
Natural and Synthetic Lactones Possessing Antitumor Activities



Abstract
:1. Introduction
2. Biological Activities and Chemistry of Natural and Synthetic Lactones
2.1. Reosorcylic Acid Lactones
2.1.1. Biological Activities of Radicicol (1)
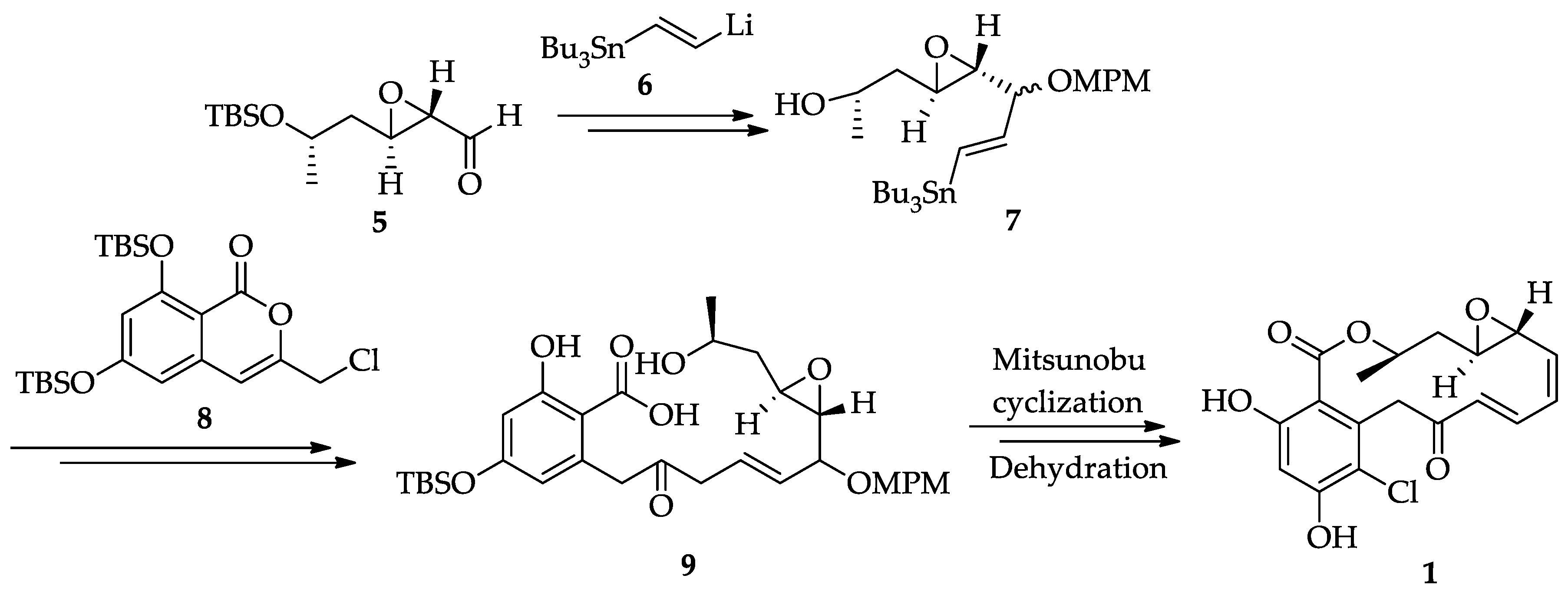
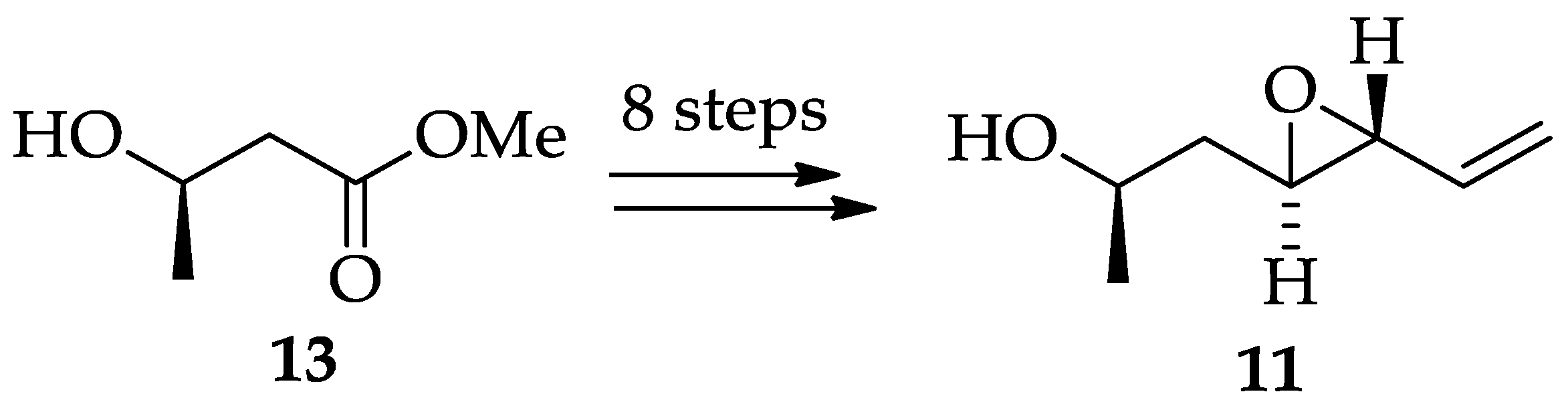
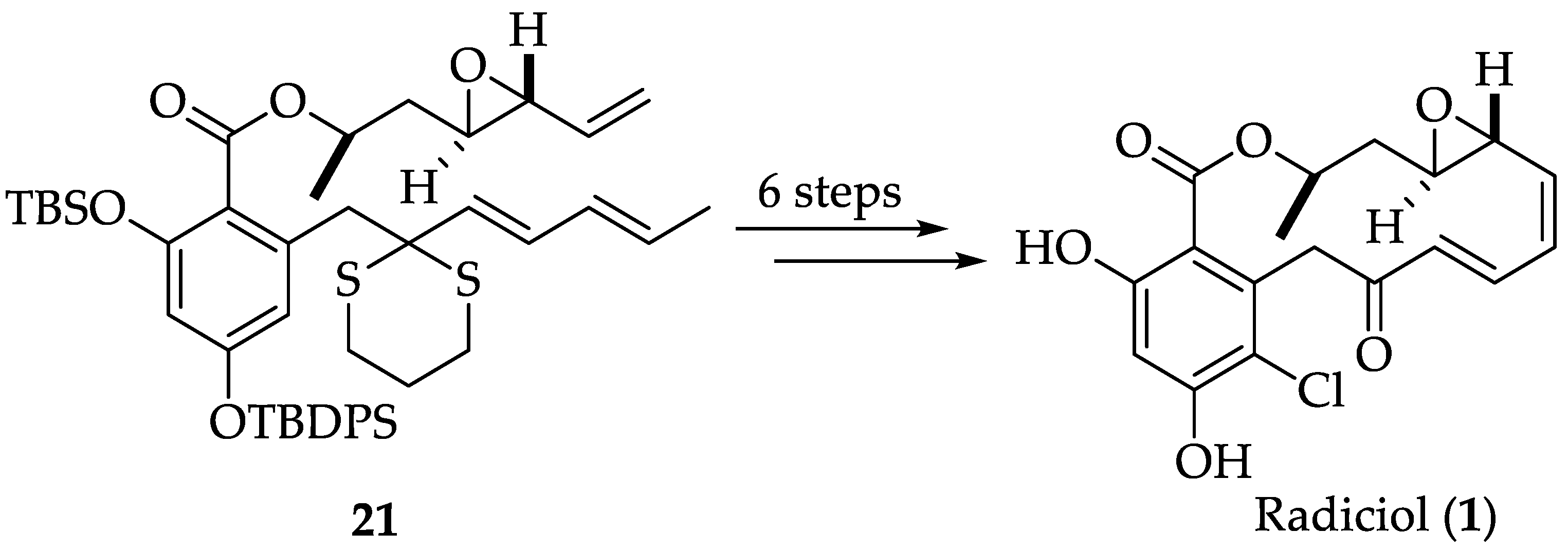
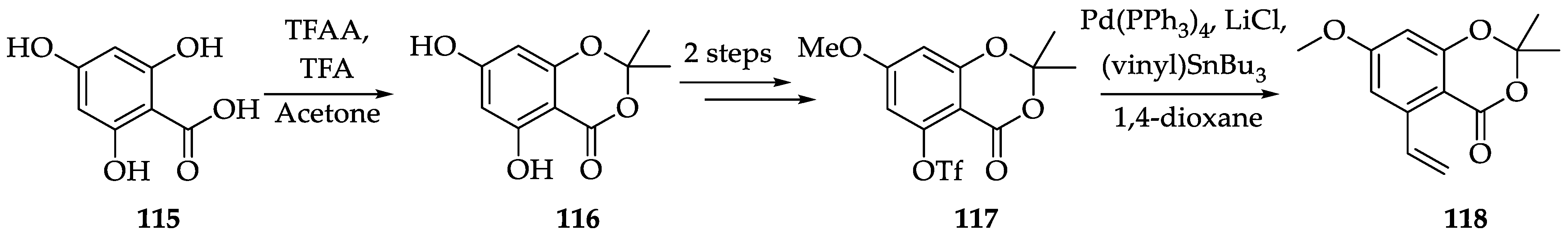
2.1.2. Chemistry of Radicicol (1)
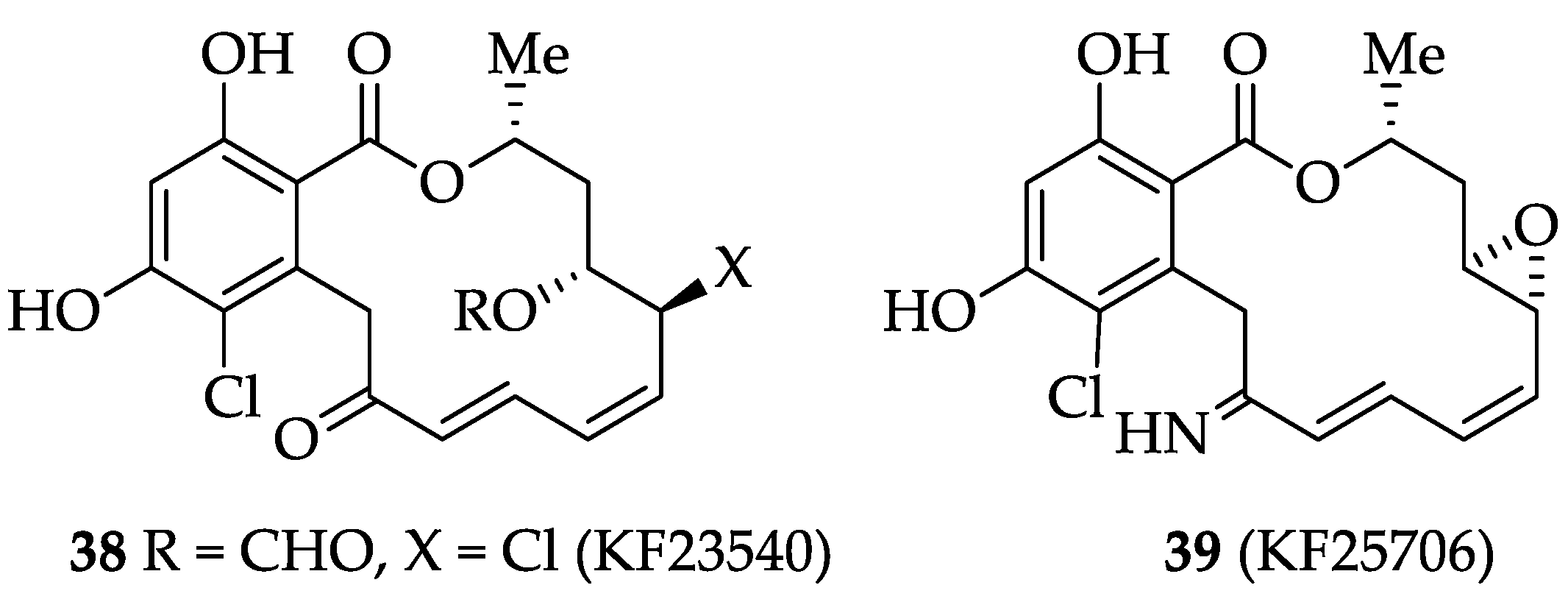
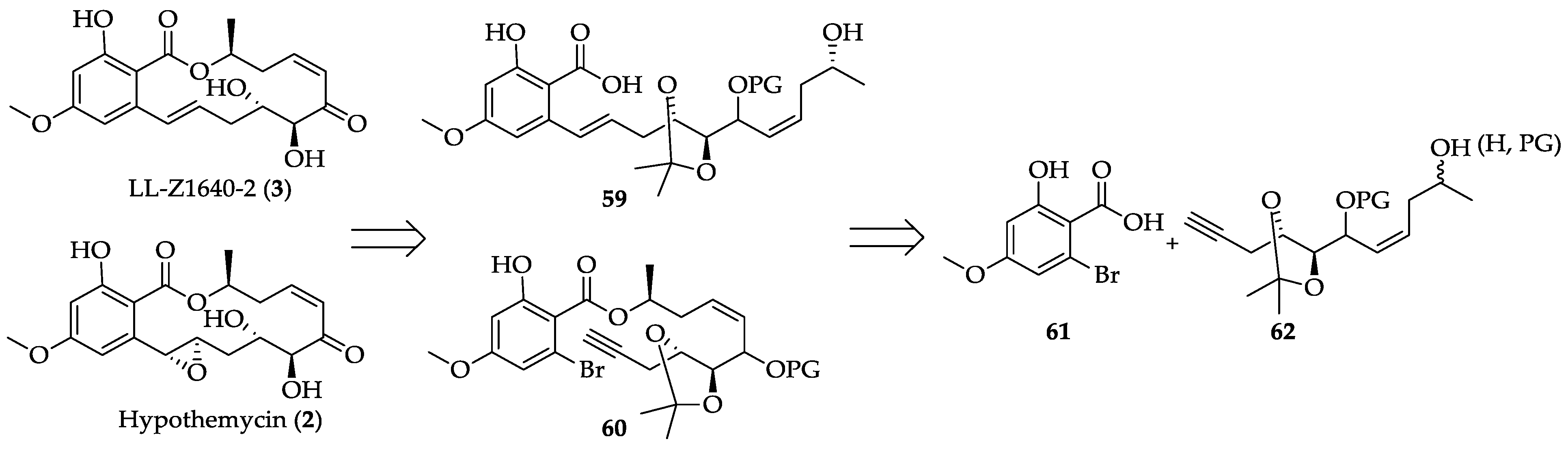
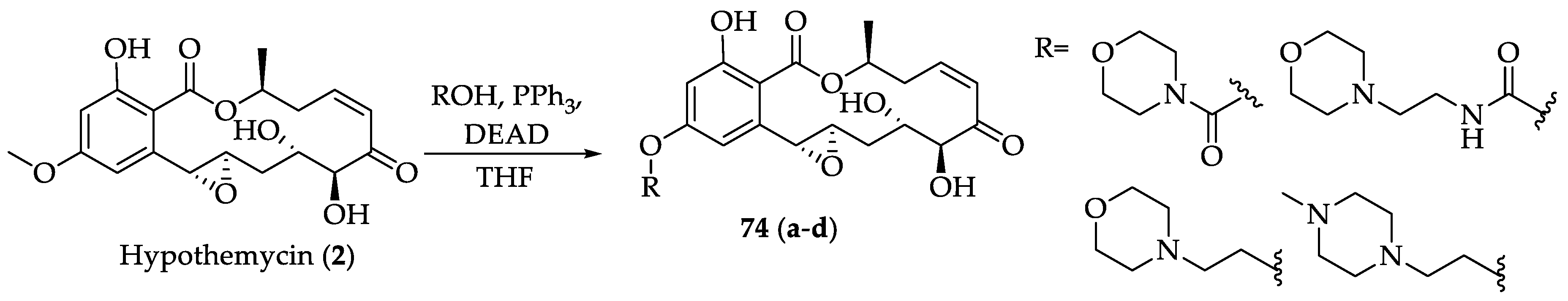
2.1.3. Biological Activities of Hypothemycin (2) and LL-Z1640-2 (3)
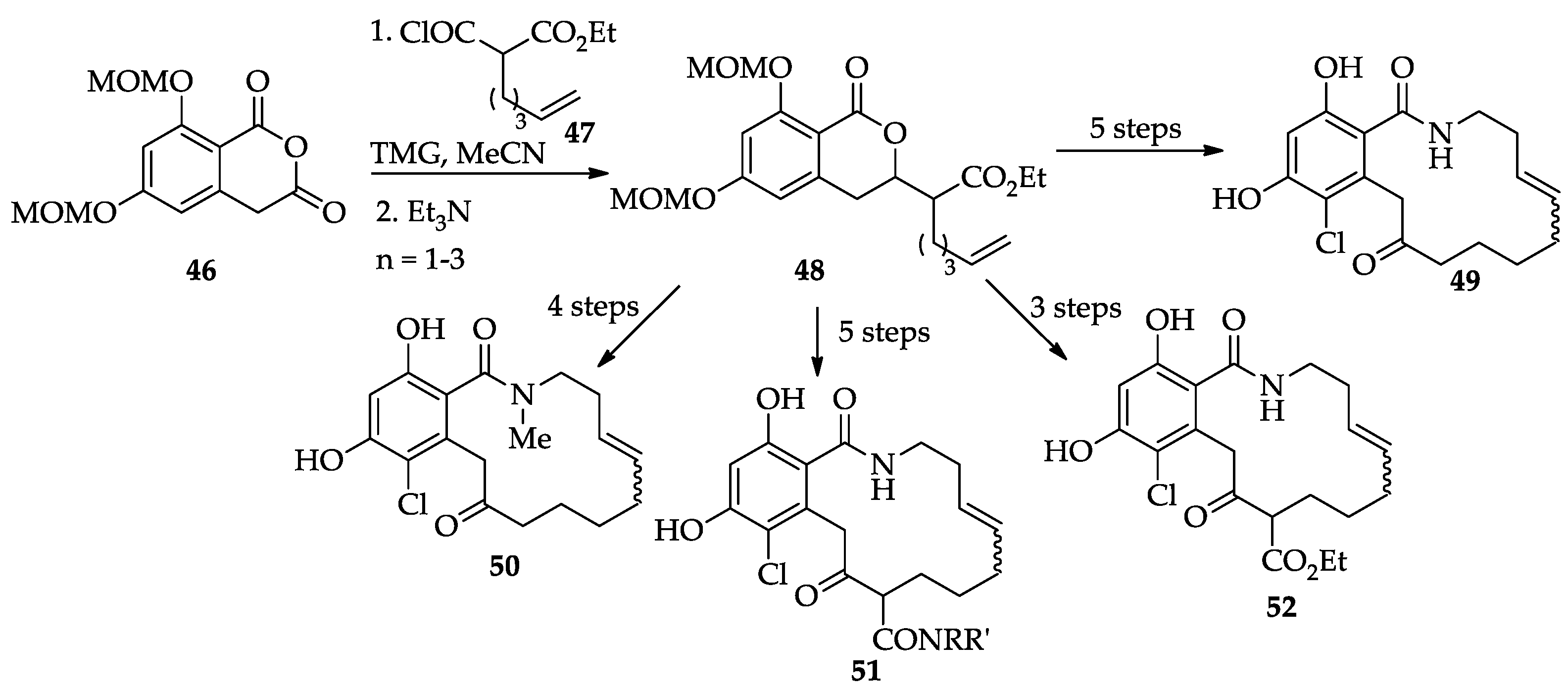
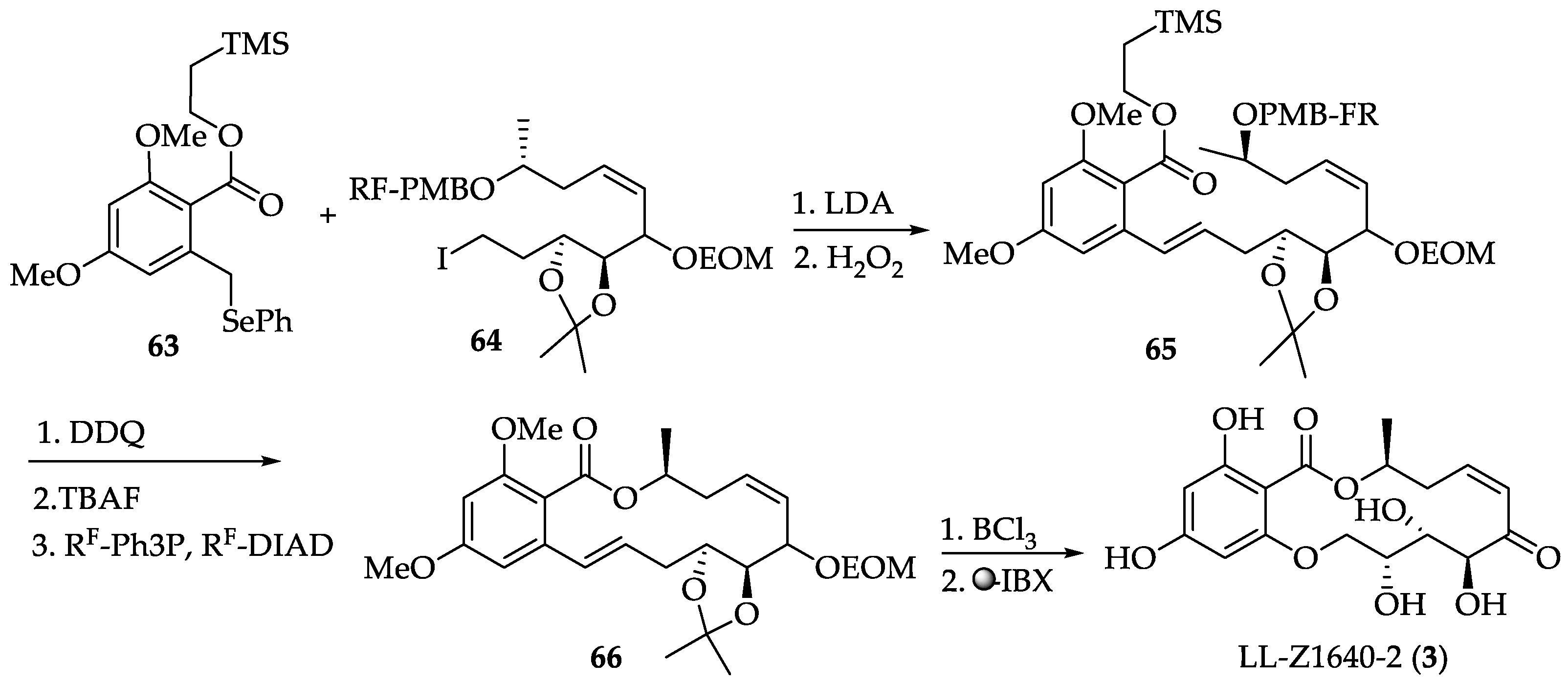
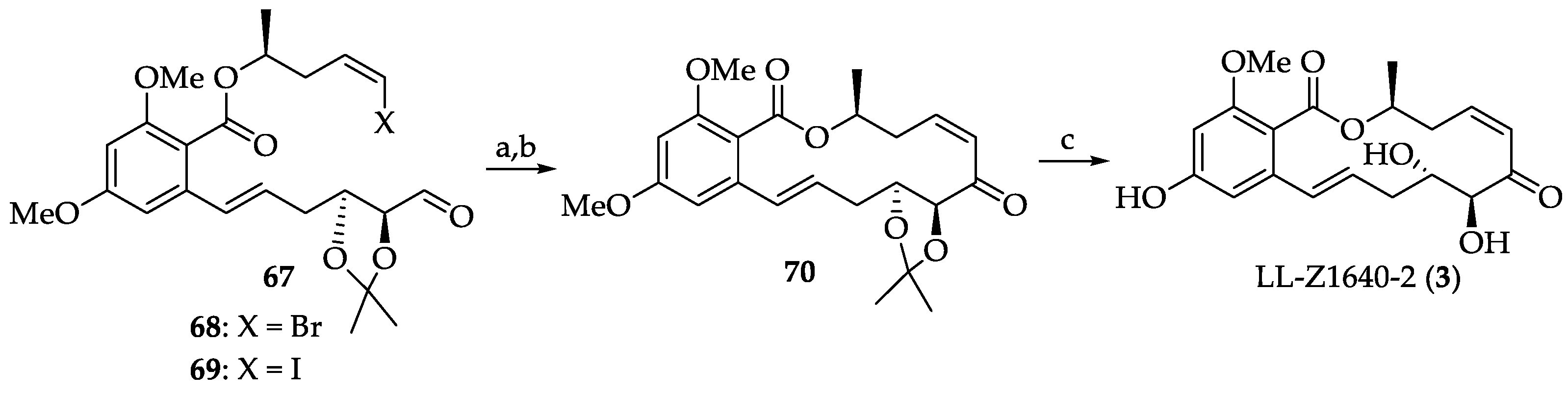
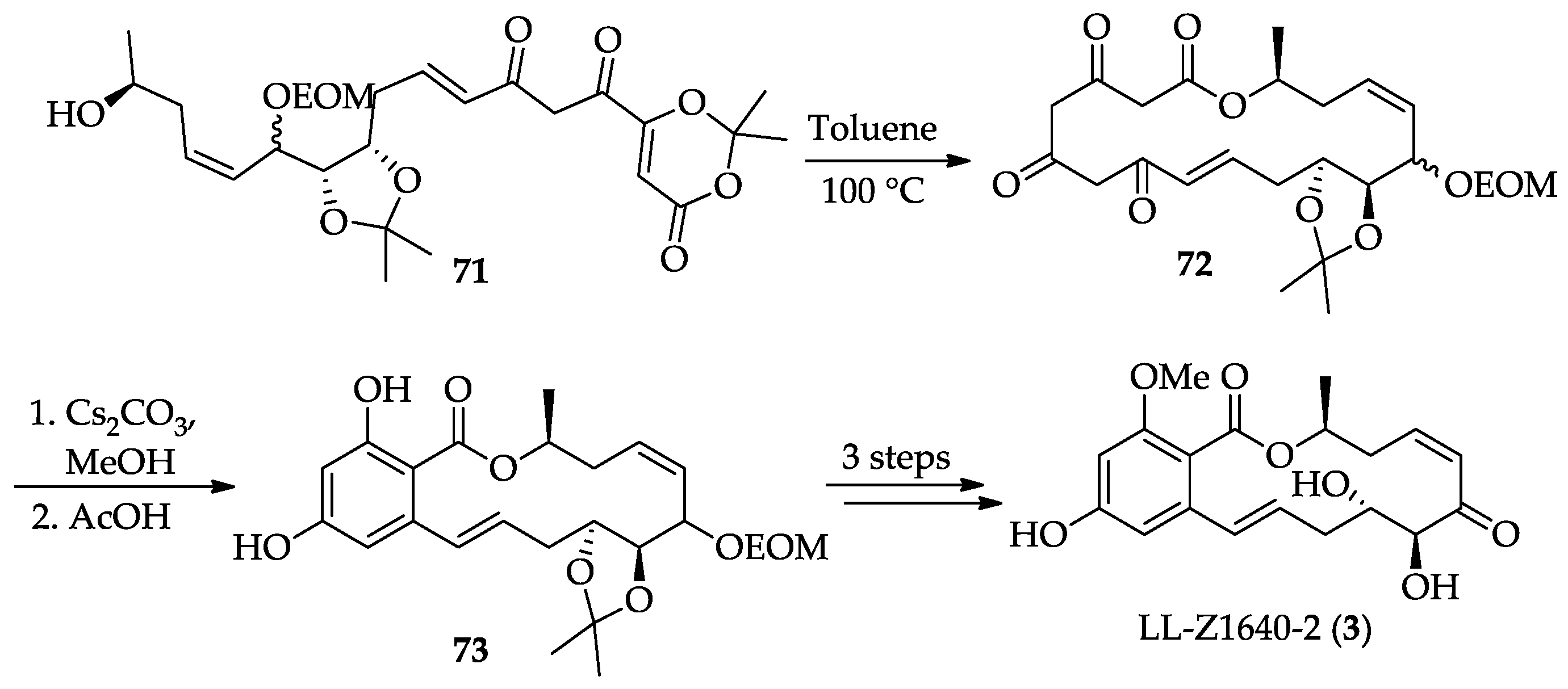
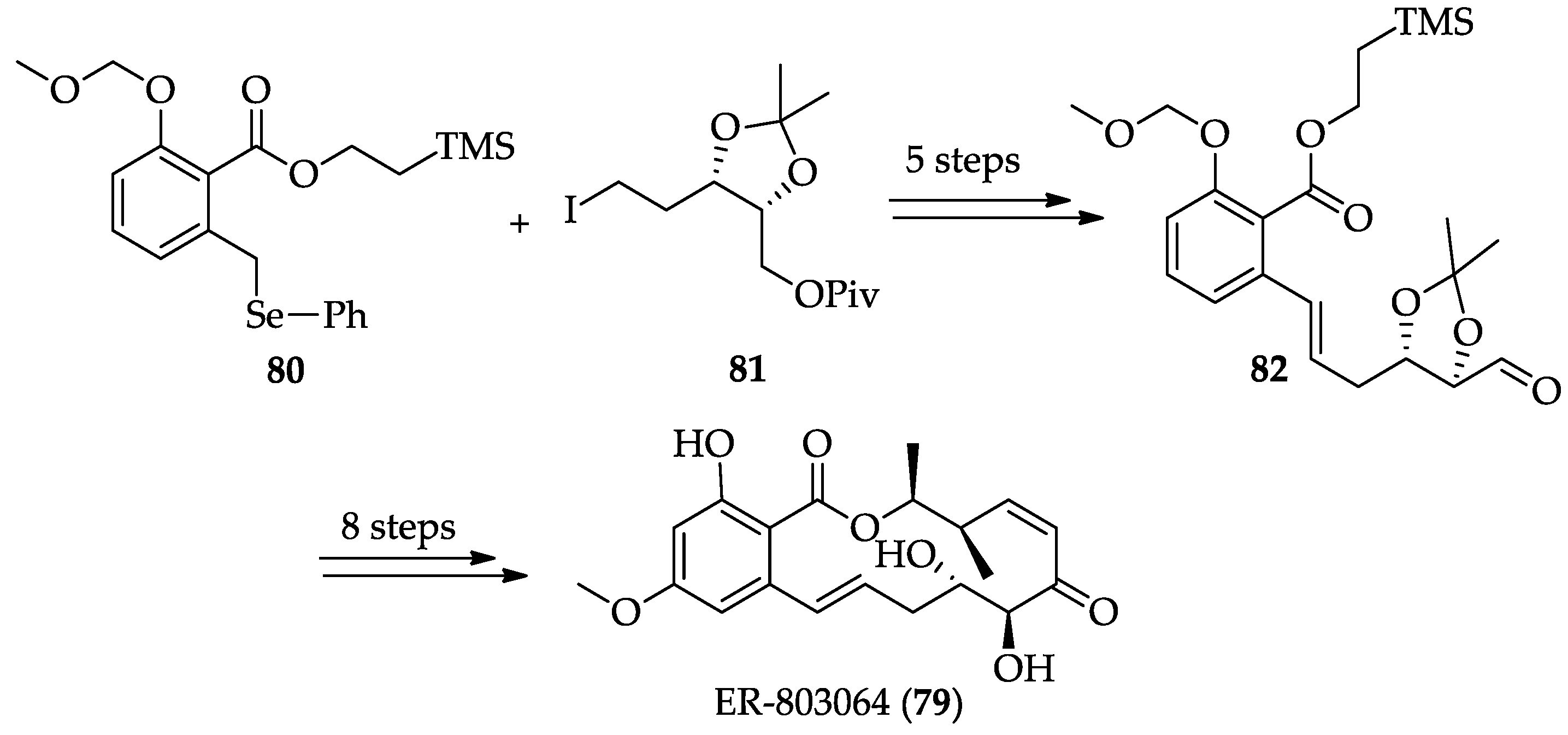
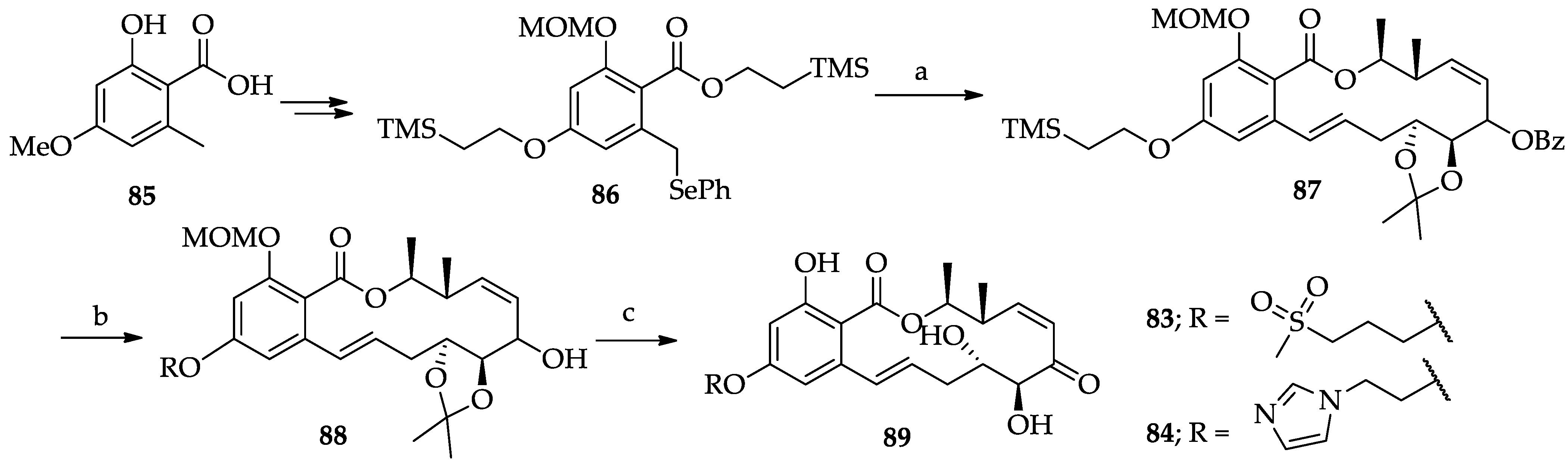
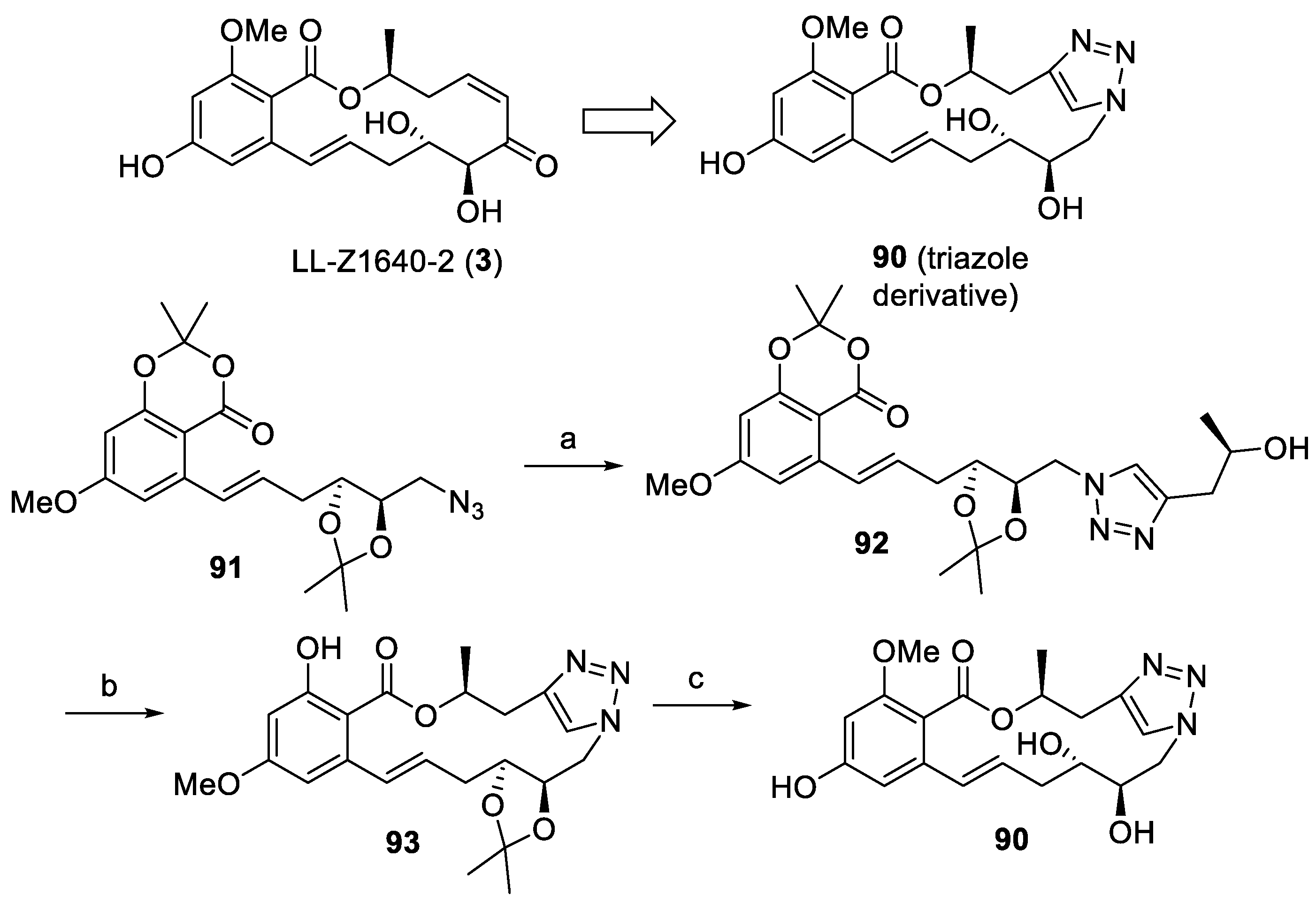
2.1.4. Chemistry of Hypothemycin (2) and LL-Z1640-2 (3)
2.1.5. Biological Activities of L-783277 (4)
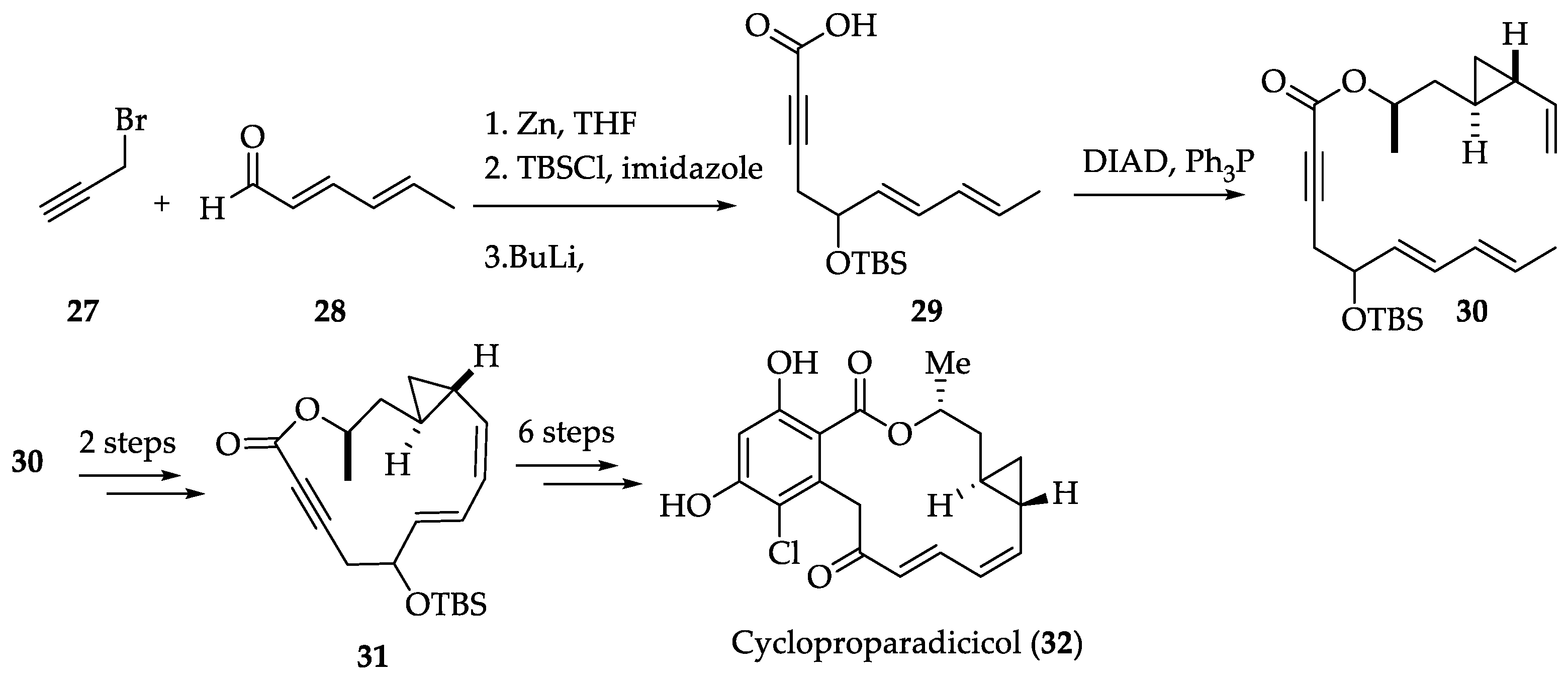
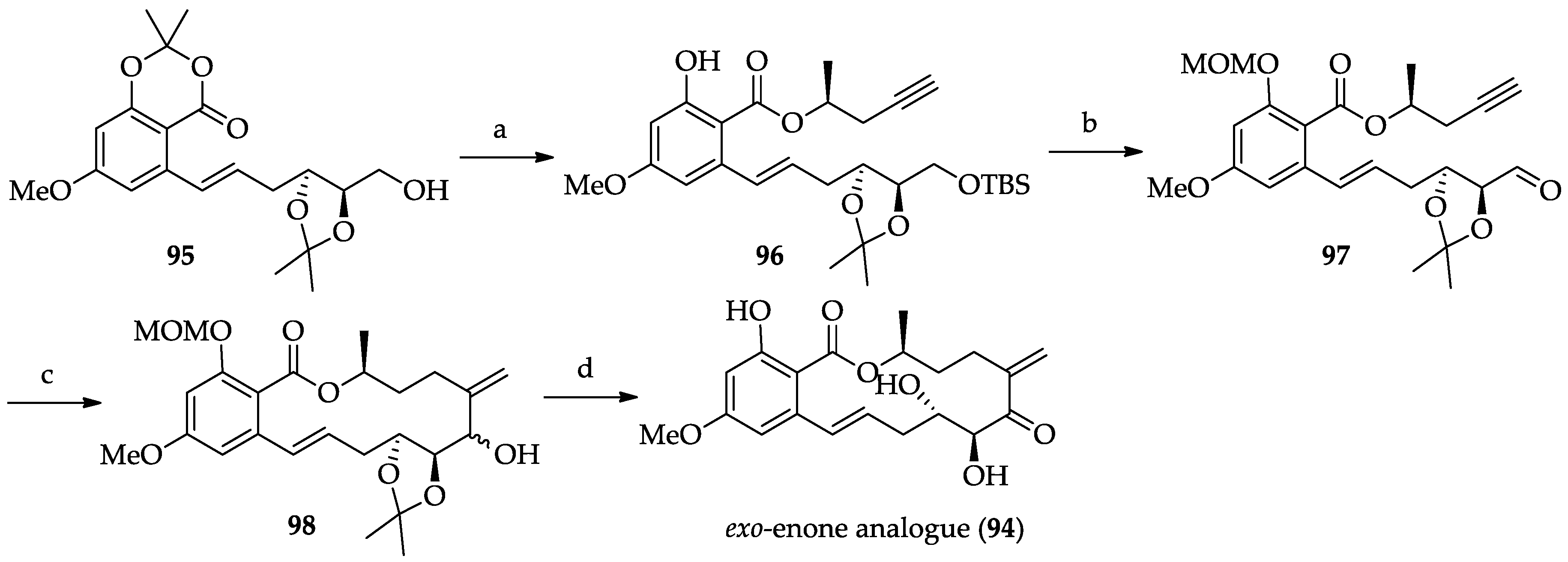
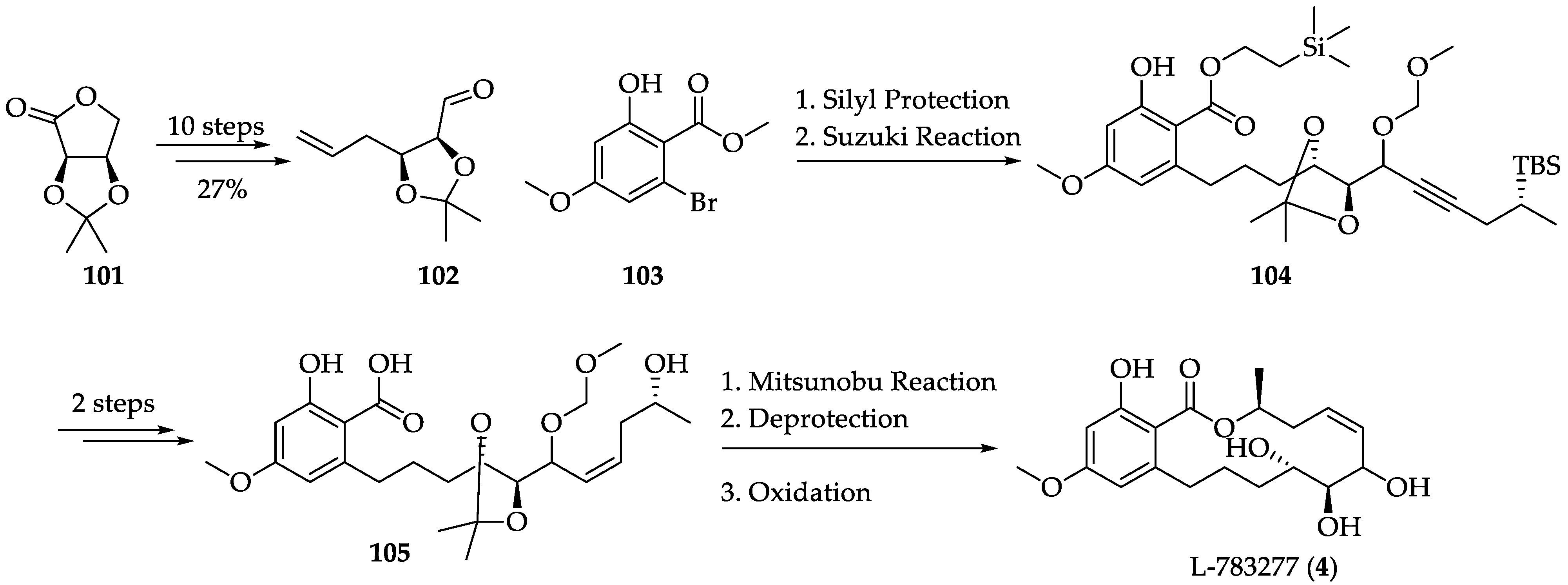
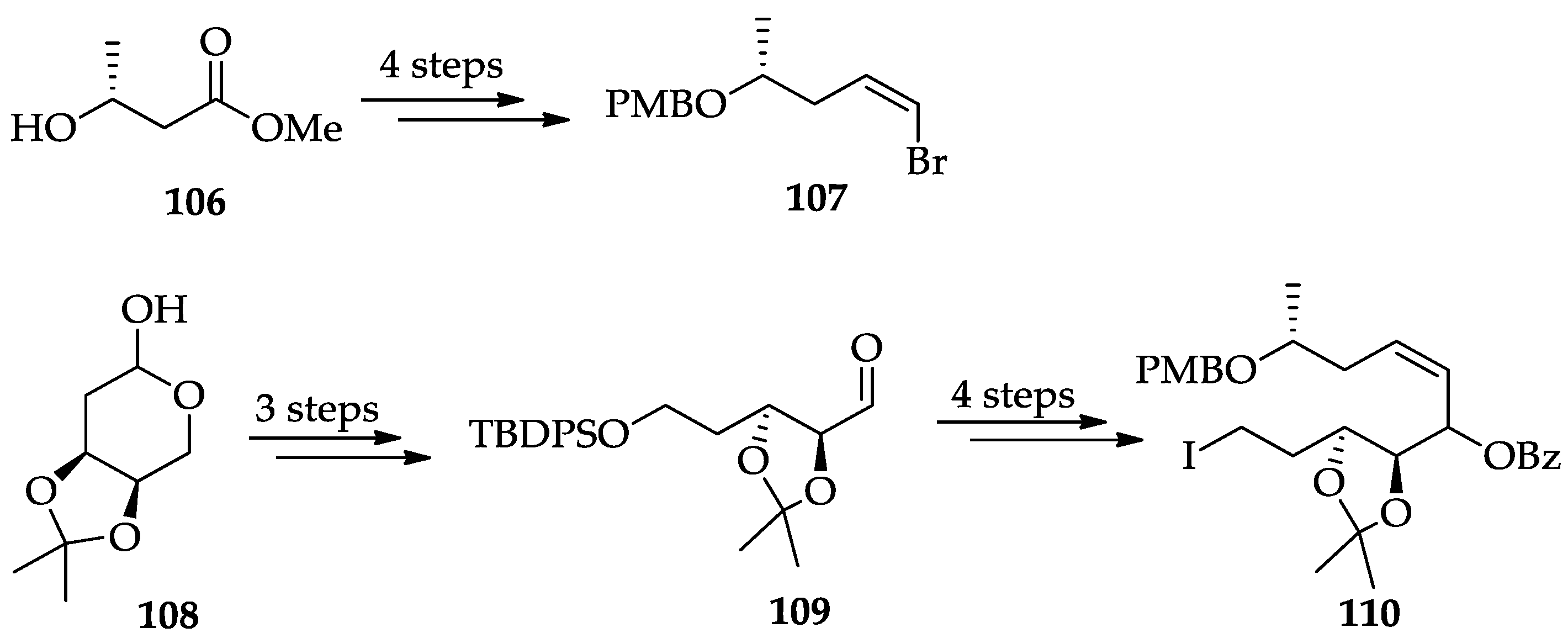
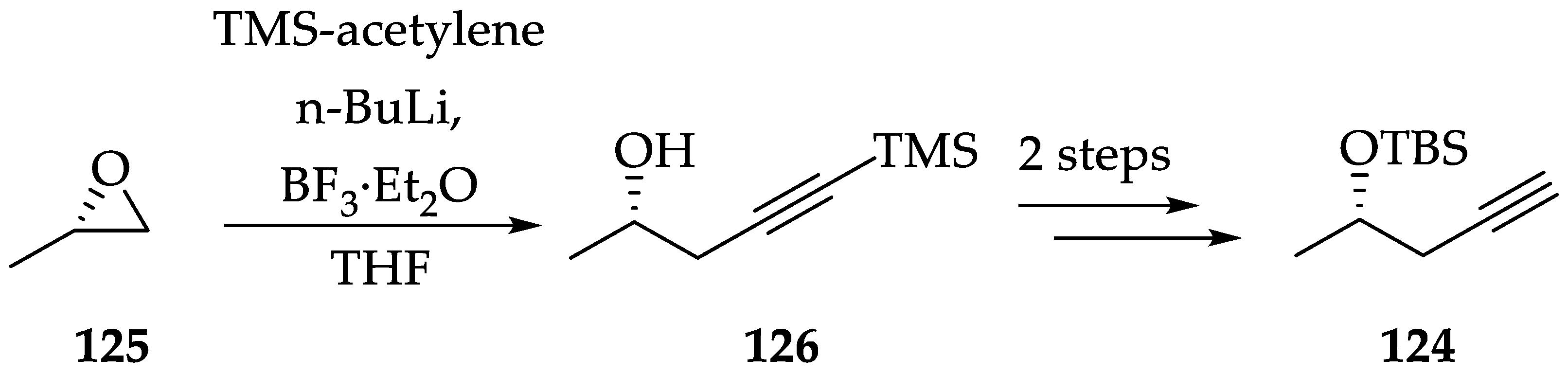
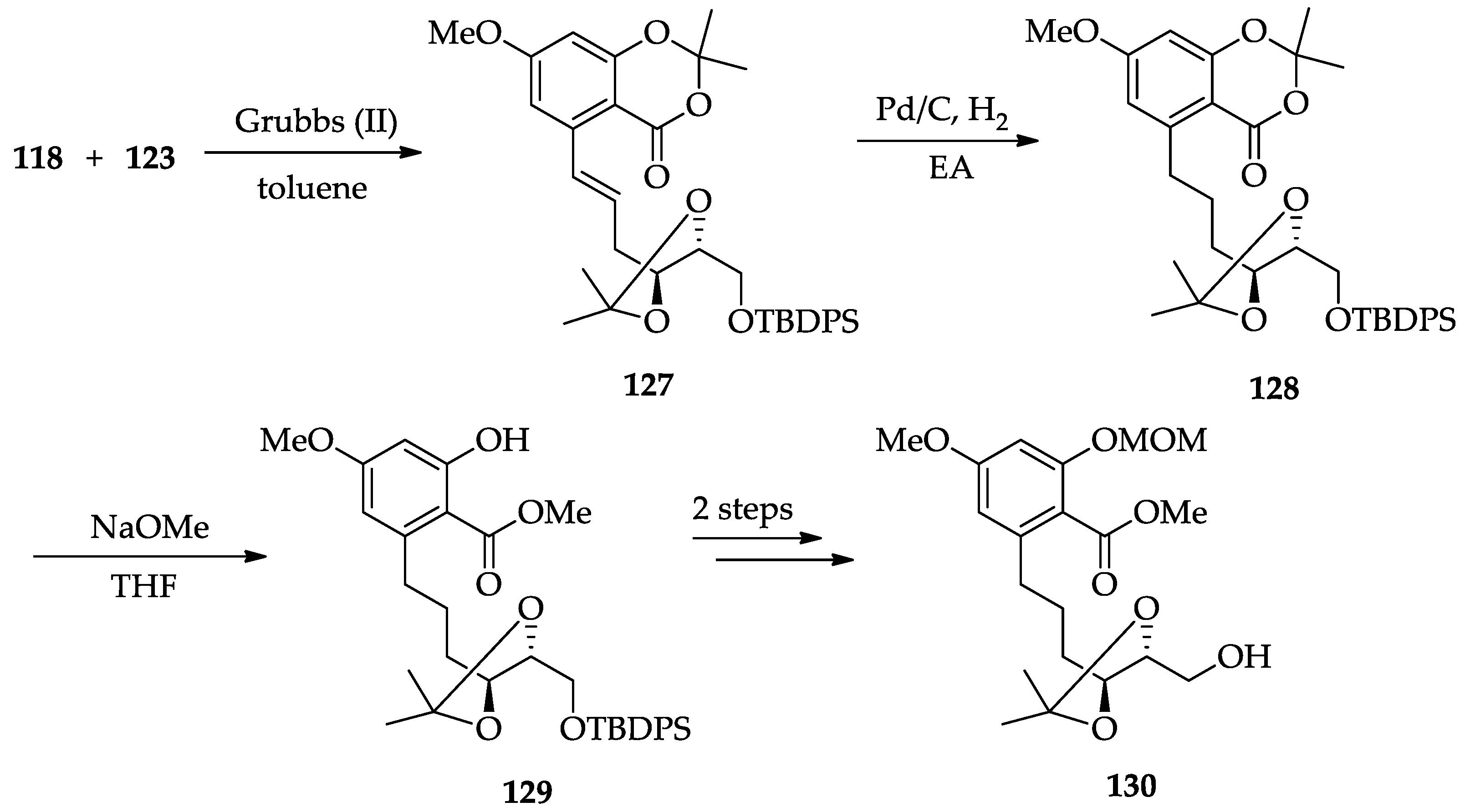
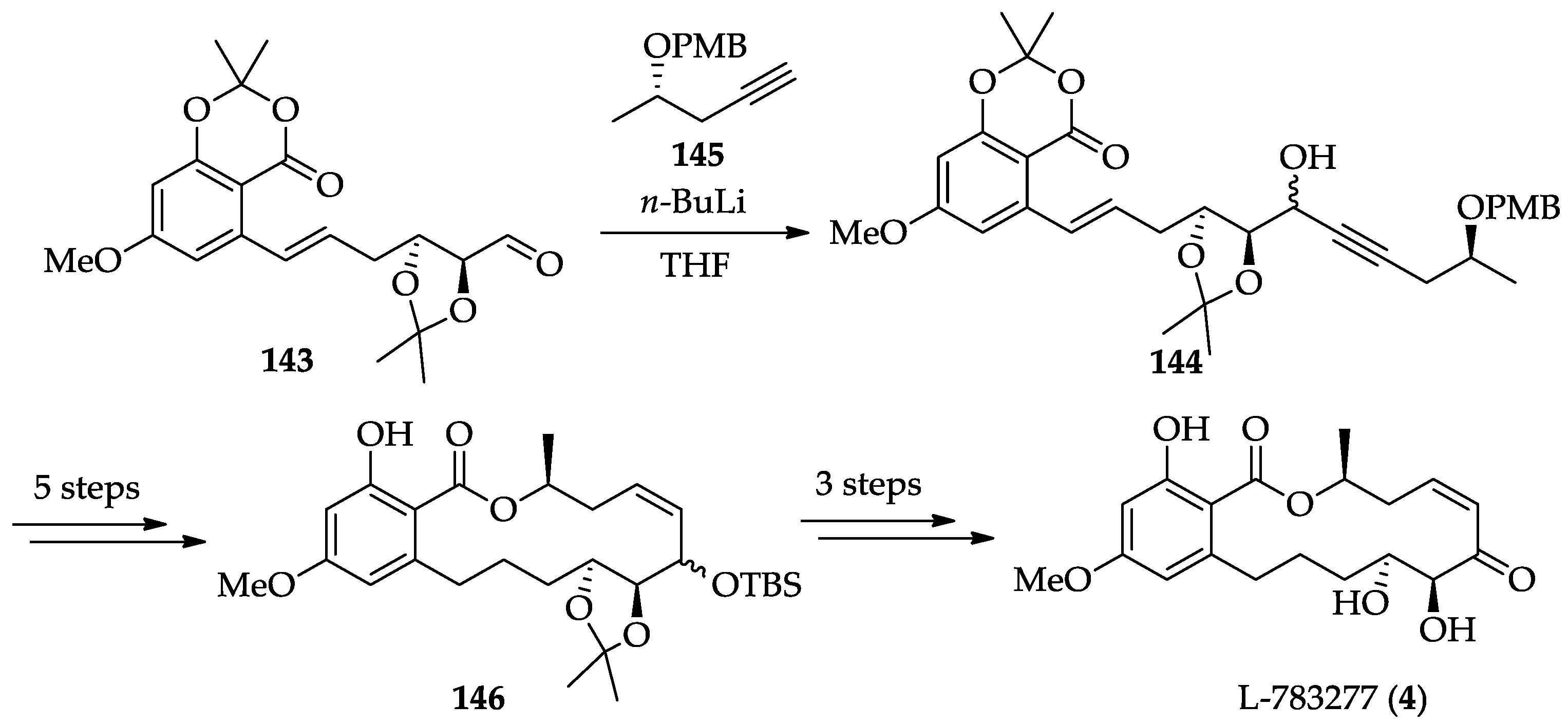
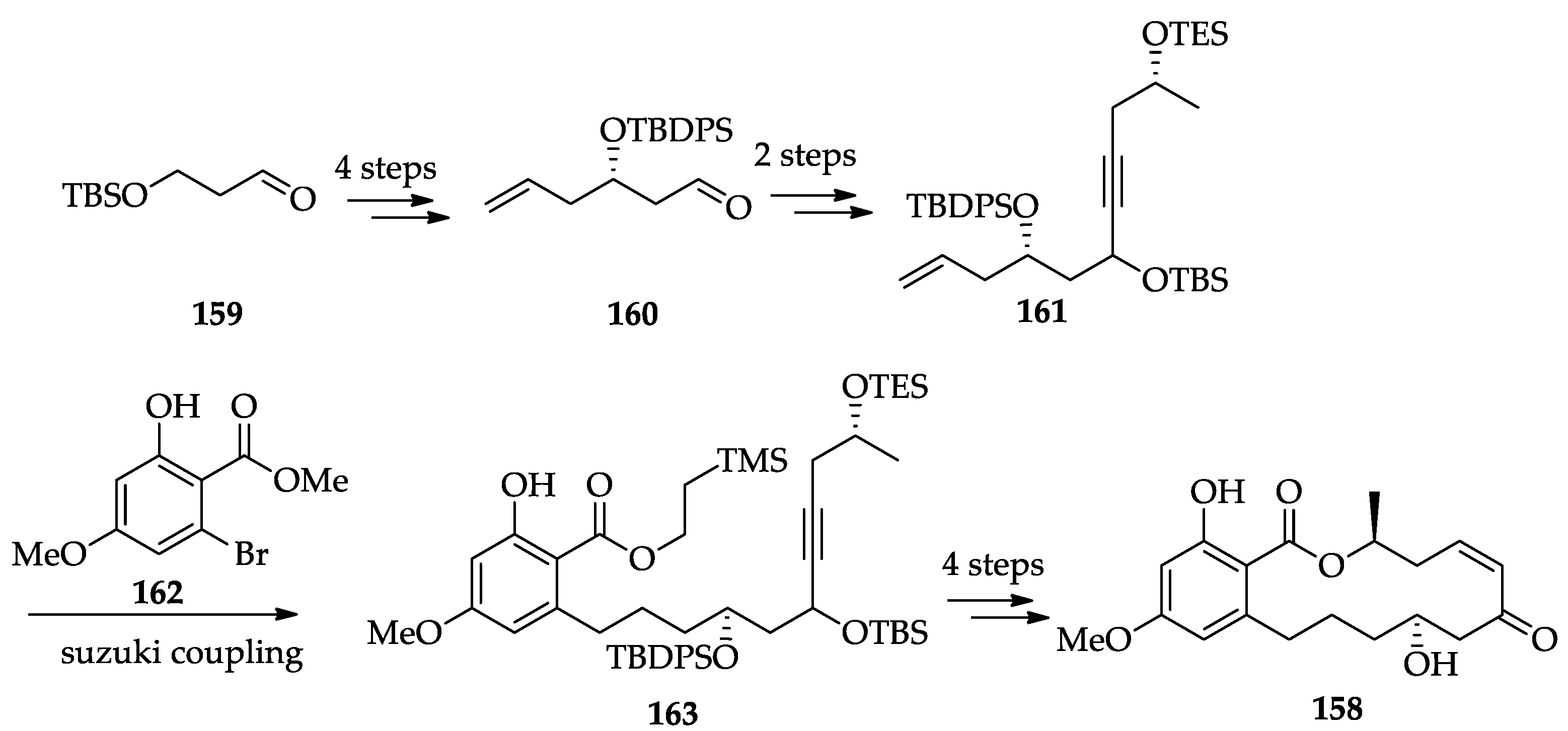
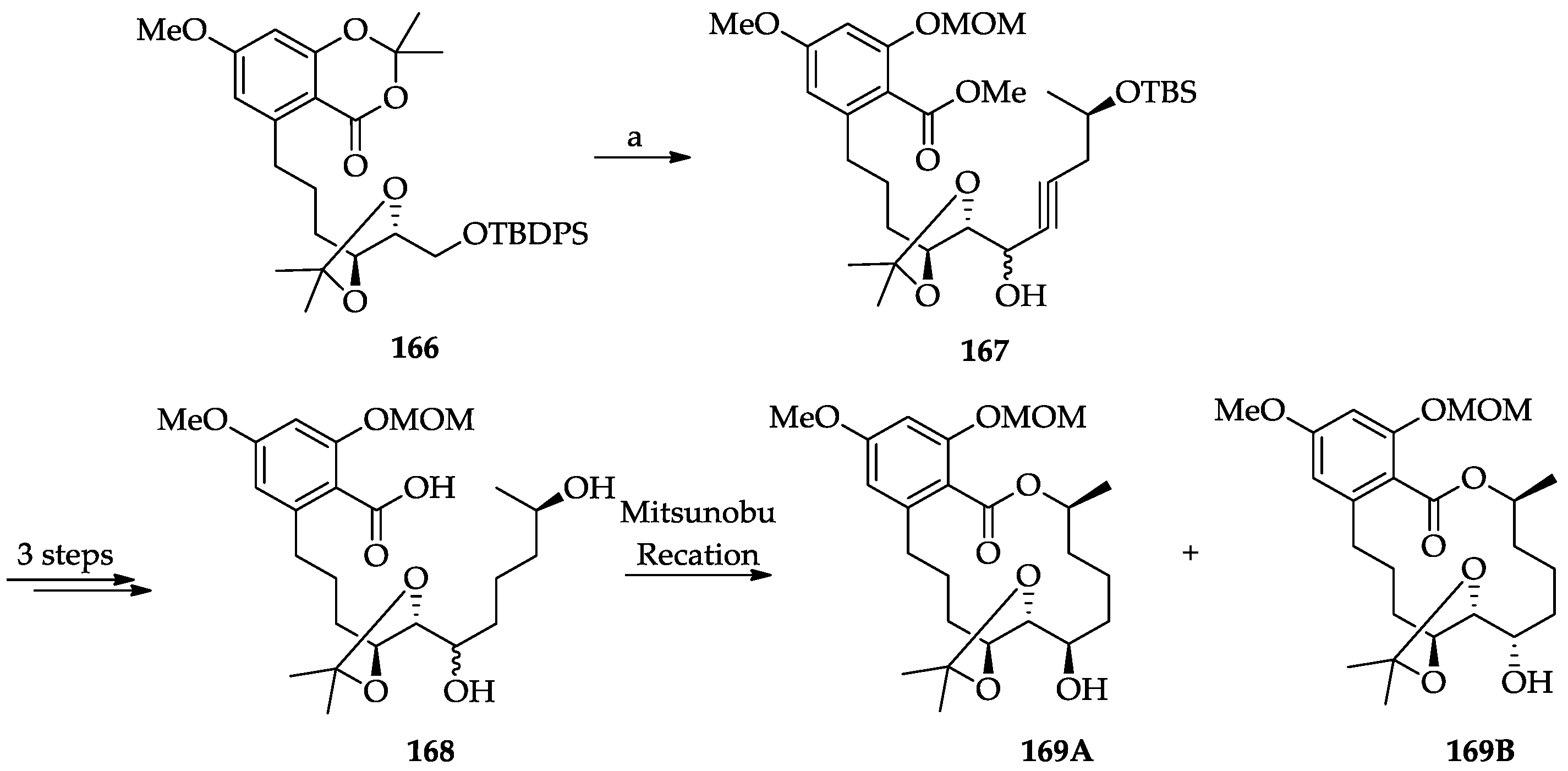
2.1.6. Chemistry of L-783277 (4)
2.2. Sesquiterpene Lactones
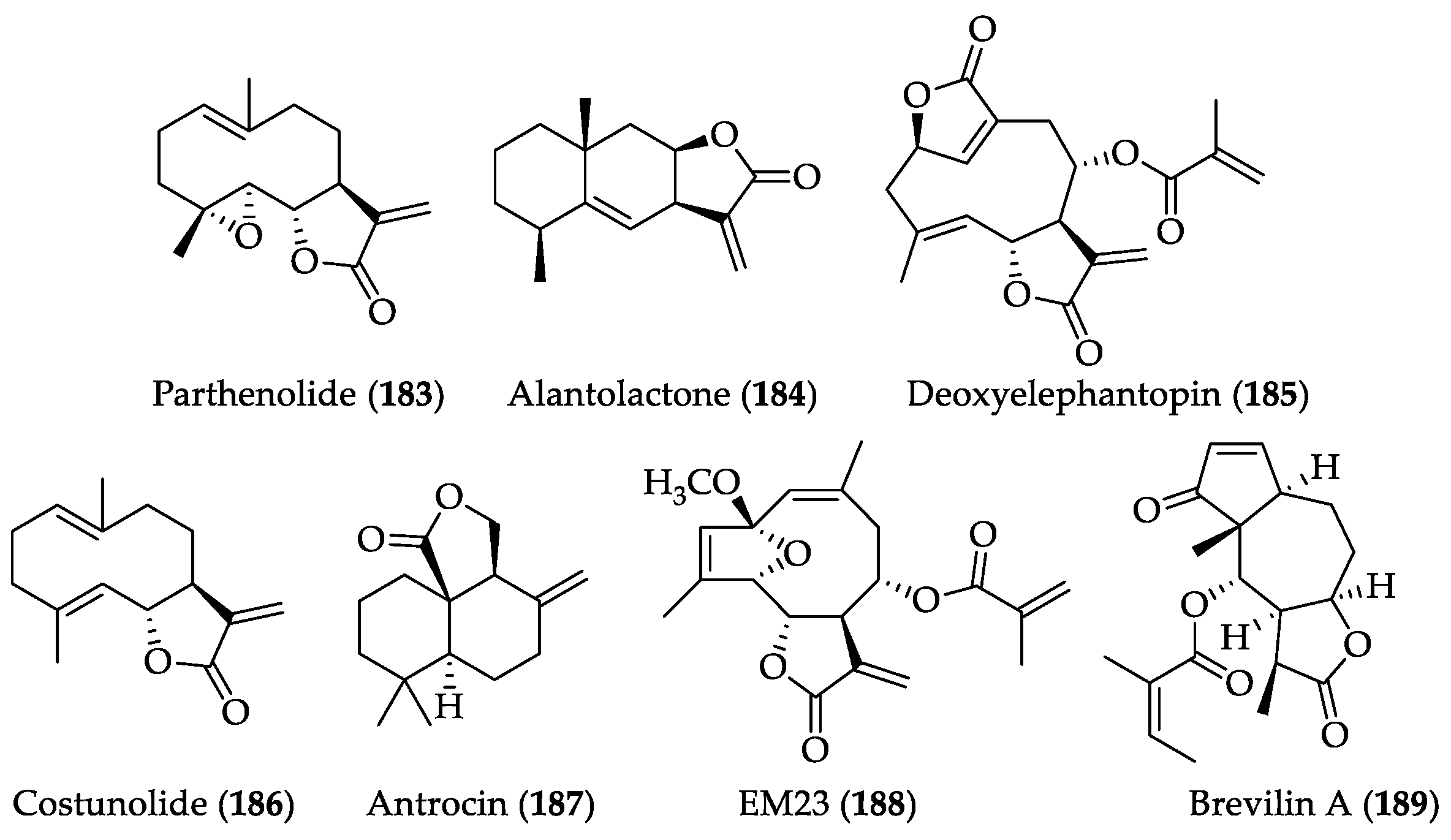
2.2.1. Biological Activities of Parthenolide (183)
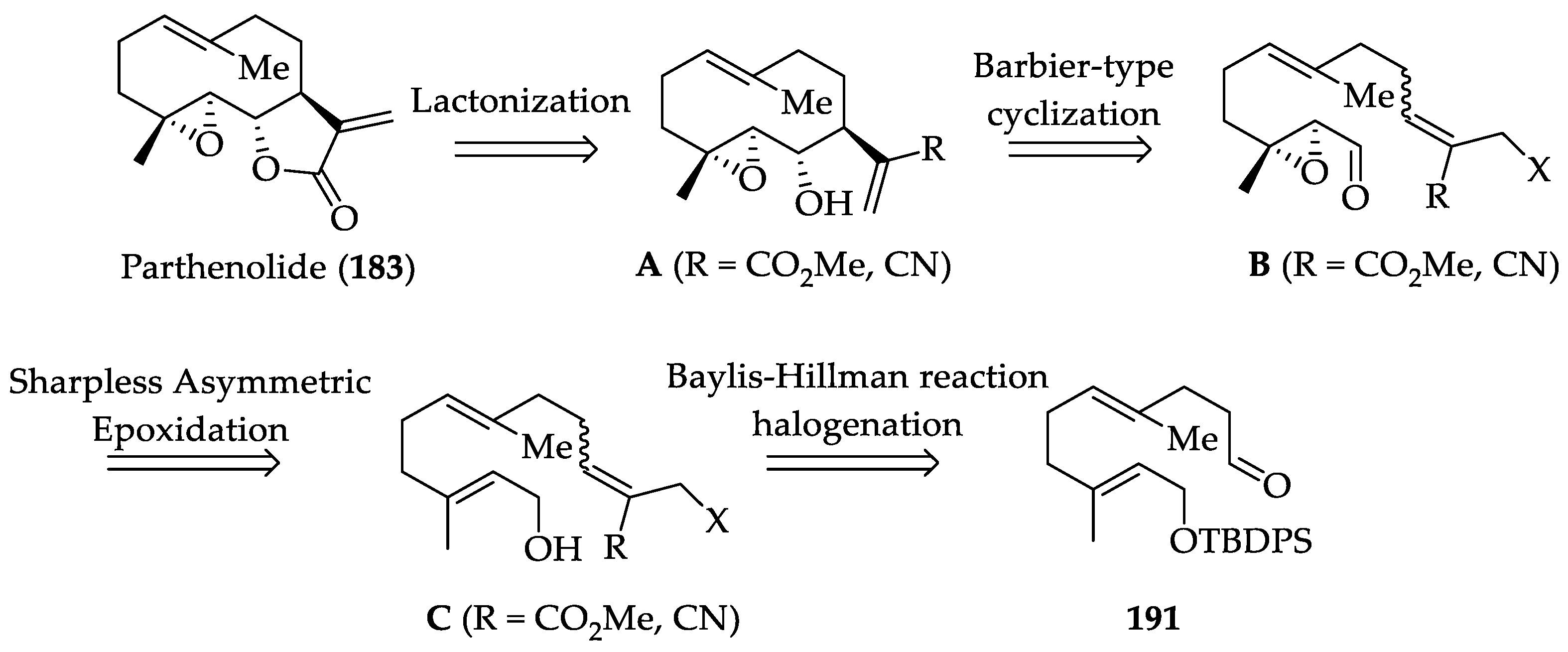
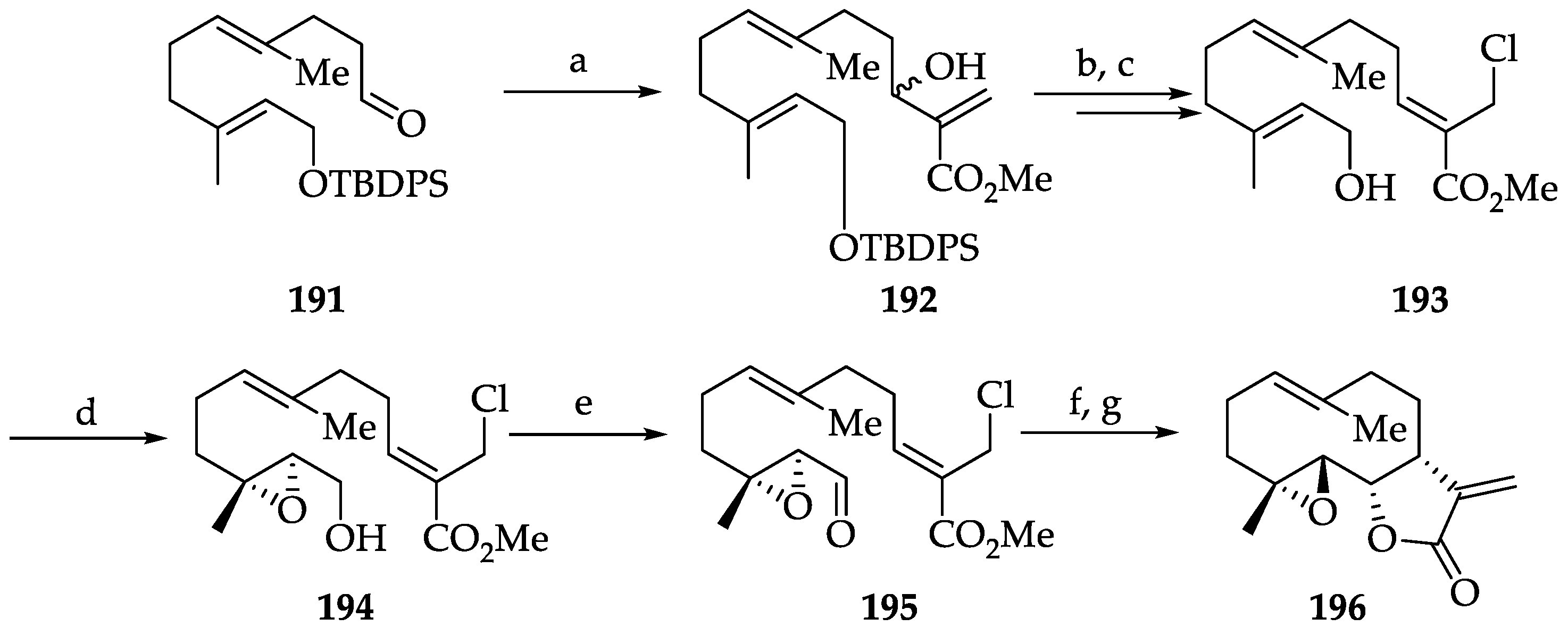
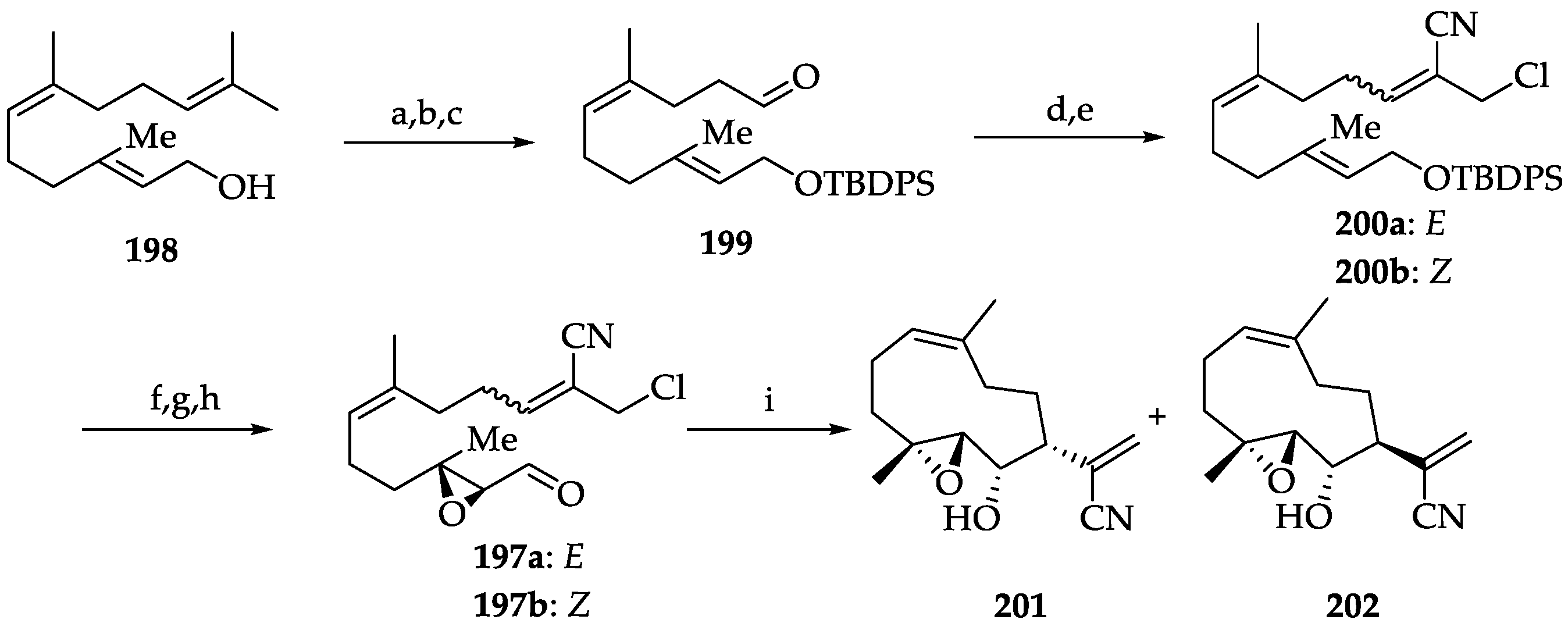
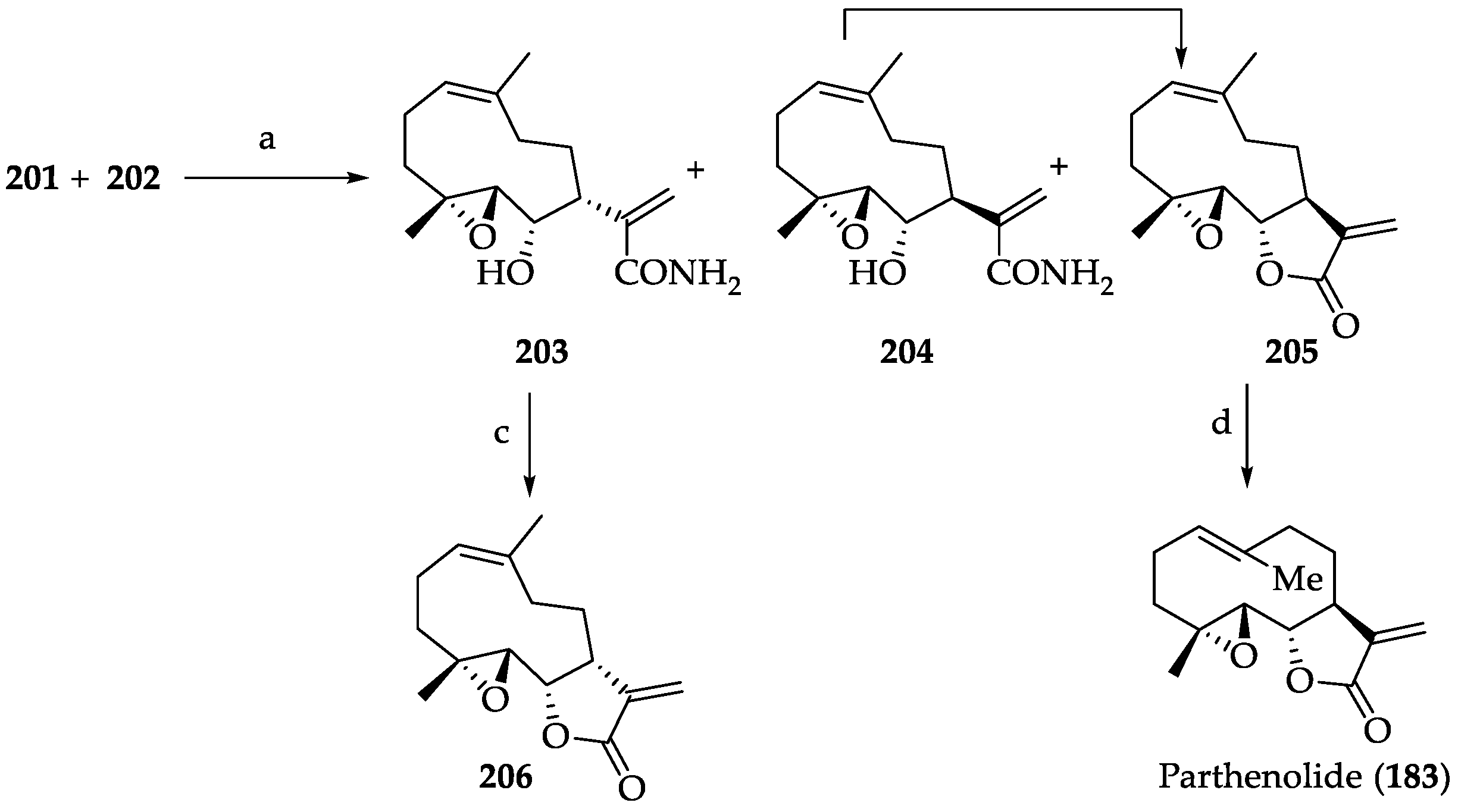
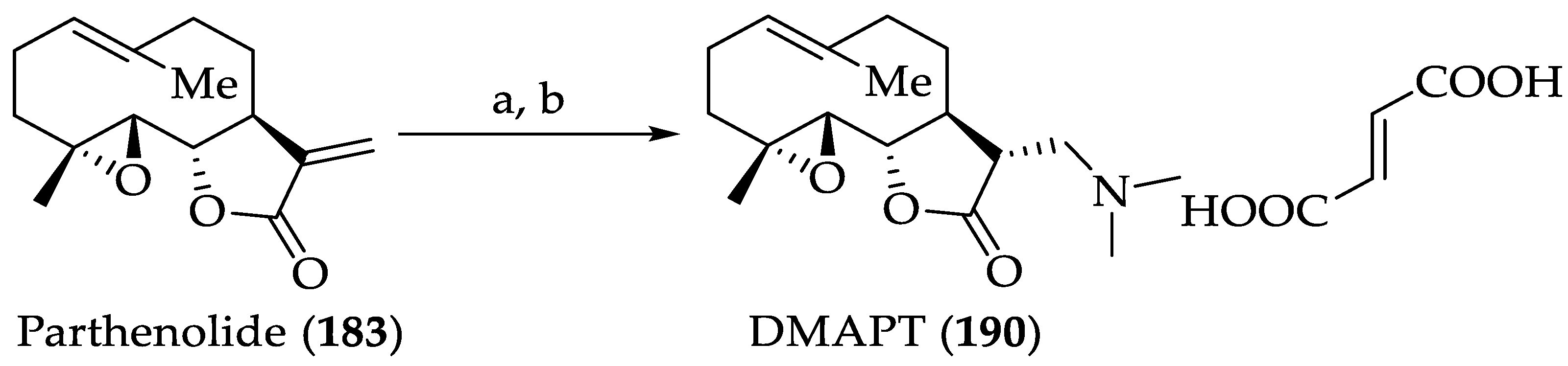
2.2.2. Chemistry of Parthenolide (183)
2.2.3. Biological Activities of Alantolactone (184)
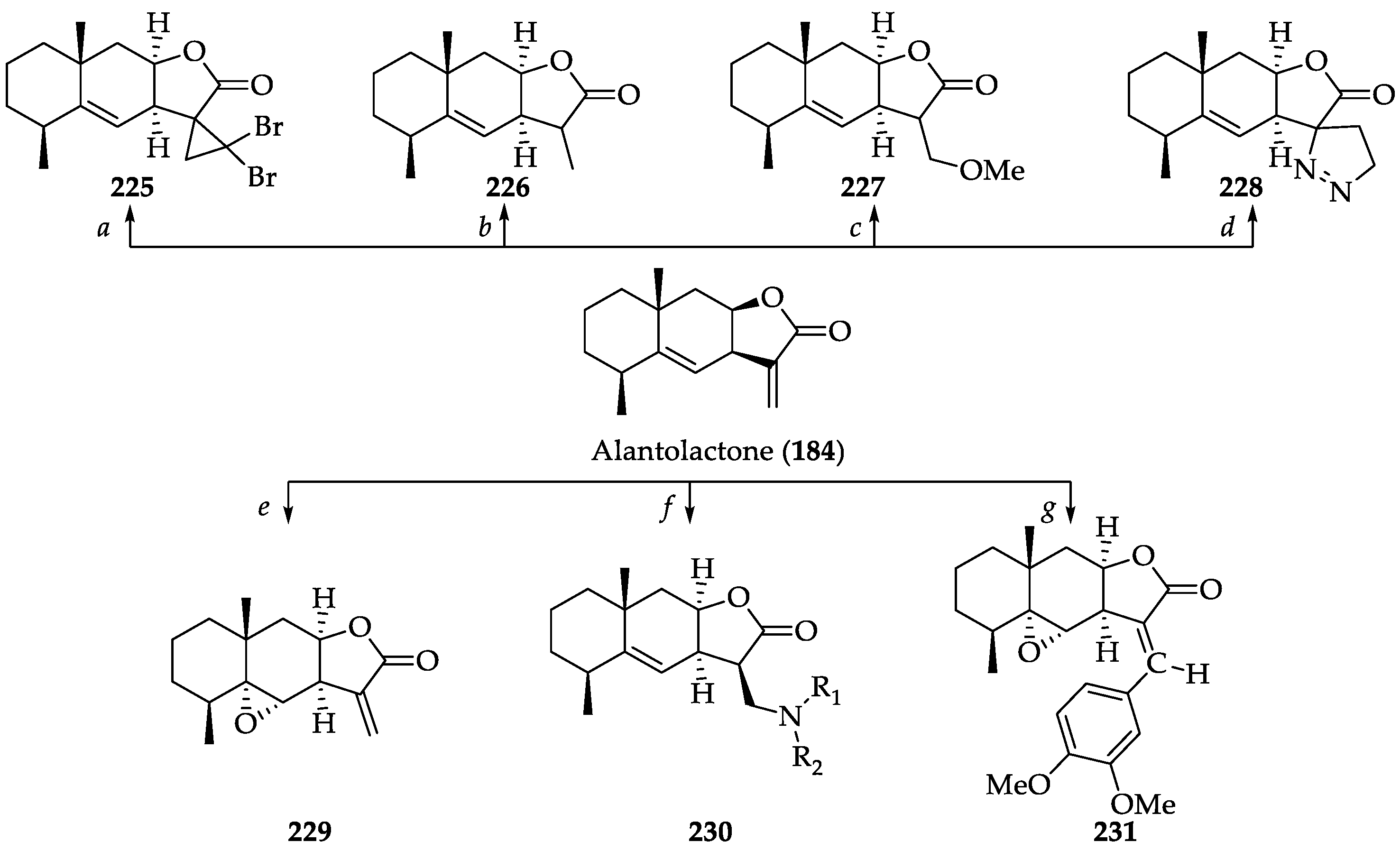
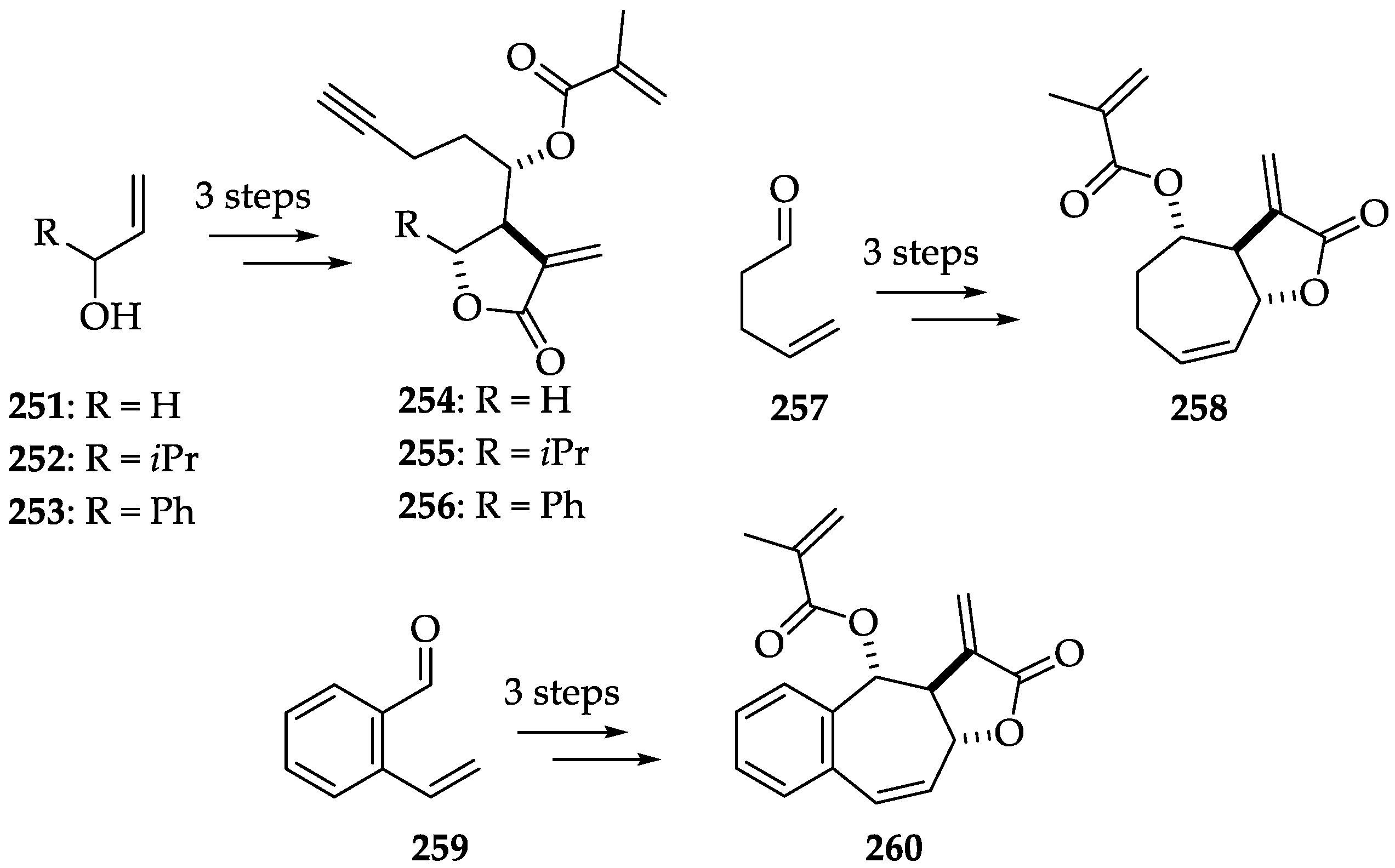
2.2.4. Chemistry of Alantolactone (184)
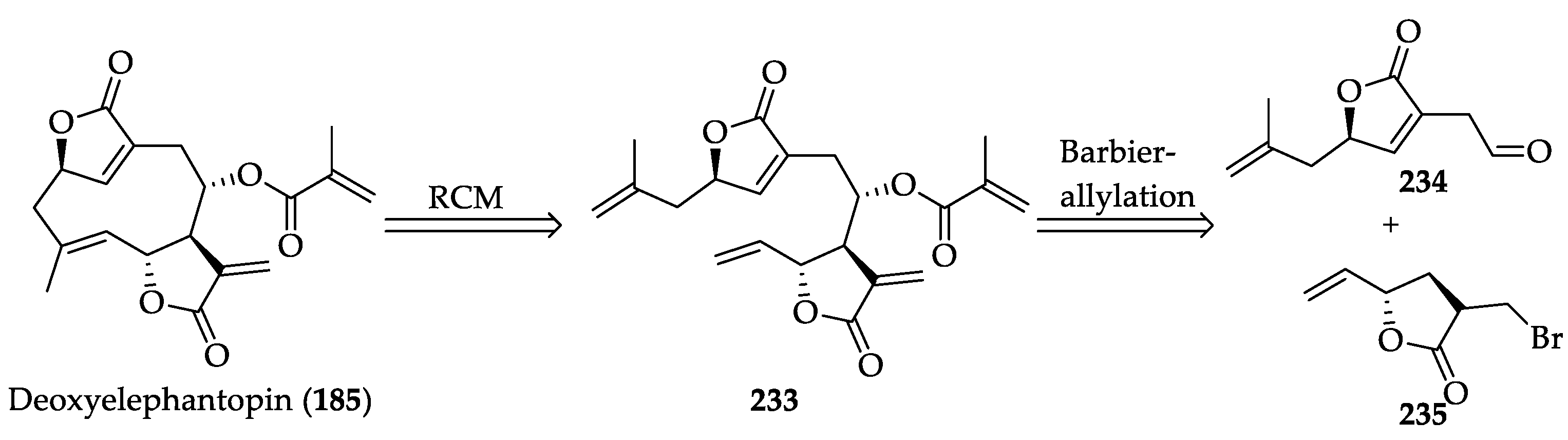
2.2.5. Biological Activities of Deoxyelephantopin (185)
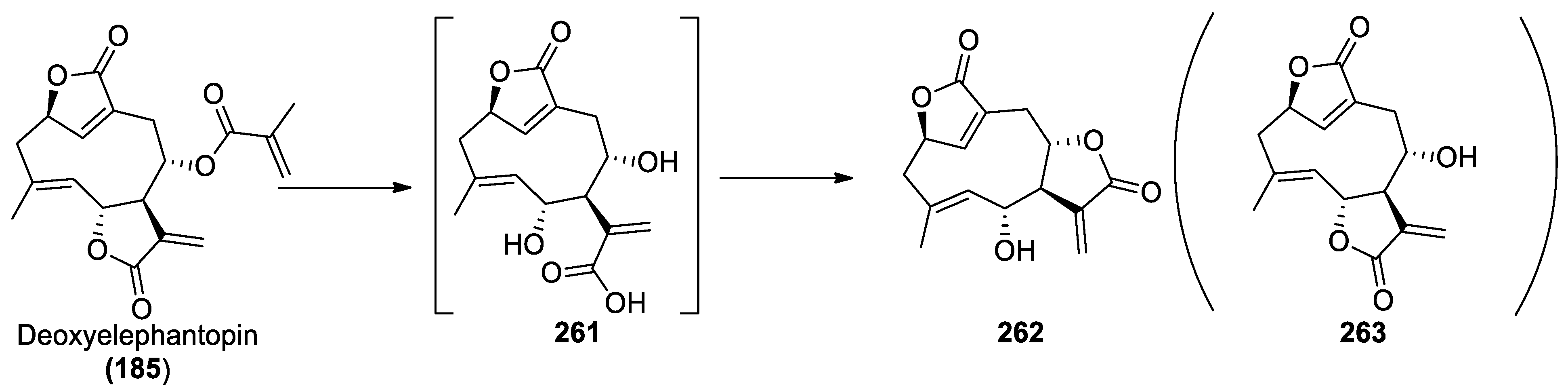
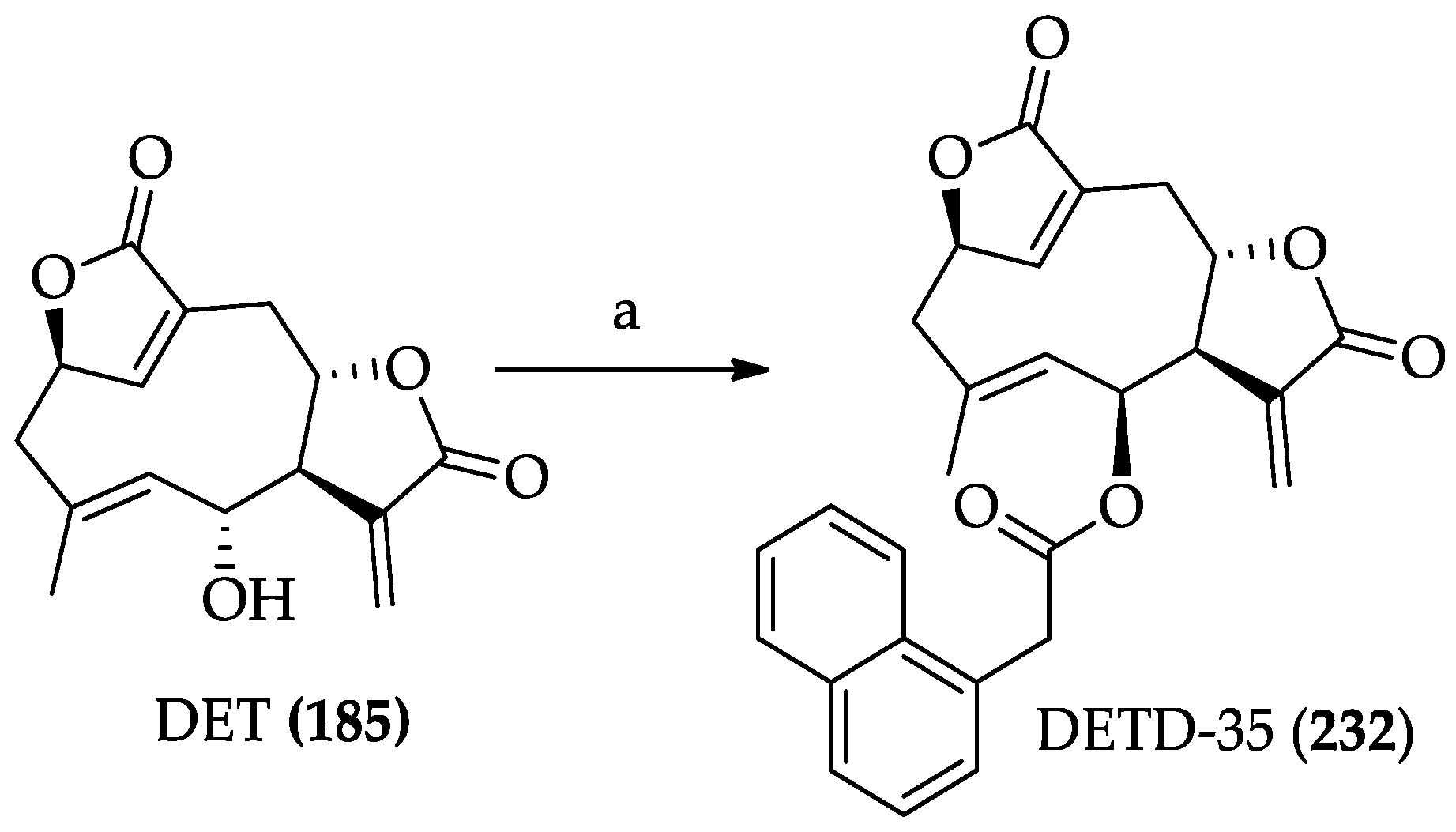
2.2.6. Chemistry of Deoxyelephantopin (185)
2.2.7. Biological Activities of Costunolide (186)
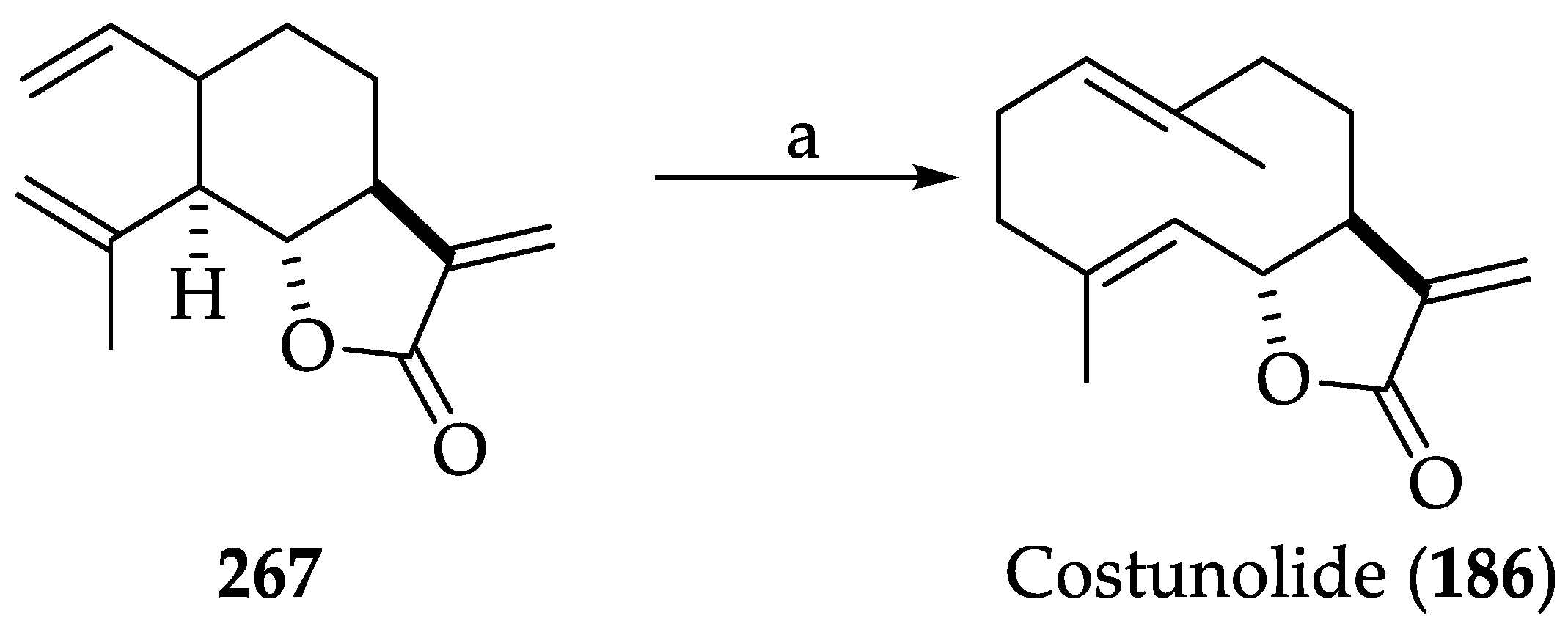
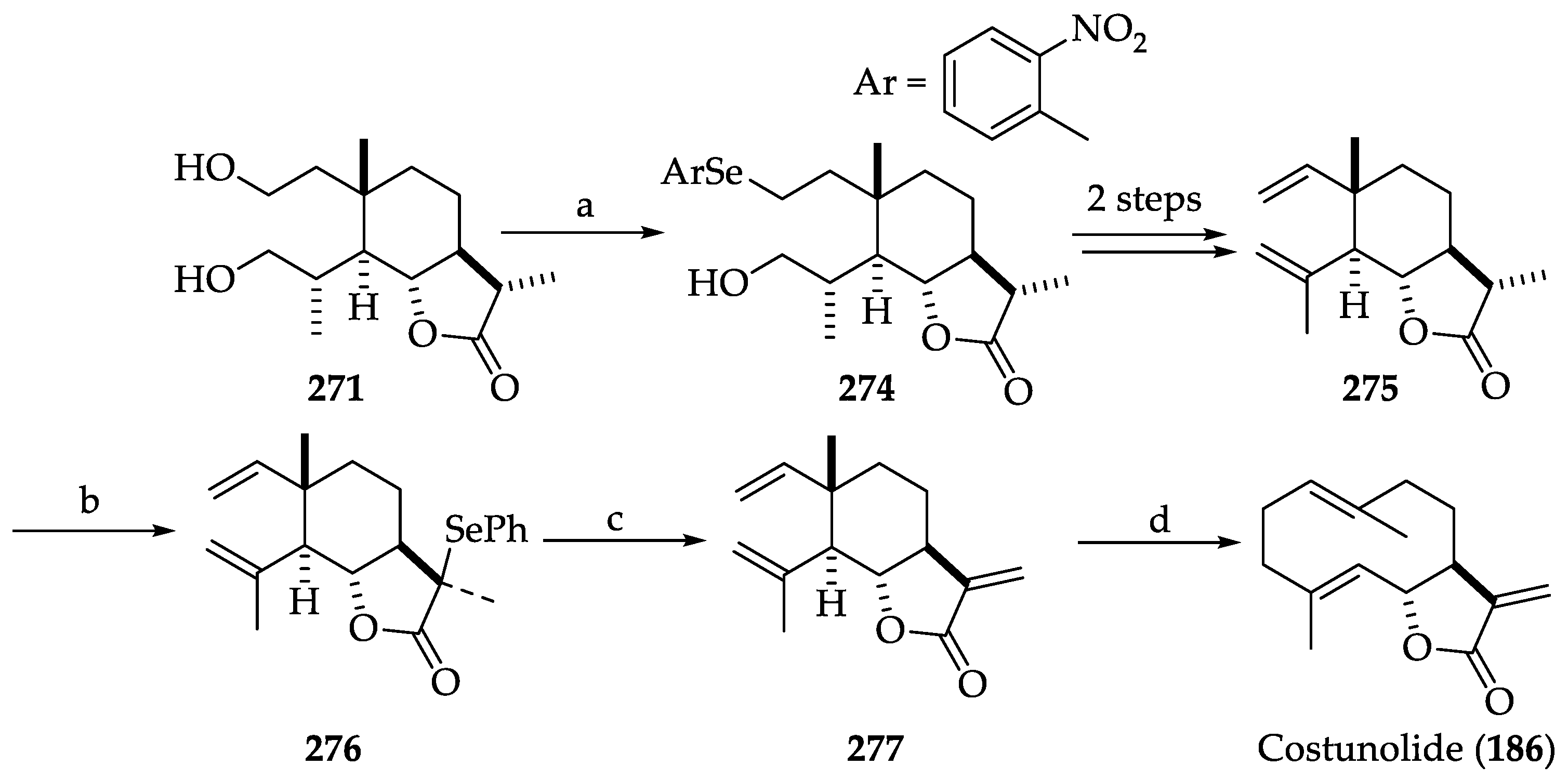
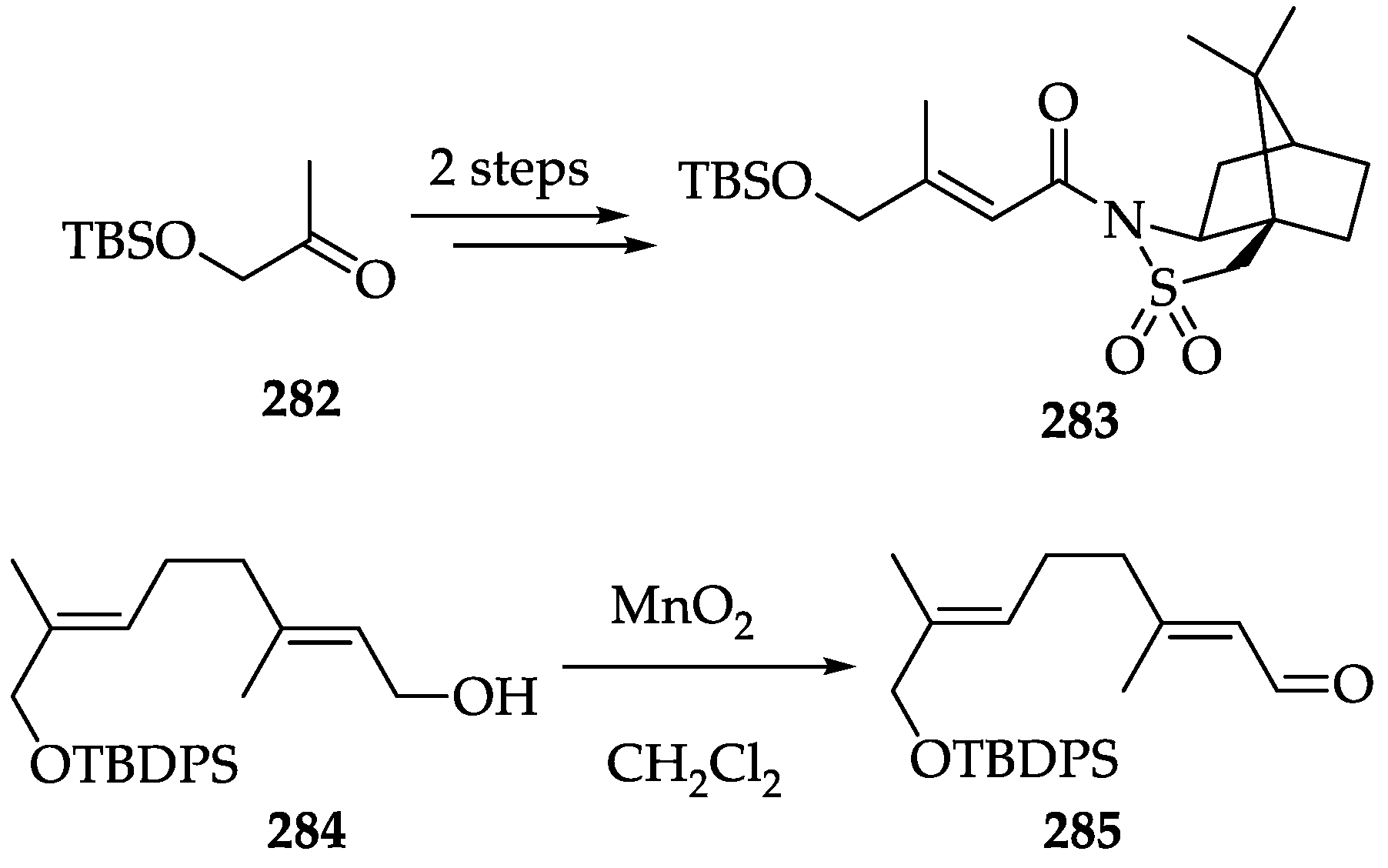
2.2.8. Chemistry of Costunolide (186)
2.2.9. Biological Activities of Antrocin (187)
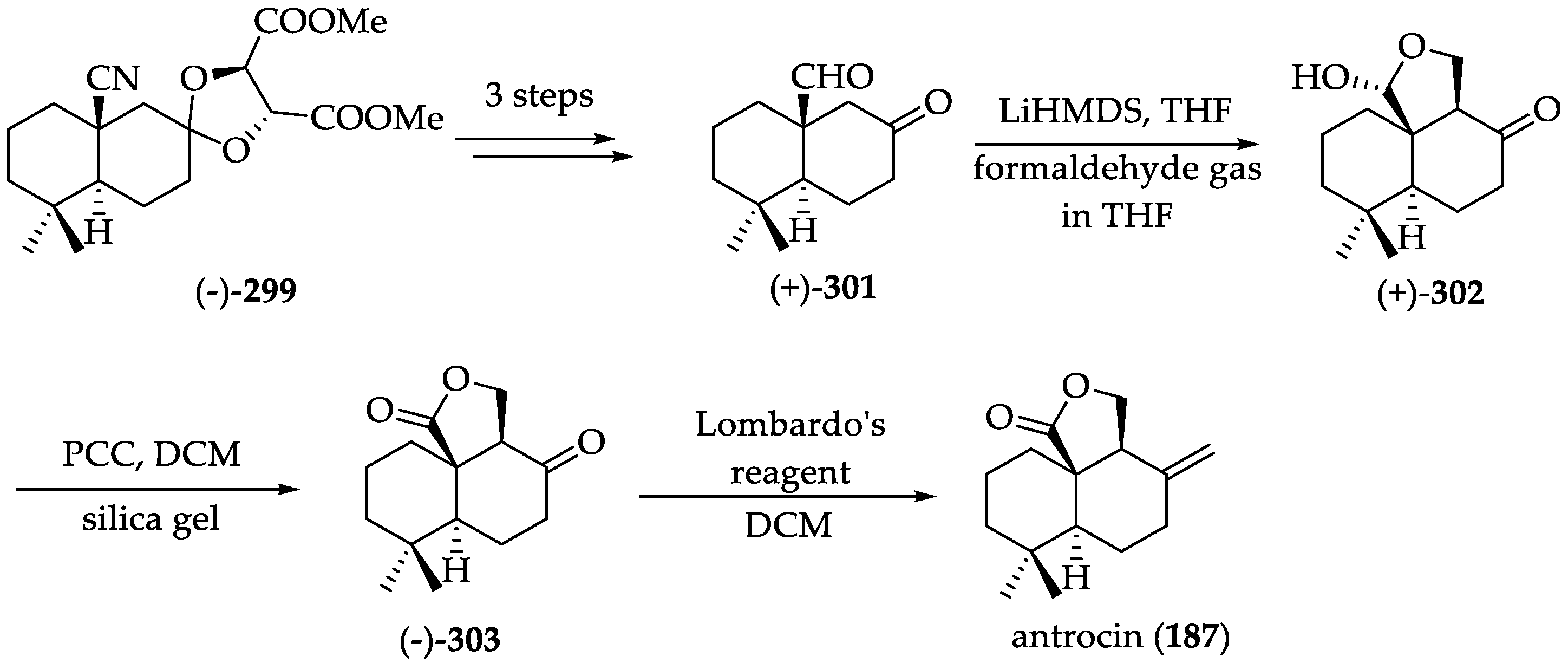
2.2.10. Chemistry of Antrocin (187)
2.2.11. Biological Activities of EM23 (188) and Brevilin A (189)
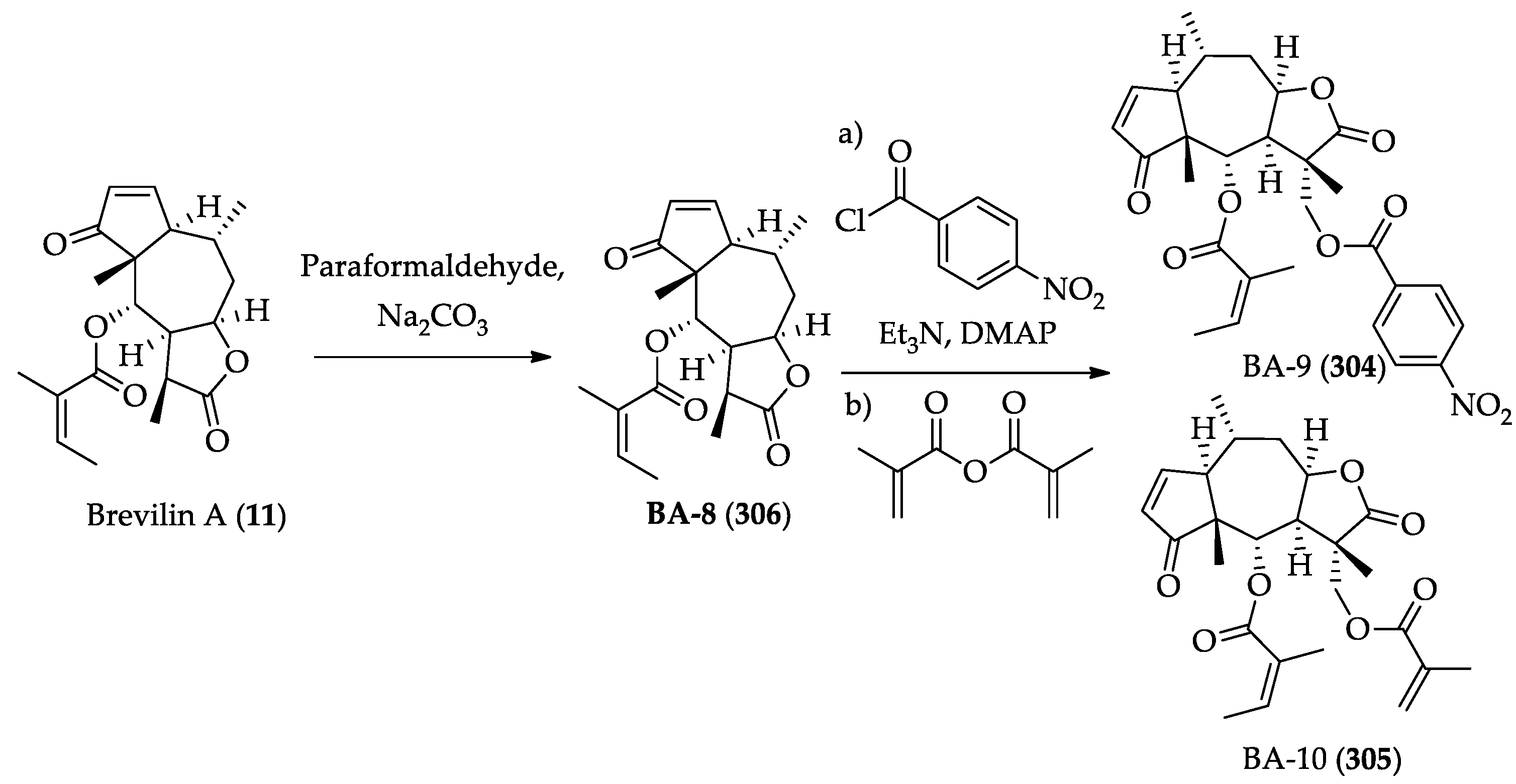
2.2.12. Chemistry of EM23 (188) and Brevilin A (189)
2.3. Diacylglycerol Lactones
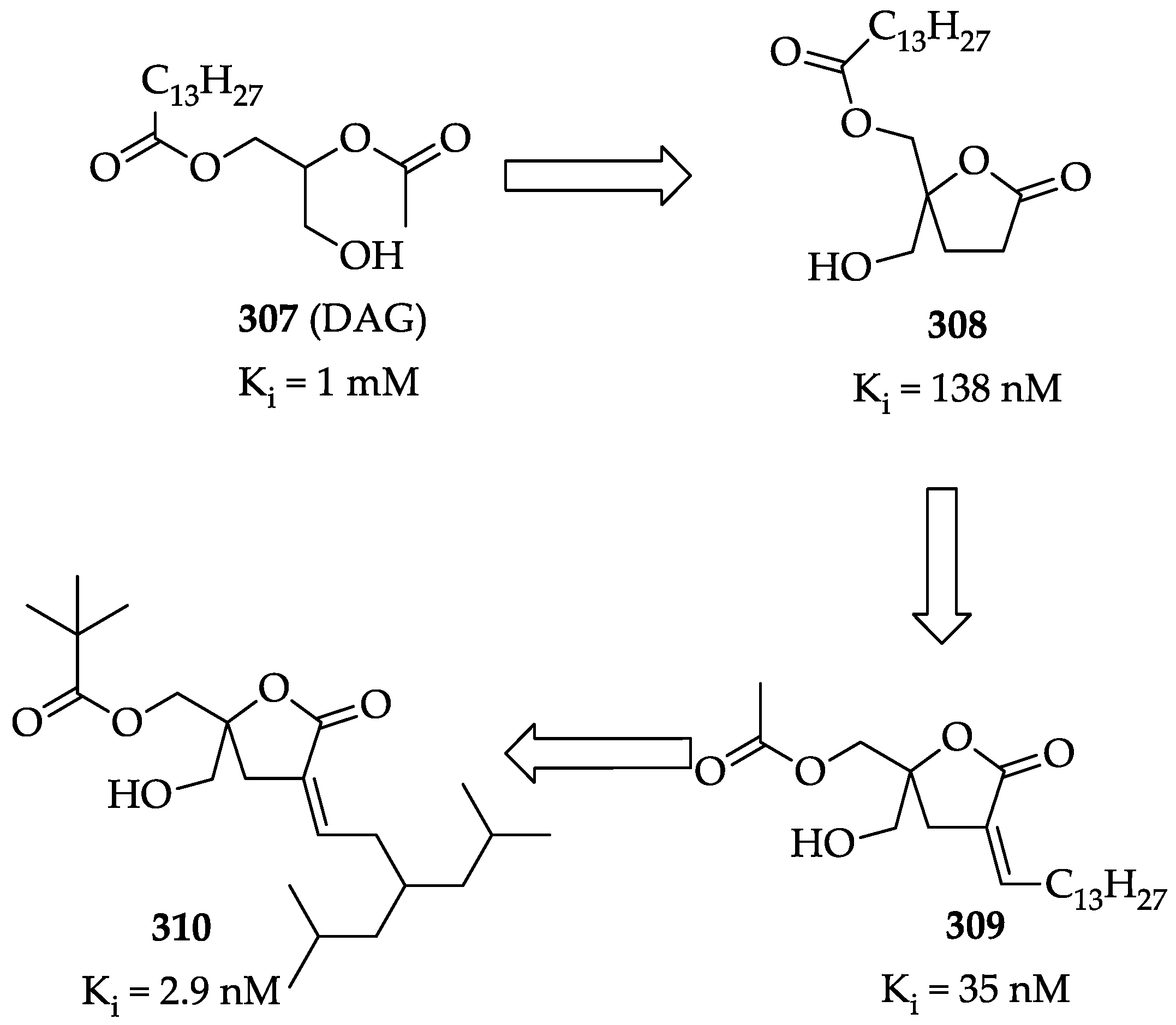
2.3.1. Biological Activities of Diacylglycerol Lactones
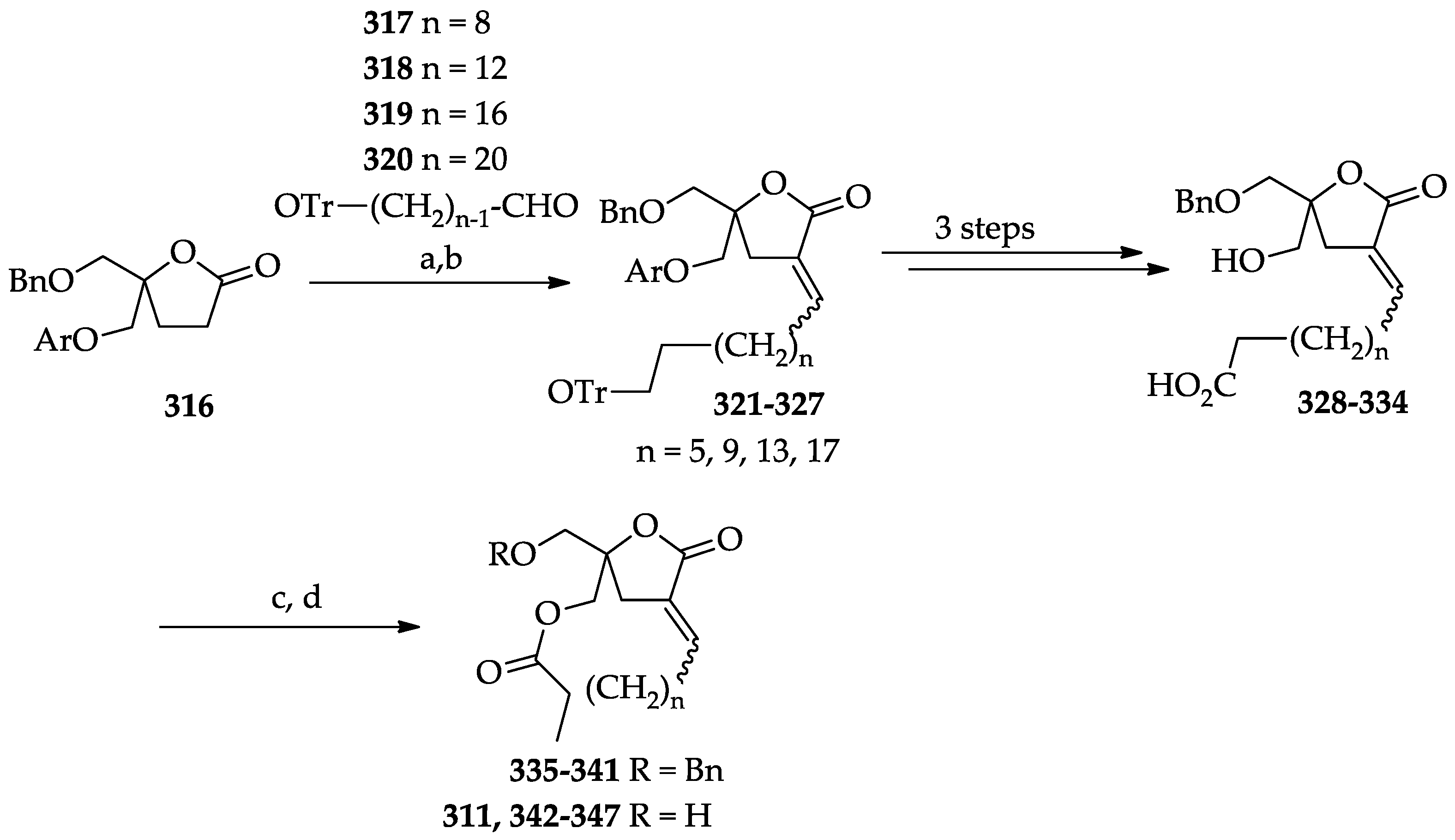
2.3.2. Chemistry of Diacylglycerol Lactones
2.4. Diterpene Lactones
2.4.1. Biological Activities of Andrographolide (373)
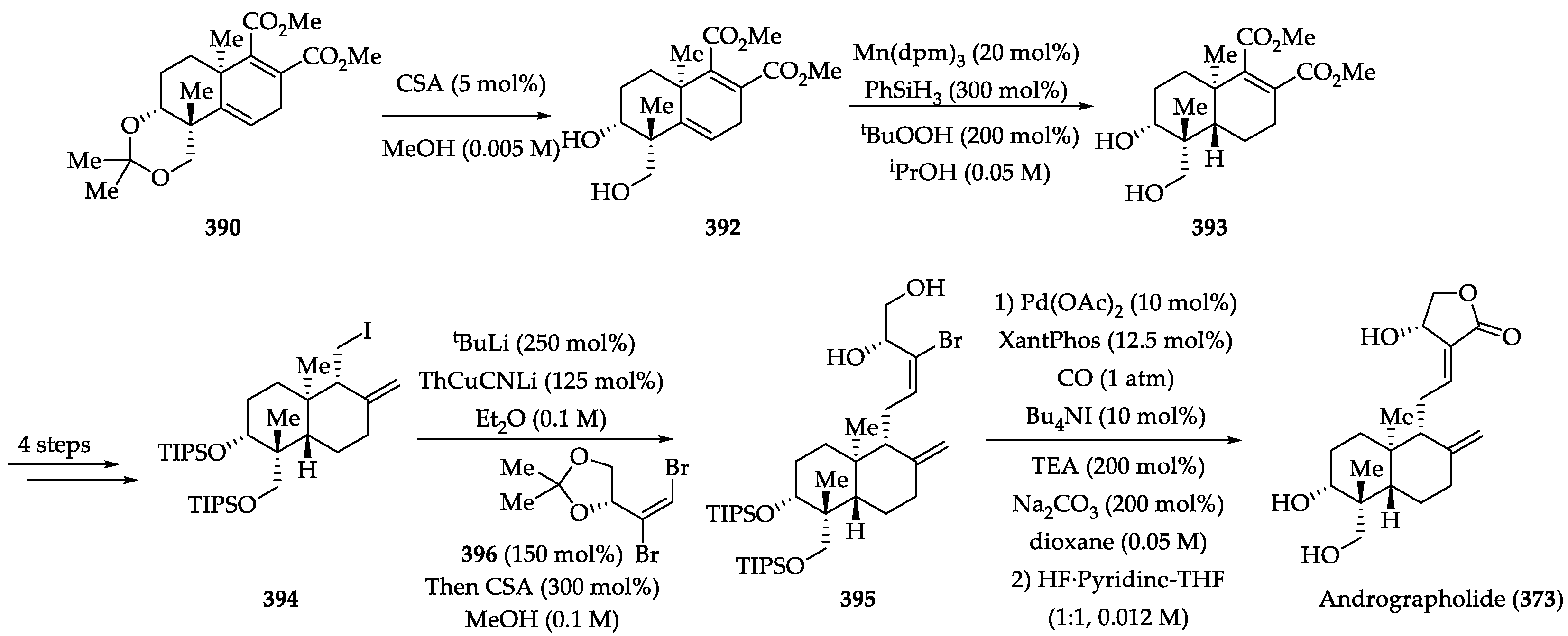
2.4.2. Chemistry of Andrographolide (373)
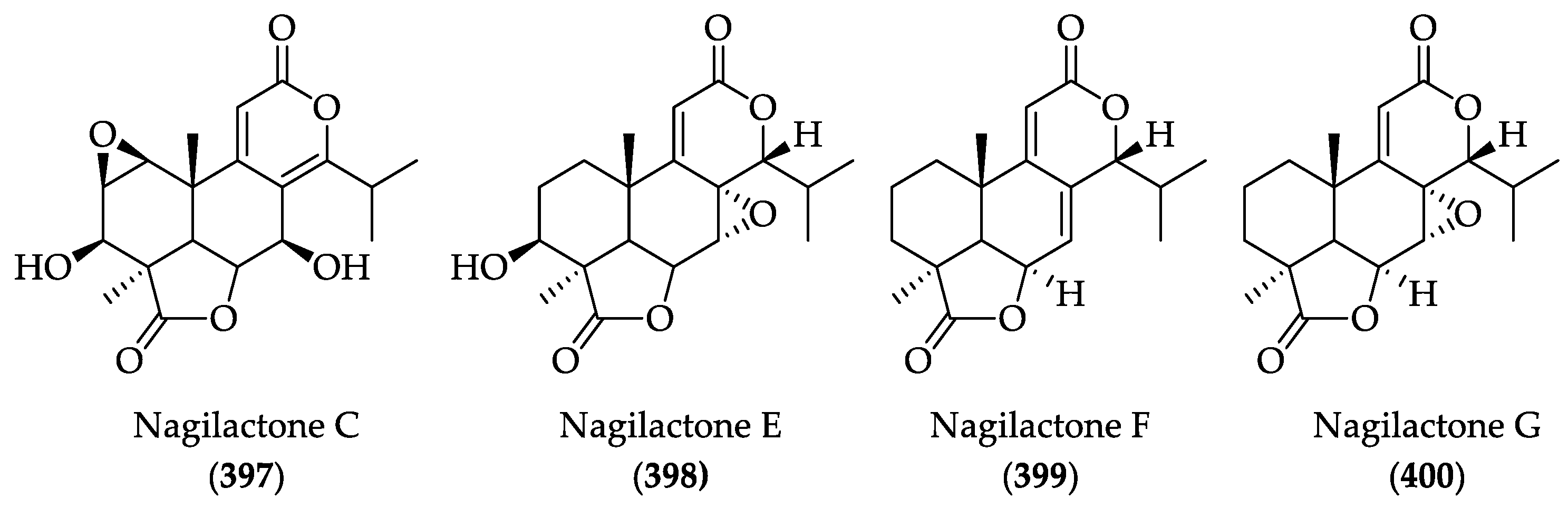
2.4.3. Anti-Cancer Activities of Nagilactones
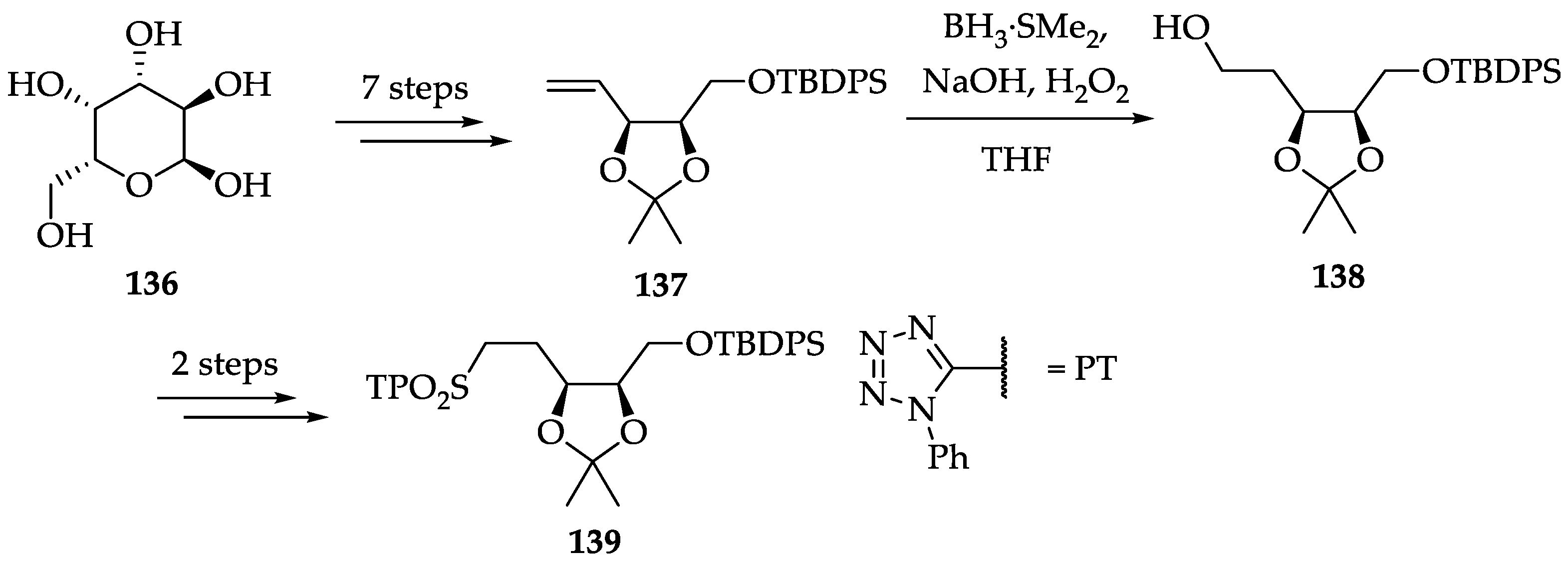
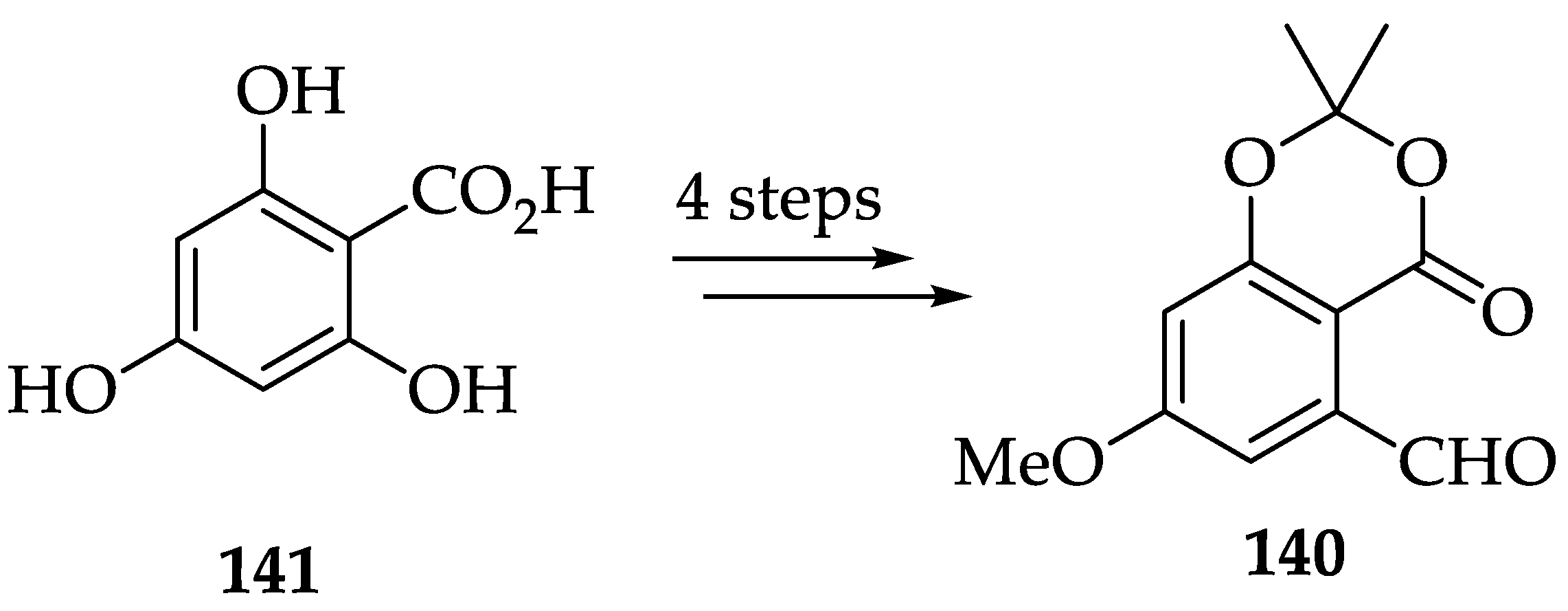
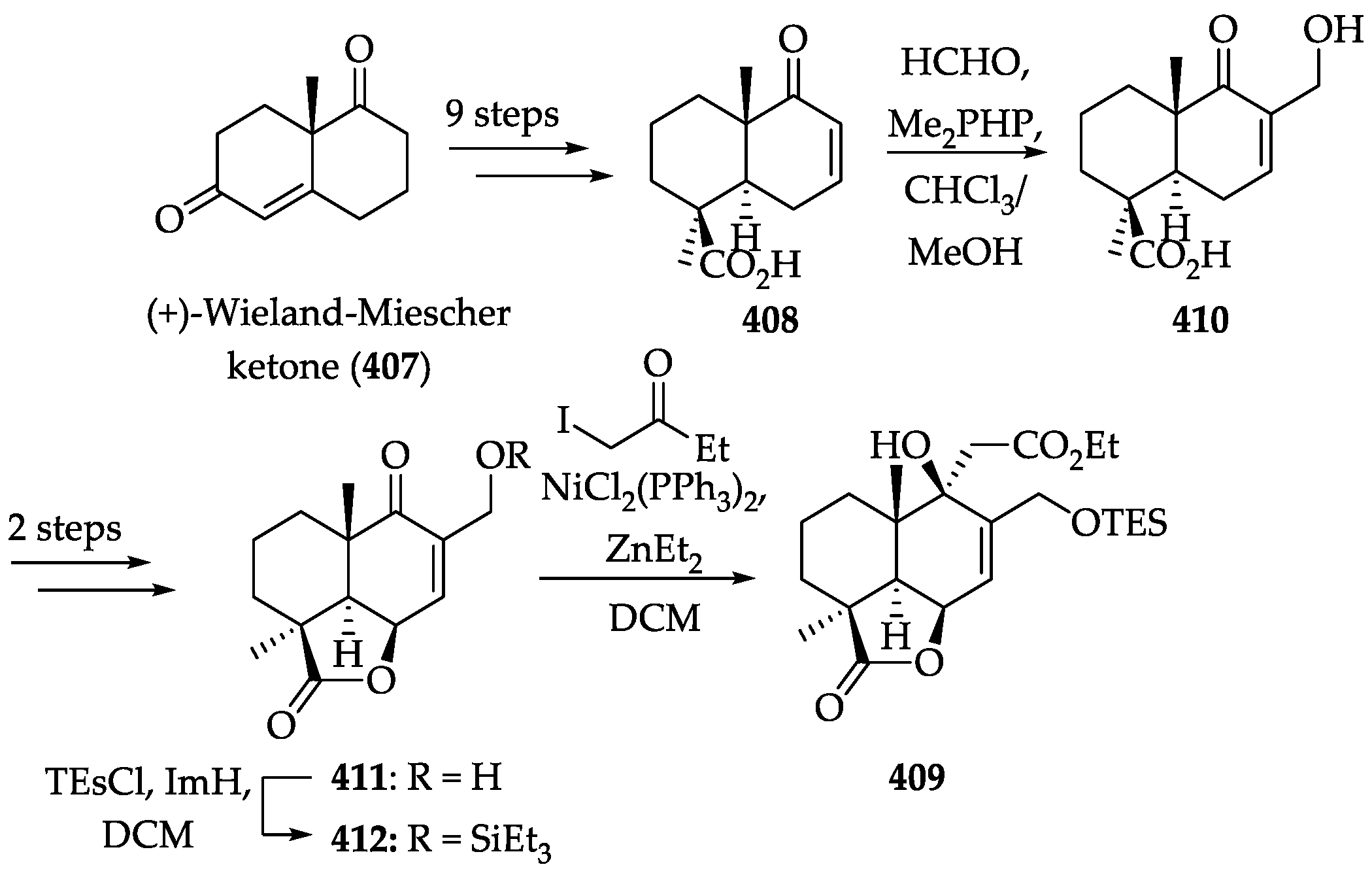
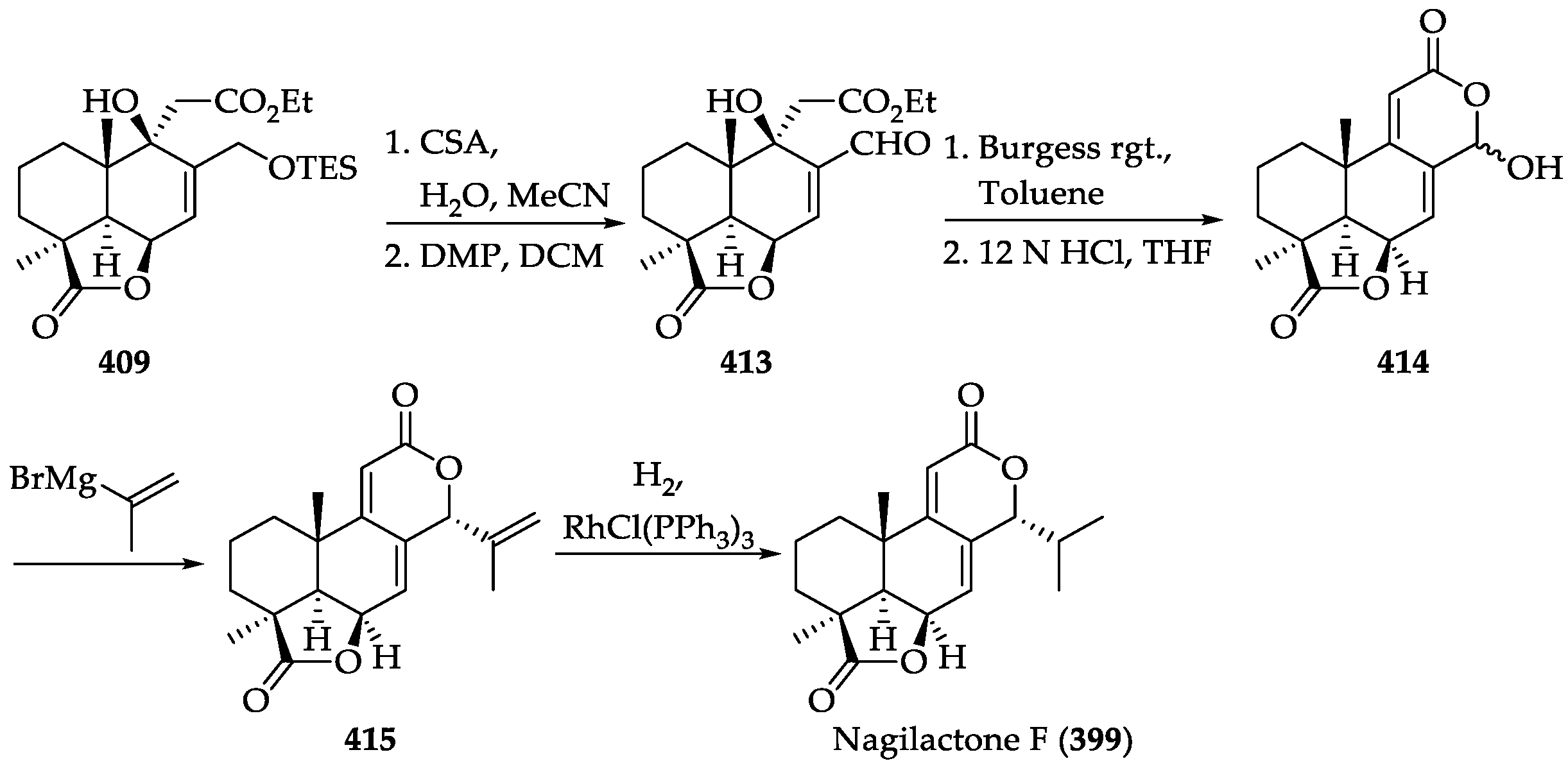
2.4.4. Chemistry of Nagilactones
3. Summary
4. Conclusions and Future Prospects
5. Materials and Methods
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangal, M.; Sagar, P.; Singh, H.; Raghava, G.P.; Agarwal, S.M. NPACT: Naturally occurring plant-based anti-cancer compound-activity-target database. Nucleic Acids Res. Spec. Publ. 2013, 41, D1124–D1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braicu, C.; Pileczki, V.; Irimie, A.; Berindan-Neagoe, I. p53siRNA therapy reduces cell proliferation, migration and induces apoptosis in triple negative breast cancer cells. Mol. Cell. Biochem. 2013, 381, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Pentassuglia, L.; Sawyer, D.B. Emerging anticancer therapeutic targets and the cardiovascular system: Is there cause for concern? Circ. Res. 2010, 106, 1022–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, Y.; de Blanco, E.J.C.; Fuchs, J.R.; Soejarto, D.D.; Burdette, J.E.; Swanson, S.M.; Kinghorn, A.D. Potential anticancer agents characterized from selected tropical plants. J. Nat. Prod. 2019, 82, 657–679. [Google Scholar] [CrossRef]
