; Li, X. The wheat GT factor TaGT2L1D negatively regulates drought tolerance and plant development. Sci. Rep. 2016, 6, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Hanashiro, I.; Itoh, K.; Kuratomi, Y.; Yamazaki, M.; Igarashi, T.; Matsugasako, J.-I.; Takeda, Y. Granule-bound starch synthase I is responsible for biosynthesis of extra-long unit chains of amylopectin in rice. Plant Cell Physiol. 2008, 49, 925–933. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Hu, Y.; Wang, C.; Liu, W.; Ma, G.; Han, Q.; Ma, D. Effects of high temperature and drought stress on the expression of gene encoding enzymes and the activity of key enzymes involved in starch biosynthesis in wheat grains. Front. Plant Sci. 2019, 10, 1414. [Google Scholar] [CrossRef] [PubMed]
- Hazard, B.; Zhang, X.; Naemeh, M.; Hamilton, M.K.; Rust, B.; Raybould, H.E.; Newman, J.W.; Martin, R.; Dubcovsky, J. Mutations in durum wheat SBEII genes affect grain yield components, quality, and fermentation responses in rats. Crop Sci. 2015, 55, 2813–2825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Hou, L.; Lu, Y.; Wu, B.; Gong, X.; Liu, M.; Wang, J.; Sun, Q.; Vierling, E.; Xu, S. Metabolic adaptation of wheat grain contributes to a stable filling rate under heat stress. J. Exp. Bot. 2018, 69, 5531–5545. [Google Scholar] [CrossRef] [Green Version]
- Labuschagne, M.; Masci, S.; Tundo, S.; Muccilli, V.; Saletti, R.; van Biljon, A. Proteomic analysis of proteins responsive to drought and low temperature stress in a hard red spring wheat cultivar. Molecules 2020, 25, 1366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nigro, D.; Blanco, A.; Anderson, O.D.; Gadaleta, A. Characterization of ferredoxin-dependent glutamine-oxoglutarate amidotransferase (Fd-GOGAT) genes and their relationship with grain protein content QTL in wheat. PLoS ONE 2014, 9, e103869. [Google Scholar] [CrossRef] [Green Version]
- Nigro, D.; Fortunato, S.; Giove, S.L.; Mangini, G.; Yacoubi, I.; Simeone, R.; Blanco, A.; Gadaleta, A. Allelic variants of glutamine synthetase and glutamate synthase genes in a collection of durum wheat and association with grain protein content. Diversity 2017, 9, 52. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.; Turano, F.J. The putative glutamate receptor 1.1 (AtGLR1. 1) functions as a regulator of carbon and nitrogen metabolism in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2003, 100, 6872–6877. [Google Scholar] [CrossRef] [Green Version]
- Philippe, F.; Verdu, I.; Morère-Le Paven, M.-C.; Limami, A.M.; Planchet, E. Involvement of Medicago truncatula glutamate receptor-like channels in nitric oxide production under short-term water deficit stress. J. Plant Physiol. 2019, 236, 1–6. [Google Scholar] [CrossRef]
- Forde, B.G.; Roberts, M.R. Glutamate receptor-like channels in plants: A role as amino acid sensors in plant defence? F1000Prime Rep. 2014, 6, 37. [Google Scholar] [CrossRef]
- Fatiukha, A.; Filler, N.; Lupo, I.; Lidzbarsky, G.; Klymiuk, V.; Korol, A.B.; Pozniak, C.; Fahima, T.; Krugman, T. Grain protein content and thousand kernel weight QTLs identified in a durum × wild emmer wheat mapping population tested in five environments. Theor. Appl. Genet. 2020, 133, 119–131. [Google Scholar] [CrossRef]
