Structural and Functional Brain Abnormalities in Mouse Models of Lafora Disease

 , ,
, ,
Abstract
:1. Introduction
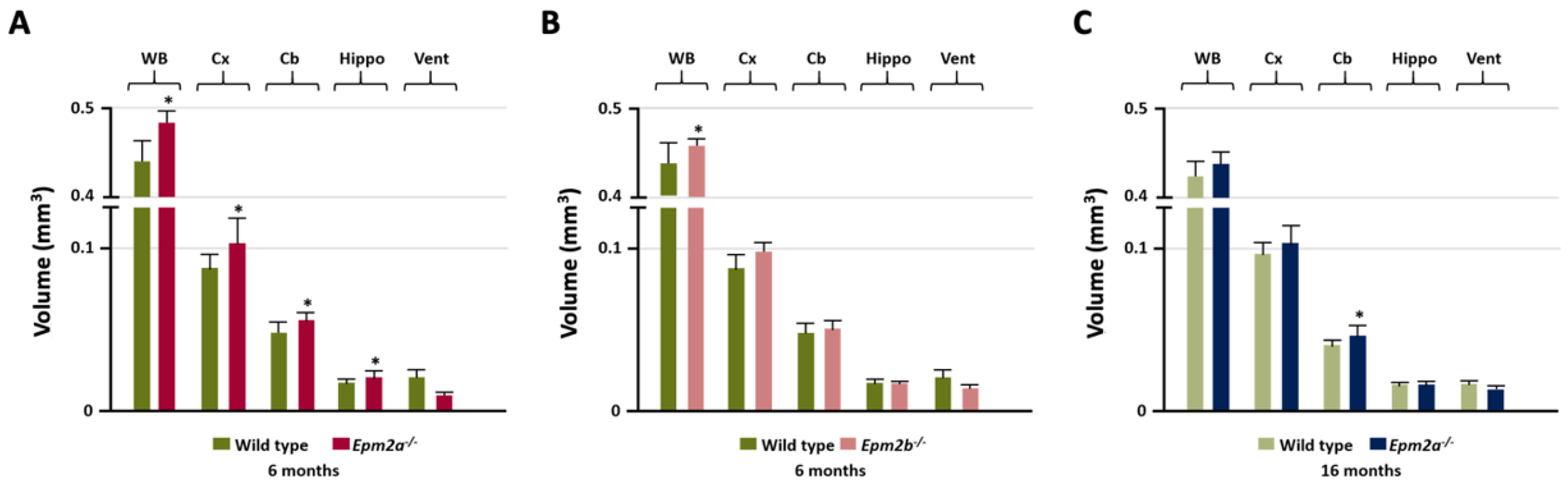
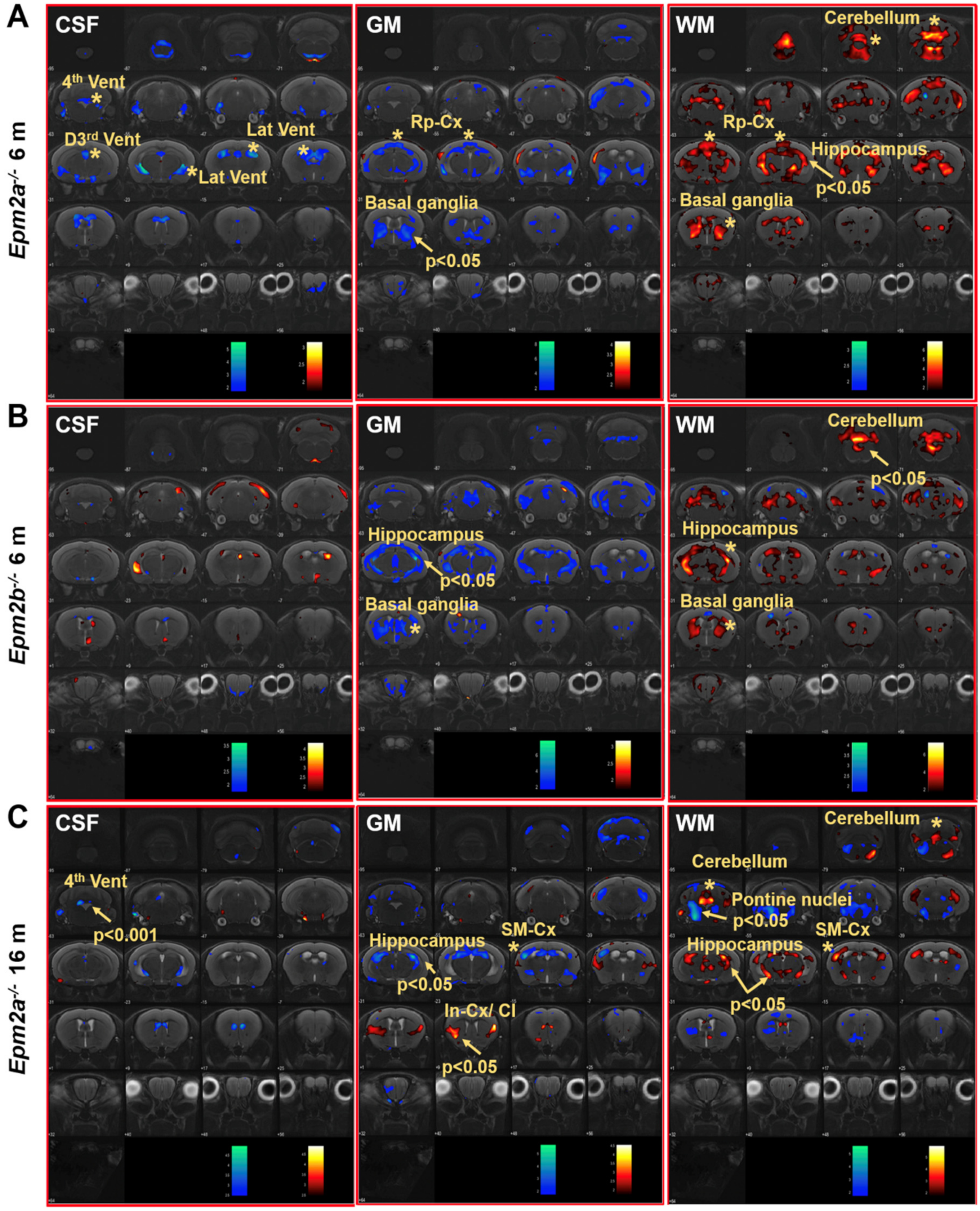
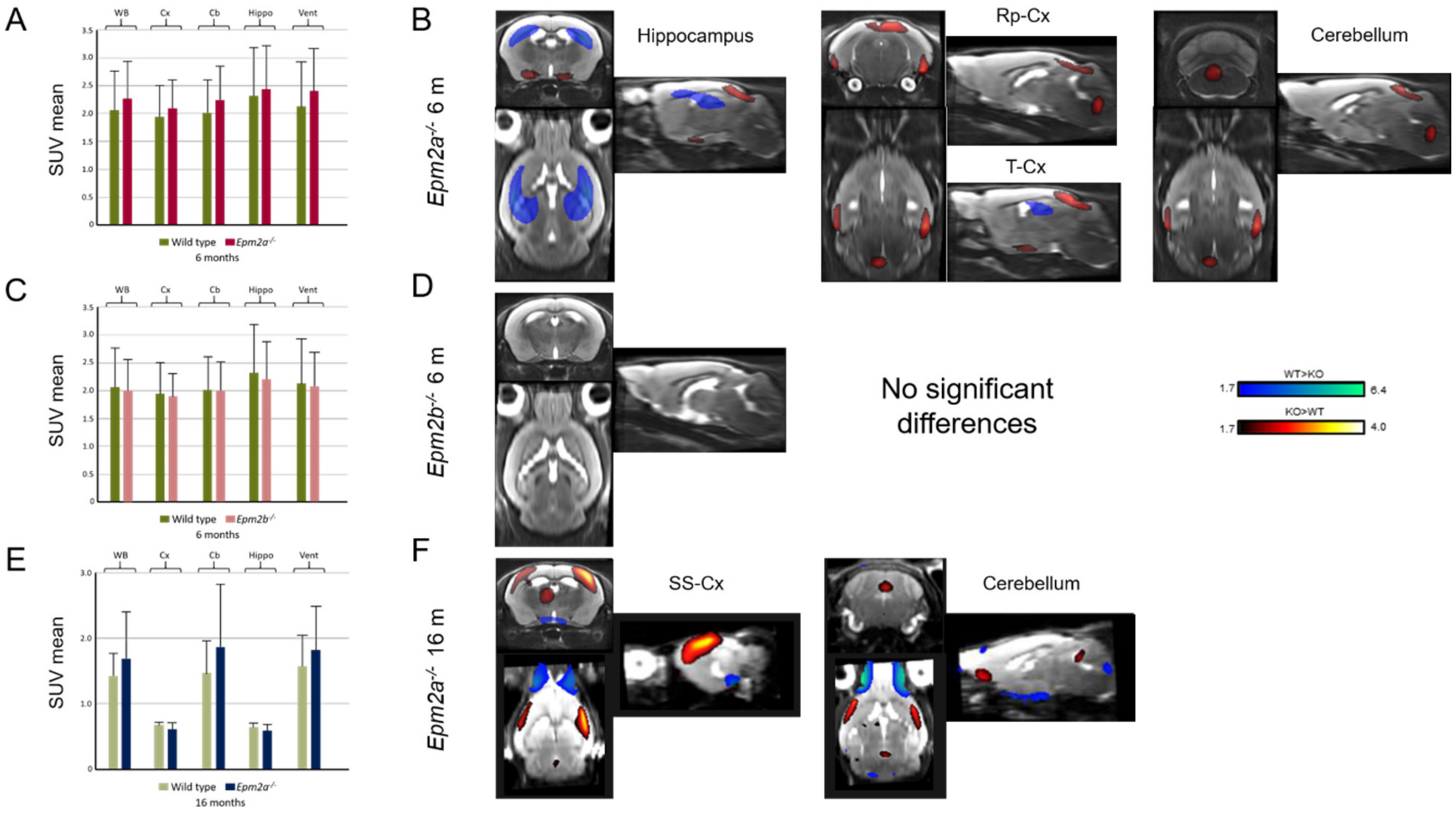
2. Results
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. In Vivo Imaging
4.3. Brain Image Analysis
4.4. Ex Vivo 1H-HRMAS MRS Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 1H-HRMAS | Proton High Resolution Magic Angle Spinning |
| Ace | Acetate |
| Ala | Alanine |
| Asp | Aspartate |
| BG | Basal Ganglia |
| BS | Brainstem |
| Cb | Cerebellum |
| Cho+GPC+PCho | Choline + Glycerylphosphorylcholine + Phosphocholine |
| Cx- | Rest of the cortex without prefrontal cortex |
| CSF | Cerebrospinal Fluid |
| CT | Computed Tomography |
| EPM2A | Epilepsy Progressive Myoclonus type 2A |
| EPM2B | Epilepsy Progressive Myoclonus type 2B |
| FDG | 2-[18F] fluoro-D-glucose |
| FEW | Family-Wise-Error |
| GABA | Gamma-aminobutyric acid |
| Glu+Gln | Glutamate + Glutamine |
| GM | Gray Matter |
| GPC | Glycerylphosphorylcholine |
| Hippo | Hippocampus |
| 1H-MRS | 1H magnetic resonance spectroscopy |
| Hyp | Hypothalamus |
| Lac | Lactate |
| mIns | myo-Inositol |
| mIns+Gly | mIns + glycine |
| MRI | Magnetic Resonance Imaging |
| NAA | N-acetylaspartate |
| NAA/Cho | NAA/choline |
| NAA/Cr | NAA /creatine |
| NAA/mIns | NAA/myo-Inositol |
| PAS | Periodic-acid-Schiff |
| PCho | Phosphocholine |
| PCr | Phosphocreatine |
| PE | Phosphoethanolamine |
| PET | Positron-Emission Tomography |
| PF-cx | Prefrontal cortex |
| ROI | Regions Of Interest |
| SD | Standard Deviation |
| SEP | Somatosensory Evoked Potentials |
| SPM | Statistical Parametric Mapping |
| SUVmean | Standard Uptake Value mean |
| tCho | total Cho |
| tCr | total Creatine (Cr+PCr) |
| Thal | Thalamus |
| VBM | Voxel-Based Morphometry |
| WM | White Matter |
References
- Berkovic, S.F.; Andermann, F.; Carpenter, S.; Wolfe, L.S. Progressive myoclonus epilepsies: Specific causes and diagnosis. N. Engl. J. Med. 1986, 315, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Berkovic, S.F.; So, N.K.; Andermann, F. Progressive myoclonus epilepsies: Clinical and neurophysiological diagnosis. J. Clin. Neurophysiol. Off. Publ. Am. Electroencephalogr. Soc. 1991, 8, 261–274. [Google Scholar] [CrossRef]