- Chamberlin, S.R.; Blucher, A.; Wu, G.; Shinto, L.; Choonoo, G.; Kulesz-Martin, M.; McWeeney, S. Natural product target network reveals potential for cancer combination therapies. Front. Pharmacol. 2019, 10, 557. [Google Scholar] [CrossRef]
- Ren, Y.; Yu, J.; Douglas Kinghorn, A. Development of anticancer agents from plant-derived sesquiterpene lactones. Curr. Med. 2016, 23, 2397–2420. [Google Scholar] [CrossRef]
- Aliarab, A.; Abroon, S.; Rasmi, Y.; Aziz, S.G.-G. Application of sesquiterpene lactone: A new promising way for cancer therapy based on anticancer activity. Biomed. Pharmacother. 2018, 106, 239–246. [Google Scholar]
- Patocka, J.; Soukup, O.; Kuca, K. Resorcylic acid lactones as the protein kinase inhibitors, naturally occuring toxins. Mini-Rev. Med. Chem. 2013, 13, 1873–1878. [Google Scholar] [CrossRef]
- Delmotte, P.; Delmotte-Plaque, J. A new antifungal substance of fungal origin. Nature 1953, 171, 344. [Google Scholar] [CrossRef]
- Zhao, A.; Lee, S.H.; Mojena, M.; Jenkins, R.G.; Patrick, D.R.; Huber, H.E.; Goetz, M.A.; Hensens, O.D.; Zink, D.L.; Vilella, D.; et al. Resorcylic acid lactones: Naturally occurring potent and selective inhibitors of MEK. J. Antibiot. 1999, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winssinger, N.; Barluenga, S. Chemistry and biology of resorcylic acid lactones. Chem. Commun. 2007, 22–36. [Google Scholar] [CrossRef] [PubMed]
- Moujir, L.; Callies, O.; Sousa, P.M.C.; Sharopov, F.; Seca, A.M.L. Applications of Sesquiterpene Lactones: A Review of Some Potential Success Cases. Appl. Sci. 2020, 10, 3001. [Google Scholar] [CrossRef]
- Victor, E.; Marquez, A.; Blumberg, P.M. Synthetic Diacylglycerols (DAG) and DAG-Lactones as Activators of Protein Kinase C (PK-C). Acc. Chem. Res. 2003, 36, 434–443. [Google Scholar]
- Dai, Y.; Chen, S.-R.; Chai, L.; Zhao, J.; Wang, Y.; Wang, Y. Overview of pharmacological activities of Andrographis paniculata and its major compound andrographolide. Crit. Rev. Food Sci. 2019, 59, S17–S29. [Google Scholar] [CrossRef]
- Bailly, C. Anticancer Activities and Mechanism of Action of Nagilactones, a Group of Terpenoid Lactones Isolated from Podocarpus Species. Nat. Prod. Bioprospect. 2020, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Schulz, S.; Hotling, S. The use of the lactone motif in chemical communication. Nat. Prod. Rep. 2015, 32, 1042–1066. [Google Scholar] [CrossRef] [Green Version]
- Dakas, P.-Y. Divergent synthesis of resorcyclic acid lactones: A privileged natural pharmacophore. Chemistry 2009, 43, 11490–11497. [Google Scholar] [CrossRef] [PubMed]
- Lagoutte, R.; Serba, C.; Winssinger, N. Synthesis of deoxyelephantopin analogues. J. Antibiot. 2018, 71, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Barluenga, S.; Wang, C.; Fontaine, J.G.; Aouadi, K.; Beebe, K.; Tsutsumi, S.; Neckers, L.; Winssinger, N. Divergent synthesis of a pochonin library targeting HSP90 and in vivo efficacy of an identified inhibitor. Angew. Chem. Int. Ed. Engl. 2008, 47, 4432–4435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, T.; Altmann, K.-H. Resorcylic acid lactones as new lead structures for kinase inhibition. C. R. Chim. 2008, 11, 1318–1335. [Google Scholar] [CrossRef]
- Schulte, T.W.; Akinaga, S.; Soga, S.; Sullivan, W.; Stensgard, B.; Toft, D.; Neckers, L.M. Antibiotic radicicol binds to the N-terminal domain of Hsp90 and shares important biologic activities with geldanamycin. Cell Stress Chaperones 1998, 3, 100–108. [Google Scholar] [CrossRef] [Green Version]
- Kwon, H.J.; Yoshida, M.; Abe, K.; Horinouchi, S.; Beppu, T. Radicicol, an agent inducing the reversal of transformed phenotypes of src-transformed fibroblasts. Biosci. Biotechnol. Biochem. 1992, 56, 538–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharp, S.Y.; Jones, K.; Workman, P. Chapter 13—HSP90 inhibitors: Targeting the cancer chaperone for combinatorial blockade of oncogenic pathways. In Cancer Drug Design and Discovery; Neidle, S., Ed.; Academic Press: New York, NY, USA, 2008; pp. 305–335. [Google Scholar]
- Lampilas, M.; Lett, R. Convergent stereospecific total synthesis of monochiral Monocillin I related macrolides. Tetrahedron Lett. 1992, 33, 773–776. [Google Scholar] [CrossRef]
- Garbaccio, R.M.; Danishefsky, S.J. Efficient asymmetric synthesis of radicicol dimethyl ether: A novel application of ring-forming olefin metathesis. Org. Lett. 2000, 2, 3127–3129. [Google Scholar] [CrossRef]
- Garbaccio, R.M.; Stachel, S.J.; Baeschlin, D.K.; Danishefsky, S.J. Concise asymmetric syntheses of radicicol and monocillin I. J. Am. Chem. Soc. 2001, 123, 10903–10908. [Google Scholar] [CrossRef]
- Barluenga, S.; Moulin, E.; Lopez, P.; Winssinger, N. Solution-and solid-phase synthesis of radicicol (monorden) and pochonin C. Chem. Eur. J. 2005, 11, 4935–4952. [Google Scholar] [CrossRef]
- Yang, Z.-Q.; Danishefsky, S.J. A concise route to benzofused macrolactones via ynolides: Cycloproparadicicol. J. Am. Chem. Soc. 2003, 125, 9602–9603. [Google Scholar] [CrossRef]
- Kitson, R.R.; Moody, C.J. Learning from nature: Advances in geldanamycin-and radicicol-based inhibitors of Hsp90. J. Org. Chem. 2013, 78, 5117–5141. [Google Scholar] [CrossRef]
- Agatsuma, T.; Ogawa, H.; Akasaka, K.; Asai, A.; Yamashita, Y.; Mizukami, T.; Akinaga, S.; Saitoh, Y. Halohydrin and oxime derivatives of radicicol: Synthesis and antitumor activities. Bioorg. Med. Chem. 2002, 10, 3445–3454. [Google Scholar] [CrossRef]
- Lei, X.; Danishefsky, S.J. Efficient synthesis of a novel resorcyclide as anticancer agent based on Hsp90 inhibition. Adv. Synth. Catal. 2008, 350, 1677–1681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutton, B.L.; Kitson, R.R.; Parry-Morris, S.; Roe, S.M.; Prodromou, C.; Moody, C.J. Synthesis of macrolactam analogues of radicicol and their binding to heat shock protein Hsp90. Org. Biomol. Chem. 2014, 12, 1328–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Day, J.E.; Sharp, S.Y.; Rowlands, M.G.; Aherne, W.; Hayes, A.; Raynaud, F.I.; Lewis, W.; Roe, S.M.; Prodromou, C.; Pearl, L.H. Targeting the Hsp90 molecular chaperone with novel macrolactams. Synthesis, structural, binding, and cellular studies. ACS Chem. Biol. 2011, 6, 1339–1347. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, H.; Omi, K.; Hojo, K.; Nishida, K.; Ômura, S.; Sugita, K. Suppression of oncogenic transformation by hypothemycin associated with accelerated cyclin D1 degradation through ubiquitin-proteasome pathway. Life Sci. 1999, 65, 381–394. [Google Scholar] [CrossRef]
- Tanaka, H.; Nishida, K.; Sugita, K.; Yoshioka, T. Antitumor Efficacy of Hypothemycin, A New Ras-signaling Inhibitor. JPN. J. Cancer Res. 1999, 90, 1139–1145. [Google Scholar] [CrossRef] [PubMed]
- Schirmer, A.; Kennedy, J.; Murli, S.; Reid, R.; Santi, D.V. Targeted covalent inactivation of protein kinases by resorcylic acid lactone polyketides. Proc. Natl. Acd. Sci. USA 2006, 103, 4234–4239. [Google Scholar] [CrossRef] [Green Version]
- Ninomiya-Tsuji, J.; Kajino, T.; Ono, K.; Ohtomo, T.; Matsumoto, M.; Shiina, M.; Mihara, M.; Tsuchiya, M.; Matsumoto, K. A resorcylic acid lactone, 5Z-7-oxozeaenol, prevents inflammation by inhibiting the catalytic activity of TAK1 MAPK kinase kinase. J. Biol. Chem. 2003, 278, 18485–18490. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Wu, W.; Fu, B.; Shi, L.; Wang, X.; Kuca, K. JNK signaling in cancer cell survival. Med. Res. Rev. 2019, 39, 2082–2104. [Google Scholar] [CrossRef]
- Takehana, K.; Sato, S.-i.; Kobayasi, T.; Maeda, T. A radicicol-related macrocyclic nonaketide compound, antibiotic LL-Z1640-2, inhibits the JNK/p38 pathways in signal-specific manner. Biochem. Biophys. Res. 1999, 257, 19–23. [Google Scholar] [CrossRef]
- Tatsuta, K.; Takano, S.; Sato, T.; Nakano, S. The first total synthesis of a macrocyclic anti-protozoan, LL-Z1640-2. Chem. Lett. 2001, 30, 172–173. [Google Scholar] [CrossRef]
- Sellès, P.; Lett, R. Convergent stereospecific synthesis of C292 (or LL-Z1640-2), and hypothemycin. Part 1. Tetrahedron Lett. 2002, 43, 4621–4625. [Google Scholar] [CrossRef]
- Dakas, P.Y.; Barluenga, S.; Totzke, F.; Zirrgiebel, U.; Winssinger, N. Modular synthesis of radicicol A and related resorcylic acid lactones, potent kinase inhibitors. Angew. Chem. Int. Ed. 2007, 46, 6899–6902. [Google Scholar] [CrossRef] [PubMed]
- LeClair, C.A.; Boxer, M.B.; Thomas, C.J.; Maloney, D.J. Total synthesis of LL-Z1640-2 utilizing a late-stage intramolecular Nozaki–Hiyama–Kishi reaction. Tetrahedron Lett. 2010, 51, 6852–6855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyatake-Ondozabal, H.; Barrett, A.G. Total synthesis of TAK-kinase inhibitor LL-Z1640-2 via consecutive macrocyclization and transannular aromatization. Org. Lett. 2010, 12, 5573–5575. [Google Scholar] [CrossRef]
- Hearn, B.R.; Sundermann, K.; Cannoy, J.; Santi, D.V. Semisynthesis and cytotoxicity of hypothemycin analogues. ChemMedChem 2007, 2, 1598–1600. [Google Scholar] [CrossRef]
- Dakas, P.Y.; Jogireddy, R.; Valot, G.; Barluenga, S.; Winssinger, N. Divergent syntheses of resorcylic acid lactones: L-783277, LL-Z1640-2, and hypothemycin. Chem. Eur. J. 2009, 15, 11490–11497. [Google Scholar] [CrossRef]
- Du, H.; Matsushima, T.; Spyvee, M.; Goto, M.; Shirota, H.; Gusovsky, F.; Chiba, K.; Kotake, M.; Yoneda, N.; Eguchi, Y. Discovery of a potent, metabolically stabilized resorcylic lactone as an anti-inflammatory lead. Bioorg. Med. Chem. Lett. 2009, 19, 6196–6199. [Google Scholar] [CrossRef]
- Shen, Y.; Du, H.; Kotake, M.; Matsushima, T.; Goto, M.; Shirota, H.; Gusovsky, F.; Li, X.; Jiang, Y.; Schiller, S. Discovery of an in vitro and in vivo potent resorcylic lactone analog of LL-Z1640-2 as anti-inflammatory lead, II. Bioorg. Med. 2010, 20, 3047–3049. [Google Scholar] [CrossRef]
- Goh, W.Y.; Chai, C.L.; Chen, A. Synthesis and Biological Studies of a Triazole Analogue of Resorcylic Acid Lactone LL-Z1640-2. Eur. J. Org. Chem. 2014, 2014, 7239–7244. [Google Scholar] [CrossRef]
- Wang, S.Q.; Goh, S.S.; Chai, C.L.; Chen, A. An efficient synthesis of an exo-enone analogue of LL-Z1640-2 and evaluation of its protein kinase inhibitory activities. Org. Biomol. Chem. 2016, 14, 639–645. [Google Scholar] [CrossRef]
- Cho, H.; Sengupta, S.; Jeon, S.S.; Hur, W.; Choi, H.G.; Seo, H.-S.; Lee, B.J.; Kim, J.H.; Chung, M.; Jeon, N.L. Identification of the first selective activin receptor-like kinase 1 inhibitor, a reversible version of L-783277. J. Med. Chem. 2017, 60, 1495–1508. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.G.; Son, J.B.; Park, D.-S.; Ham, Y.J.; Hah, J.-M.; Sim, T. An efficient and enantioselective total synthesis of naturally occurring L-783277. Tetrahedron Lett. 2010, 51, 4942–4946. [Google Scholar] [CrossRef]
- Han, Y.; Sengupta, S.; Lee, B.J.; Cho, H.; Kim, J.; Choi, H.G.; Dash, U.; Kim, J.H.; Kim, N.D.; Kim, J.H.; et al. Identification of a Unique Resorcylic Acid Lactone Derivative That Targets Both Lymphangiogenesis and Angiogenesis. J. Med. Chem. 2019, 62, 9141–9160. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, T.; Altmann, K.-H. Total synthesis of the resorcylic lactone-based kinase inhibitor L-783277. Synlett 2008, 2008, 1500–1504. [Google Scholar]
- Goumans, M.J.; Valdimarsdottir, G.; Itoh, S.; Rosendahl, A.; Sideras, P.; ten Dijke, P. Balancing the activation state of the endothelium via two distinct TGF-β type I receptors. EMBO J. 2002, 21, 1743–1753. [Google Scholar] [CrossRef]
- Boergermann, J.; Kopf, J.; Yu, P.; Knaus, P. Dorsomorphin and LDN-193189 inhibit BMP-mediated Smad, p38 and Akt signalling in C2C12 cells. Int. J. Biochem. Cell Biol. 2010, 42, 1802–1807. [Google Scholar] [CrossRef]
- González-Núñez, M.; Muñoz-Félix, J.M.; López-Novoa, J.M. The ALK-1/Smad1 pathway in cardiovascular physiopathology. A new target for therapy? Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 1492–1510. [Google Scholar] [CrossRef] [Green Version]
- Roberts, N.; Kloos, B.; Cassella, M.; Podgrabinska, S.; Persaud, K.; Wu, Y.; Pytowski, B.; Skobe, M. Inhibition of VEGFR-3 activation with the antagonistic antibody more potently suppresses lymph node and distant metastases than inactivation of VEGFR-2. Cancer Res. 2006, 66, 2650–2657. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, Y.; Kitadai, Y.; Bucana, C.D.; Cleary, K.R.; Ellis, L.M. Expression of vascular endothelial growth factor and its receptor, KDR, correlates with vascularity, metastasis, and proliferation of human colon cancer. Cancer Res. 1995, 55, 3964–3968. [Google Scholar]
- Scavelli, C.; Vacca, A.; Di Pietro, G.; Dammacco, F.; Ribatti, D. Crosstalk between angiogenesis and lymphangiogenesis in tumor progression. Leukemia 2004, 18, 1054–1058. [Google Scholar] [CrossRef]
- Lee, Y.T.; Lim, S.H.; Lee, B.; Kang, I.; Yeo, E.-J. Compound C inhibits B16-F1 tumor growth in a Syngeneic Mouse Model via the blockage of cell cycle progression and angiogenesis. Cancers 2019, 11, 823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, C.R.; Bryant, J.D. D-(R)-Glyceraldehyde Acetonide: 1,3-Dioxolane-4-carboxaldehyde, 2,2-dimethyl-,(R)-. Organic Synth. 2003, 72, 6. [Google Scholar]
- Chakraborty, J.; Ghosh, A.; Nanda, S. Asymmetric total syntheses of naturally occurring α, β-enone-containing RALs, L-783290 and L-783277 through intramolecular base-mediated macrolactonization reaction. Org. Biomol. Chem. 2020, 18, 2331–2345. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.; Willis, A.C.; Banwell, M.G. A chemoenzymatic and enantioselective total synthesis of the resorcylic acid lactone L-783,290, the trans-isomer of L-783,277. Tetrahedron Lett. 2010, 51, 1044–1047. [Google Scholar] [CrossRef]
- Liniger, M.; Neuhaus, C.; Hofmann, T.; Fransioli-Ignazio, L.; Jordi, M.; Drueckes, P.; Trappe, J.r.; Fabbro, D.; Altmann, K.-H. Kinase inhibition by deoxy analogues of the resorcylic lactone L-783277. ACS Med. Chem. Lett. 2011, 2, 22–27. [Google Scholar] [CrossRef] [Green Version]
- Aharoni, A.; Giri, A.P.; Deuerlein, S.; Griepink, F.; de Kogel, W.-J.; Verstappen, F.W.; Verhoeven, H.A.; Jongsma, M.A.; Schwab, W.; Bouwmeester, H.J. Terpenoid metabolism in wild-type and transgenic Arabidopsis plants. Plant Cell. 2003, 15, 2866–2884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pichersky, E.; Noel, J.P.; Dudareva, N. Biosynthesis of plant volatiles: Nature’s diversity and ingenuity. Science 2006, 311, 808–811. [Google Scholar] [CrossRef] [Green Version]
- Fraga, B.M. Natural sesquiterpenoids. Nat. Prod. Rep. 2008, 25, 1180–1209. [Google Scholar] [CrossRef]
- Chadwick, M.; Trewin, H.; Gawthrop, F.; Wagstaff, C. Sesquiterpenoids lactones: Benefits to plants and people. Int. J. Mol. Sci. 2013, 14, 12780–12805. [Google Scholar] [CrossRef] [Green Version]
- Harborne, J. The Biology and Chemistry of the Compositae; Heywood, V.H., Harborne, J.B., Turner, B.L., Eds.; Academic Press: London, UK, 1977; Volume 2. [Google Scholar]
- Maries, R.J.; Pazos-Sanou, L.; Compadre, C.M.; Pezzuto, J.M.; Bloszyk, E.; Arnason, J.T. Sesquiterpene Lactones Revisited. In Phytochemistry of Medicinal Plants (Recent Advances in Phytochemistry), Proceedings of the Phytochemical Society of North America; Arnason, J.T., Mata, R., Romeo, J.T., Eds.; Springer: Boston, MA, USA, 1995; Volume 29. [Google Scholar]
- Schmidt, T.J. Structure-activity relationships of sesquiterpene lactones. In Studies in Natural Products Chemistry; Elsevier: Amsterdam, The Netherlands, 2006; Volume 33, pp. 309–392. [Google Scholar]
- Smolinski, A.T.; Pestka, J.J. Comparative effects of the herbal constituent parthenolide (Feverfew) on lipopolysaccharide-induced inflammatory gene expression in murine spleen and liver. J. Inflamm. 2005, 2, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Mathema, V.B.; Koh, Y.-S.; Thakuri, B.C.; Sillanpää, M. Parthenolide, a sesquiterpene lactone, expresses multiple anti-cancer and anti-inflammatory activities. Inflammation 2012, 35, 560–565. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Ong, C.-N.; Shen, H.-M. Involvement of proapoptotic Bcl-2 family members in parthenolide-induced mitochondrial dysfunction and apoptosis. Cancer Lett. 2004, 211, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Pajak, B.; Gajkowska, B.; Orzechowski, A. Molecular basis of parthenolide-dependent proapoptotic activity in cancer cells. Folia Histochem. Cytobiol. 2008, 46, 129–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Clair, D.K.S.; Xu, Y.; Crooks, P.A.; Clair, W.H.S. A NADPH oxidase–dependent redox signaling pathway mediates the selective radiosensitization effect of parthenolide in prostate cancer cells. Cancer Res. 2010, 70, 2880–2890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Zhou, Y.; Cai, Y.; Shui, C.; Liu, W.; Wang, X.; Jiang, J.; Zeng, D.; Gui, C.; Sun, R. Parthenolide Inhibits the Proliferation of MDA-T32 Papillary Thyroid Carcinoma Cells in Vitro and in Mouse Tumor Xenografts and Activates Autophagy and Apoptosis by Downregulation of the Mammalian Target of Rapamycin (mTOR)/PI3K/AKT Signaling Pathway. Med. Sci. Mon. Int. Med. J. Exp. Clin. Res. 2019, 25, 5054. [Google Scholar] [CrossRef]
- Sweeney, C.J.; Mehrotra, S.; Sadaria, M.R.; Kumar, S.; Shortle, N.H.; Roman, Y.; Sheridan, C.; Campbell, R.A.; Murry, D.J.; Badve, S. The sesquiterpene lactone parthenolide in combination with docetaxel reduces metastasis and improves survival in a xenograft model of breast cancer. Mol. Cancer Ther. 2005, 4, 1004–1012. [Google Scholar] [CrossRef] [Green Version]
- Curry, E.A.; Murry, D.J.; Yoder, C.; Fife, K.; Armstrong, V.; Nakshatri, H.; O’Connell, M.; Sweeney, C.J. Phase I dose escalation trial of feverfew with standardized doses of parthenolide in patients with cancer. Investig. New Drugs. 2004, 22, 299–305. [Google Scholar] [CrossRef]
- Kwok, B.H.; Koh, B.; Ndubuisi, M.I.; Elofsson, M.; Crews, C.M. The anti-inflammatory natural product parthenolide from the medicinal herb Feverfew directly binds to and inhibits IκB kinase. Chem. Biol. 2001, 8, 759–766. [Google Scholar] [CrossRef] [Green Version]
- Carlisi, D.; D’Anneo, A.; Angileri, L.; Lauricella, M.; Emanuele, S.; Santulli, A.; Vento, R.; Tesoriere, G. Parthenolide sensitizes hepatocellular carcinoma cells to TRAIL by inducing the expression of death receptors through inhibition of STAT3 activation. J. Cell. Physiol. 2011, 226, 1632–1641. [Google Scholar] [CrossRef]
- Jafari, N.; Nazeri, S.; Enferadi, S.T. Parthenolide reduces metastasis by inhibition of vimentin expression and induces apoptosis by suppression elongation factor α− 1 expression. Phytomedicine 2018, 41, 67–73. [Google Scholar] [CrossRef]
- Kim, S.L.; Park, Y.R.; Lee, S.T.; Kim, S.-W. Parthenolide suppresses hypoxia-inducible factor-1α signaling and hypoxia induced epithelial-mesenchymal transition in colorectal cancer. Int. J. Oncol. 2017, 51, 1809–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, M.; Bi, H.; Yan, Y.; Huang, W.; Zhang, G.; Zhang, G.; Tang, S.; Liu, Y.; Zhang, L.; Ma, J. Parthenolide suppresses non-small cell lung cancer GLC-82 cells growth via B-Raf/MAPK/Erk pathway. Oncotarget 2017, 8, 23436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berdan, C.A.; Ho, R.; Lehtola, H.S.; To, M.; Hu, X.; Huffman, T.R.; Petri, Y.; Altobelli, C.R.; Demeulenaere, S.G.; Olzmann, J.A. Parthenolide Covalently targets and inhibits focal adhesion kinase in breast cancer cells. Cell Chem. Biol. 2019, 26, 1027–1035. [Google Scholar] [CrossRef] [PubMed]
- Neelakantan, S.; Nasim, S.; Guzman, M.L.; Jordan, C.T.; Crooks, P.A. Aminoparthenolides as novel anti-leukemic agents: Discovery of the NF-κB inhibitor, DMAPT (LC-1). Bioorg. Med. Chem. Lett. 2009, 19, 4346–4349. [Google Scholar] [CrossRef]
- Ghantous, A.; Sinjab, A.; Herceg, Z.; Darwiche, N. Parthenolide: From plant shoots to cancer roots. Drug Discov. Today. 2013, 18, 894–905. [Google Scholar] [CrossRef]
- Long, J.; Zhang, S.-F.; Wang, P.-P.; Zhang, X.-M.; Yang, Z.-J.; Zhang, Q.; Chen, Y. Total syntheses of parthenolide and its analogues with macrocyclic stereocontrol. J. Med. Chem. 2014, 57, 7098–7112. [Google Scholar] [CrossRef]
- Yang, H.; Gao, Y.; Qiao, X.; Xie, L.; Xu, X. Concise total synthesis of (−)-8-epigrosheimin. Org. Lett. 2011, 13, 3670–3673. [Google Scholar] [CrossRef]
- Sen, S.E.; Garvin, G.M. Synthesis of (2E, 6E)-[10-3H] farnesol and (2E6E)-[10-3H] farnesal for insect dehydrogenase studies. J. Labelled Comp. Radiopharm. 1995, 36, 1063–1069. [Google Scholar] [CrossRef]
- Still, W.C.; Galynker, I. Chemical consequences of conformation in macrocyclic compounds: An effective approach to remote asymmetric induction. Tetrahedron 1981, 37, 3981–3996. [Google Scholar] [CrossRef]
- Snyder, S.A.; Treitler, D.S.; Brucks, A.P. Simple reagents for direct halonium-induced polyene cyclizations. J. Am. Chem. Soc. 2010, 132, 14303–14314. [Google Scholar] [CrossRef]
- Foo, K.; Usui, I.; Götz, D.C.; Werner, E.W.; Holte, D.; Baran, P.S. Scalable, enantioselective synthesis of germacrenes and related sesquiterpenes inspired by terpene cyclase phase logic. Angew. Chem. Int. Ed. 2012, 51, 11491–11495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolev, J.N.; O’Dwyer, K.M.; Jordan, C.T.; Fasan, R. Discovery of Potent Parthenolide-Based Antileukemic Agents Enabled by Late-Stage P450-Mediated C H Functionalization. ACS Chem. Biol. 2014, 9, 164–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
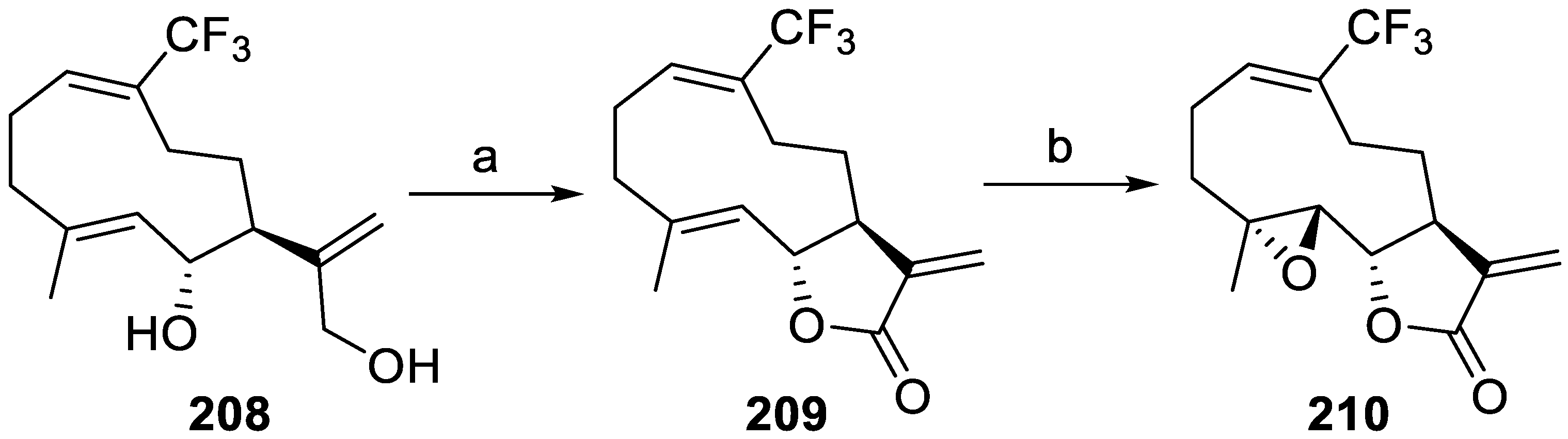
- Yang, Z.-J.; Ge, W.-Z.; Li, Q.-Y.; Lu, Y.; Gong, J.-M.; Kuang, B.-J.; Xi, X.; Wu, H.; Zhang, Q.; Chen, Y. Syntheses and Biological Evaluation of Costunolide, Parthenolide, and Their Fluorinated Analogues. J. Med. Chem. 2015, 58, 7007–7020. [Google Scholar] [CrossRef] [PubMed]
- PR, P.S.M. Swallow S. Gouverneur V. Chem. Soc. Rev. 2008, 37, 320. [Google Scholar]
- Hagmann, W.K. The many roles for fluorine in medicinal chemistry. J. Med. Chem. 2008, 51, 4359–4369. [Google Scholar] [CrossRef]
- Nagib, D.A.; MacMillan, D.W. Trifluoromethylation of arenes and heteroarenes by means of photoredox catalysis. Nature 2011, 480, 224–228. [Google Scholar] [CrossRef]
- Ding, Y.; Wang, H.; Niu, J.; Luo, M.; Gou, Y.; Miao, L.; Zou, Z.; Cheng, Y. Induction of ROS overload by alantolactone prompts oxidative DNA damage and apoptosis in colorectal cancer cells. Int. J. Mol. Sci. 2016, 17, 558. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.; Xia, D.; Bian, Y.; Sun, Y.; Zhu, F.; Pan, B.; Niu, M.; Zhao, K.; Wu, Q.; Qiao, J. Alantolactone induces G1 phase arrest and apoptosis of multiple myeloma cells and overcomes bortezomib resistance. Apoptosis 2015, 20, 1122–1133. [Google Scholar] [CrossRef]
- Chun, J.; Li, R.-J.; Cheng, M.-S.; Kim, Y.S. Alantolactone selectively suppresses STAT3 activation and exhibits potent anticancer activity in MDA-MB-231 cells. Cancer Lett. 2015, 357, 393–403. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.R.; Cai, Q.Y.; Gao, Y.G.; Luan, X.; Guan, Y.Y.; Lu, Q.; Sun, P.; Zhao, M.; Fang, C. Alantolactone, a sesquiterpene lactone, inhibits breast cancer growth by antiangiogenic activity via blocking VEGFR2 signaling. Phytother. Res. 2018, 32, 643–650. [Google Scholar] [CrossRef]
- Kang, X.; Wang, H.; Li, Y.; Xiao, Y.; Zhao, L.; Zhang, T.; Zhou, S.; Zhou, X.; Li, Y.; Shou, Z. Alantolactone induces apoptosis through ROS-mediated AKT pathway and inhibition of PINK1-mediated mitophagy in human HepG2 cells. Artif. Cells Nanomed. Biotechnol. 2019, 47, 1961–1970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Yu, Z.; Wang, C.; Cheng, W.; Tian, X.; Huo, X.; Wang, Y.; Sun, C.; Feng, L.; Xing, J. Alantolactone, a natural sesquiterpene lactone, has potent antitumor activity against glioblastoma by targeting IKKβ kinase activity and interrupting NF-κB/COX-2-mediated signaling cascades. J. Exp. Clin. Cancer Res. 2017, 36, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.A.; Cohen, N. The stereoselective total synthesis of alantolactone. J. Am. Chem. Soc. 1965, 87, 2773–2774. [Google Scholar] [CrossRef]
- Stojanović-Radić, Z.; Čomić, L.; Radulović, N.; Blagojević, P.; Denić, M.; Miltojević, A.; Rajković, J.; Mihajilov-Krstev, T. Antistaphylococcal activity of Inula helenium L. root essential oil: Eudesmane sesquiterpene lactones induce cell membrane damage. Eur. J. Clin. Microbiol. Infect. Dis. 2012, 31, 1015–1025. [Google Scholar] [CrossRef]
- Li, Y.; Ni, Z.-Y.; Zhu, M.-C.; Dong, M.; Wang, S.-M.; Shi, Q.-W.; Zhang, M.-L.; Wang, Y.-F.; Huo, C.-H.; Kiyota, H. Antitumour activities of sesquiterpene lactones from Inula helenium and Inula japonica. Z. Naturforsch. C. 2012, 67, 375–380. [Google Scholar] [CrossRef]
- Chun, J.; Choi, R.J.; Khan, S.; Lee, D.-S.; Kim, Y.-C.; Nam, Y.-J.; Lee, D.-U.; Kim, Y.S. Alantolactone suppresses inducible nitric oxide synthase and cyclooxygenase-2 expression by down-regulating NF-κB, MAPK and AP-1 via the MyD88 signaling pathway in LPS-activated RAW 264.7 cells. Int. Immunopharmacol. 2012, 14, 375–383. [Google Scholar] [CrossRef]
- Ketai, W.; Huitao, L.; Yunkun, Z.; Xingguo, C.; Zhide, H.; Yucheng, S.; Xiao, M. Separation and determination of alantolactone and isoalantolactone in traditional Chinese herbs by capillary electrophoresis. Talanta 2000, 52, 1001–1005. [Google Scholar] [CrossRef]
- Johnson, W.S.; Lunn, W.H.; Fitzi, K. Cationic Cyclizations Involving Olefinic Bonds. IV. 1 The Butenylcyclohexenol System. J. Am. Chem. Soc. 1964, 86, 1972–1978. [Google Scholar] [CrossRef]
- Bowden, K.; Heilbron, I.M.; Jones, E.R.H.; Weedon, B.C.L. 13. Researches on acetylenic compounds. Part I. The preparation of acetylenic ketones by oxidation of acetylenic carbinols and glycols. J. Am. Chem. Soc. 1946, 39–45. [Google Scholar] [CrossRef]
- Stork, G.; Brizzolara, A.; Landesman, H.; Szmuszkovicz, J.; Terrell, R. The enamine alkylation and acylation of carbonyl compounds. J. Am. Chem. Soc. 1963, 85, 207–222. [Google Scholar] [CrossRef]
- Nickon, A.; Bagli, J.F. Reactivity and Geometry in Allylic Systems. I. Stereochemistry of Photosensitized Oxygenation of Monoölefins1, 2. J. Am. Chem. Soc. 1961, 83, 1498–1508. [Google Scholar] [CrossRef]
- Benešová, V.; Herout, V.; Šorm, F. On terpenes. CXXIV. Structure of telekin and isotelekin, new sesquiterpenic lactones from Telekia speciosa (SCHREB) BAUMG. Collect. Czech. Chem. Commun. 1961, 26, 1350–1357. [Google Scholar] [CrossRef]
- Kaur, R.; Chahal, K. Isolation, Chemical Transformation, and Antifungal Potential of Sesquiterpene Lactones from Inula Racemosa. Chem. Nat. Compd. 2020, 56, 207–212. [Google Scholar] [CrossRef]
- Kulyyasov, A.; Seitembetov, T.; Turdybekov, K.; Adekenov, S. Epoxidation of alantolactone and isoalantolactone. Chem. Nat. Compd. 1996, 32, 869–872. [Google Scholar] [CrossRef]
- Lawrence, N.J.; McGown, A.T.; Nduka, J.; Hadfield, J.A.; Pritchard, R.G. Cytotoxic michael-type amine adducts of α-methylene lactones alantolactone and isoalantolactone. Bioorg. Med. Chem. Lett. 2001, 11, 429–431. [Google Scholar] [CrossRef]
- Shul’ts, E.; Belovodskii, A.; Shakirov, M.; Tolstikov, G. Synthetic transformations of sesquiterpene lactones 6. Alantolactone and isoalantolactone derivatives in the Heck reaction. Russ. Chem. Bull. 2012, 61, 1975–1985. [Google Scholar] [CrossRef]
- Kabeer, F.A.; Prathapan, R. Phytopharımacological Profile of Elephantopus scaber. Pharmacologia 2014, 5, 272–285. [Google Scholar] [CrossRef] [Green Version]
- Beeran, A.A.; Maliyakkal, N.; Rao, C.M.; Udupa, N. The enriched fraction of Elephantopus scaber Triggers apoptosis and inhibits multi-drug resistance transporters in human epithelial cancer cells. Pharmacogn. Mag. 2015, 11, 257. [Google Scholar] [PubMed] [Green Version]
- Yan, G.R.; Tan, Z.; Wang, Y.; Xu, M.L.; