
Hedgehog Signaling in Colorectal Cancer: All in the Stroma?


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
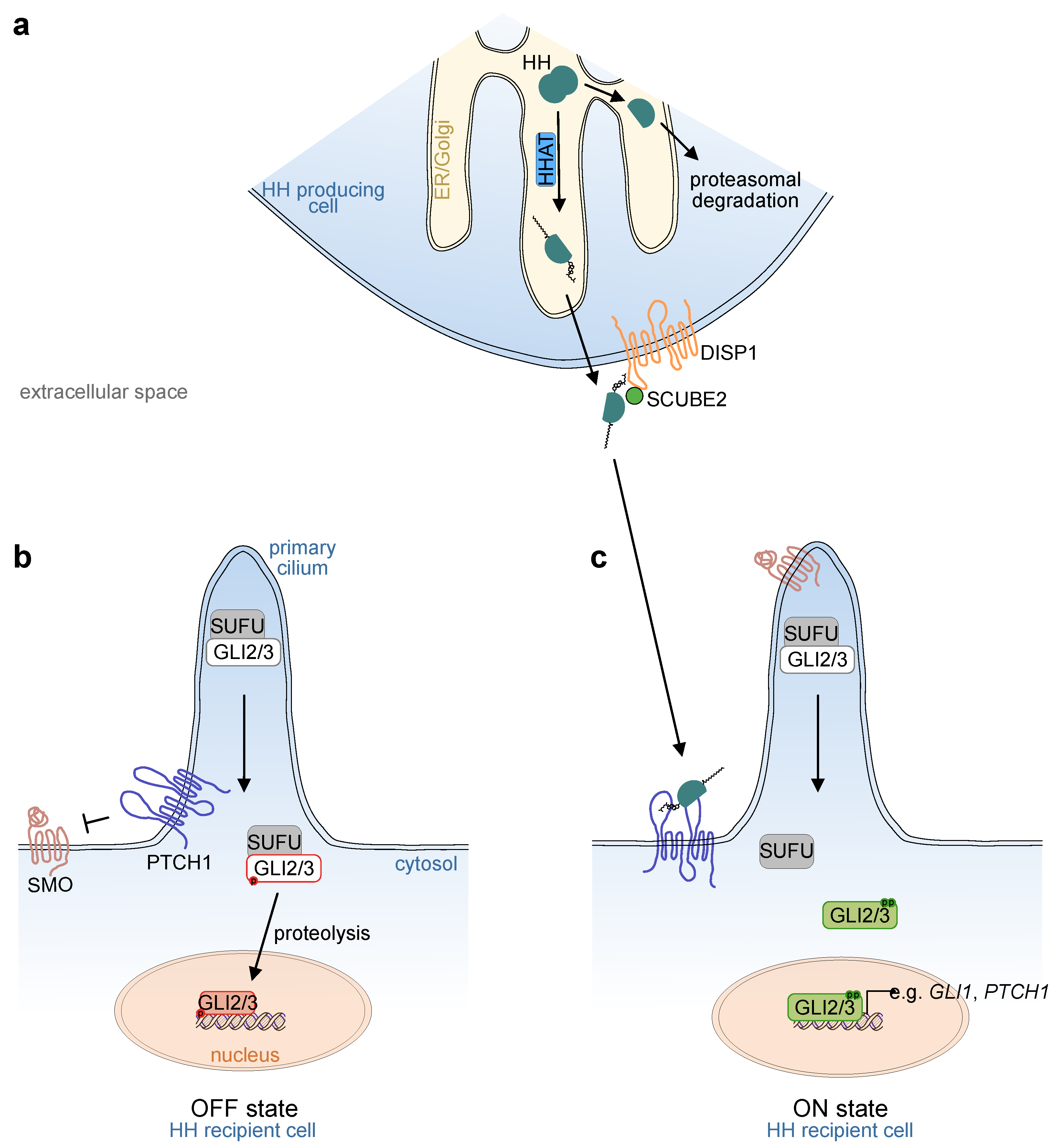
2. Molecular Basis of Hedgehog Signaling
2.1. Ligand Production and Secretion
2.2. Reception of the Ligand
2.3. Downstream Signal Transduction
2.4. Canonical vs. Non-Canonical Hedgehog Signaling
3. Intestinal Hedgehog Signaling in Development and Homeostasis
3.1. Importance of the Intestinal Stroma for Epithelial Maintenance
3.2. The Role of Hedgehog Signaling for Intestinal Development: A Paracrine Requirement
3.3. Response to Hh Ligands: Cell Types and Activated Target Genes
3.4. Hedgehog I s Critical for Epithelial Regeneration
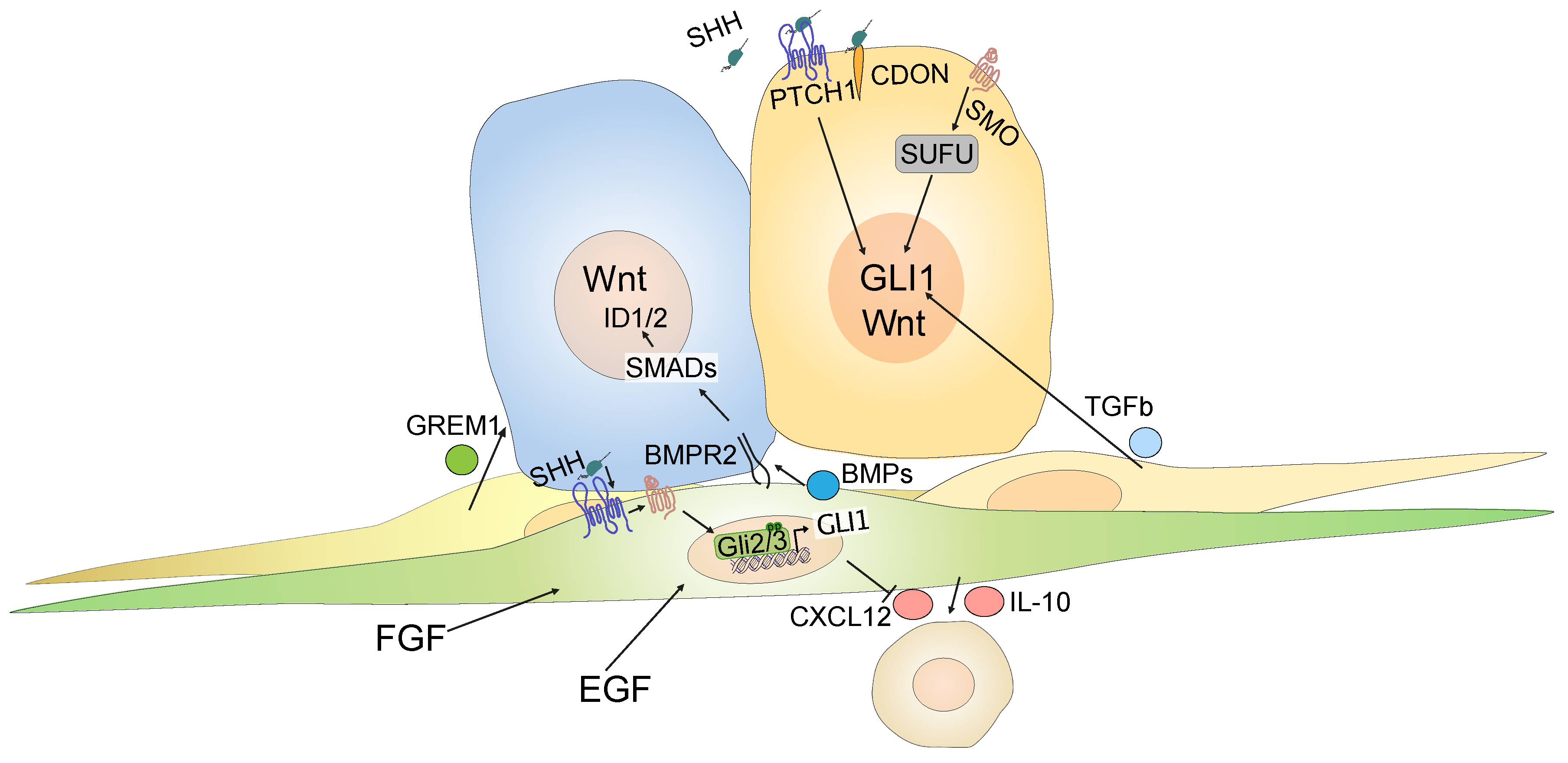
4. Stromal Hedgehog Signaling in Colorectal Cancer
4.1. Epidemiology and Genetic Background of Colorectal Cancer
4.2. Wnt Signaling as the Major Oncogenic Pathway in Colorectal Cancer
4.3. Crosstalk between Signaling Pathways in Colorectal Cancer
4.4. Epithelial and Non-Canonical Hedgehog Signaling in Colorectal Cancer
4.5. Failure of Hedgehog Inhibition in a Clinical Trial
5. Future Directions for Targeting Hedgehog Signaling in Colorectal Cancer
6. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nüsslein-Volhard, C.; Wieschaus, E. Mutations Affecting Segment Number and Polarity in Drosophila. Nature 1980, 287, 795–801. [Google Scholar] [CrossRef]
- The Nobel Prize in Physiology or Medicine. 1995. Available online: https://www.nobelprize.org/prizes/medicine/1995/nusslein-volhard/facts/ (accessed on 12 July 2020).