- Quraishi, U.M.; Abrouk, M.; Murat, F.; Pont, C.; Foucrier, S.; Desmaizieres, G.; Confolent, C.; Riviere, N.; Charmet, G.; Paux, E. Cross-genome map based dissection of a nitrogen use efficiency ortho-metaQTL in bread wheat unravels concerted cereal genome evolution. Plant J. 2011, 65, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Blanco, A.; Mangini, G.; Giancaspro, A.; Giove, S.; Colasuonno, P.; Simeone, R.; Signorile, A.; De Vita, P.; Mastrangelo, A.; Cattivelli, L. Relationships between grain protein content and grain yield components through quantitative trait locus analyses in a recombinant inbred line population derived from two elite durum wheat cultivars. Mol. Breed. 2012, 30, 79–92. [Google Scholar] [CrossRef]
- Colasuonno, P.; Marcotuli, I.; Blanco, A.; Maccaferri, M.; Condorelli, G.E.; Tuberosa, R.; Parada, R.; de Camargo, A.C.; Schwember, A.R.; Gadaleta, A. Carotenoid pigment content in durum wheat (Triticum turgidum L. var durum): An overview of quantitative trait loci and candidate genes. Front. Plant Sci. 2019, 10, 1347. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Able, A.J.; Able, J.A. Transgenerational effects of water-deficit and heat stress on germination and seedling vigour—New insights from durum wheat microRNAs. Plants 2020, 9, 189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Shahid, M.Q.; Wu, J.; Wang, L.; Liu, X.; Lu, Y. Comparative small RNA analysis of pollen development in autotetraploid and diploid rice. Int. J. Mol. Sci. 2016, 17, 499. [Google Scholar] [CrossRef]
- Li, W.; Jia, Y.; Liu, F.; Wang, F.; Fan, F.; Wang, J.; Zhu, J.; Xu, Y.; Zhong, W.; Yang, J. Integration analysis of small RNA and degradome sequencing reveals microRNAs responsive to Dickeya zeae in resistant rice. Int. J. Mol. 2019, 20, 222. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Able, A.J.; Able, J.A. Genotypic water-deficit stress responses in durum wheat: Association between physiological traits, microRNA regulatory modules and yield components. Funct. Plant Biol. 2017, 44, 538–551. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Terms | Abbreviations | Description |
|---|---|---|
| Four treatment groups | CG | Control group |
| WS | Pre-anthesis water deficit group | |
| HS | Post-anthesis heat stress group | |
| WH | Pre-anthesis water deficit plus post anthesis heat stress group | |
| Two sample types | TG | Developing grains from the stress-tolerant genotype DBA Aurora |
| SG | Developing grains from the stress-sensitive genotype L6 | |
| Five time-points | 5, 15, 25, 35, 45 DPA | Sampling time-points; DPA represents days post anthesis. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, H.; Able, A.J.; Able, J.A. Multi-Omics Analysis of Small RNA, Transcriptome, and Degradome in T. turgidum—Regulatory Networks of Grain Development and Abiotic Stress Response. Int. J. Mol. Sci. 2020, 21, 7772. https://doi.org/10.3390/ijms21207772
Liu H, Able AJ, Able JA. Multi-Omics Analysis of Small RNA, Transcriptome, and Degradome in T. turgidum—Regulatory Networks of Grain Development and Abiotic Stress Response. International Journal of Molecular Sciences. 2020; 21(20):7772. https://doi.org/10.3390/ijms21207772
Chicago/Turabian StyleLiu, Haipei, Amanda J. Able, and Jason A. Able. 2020. "Multi-Omics Analysis of Small RNA, Transcriptome, and Degradome in T. turgidum—Regulatory Networks of Grain Development and Abiotic Stress Response" International Journal of Molecular Sciences 21, no. 20: 7772. https://doi.org/10.3390/ijms21207772
APA StyleLiu, H., Able, A. J., & Able, J. A. (2020). Multi-Omics Analysis of Small RNA, Transcriptome, and Degradome in T. turgidum—Regulatory Networks of Grain Development and Abiotic Stress Response. International Journal of Molecular Sciences, 21(20), 7772. https://doi.org/10.3390/ijms21207772