- Minassian, B.A.; Lee, J.R.; Herbrick, J.A.; Huizenga, J.; Soder, S.; Mungall, A.J.; Dunham, I.; Gardner, R.; Fong, C.Y.; Carpenter, S.; et al. Mutations in a gene encoding a novel protein tyrosine phosphatase cause progressive myoclonus epilepsy. Nat. Genet. 1998, 20, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Serratosa, J.M.; Gomez-Garre, P.; Gallardo, M.E.; Anta, B.; de Bernabe, D.B.; Lindhout, D.; Augustijn, P.B.; Tassinari, C.A.; Malafosse, R.M.; Topcu, M.; et al. A novel protein tyrosine phosphatase gene is mutated in progressive myoclonus epilepsy of the Lafora type (EPM2). Hum. Mol. Genet. 1999, 8, 345–352. [Google Scholar] [CrossRef]
- Ganesh, S.; Agarwala, K.; Ueda, K.; Akagi, T.; Shoda, K.; Usui, T.; Hashikawa, T.; Osada, H.; Delgado-Escueta, A.; Yamakawa, K. Laforin, defective in the progressive myoclonus epilepsy of Lafora type, is a dual-specificity phosphatase associated with polyribosomes. Hum. Mol. Genet. 2000, 9, 2251–2261. [Google Scholar] [CrossRef] [Green Version]
- Chan, E.M.; Young, E.J.; Ianzano, L.; Munteanu, I.; Zhao, X.; Christopoulos, C.C.; Avanzini, G.; Elia, M.; Ackerley, C.A.; Jovic, N.J.; et al. Mutations in NHLRC1 cause progressive myoclonus epilepsy. Nat. Genet. 2003, 35, 125–127. [Google Scholar] [CrossRef]
- Gentry, M.S.; Worby, C.A.; Dixon, J.E. Insights into Lafora disease: Malin is an E3 ubiquitin ligase that ubiquitinates and promotes the degradation of laforin. Proc. Natl. Acad. Sci. USA 2005, 102, 8501–8506. [Google Scholar] [CrossRef] [Green Version]
- Lafora, G. The presence of amyloid bodies in the inner part of ganglion cells; also an article on the study of amyloid substances in the nervous system. Virchows Arch. Pathol. Anat. Physiol. Klin. Med. 1911, 205, 295–303. [Google Scholar] [CrossRef]
- Harriman, D.G.; Millar, J.H.; Stevenson, A.C. Progressive familial myoclonic epilepsy in three families: Its clinical features and pathological basis. Brain 1955, 78, 325–349. [Google Scholar] [CrossRef]
- Carpenter, S.; Karpati, G. Ultrastructural findings in Lafora disease. Ann. Neurol. 1981, 10, 63–64. [Google Scholar] [CrossRef]
- Villanueva, V.; Alvarez-Linera, J.; Gomez-Garre, P.; Gutierrez, J.; Serratosa, J.M. MRI volumetry and proton MR spectroscopy of the brain in Lafora disease. Epilepsia 2006, 47, 788–792. [Google Scholar] [CrossRef] [PubMed]
- Jennesson, M.; Milh, M.; Villeneuve, N.; Guedj, E.; Marie, P.Y.; Vignal, J.P.; Raffo, E.; Vespignani, H.; Mancini, J.; Maillard, L. Posterior glucose hypometabolism in Lafora disease: Early and late FDG-PET assessment. Epilepsia 2010, 51, 708–711. [Google Scholar] [CrossRef] [PubMed]
- Casciato, S.; Gambardella, S.; Mascia, A.; Quarato, P.P.; D’Aniello, A.; Ackurina, Y.; Albano, V.; Fornai, F.; Scala, S.; Di Gennaro, G. Severe and rapidly-progressive Lafora disease associated with NHLRC1 mutation: A case report. Int. J. Neurosci. 2017, 127, 1150–1153. [Google Scholar] [CrossRef] [PubMed]
- Corkill, R.G.; Hardie, R.J. An unusual case of Lafora body disease. Eur. J. Neurol. 1999, 6, 245–247. [Google Scholar] [CrossRef] [PubMed]
- Caballero, P. Progressive myoclonus epilepsies: Description of a case of Lafora disease with autopsy. Neurologia 2013, 28, 584–586. [Google Scholar]
- Fernández-Torre, J.L.; Kaplan, P.W.; Rebollo, M.; Gutiérrez, A.; Hernández-Hernández, M.A.; Vázquez-Higuera, J.L. Ambulatory non-convulsive status epilepticus evolving into a malignant form. Epileptic Disord. 2012, 14, 41–50. [Google Scholar] [CrossRef]
- Potes, T.; Galicchio, S.; Rosso, B.; Besocke, G.; García, M.D.C.; Avalos, J.C. Progressive myoclonic epilepsy secondary to Lafora’s body disease. Medicina 2018, 78, 436–439. [Google Scholar]
- Baykan, B.; Striano, P.; Gianotti, S.; Bebek, N.; Gennaro, E.; Gurses, C.; Zara, F. Late-onset and slow-progressing Lafora disease in four siblings with EPM2B mutation. Epilepsia 2005, 46, 1695–1697. [Google Scholar] [CrossRef]
- Ganesh, S.; Shoda, K.; Amano, K.; Uchiyama, A.; Kumada, S.; Moriyama, N.; Hirose, S.; Yamakawa, K. Mutation screening for Japanese Lafora’s disease patients: Identification of novel sequence variants in the coding and upstream regulatory regions of EPM2A gene. Mol. Cell Probes 2001, 15, 281–289. [Google Scholar] [CrossRef]
- Widdess-Walsh, P.; Prayson, R.A.; Cohen, B.; Lachhwani, D. A 12-year-old girl with seizures and dementia. Brain Pathol. 2007, 464–465, 474. [Google Scholar] [CrossRef]
- Striano, P.; Zara, F.; Turnbull, J.; Girard, J.M.; Ackerley, C.A.; Cervasio, M.; De Rosa, G.; Del Basso-De Caro, M.L.; Striano, S.; Minassian, B.A. Typical progression of myoclonic epilepsy of the Lafora type: A case report. Nat. Clin. Pract. Neurol. 2008, 4, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Striano, P.; Ackerley, C.A.; Cervasio, M.; Girard, J.M.; Turnbull, J.; Del Basso-De Caro, M.L.; Striano, S.; Zara, F.; Minassian, B.A. 22-year-old girl with status epilepticus and progressive neurological symptoms. Brain Pathol. Zur. Switz. 2009, 19, 727–730. [Google Scholar] [CrossRef]
- Alisauskaite, N.; Beckmann, K.; Dennler, M.; Zolch, N. Brain proton magnetic resonance spectroscopy findings in a Beagle dog with genetically confirmed Lafora disease. J. Vet. Intern. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, H.; Katsumi, Y.; Nakamura, M.; Ikeda, A.; Fukuyama, H.; Kimura, J.; Shibasaki, H. Cerebral blood flow and metabolism in Lafora disease. Rinsho Shinkeigaku 1995, 35, 175–179. [Google Scholar]
- Al Otaibi, S.F.; Minassian, B.A.; Ackerley, C.A.; Logan, W.J.; Weiss, S. Unusual presentation of Lafora’s disease. J. Child. Neurol. 2003, 18, 499–501. [Google Scholar] [CrossRef] [Green Version]
- Kato, Z.; Yasuda, K.; Ishii, K.; Takagi, H.; Mizuno, S.; Shimozawa, N.; Kondo, N. Glucose metabolism evaluated by positron emission tomography in Lafora disease. Pediatr. Int. Off. J. Jpn. Pediatr. Soc. 1999, 41, 689–692. [Google Scholar] [CrossRef]
- Kumada, S.; Kubota, M.; Hayashi, M.; Uchiyama, A.; Kurata, K.; Kagamihara, Y. Fixation-sensitive myoclonus in Lafora disease. Neurology 2006, 66, 1574–1576. [Google Scholar] [CrossRef]
- Muccioli, L.; Farolfi, A.; Pondrelli, F.; d’Orsi, G.; Michelucci, R.; Freri, E.; Canafoglia, L.; Licchetta, L.; Toni, F.; Bonfiglioli, R.; et al. FDG-PET assessment and metabolic patterns in Lafora disease. Eur. J. Nucl. Med. Mol. Imaging 2019. [Google Scholar] [CrossRef] [PubMed]
- Shandal, V.; Veenstra, A.L.; Behen, M.; Sundaram, S.; Chugani, H. Long-term outcome in children with intractable epilepsy showing bilateral diffuse cortical glucose hypometabolism pattern on positron emission tomography. J. Child. Neurol. 2012, 27, 39–45. [Google Scholar] [CrossRef] [Green Version]
- Pichiecchio, A.; Veggiotti, P.; Cardinali, S.; Longaretti, F.; Poloni, G.U.; Uggetti, C. Lafora disease: Spectroscopy study correlated with neuropsychological findings. Eur. J. Paediatr. Neurol. 2008, 12, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Altindag, E.; Kara, B.; Baykan, B.; Terzibasioglu, E.; Sencer, S.; Onat, L.; Sirvanci, M. MR spectroscopy findings in Lafora disease. J. Neuroimaging 2009, 19, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, S.; Delgado-Escueta, A.V.; Sakamoto, T.; Avila, M.R.; Machado-Salas, J.; Hoshii, Y.; Akagi, T.; Gomi, H.; Suzuki, T.; Amano, K.; et al. Targeted disruption of the Epm2a gene causes formation of Lafora inclusion bodies, neurodegeneration, ataxia, myoclonus epilepsy and impaired behavioral response in mice. Hum. Mol. Genet. 2002, 11, 1251–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Criado, O.; Aguado, C.; Gayarre, J.; Duran-Trio, L.; Garcia-Cabrero, A.M.; Vernia, S.; San Millan, B.; Heredia, M.; Roma-Mateo, C.; Mouron, S.; et al. Lafora bodies and neurological defects in malin-deficient mice correlate with impaired autophagy. Hum. Mol. Genet. 2012, 21, 1521–1533. [Google Scholar] [CrossRef] [PubMed]
- Knecht, E.; Criado-García, O.; Aguado, C.; Gayarre, J.; Duran-Trio, L.; Garcia-Cabrero, A.M.; Vernia, S.; San Millán, B.; Heredia, M.; Romá-Mateo, C.; et al. Malin knockout mice support a primary role of autophagy in the pathogenesis of Lafora disease. Autophagy 2012, 8, 701–703. [Google Scholar] [CrossRef] [Green Version]
- García-Cabrero, A.M.; Sánchez-Elexpuru, G.; Serratosa, J.M.; Sánchez, M.P. Enhanced sensitivity of laforin- and malin-deficient mice to the convulsant agent pentylenetetrazole. Front. Neurosci. 2014, 8, 291. [Google Scholar] [CrossRef] [Green Version]