- Chen, X.; Tukachinsky, H.; Huang, C.-H.; Jao, C.; Chu, Y.-R.; Tang, H.-Y.; Mueller, B.; Schulman, S.; Rapoport, T.A.; Salic, A. Processing and Turnover of the Hedgehog Protein in the Endoplasmic Reticulum. J. Cell Biol. 2011, 192, 825–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, E.T.; Cleverdon, E.R.; Ogden, S.K. Dispatching Sonic Hedgehog: Molecular Mechanisms Controlling Deployment. Trends Cell Biol. 2019, 29, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Erkner, A.; Gong, R.; Yao, S.; Taipale, J.; Basler, K.; Beachy, P.A. Hedgehog-Mediated Patterning of the Mammalian Embryo Requires Transporter-like Function of Dispatched. Cell 2002, 111, 63–75. [Google Scholar] [CrossRef] [Green Version]
- Burke, R.; Nellen, D.; Bellotto, M.; Hafen, E.; Senti, K.-A.; Dickson, B.J.; Basler, K. Dispatched, a Novel Sterol-Sensing Domain Protein Dedicated to the Release of Cholesterol-Modified Hedgehog from Signaling Cells. Cell 1999, 99, 803–815. [Google Scholar] [CrossRef] [Green Version]
- Cannac, F.; Qi, C.; Falschlunger, J.; Hausmann, G.; Basler, K.; Korkhov, V.M. Cryo-EM Structure of the Hedgehog Release Protein Dispatched. Sci. Adv. 2020, 6, eaay7928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wierbowski, B.M.; Petrov, K.; Aravena, L.; Gu, G.; Xu, Y.; Salic, A. Hedgehog Pathway Activation Requires Coreceptor-Catalyzed, Lipid-Dependent Relay of the Sonic Hedgehog Ligand. Dev. Cell 2020, 55, 450–467. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Skacel, M.; Adams, J.C. Association of Loss of Epithelial Syndecan-1 with Stage and Local Metastasis of Colorectal Adenocarcinomas: An Immunohistochemical Study of Clinically Annotated Tumors. BMC Cancer 2008, 8, 185. [Google Scholar] [CrossRef] [Green Version]
- Filmus, J.; Capurro, M.; Rast, J. Glypicans. Genome Biol. 2008, 9, 224. [Google Scholar] [CrossRef]
- McGough, I.J.; Vecchia, L.; Bishop, B.; Malinauskas, T.; Beckett, K.; Joshi, D.; O’Reilly, N.; Siebold, C.; Jones, E.Y.; Vincent, J.-P. Glypicans Shield the Wnt Lipid Moiety to Enable Signalling at a Distance. Nature 2020, 585, 85–90. [Google Scholar] [CrossRef]
- Bellaiche, Y.; The, I.; Perrimon, N. Tout-Velu Is a Drosophila Homologue of the Putative Tumour Suppressor EXT-1 and Is Needed for Hh Diffusion. Nature 1998, 394, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Belenkaya, T.Y.; Wang, B.; Lin, X. Drosophila Glypicans Control the Cell-to-Cell Movement of Hedgehog by a Dynamin-Independent Process. Development 2004, 131, 601–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, W.; Roelink, H. Loss of the Heparan Sulfate Proteoglycan Glypican5 Facilitates Long-Range Sonic Hedgehog Signaling. Stem Cells 2019, 37, 899–909. [Google Scholar] [CrossRef] [PubMed]
- Vicente, C.M.; da Silva, D.A.; Sartorio, P.V.; Silva, T.D.; Saad, S.S.; Nader, H.B.; Forones, N.M.; Toma, L. Heparan Sulfate Proteoglycans in Human Colorectal Cancer. Anal. Cell Pathol. 2018, 2018. [Google Scholar] [CrossRef] [Green Version]
- Kong, J.H.; Siebold, C.; Rohatgi, R. Biochemical Mechanisms of Vertebrate Hedgehog Signaling. Development 2019, 146. [Google Scholar] [CrossRef] [Green Version]
- Sénicourt, B.; Boudjadi, S.; Carrier, J.C.; Beaulieu, J.-F. Neoexpression of a Functional Primary Cilium in Colorectal Cancer Cells. Heliyon 2016, 2, e00109. [Google Scholar] [CrossRef] [Green Version]
- Saqui-Salces, M.; Dowdle, W.E.; Reiter, J.F.; Merchant, J.L. A High-Fat Diet Regulates Gastrin and Acid Secretion through Primary Cilia. FASEB J. 2012, 26, 3127–3139. [Google Scholar] [CrossRef] [Green Version]
- Gong, X.; Qian, H.; Cao, P.; Zhao, X.; Zhou, Q.; Lei, J.; Yan, N. Structural Basis for the Recognition of Sonic Hedgehog by Human Patched1. Science 2018, 361. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Bulkley, D.P.; Xin, Y.; Roberts, K.J.; Asarnow, D.E.; Sharma, A.; Myers, B.R.; Cho, W.; Cheng, Y.; Beachy, P.A. Structural Basis for Cholesterol Transport-like Activity of the Hedgehog Receptor Patched. Cell 2018, 175, 1352–1364.e14. [Google Scholar] [CrossRef] [Green Version]
- Huang, P.; Nedelcu, D.; Watanabe, M.; Jao, C.; Kim, Y.; Liu, J.; Salic, A. Cellular Cholesterol Directly Activates Smoothened in Hedgehog Signaling. Cell 2016, 166, 1176–1187.e14. [Google Scholar] [CrossRef] [Green Version]
- Luchetti, G.; Sircar, R.; Kong, J.H.; Nachtergaele, S.; Sagner, A.; Byrne, E.F.; Covey, D.F.; Siebold, C.; Rohatgi, R. Cholesterol Activates the G-Protein Coupled Receptor Smoothened to Promote Hedgehog Signaling. eLife 2016, 5, e20304. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Zheng, S.; Wierbowski, B.M.; Kim, Y.; Nedelcu, D.; Aravena, L.; Liu, J.; Kruse, A.C.; Salic, A. Structural Basis of Smoothened Activation in Hedgehog Signaling. Cell 2018, 174, 312–324.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deshpande, I.; Liang, J.; Hedeen, D.; Roberts, K.J.; Zhang, Y.; Ha, B.; Latorraca, N.R.; Faust, B.; Dror, R.O.; Beachy, P.A.; et al. Smoothened Stimulation by Membrane Sterols Drives Hedgehog Pathway Activity. Nature 2019, 571, 284–288. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.H.; Young, C.B.; Pusapati, G.V.; Patel, C.B.; Ho, S.; Krishnan, A.; Lin, J.-H.I.; Devine, W.; Moreau de Bellaing, A.; Athni, T.S.; et al. A Membrane-Tethered Ubiquitination Pathway Regulates Hedgehog Signaling and Heart Development. Dev. Cell 2020, 55, 432–449.e12. [Google Scholar] [CrossRef]
- Pusapati, G.V.; Kong, J.H.; Patel, B.B.; Krishnan, A.; Sagner, A.; Kinnebrew, M.; Briscoe, J.; Aravind, L.; Rohatgi, R. CRISPR Screens Uncover Genes That Regulate Target Cell Sensitivity to the Morphogen Sonic Hedgehog. Dev. Cell 2018, 44, 113–129.e8. [Google Scholar] [CrossRef] [Green Version]
- Martinelli, D.C.; Fan, C.-M. Gas1 Extends the Range of Hedgehog Action by Facilitating Its Signaling. Genes Dev. 2007, 21, 1231–1243. [Google Scholar] [CrossRef] [Green Version]
- Allen, B.L.; Song, J.Y.; Izzi, L.; Althaus, I.W.; Kang, J.-S.; Charron, F.; Krauss, R.S.; McMahon, A.P. Overlapping Roles and Collective Requirement for the Coreceptors GAS1, CDO, and BOC in SHH Pathway Function. Dev. Cell 2011, 20, 775–787. [Google Scholar] [CrossRef] [Green Version]
- Allen, B.L.; Tenzen, T.; McMahon, A.P. The Hedgehog-Binding Proteins Gas1 and Cdo Cooperate to Positively Regulate Shh Signaling during Mouse Development. Genes Dev. 2007, 21, 1244–1257. [Google Scholar] [CrossRef] [Green Version]
- Tenzen, T.; Allen, B.L.; Cole, F.; Kang, J.-S.; Krauss, R.S.; McMahon, A.P. The Cell Surface Membrane Proteins Cdo and Boc Are Components and Targets of the Hedgehog Signaling Pathway and Feedback Network in Mice. Dev. Cell 2006, 10, 647–656. [Google Scholar] [CrossRef] [Green Version]
- Seppala, M.; Depew, M.J.; Martinelli, D.C.; Fan, C.-M.; Sharpe, P.T.; Cobourne, M.T. Gas1 Is a Modifier for Holoprosencephaly and Genetically Interacts with Sonic Hedgehog. J. Clin. Investig. 2007, 117, 1575–1584. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Lee, J.; Seppala, M.; Cobourne, M.T.; Kim, S.-H. Ptch2/Gas1 and Ptch1/Boc Differentially Regulate Hedgehog Signalling in Murine Primordial Germ Cell Migration. Nat. Commun. 2020, 11, 1994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niewiadomski, P.; Kong, J.H.; Ahrends, R.; Ma, Y.; Humke, E.W.; Khan, S.; Teruel, M.N.; Novitch, B.G.; Rohatgi, R. Gli Protein Activity Is Controlled by Multisite Phosphorylation in Vertebrate Hedgehog Signaling. Cell Rep. 2014, 6, 168–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.; Wang, B.; Cho, Y.S.; Zhu, J.; Wu, J.; Chen, Y.; Jiang, J. Phosphorylation of Ci/Gli by Fused Family Kinases Promotes Hedgehog Signaling. Dev. Cell 2019, 50, 610–626.e4. [Google Scholar] [CrossRef] [PubMed]
- Svärd, J.; Henricson, K.H.; Persson-Lek, M.; Rozell, B.; Lauth, M.; Bergström, Å.; Ericson, J.; Toftgård, R.; Teglund, S. Genetic Elimination of Suppressor of Fused Reveals an Essential Repressor Function in the Mammalian Hedgehog Signaling Pathway. Dev. Cell 2006, 10, 187–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoelzl, M.A.; Heby-Henricson, K.; Gerling, M.; Dias, J.M.; Kuiper, R.V.; Trünkle, C.; Bergström, Å.; Ericson, J.; Toftgård, R.; Teglund, S. Differential Requirement of SUFU in Tissue Development Discovered in a Hypomorphic Mouse Model. Dev. Biol. 2017, 429, 132–146. [Google Scholar] [CrossRef]
- Cherry, A.L.; Finta, C.; Karlström, M.; Jin, Q.; Schwend, T.; Astorga-Wells, J.; Zubarev, R.A.; Del Campo, M.; Criswell, A.R.; de Sanctis, D.; et al. Structural Basis of SUFU–GLI Interaction in Human Hedgehog Signalling Regulation. Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 2563–2579. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Shen, L.; Law, K.; Zhang, Z.; Liu, X.; Hua, H.; Li, S.; Huang, H.; Yue, S.; Hui, C.; et al. Suppressor of Fused Chaperones Gli Proteins to Generate Transcriptional Responses to Sonic Hedgehog Signaling. Mol. Cell. Biol. 2017, 37. [Google Scholar] [CrossRef] [Green Version]
- Breslow, D.K.; Hoogendoorn, S.; Kopp, A.R.; Morgens, D.W.; Vu, B.K.; Kennedy, M.C.; Han, K.; Li, A.; Hess, G.T.; Bassik, M.C.; et al. A CRISPR-Based Screen for Hedgehog Signaling Provides Insights into Ciliary Function and Ciliopathies. Nat. Genet. 2018, 50, 460–471. [Google Scholar] [CrossRef]
- Kinzler, K.W.; Vogelstein, B. The GLI Gene Encodes a Nuclear Protein Which Binds Specific Sequences in the Human Genome. Mol. Cell. Biol. 1990, 10, 634–642. [Google Scholar] [CrossRef] [Green Version]
- Hallikas, O.; Palin, K.; Sinjushina, N.; Rautiainen, R.; Partanen, J.; Ukkonen, E.; Taipale, J. Genome-Wide Prediction of Mammalian Enhancers Based on Analysis of Transcription-Factor Binding Affinity. Cell 2006, 124, 47–59. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, H.; Hui, C.; Nakafuku, M.; Kondoh, H. A Binding Site for Gli Proteins Is Essential for HNF-3beta Floor Plate Enhancer Activity in Transgenics and Can Respond to Shh in Vitro. Development 1997, 124, 1313–1322. [Google Scholar] [PubMed]
- Aberger, F.; Ruiz i Altaba, A. Context-Dependent Signal Integration by the GLI Code: The Oncogenic Load, Pathways, Modifiers and Implications for Cancer Therapy. Semin. Cell Dev. Biol. 2014, 33, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Palma, V.; Altaba, A.R. i Hedgehog-GLI Signaling Regulates the Behavior of Cells with Stem Cell Properties in the Developing Neocortex. Development 2004, 131, 337–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brewster, R.; Mullor, J.L.; Altaba, A.R. i Gli2 Functions in FGF Signaling during Antero-Posterior Patterning. Development 2000, 127, 4395–4405. [Google Scholar]
- Pietrobono, S.; Gagliardi, S.; Stecca, B. Non-Canonical Hedgehog Signaling Pathway in Cancer: Activation of GLI Transcription Factors Beyond Smoothened. Front. Genet. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barker, N.; van Es, J.H.; Kuipers, J.; Kujala, P.; van den Born, M.; Cozijnsen, M.; Haegebarth, A.; Korving, J.; Begthel, H.; Peters, P.J.; et al. Identification of Stem Cells in Small Intestine and Colon by Marker Gene Lgr5. Nature 2007, 449, 1003–1007. [Google Scholar] [CrossRef]
- Sato, T.; van Es, J.H.; Snippert, H.J.; Stange, D.E.; Vries, R.G.; van den Born, M.; Barker, N.; Shroyer, N.F.; van de Wetering, M.; Clevers, H. Paneth Cells Constitute the Niche for Lgr5 Stem Cells in Intestinal Crypts. Nature 2010, 469, 415–418. [Google Scholar] [CrossRef] [Green Version]
- Degirmenci, B.; Valenta, T.; Dimitrieva, S.; Hausmann, G.; Basler, K. GLI1-Expressing Mesenchymal Cells Form the Essential Wnt-Secreting Niche for Colon Stem Cells. Nature 2018, 558, 449–453. [Google Scholar] [CrossRef]
- Walton, K.D.; Whidden, M.; Kolterud, Å.; Shoffner, S.K.; Czerwinski, M.J.; Kushwaha, J.; Parmar, N.; Chandhrasekhar, D.; Freddo, A.M.; Schnell, S.; et al. Villification in the Mouse: Bmp Signals Control Intestinal Villus Patterning. Development 2016, 143, 427–436. [Google Scholar] [CrossRef] [Green Version]
- Farrall, A.L.; Riemer, P.; Leushacke, M.; Sreekumar, A.; Grimm, C.; Herrmann, B.G.; Morkel, M. Wnt and BMP Signals Control Intestinal Adenoma Cell Fates. Int. J. Cancer 2012, 131, 2242–2252. [Google Scholar] [CrossRef]
- Ramalho-Santos, M.; Melton, D.A.; McMahon, A.P. Hedgehog Signals Regulate Multiple Aspects of Gastrointestinal Development. Development 2000, 127, 2763–2772. [Google Scholar] [PubMed]
- Litingtung, Y.; Lei, L.; Westphal, H.; Chiang, C. Sonic Hedgehog Is Essential to Foregut Development. Nat. Genetics 1998, 20, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Huang, Z.; Mo, R. Gli3 Null Mice Display Glandular Overgrowth of the Developing Stomach. Dev. Dyn. 2005, 234, 984–991. [Google Scholar] [CrossRef] [PubMed]
- Park, H.L.; Bai, C.; Platt, K.A.; Matise, M.P.; Beeghly, A.; Hui, C.C.; Nakashima, M.; Joyner, A.L. Mouse Gli1 Mutants Are Viable but Have Defects in SHH Signaling in Combination with a Gli2 Mutation. Development 2000, 127, 1593–1605. [Google Scholar] [PubMed]
- Kolterud, A.; Grosse, A.S.; Zacharias, W.J.; Walton, K.D.; Kretovich, K.E.; Madison, B.B.; Waghray, M.; Ferris, J.E.; Hu, C.; Merchant, J.L.; et al. Paracrine Hedgehog Signaling in Stomach and Intestine: New Roles for Hedgehog in Gastrointestinal Patterning. Gastroenterology 2009, 137, 618–628. [Google Scholar] [CrossRef] [Green Version]
- Varnat, F.; Heggeler, B.B.-T.; Grisel, P.; Boucard, N.; Corthésy-Theulaz, I.; Wahli, W.; Desvergne, B. PPARbeta/Delta Regulates Paneth Cell Differentiation via Controlling the Hedgehog Signaling Pathway. Gastroenterology 2006, 131, 538–553. [Google Scholar] [CrossRef]
- Shyer, A.E.; Huycke, T.R.; Lee, C.; Mahadevan, L.; Tabin, C.J. Bending Gradients: How the Intestinal Stem Cell Gets Its Home. Cell 2015, 161, 569–580. [Google Scholar] [CrossRef] [Green Version]
- Walton, K.D.; Kolterud, Å.; Czerwinski, M.J.; Bell, M.J.; Prakash, A.; Kushwaha, J.; Grosse, A.S.; Schnell, S.; Gumucio, D.L. Hedgehog-Responsive Mesenchymal Clusters Direct Patterning and Emergence of Intestinal Villi. Proc. Natl. Aacd. Sci. USA 2012, 109, 15817–15822. [Google Scholar] [CrossRef] [Green Version]
- Van Dop, W.A.; Uhmann, A.; Wijgerde, M.; Sleddens–Linkels, E.; Heijmans, J.; Offerhaus, G.J.; van den Bergh Weerman, M.A.; Boeckxstaens, G.E.; Hommes, D.W.; Hardwick, J.C.; et al. Depletion of the Colonic Epithelial Precursor Cell Compartment Upon Conditional Activation of the Hedgehog Pathway. Gastroenterology 2009, 136, 2195–2203.e7. [Google Scholar] [CrossRef]
- Gerling, M.; Büller, N.V.J.A.; Kirn, L.M.; Joost, S.; Frings, O.; Englert, B.; Bergström, Å.; Kuiper, R.V.; Blaas, L.; Wielenga, M.C.B.; et al. Stromal Hedgehog Signalling Is Downregulated in Colon Cancer and Its Restoration Restrains Tumour Growth. Nat. Commun. 2016, 7, 12321. [Google Scholar] [CrossRef]
- Van Dop, W.A.; Heijmans, J.; Büller, N.V.J.A.; Snoek, S.A.; Rosekrans, S.L.; Wassenberg, E.A.; van den Bergh Weerman, M.A.; Lanske, B.; Clarke, A.R.; Winton, D.J.; et al. Loss of Indian Hedgehog Activates Multiple Aspects of a Wound Healing Response in the Mouse Intestine. Gastroenterology 2010, 139, 1665–1676.e10. [Google Scholar] [CrossRef] [PubMed]
- Madison, B.B.; McKenna, L.B.; Dolson, D.; Epstein, D.J.; Kaestner, K.H. FoxF1 and FoxL1 Link Hedgehog Signaling and the Control of Epithelial Proliferation in the Developing Stomach and Intestine. J. Biol. Chem. 2009, 284, 5936–5944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madison, B.B.; Braunstein, K.; Kuizon, E.; Portman, K.; Qiao, X.T.; Gumucio, D.L. Epithelial Hedgehog Signals Pattern the Intestinal Crypt-Villus Axis. Development 2005, 132, 279–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zacharias, W.J.; Madison, B.B.; Kretovich, K.E.; Walton, K.D.; Richards, N.; Udager, A.M.; Li, X.; Gumucio, D.L. Hedgehog Signaling Controls Homeostasis of Adult Intestinal Smooth Muscle. Dev. Biol. 2011, 355, 152–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Büller, N.V.J.A.; Rosekrans, S.L.; Metcalfe, C.; Heijmans, J.; van Dop, W.A.; Fessler, E.; Jansen, M.; Ahn, C.; Vermeulen, J.L.M.; Westendorp, B.F.; et al. Stromal Indian Hedgehog Signaling Is Required for Intestinal Adenoma Formation in Mice. Gastroenterology 2015, 148, 170–180.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valenta, T.; Degirmenci, B.; Moor, A.E.; Herr, P.; Zimmerli, D.; Moor, M.B.; Hausmann, G.; Cantù, C.; Aguet, M.; Basler, K. Wnt Ligands Secreted by Subepithelial Mesenchymal Cells Are Essential for the Survival of Intestinal Stem Cells and Gut Homeostasis. Cell Rep. 2016, 15, 911–918. [Google Scholar] [CrossRef] [Green Version]
- Altmann, G.G. Morphological Observations on Mucus-Secreting Nongoblet Cells in the Deep Crypts of the Rat Ascending Colon. Am. J. Anat. 1983, 167, 95–117. [Google Scholar] [CrossRef] [PubMed]
- Qi, Z.; Li, Y.; Zhao, B.; Xu, C.; Liu, Y.; Li, H.; Zhang, B.; Wang, X.; Yang, X.; Xie, W.; et al. BMP Restricts Stemness of Intestinal Lgr5 + Stem Cells by Directly Suppressing Their Signature Genes. Nat. Commun. 2017, 8, 13824. [Google Scholar] [CrossRef]
- Zacharias, W.J.; Li, X.; Madison, B.B.; Kretovich, K.; Kao, J.Y.; Merchant, J.L.; Gumucio, D.L. Hedgehog Is an Anti-Inflammatory Epithelial Signal for the Intestinal Lamina Propria. Gastroenterology 2010, 138, 2368–2377.e4. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.J.; Rothenberg, M.E.; Seeley, E.S.; Zimdahl, B.; Kawano, S.; Lu, W.-J.; Shin, K.; Sakata-Kato, T.; Chen, J.K.; Diehn, M.; et al. Control of Inflammation by Stromal Hedgehog Pathway Activation Restrains Colitis. Proc. Natl. Acad. Sci. USA 2016. [Google Scholar] [CrossRef] [Green Version]
- Westendorp, B.F.; Büller, N.V.J.A.; Karpus, O.N.; van Dop, W.A.; Koster, J.; Versteeg, R.; Koelink, P.J.; Snel, C.Y.; Meisner, S.; Roelofs, J.J.T.H.; et al. Indian Hedgehog Suppresses a Stromal Cell-Driven Intestinal Immune Response. Cell Mol. Gastroenterol. Hepatol. 2018, 5, 67–82.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lees, C.W.; Zacharias, W.J.; Tremelling, M.; Noble, C.L.; Nimmo, E.R.; Tenesa, A.; Cornelius, J.; Torkvist, L.; Kao, J.; Farrington, S.; et al. Analysis of Germline GLI1 Variation Implicates Hedgehog Signalling in the Regulation of Intestinal Inflammatory Pathways. PLoS Med. 2008, 5, e239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razumilava, N.; Gumucio, D.L.; Samuelson, L.C.; Shah, Y.M.; Nusrat, A.; Merchant, J.L. Indian Hedgehog Suppresses Intestinal Inflammation. Cell Mol. Gastroenterol. Hepatol. 2017, 5, 63–64. [Google Scholar] [CrossRef] [Green Version]
- Kosinski, C.; Stange, D.E.; Xu, C.; Chan, A.S.; Ho, C.; Yuen, S.T.; Mifflin, R.C.; Powell, D.W.; Clevers, H.; Leung, S.Y.; et al. Indian Hedgehog Regulates Intestinal Stem Cell Fate Through Epithelial−Mesenchymal Interactions During Development. Gastroenterology 2010, 139, 893–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haramis, A.-P.G.; Begthel, H.; Born, M.; van Es, J.; Jonkheer, S.; Offerhaus, G.J.A.; Clevers, H. De Novo Crypt Formation and Juvenile Polyposis on BMP Inhibition in Mouse Intestine. Science 2004, 303, 1684–1686. [Google Scholar] [CrossRef] [Green Version]
- Davis, H.; Irshad, S.; Bansal, M.; Rafferty, H.; Boitsova, T.; Bardella, C.; Jaeger, E.; Lewis, A.; Freeman-Mills, L.; Giner, F.C.; et al. Aberrant Epithelial GREM1 Expression Initiates Colonic Tumorigenesis from Cells Outside the Stem Cell Niche. Nat. Med. 2014. [Google Scholar] [CrossRef]
- Kosinski, C.; Li, V.S.W.; Chan, A.S.Y.; Zhang, J.; Ho, C.; Tsui, W.Y.; Chan, T.L.; Mifflin, R.C.; Powell, D.W.; Yuen, S.T.; et al. Gene Expression Patterns of Human Colon Tops and Basal Crypts and BMP Antagonists as Intestinal Stem Cell Niche Factors. Proc. Nalt. Acad. Sci. USA 2007, 104, 15418–15423. [Google Scholar] [CrossRef] [Green Version]
- Benayoun, B.A.; Caburet, S.; Veitia, R.A. Forkhead Transcription Factors: Key Players in Health and Disease. Trends Genetics 2011, 27, 224–232. [Google Scholar] [CrossRef]
- Kaestner, K.H. The Intestinal Stem Cell Niche: A Central Role for Foxl1-Expressing Subepithelial Telocytes. Cell Mol. Gastroenterol. Hepatol. 2019, 8, 111–117. [Google Scholar] [CrossRef] [Green Version]
- Shoshkes-Carmel, M.; Wang, Y.J.; Wangensteen, K.J.; Tóth, B.; Kondo, A.; Massasa, E.E.; Itzkovitz, S.; Kaestner, K.H. Subepithelial Telocytes Are an Important Source of Wnts That Supports Intestinal Crypts. Nature 2018, 557, 242–246. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.M.; Wei, C.; Ensor, J.E.; Smolenski, D.J.; Amos, C.I.; Levin, B.; Berry, D.A. Meta-Analyses of Colorectal Cancer Risk Factors. Cancer Causes Control. 2013, 24, 1207–1222. [Google Scholar] [CrossRef] [PubMed]
- Pleguezuelos-Manzano, C.; Puschhof, J.; Rosendahl Huber, A.; van Hoeck, A.; Wood, H.M.; Nomburg, J.; Gurjao, C.; Manders, F.; Dalmasso, G.; Stege, P.B.; et al. Mutational Signature in Colorectal Cancer Caused by Genotoxic Pks+ E. Coli. Nature 2020, 580, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Vasen, H.F.A.; Mecklin, J.-P.; Meera Khan, P.; Lynch, H.T. The International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC). Dis. Colon Rectum 1991, 34, 424–425. [Google Scholar] [CrossRef] [PubMed]
- Soravia, C.; Berk, T.; Madlensky, L.; Mitri, A.; Cheng, H.; Gallinger, S.; Cohen, Z.; Bapat, B. Genotype-Phenotype Correlations in Attenuated Adenomatous Polyposis Coli. Am. J. Hum. Genet. 1998, 62, 1290–1301. [Google Scholar] [CrossRef] [Green Version]
- Bienz, M.; Clevers, H. Linking Colorectal Cancer to Wnt Signaling. Cell 2000, 103, 311–320. [Google Scholar] [CrossRef] [Green Version]
- Segditsas, S.; Tomlinson, I. Colorectal Cancer and Genetic Alterations in the Wnt Pathway. Oncogene 2006, 25, 7531–7537. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Network Comprehensive Molecular Characterization of Human Colon and Rectal Cancer. Nature 2012, 487, 330–337. [CrossRef] [Green Version]
- Morikawa, T.; Kuchiba, A.; Yamauchi, M.; Meyerhardt, J.A.; Shima, K.; Nosho, K.; Chan, A.T.; Giovannucci, E.; Fuchs, C.S.; Ogino, S. Association of CTNNB1 (β-Catenin) Alterations, Body Mass Index, and Physical Activity With Survival in Patients With Colorectal Cancer. JAMA 2011, 305, 1685–1694. [Google Scholar] [CrossRef] [Green Version]
- Seshagiri, S.; Stawiski, E.W.; Durinck, S.; Modrusan, Z.; Storm, E.E.; Conboy, C.B.; Chaudhuri, S.; Guan, Y.; Janakiraman, V.; Jaiswal, B.S.; et al. Recurrent R-Spondin Fusions in Colon Cancer. Nature 2012, 488, 660–664. [Google Scholar] [CrossRef]
- Amado, R.G.; Wolf, M.; Peeters, M.; Cutsem, E.V.; Siena, S.; Freeman, D.J.; Juan, T.; Sikorski, R.; Suggs, S.; Radinsky, R.; et al. Wild-Type KRAS Is Required for Panitumumab Efficacy in Patients with Metastatic Colorectal Cancer. J. Clin. Oncol. 2008, 9, 1626–1634. [Google Scholar] [CrossRef] [PubMed]
- Kopetz, S.; Grothey, A.; Yaeger, R.; Van Cutsem, E.; Desai, J.; Yoshino, T.; Wasan, H.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E–Mutated Colorectal Cancer. N. Eng. J. Med. 2019, 381, 1632–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaughn, C.P.; ZoBell, S.D.; Furtado, L.V.; Baker, C.L.; Samowitz, W.S. Frequency of KRAS, BRAF, and NRAS Mutations in Colorectal Cancer. Genes Chrom. Cancer 2011, 50, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, K.; Stadler, Z.K.; Cercek, A.; Mendelsohn, R.B.; Shia, J.; Segal, N.H.; Diaz, L.A. Immunotherapy in Colorectal Cancer: Rationale, Challenges and Potential. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 361–375. [Google Scholar] [CrossRef] [PubMed]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The Consensus Molecular Subtypes of Colorectal Cancer. Nat. Med. 2015. [Google Scholar] [CrossRef] [PubMed]
- Kuhnert, F.; Davis, C.R.; Wang, H.-T.; Chu, P.; Lee, M.; Yuan, J.; Nusse, R.; Kuo, C.J. Essential Requirement for Wnt Signaling in Proliferation of Adult Small Intestine and Colon Revealed by Adenoviral Expression of Dickkopf-1. Proc. Natl. Acad. Sci. USA 2004, 101, 266–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barker, N.; Ridgway, R.A.; van Es, J.H.; van de Wetering, M.; Begthel, H.; van den Born, M.; Danenberg, E.; Clarke, A.R.; Sansom, O.J.; Clevers, H. Crypt Stem Cells as the Cells-of-Origin of Intestinal Cancer. Nature 2008, 457, 608–611. [Google Scholar] [CrossRef]
- Schwitalla, S.; Fingerle, A.A.; Cammareri, P.; Nebelsiek, T.; Göktuna, S.I.; Ziegler, P.K.; Canli, O.; Heijmans, J.; Huels, D.J.; Moreaux, G.; et al. Intestinal Tumorigenesis Initiated by Dedifferentiation and Acquisition of Stem-Cell-like Properties. Cell 2013, 152, 25–38. [Google Scholar] [CrossRef] [Green Version]
- Cortina, C.; Turon, G.; Stork, D.; Hernando-Momblona, X.; Sevillano, M.; Aguilera, M.; Tosi, S.; Merlos-Suárez, A.; Stephan-Otto Attolini, C.; Sancho, E.; et al. A Genome Editing Approach to Study Cancer Stem Cells in Human Tumors. EMBO Mol. Med. 2017, 9, 869–879. [Google Scholar] [CrossRef]
- De Sousa e Melo, F.; Kurtova, A.V.; Harnoss, J.M.; Kljavin, N.; Hoeck, J.D.; Hung, J.; Anderson, J.E.; Storm, E.E.; Modrusan, Z.; Koeppen, H.; et al. A Distinct Role for Lgr5+ Stem Cells in Primary and Metastatic Colon Cancer. Nature 2017, 543, 676–680. [Google Scholar] [CrossRef]
- Van den Brink, G.R. Hedgehog Signaling in Development and Homeostasis of the Gastrointestinal Tract. Physiol. Rev. 2007, 87. [Google Scholar] [CrossRef] [PubMed]
- Van den Brink, G.R.; Bleuming, S.A.; Hardwick, J.C.H.; Schepman, B.L.; Offerhaus, G.J.; Keller, J.J.; Nielsen, C.; Gaffield, W.; van Deventer, S.J.H.; Roberts, D.J.; et al. Indian Hedgehog Is an Antagonist of Wnt Signaling in Colonic Epithelial Cell Differentiation. Nat. Genet. 2004, 36, 277–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serviss, J.T.; Andrews, N.; Andersson, A.B.; Dzwonkowska, E.; Heijboer, R.; Geyer, N.; Gerling, M.; Enge, M. Unsupervised Cell Interaction Profiling Reveals Major Architectural Differences between Small Intestinal and Colonic Epithelial Crypts. bioRxiv 2020. [Google Scholar] [CrossRef]
- Lewis, A.; Segditsas, S.; Deheragoda, M.; Pollard, P.; Jeffery, R.; Nye, E.; Lockstone, H.; Davis, H.; Clark, S.; Stamp, G.; et al. Severe Polyposis in Apc1322T Mice Is Associated with Submaximal Wnt Signalling and Increased Expression of the Stem Cell Marker Lgr5. Gut 2010, 59, 1680–1686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robanus-Maandag, E.C.; Koelink, P.J.; Breukel, C.; Salvatori, D.C.F.; Jagmohan-Changur, S.C.; Bosch, C.A.J.; Verspaget, H.W.; Devilee, P.; Fodde, R.; Smits, R. A New Conditional Apc-Mutant Mouse Model for Colorectal Cancer. Carcinogenesis 2010, 31, 946–952. [Google Scholar] [CrossRef]
- Su, L.-K.; Kinzler, K.W.; Vogelstein, B.; Preisinger, A.C.; Moser, A.R.; Luongo, C.; Gould, K.A.; Dove, W.F. Multiple Intestinal Neoplasia Caused by a Mutation in the Murine Homolog of the APC Gene. Science 1992, 256, 668–670. [Google Scholar] [CrossRef]
- Dow, L.E.; O’Rourke, K.P.; Simon, J.; Tschaharganeh, D.F.; van Es, J.H.; Clevers, H.; Lowe, S.W. Apc Restoration Promotes Cellular Differentiation and Reestablishes Crypt Homeostasis in Colorectal Cancer. Cell 2015, 161, 1539–1552. [Google Scholar] [CrossRef] [Green Version]
- Neufert, C.; Becker, C.; Neurath, M.F. An Inducible Mouse Model of Colon Carcinogenesis for the Analysis of Sporadic and Inflammation-Driven Tumor Progression. Nat. Protocols 2007, 2, 1998–2004. [Google Scholar] [CrossRef]
- Karim, B.O.; Huso, D.L. Mouse Models for Colorectal Cancer. Am. J. Cancer Res. 2013, 3, 240–250. [Google Scholar]
- Hahn, H.; Christiansen, J.; Wicking, C.; Zaphiropoulos, P.G.; Chidambaram, A.; Gerrard, B.; Vorechovsky, I.; Bale, A.E.; Toftgard, R.; Dean, M.; et al. A Mammalian Patched Homolog Is Expressed in Target Tissues of Sonic Hedgehog and Maps to a Region Associated with Developmental Abnormalities. J. Biol. Chem. 1996, 271, 12125–12128. [Google Scholar] [CrossRef] [Green Version]
- Gailani, M.R.; Ståhle-Bäckdahl, M.; Leffell, D.J.; Glyn, M.; Zaphiropoulos, P.G.; Undén, A.B.; Dean, M.; Brash, D.E.; Bale, A.E.; Toftgård, R. The Role of the Human Homologue of Drosophila Patched in Sporadic Basal Cell Carcinomas. Nat. Genetics 1996, 14, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Berman, D.M.; Karhadkar, S.S.; Maitra, A.; Montes De Oca, R.; Gerstenblith, M.R.; Briggs, K.; Parker, A.R.; Shimada, Y.; Eshleman, J.R.; Watkins, D.N.; et al. Widespread Requirement for Hedgehog Ligand Stimulation in Growth of Digestive Tract Tumours. Nature 2003, 425, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Chatel, G.; Ganeff, C.; Boussif, N.; Delacroix, L.; Briquet, A.; Nolens, G.; Winkler, R. Hedgehog Signaling Pathway Is Inactive in Colorectal Cancer Cell Lines. Int. J. Cancer 2007, 121, 2622–2627. [Google Scholar] [CrossRef] [PubMed]
- Akiyoshi, T.; Nakamura, M.; Koga, K.; Nakashima, H.; Yao, T.; Tsuneyoshi, M.; Tanaka, M.; Katano, M. Gli1, Downregulated in Colorectal Cancers, Inhibits Proliferation of Colon Cancer Cells Involving Wnt Signalling Activation. Gut 2006, 55, 991–999. [Google Scholar] [CrossRef] [Green Version]
- Ricci-Vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; De Maria, R. Identification and Expansion of Human Colon-Cancer-Initiating Cells. Nature 2007, 445, 111–115. [Google Scholar] [CrossRef]
- Varnat, F.; Duquet, A.; Malerba, M.; Zbinden, M.; Mas, C.; Gervaz, P.; Ruiz i Altaba, A. Human Colon Cancer Epithelial Cells Harbour Active HEDGEHOG-GLI Signalling That Is Essential for Tumour Growth, Recurrence, Metastasis and Stem Cell Survival and Expansion. EMBO Mol. Med. 2009, 1, 338–351. [Google Scholar] [CrossRef]
- Wu, C.; Zhu, X.; Liu, W.; Ruan, T.; Tao, K. Hedgehog Signaling Pathway in Colorectal Cancer: Function, Mechanism, and Therapy. Onco Targets Ther. 2017, 10, 3249–3259. [Google Scholar] [CrossRef] [Green Version]
- Oniscu, A.; James, R.M.; Morris, R.G.; Bader, S.; Malcomson, R.D.G.; Harrison, D.J. Expression of Sonic Hedgehog Pathway Genes Is Altered in Colonic Neoplasia. J. Pathol. 2004, 203, 909–917. [Google Scholar] [CrossRef]
- Calon, A.; Espinet, E.; Palomo-Ponce, S.; Tauriello, D.V.F.; Iglesias, M.; Céspedes, M.V.; Sevillano, M.; Nadal, C.; Jung, P.; Zhang, X.H.-F.; et al. Dependency of Colorectal Cancer on a TGF-β-Driven Program in Stromal Cells for Metastasis Initiation. Cancer Cell 2012, 22, 571–584. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, K.; Shimada, M.; Miyamoto, H.; Higashijima, J.; Miyatani, T.; Nishioka, M.; Kurita, N.; Iwata, T.; Uehara, H. Sonic Hedgehog Relates to Colorectal Carcinogenesis. J. Gastroenterol. 2009, 44, 1113. [Google Scholar] [CrossRef]
- Buczacki, S.J.A.; Popova, S.; Biggs, E.; Koukorava, C.; Buzzelli, J.; Vermeulen, L.; Hazelwood, L.; Francies, H.; Garnett, M.J.; Winton, D.J. Itraconazole Targets Cell Cycle Heterogeneity in Colorectal Cancer. J. Exp. Med. 2018, 215, 1891–1912. [Google Scholar] [CrossRef] [PubMed]
- Lauth, M.; Bergström, A.; Shimokawa, T.; Toftgård, R. Inhibition of GLI-Mediated Transcription and Tumor Cell Growth by Small-Molecule Antagonists. Proc. Natl. Acad. Sci. USA 2007, 104, 8455–8460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regan, J.L.; Schumacher, D.; Staudte, S.; Steffen, A.; Haybaeck, J.; Keilholz, U.; Schweiger, C.; Golob-Schwarzl, N.; Mumberg, D.; Henderson, D.; et al. Non-Canonical Hedgehog Signaling Is a Positive Regulator of the WNT Pathway and Is Required for the Survival of Colon Cancer Stem Cells. Cell Rep. 2017, 21, 2813–2828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delloye-Bourgeois, C.; Gibert, B.; Rama, N.; Delcros, J.-G.; Gadot, N.; Scoazec, J.-Y.; Krauss, R.; Bernet, A.; Mehlen, P. Sonic Hedgehog Promotes Tumor Cell Survival by Inhibiting CDON Pro-Apoptotic Activity. PLoS Biol. 2013, 11, e1001623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.-A.; Chen, Y.-F.; Bao, Y.; Mahara, S.; Yatim, S.M.J.M.; Oguz, G.; Lee, P.L.; Feng, M.; Cai, Y.; Tan, E.Y.; et al. Hypoxic Tumor Microenvironment Activates GLI2 via HIF-1α and TGF-Β2 to Promote Chemoresistance in Colorectal Cancer. Proc. Natl. Acad. Sci. USA 2018, 115, E5990–E5999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Axelson, M.; Liu, K.; Jiang, X.; He, K.; Wang, J.; Zhao, H.; Kufrin, D.; Palmby, T.; Dong, Z.; Russell, A.M.; et al. Food and Drug Administration Approval: Vismodegib for Recurrent, Locally Advanced, or Metastatic Basal Cell Carcinoma. Clin. Cancer Res. 2013, 19, 2289–2293. [Google Scholar] [CrossRef] [Green Version]
- Berlin, J.; Bendell, J.C.; Hart, L.L.; Firdaus, I.; Gore, I.; Hermann, R.C.; Mulcahy, M.F.; Zalupski, M.M.; Mackey, H.M.; Yauch, R.L.; et al. A Randomized Phase II Trial of Vismodegib versus Placebo with FOLFOX or FOLFIRI and Bevacizumab in Patients with Previously Untreated Metastatic Colorectal Cancer. Clin. Cancer Res. 2013, 19, 258–267. [Google Scholar] [CrossRef] [Green Version]
- Catenacci, D.V.T.; Junttila, M.R.; Karrison, T.; Bahary, N.; Horiba, M.N.; Nattam, S.R.; Marsh, R.; Wallace, J.; Kozloff, M.; Rajdev, L.; et al. Randomized Phase Ib/II Study of Gemcitabine Plus Placebo or Vismodegib, a Hedgehog Pathway Inhibitor, in Patients with Metastatic Pancreatic Cancer. J. Clin. Oncol. 2015, 33, 4284–4292. [Google Scholar] [CrossRef]
- De Jesus-Acosta, A.; Sugar, E.A.; O’Dwyer, P.J.; Ramanathan, R.K.; Von Hoff, D.D.; Rasheed, Z.; Zheng, L.; Begum, A.; Anders, R.; Maitra, A.; et al. Phase 2 Study of Vismodegib, a Hedgehog Inhibitor, Combined with Gemcitabine and Nab-Paclitaxel in Patients with Untreated Metastatic Pancreatic Adenocarcinoma. Br. J. Cancer 2020, 122, 498–505. [Google Scholar] [CrossRef]
- Belani, C.P.; Dahlberg, S.E.; Rudin, C.M.; Fleisher, M.; Chen, H.X.; Takebe, N.; Velasco, M.R.; Tester, W.J.; Sturtz, K.; Hann, C.L.; et al. Vismodegib or Cixutumumab in Combination with Standard Chemotherapy for Patients with Extensive-Stage Small Cell Lung Cancer: A Trial of the ECOG-ACRIN Cancer Research Group (E1508). Cancer 2016, 122, 2371–2378. [Google Scholar] [CrossRef]
- Kaye, S.B.; Fehrenbacher, L.; Holloway, R.; Amit, A.; Karlan, B.; Slomovitz, B.; Sabbatini, P.; Fu, L.; Yauch, R.L.; Chang, I.; et al. A Phase II, Randomized, Placebo-Controlled Study of Vismodegib as Maintenance Therapy in Patients with Ovarian Cancer in Second or Third Complete Remission. Clin. Cancer Res. 2012, 18, 6509–6518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robbins, D.J.; Fei, D.L.; Riobo, N.A. The Hedgehog Signal Transduction Network. Sci. Signal. 2012, 5, re6. [Google Scholar] [CrossRef] [Green Version]
- Casali, A.; Struhl, G. Reading the Hedgehog Morphogen Gradient by Measuring the Ratio of Bound to Unbound Patched Protein. Nature 2004, 431, 76–80. [Google Scholar] [CrossRef]
- Mathew, E.; Zhang, Y.; Holtz, A.M.; Kane, K.T.; Song, J.Y.; Allen, B.L.; Pasca di Magliano, M. Dosage-Dependent Regulation of Pancreatic Cancer Growth and Angiogenesis by Hedgehog Signaling. Cell Rep. 2014, 9, 484–494. [Google Scholar] [CrossRef] [Green Version]
- Hong, K.D.; Lee, Y.; Kim, B.-H.; Lee, S.I.; Moon, H.Y. Expression of GLI1 Correlates with Expression of Lymphangiogenesis Proteins, Vascular Endothelial Growth Factor C and Vascular Endothelial Growth Factor Receptor 3, in Colorectal Cancer. Am. Surg. 2013, 79, 198–204. [Google Scholar] [CrossRef]
- Salovaara, R.; Roth, S.; Loukola, A.; Launonen, V.; Sistonen, P.; Avizienyte, E.; Kristo, P.; Järvinen, H.; Souchelnytskyi, S.; Sarlomo-Rikala, M.; et al. Frequent Loss of SMAD4/DPC4 Protein in Colorectal Cancers. Gut 2002, 51, 56–59. [Google Scholar] [CrossRef] [PubMed]
- Voorneveld, P.W.; Kodach, L.L.; Jacobs, R.J.; Liv, N.; Zonnevylle, A.C.; Hoogenboom, J.P.; Biemond, I.; Verspaget, H.W.; Hommes, D.W.; de Rooij, K.; et al. Loss of SMAD4 Alters BMP Signaling to Promote Colorectal Cancer Cell Metastasis via Activation of Rho and ROCK. Gastroenterology 2014, 147, 196–208.e13. [Google Scholar] [CrossRef] [PubMed]
- Miyaki, M.; Iijima, T.; Konishi, M.; Sakai, K.; Ishii, A.; Yasuno, M.; Hishima, T.; Koike, M.; Shitara, N.; Iwama, T.; et al. Higher Frequency of Smad4 Gene Mutation in Human Colorectal Cancer with Distant Metastasis. Oncogene 1999, 18, 3098–3103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, S.; Sugiura, T. Characterization of Human Bone Morphogenetic Protein (BMP)-4 and -7 Gene Promoters: Activation of BMP Promoters by Gli, a Sonic Hedgehog Mediator. Bone 2001, 29, 54–61. [Google Scholar] [CrossRef]
- Andre, T.; Shiu, K.; Kim, W.; Jensen, V.; Jensen, H.; Punt, J.A.; Smith, M.; Garcia-Carbonero, R.-G.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab Versus Chemotherapy for Microsatellite Instability-High/Mismatch Repair Deficient Metastatic Colorectal Cancer: The Phase 3 KEYNOTE-177 Study. J. Clin. Oncol. 2020, 38. [Google Scholar] [CrossRef]
- Grund-Gröschke, S.; Stockmaier, G.; Aberger, F. Hedgehog/GLI Signaling in Tumor Immunity—New Therapeutic Opportunities and Clinical Implications. Cell Commun. Signal. 2019, 17, 172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guy, C.D.; Suzuki, A.; Zdanowicz, M.; Abdelmalek, M.F.; Burchette, J.; Unalp, A.; Diehl, A.M.; Crn, N. Hedgehog Pathway Activation Parallels Histologic Severity of Injury and Fibrosis in Human Nonalcoholic Fatty Liver Disease. Hepatology 2012, 55, 1711–1721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michelotti, G.A.; Xie, G.; Swiderska, M.; Choi, S.S.; Karaca, G.; Krüger, L.; Premont, R.; Yang, L.; Syn, W.-K.; Metzger, D.; et al. Smoothened Is a Master Regulator of Adult Liver Repair. J. Clin. Inv. 2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Kugler, M.C.; Loomis, C.A.; Samdani, R.; Zhao, Z.; Chen, G.J.; Brandt, J.P.; Brownell, I.; Joyner, A.L.; Rom, W.N.; et al. Hedgehog Signaling in Neonatal and Adult Lung. Am. J. Respir. Cell Mol. Biol. 2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Dam, P.-J.; van der Stok, E.P.; Teuwen, L.-A.; Van den Eynden, G.G.; Illemann, M.; Frentzas, S.; Majeed, A.W.; Eefsen, R.L.; Coebergh van den Braak, R.R.J.; Lazaris, A.; et al. International Consensus Guidelines for Scoring the Histopathological Growth Patterns of Liver Metastasis. Br. J. Cancer 2017, 117, 1427–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández Moro, C.; Bozóky, B.; Gerling, M. Growth Patterns of Colorectal Cancer Liver Metastases and Their Impact on Prognosis: A Systematic Review. BMJ Open Gastroenterol. 2018, 5, e000217. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Geyer, N.; Gerling, M. Hedgehog Signaling in Colorectal Cancer: All in the Stroma? Int. J. Mol. Sci. 2021, 22, 1025. https://doi.org/10.3390/ijms22031025
Geyer N, Gerling M. Hedgehog Signaling in Colorectal Cancer: All in the Stroma? International Journal of Molecular Sciences. 2021; 22(3):1025. https://doi.org/10.3390/ijms22031025
Chicago/Turabian StyleGeyer, Natalie, and Marco Gerling. 2021. "Hedgehog Signaling in Colorectal Cancer: All in the Stroma?" International Journal of Molecular Sciences 22, no. 3: 1025. https://doi.org/10.3390/ijms22031025
APA StyleGeyer, N., & Gerling, M. (2021). Hedgehog Signaling in Colorectal Cancer: All in the Stroma? International Journal of Molecular Sciences, 22(3), 1025. https://doi.org/10.3390/ijms22031025