- Ganesh, S.; Agarwala, K.L.; Amano, K.; Suzuki, T.; Delgado-Escueta, A.V.; Yamakawa, K. Regional and developmental expression of Epm2a gene and its evolutionary conservation. Biochem. Biophys. Res. Commun. 2001, 283, 1046–1053. [Google Scholar] [CrossRef]
- Berthier, A.; Paya, M.; Garcia-Cabrero, A.M.; Ballester, M.I.; Heredia, M.; Serratosa, J.M.; Sanchez, M.P.; Sanz, P. Pharmacological Interventions to Ameliorate Neuropathological Symptoms in a Mouse Model of Lafora Disease. Mol. Neurobiol. 2016, 53, 1296–1309. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Abad, C.; Gomez-Garre, P.; Gutierrez-Delicado, E.; Saygi, S.; Michelucci, R.; Tassinari, C.A.; Rodriguez de Cordoba, S.; Serratosa, J.M. Lafora disease due to EPM2B mutations: a clinical and genetic study. Neurology 2005, 64, 982–986. [Google Scholar] [CrossRef]
- Garcia-Cabrero, A.M.; Marinas, A.; Guerrero, R.; Rodriguez de Cordoba, S.; Serratosa, J.M.; Sanchez, M.P. Laforin and Malin Deletions in Mice Produce Similar Neurologic Impairments. J. Neuropathol. Exp. Neurol. 2012, 71, 413–421. [Google Scholar] [CrossRef]
- Moffett, J.R.; Ross, B.; Arun, P.; Madhavarao, C.N.; Namboodiri, A.M. N-Acetylaspartate in the CNS: From neurodiagnostics to neurobiology. Prog. Neurobiol. 2007, 81, 89–131. [Google Scholar] [CrossRef] [Green Version]
- Simister, R.J.; McLean, M.A.; Barker, G.J.; Duncan, J.S. Proton MRS reveals frontal lobe metabolite abnormalities in idiopathic generalized epilepsy. Neurology 2003, 61, 897–902. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Elexpuru, G.; Serratosa, J.M.; Sanchez, M.P. Sodium selenate treatment improves symptoms and seizure susceptibility in a malin-deficient mouse model of Lafora disease. Epilepsia 2017, 58, 467–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vernia, S.; Heredia, M.; Criado, O.; Rodriguez de Cordoba, S.; Garcia-Roves, P.M.; Cansell, C.; Denis, R.; Luquet, S.; Foufelle, F.; Ferre, P.; et al. Laforin, a dual specificity phosphatase involved in Lafora disease, regulates insulin response and whole-body energy balance in mice. Hum. Mol. Genet. 2011, 20, 2571–2584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villalba-Orero, M.; Sánchez-Elexpuru, G.; López-Olañeta, M.; Campuzano, O.; Bello-Arroyo, E.; García-Pavía, P.; Serratosa, J.M.; Brugada, R.; Sánchez, M.P.; Lara-Pezzi, E. Lafora Disease Is an Inherited Metabolic Cardiomyopathy. J. Am. Coll. Cardiol. 2017, 69, 3007–3009. [Google Scholar] [CrossRef] [PubMed]
- Abella, M.; Vaquero, J.; Sisniega, A.; Pascau, J.; Udias, A.; Garcia, V.; Vidal, I.; Desco, M. Software architecture for multi-bed FDK-based reconstruction in X-ray CT scanners. Comput. Methods Progr. Biomed. 2012, 107, 218–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascau, J.; Gispert, J.; Soto-Montenegro, M.; Rodriguez-Ruano, A.; Garcia-Vazquez, V.; Udias, A.; Vaquero, J.; Desco, M. Small-animal PET registration method with intrinsic validation designed for large datasets. In Proceedings of the 2007 IEEE Nuclear Science Symposium Conference Record, Honolulu, HI, USA, 26 October–3 November 2007; Volume 5, pp. 3751–3753. [Google Scholar]
- Provencher, S.W. Automatic quantitation of localized in vivo 1H spectra with LCModel. NMR Biomed. 2001, 14, 260–264. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| MRI | PET | ||||
|---|---|---|---|---|---|
| Manual (mm3) | CSF | GM | WM | SPM | |
| 6 months | Epm2a−/− /Epm2b−/− | Epm2a−/− /Epm2b−/− | Epm2a−/− /Epm2b−/− | Epm2a−/− /Epm2b−/− | Epm2a−/− /Epm2b−/− |
| Cortex | ↑/− | ||||
| Sensorimotor cortex | |||||
| Hippocampus | ↑/− | ∇/↓ | ↑/Δ | ↓/− | |
| Basal ganglia | ↓/− | ||||
| Temporal cortex | ↑/− | ||||
| Retrosplenial cortex | ∇/− | Δ/− | ↑/− | ||
| Ventricles | ∇/− | ||||
| Cerebellum | ↑/− | Δ/↑ | |||
| Whole brain | ↑/↑ | ||||
| 16 months | Epm2a−/− | Epm2a−/− | Epm2a−/− | Epm2a−/− | Epm2a−/− |
| Cortex | |||||
| Sensorimotor cortex | ↓ | Δ | ↑ | ||
| Hippocampus | ↓ | ↑ | |||
| Basal ganglia | |||||
| Insular-cx/claustrum | ↑ | ||||
| Temporal cortex | |||||
| Retrosplenial cortex | |||||
| Ventricles | ↓↓↓ 4th | ||||
| Cerebellum | ↑ | ↑ | ↑ | ||
| Whole Brain | |||||
| [Ace]/[tCr] | [Ala]/[tCr] | [Asp]/[tCr] | [GABA]/[tCr] | [GPC]/[tCr] | [Lac/tCr] | |
| Epm2a−/−/Epm2b−/− | Epm2a−/−/Epm2b−/− | Epm2a−/−/Epm2b−/− | Epm2a−/−/Epm2b−/− | Epm2a−/−/Epm2b−/− | Epm2a−/−/Epm2b−/− | |
| PF-cortex | −/↑ | −/↑↑ | ↑/↑↑ | |||
| Cortex - | ↑/− | ↑/↑ | ↑/↑ | |||
| Hippocampus | ↑↑/− | ↑↑/↑ | −/↑ | |||
| Basal ganglia | ↑↑/− | ↑/↑ | ||||
| Thalamus | ↑/− | |||||
| Hypothalamus | ↑/↑↑ | |||||
| Brainstem | ↑↑/↑ | ↓/↓ | −/↓ | |||
| Cerebellum | ↑/− | ↑/− | ↑/− | |||
| [mIns]/[tCr] | [NAA]/[tCr] | [PCho]/[tCr] | [Glu+Gln]/[tCr] | [mIns+Gly]/[tCr] | [Cho+GPC+PCho]/[tCr] | |
| Epm2a−/−/Epm2b−/− | Epm2a−/−/Epm2b−/− | Epm2a−/−/Epm2b−/− | Epm2a−/−/Epm2b−/− | Epm2a−/−/Epm2b−/− | Epm2a−/−/Epm2b−/− | |
| PF-cortex | ↑/↑ | ↑/− | ||||
| Cortex- | ↑/− | ↑↑/↑ | ||||
| Hippocampus | ↓/↓ | ↓↓/↓ | ↑/− | |||
| Basal ganglia | ↓/↓↓ | ↓/↓ | −/↓ | |||
| Thalamus | ↑↑/− | ↓/↓↓ | ↓/- | ↑/− | ||
| Hypothalamus | −/↓↓↓ | |||||
| Brainstem | ↑/− | ↓↓/↓↓↓ | ↓/↓↓ | |||
| Cerebellum | ↓↓/↓↓↓ | ↓/− |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burgos, D.F.; Cussó, L.; Sánchez-Elexpuru, G.; Calle, D.; Perpinyà, M.B.; Desco, M.; Serratosa, J.M.; Sánchez, M.P. Structural and Functional Brain Abnormalities in Mouse Models of Lafora Disease. Int. J. Mol. Sci. 2020, 21, 7771. https://doi.org/10.3390/ijms21207771
Burgos DF, Cussó L, Sánchez-Elexpuru G, Calle D, Perpinyà MB, Desco M, Serratosa JM, Sánchez MP. Structural and Functional Brain Abnormalities in Mouse Models of Lafora Disease. International Journal of Molecular Sciences. 2020; 21(20):7771. https://doi.org/10.3390/ijms21207771
Chicago/Turabian StyleBurgos, Daniel F., Lorena Cussó, Gentzane Sánchez-Elexpuru, Daniel Calle, Max Bautista Perpinyà, Manuel Desco, José M. Serratosa, and Marina P. Sánchez. 2020. "Structural and Functional Brain Abnormalities in Mouse Models of Lafora Disease" International Journal of Molecular Sciences 21, no. 20: 7771. https://doi.org/10.3390/ijms21207771
APA StyleBurgos, D. F., Cussó, L., Sánchez-Elexpuru, G., Calle, D., Perpinyà, M. B., Desco, M., Serratosa, J. M., & Sánchez, M. P. (2020). Structural and Functional Brain Abnormalities in Mouse Models of Lafora Disease. International Journal of Molecular Sciences, 21(20), 7771. https://doi.org/10.3390/ijms21207771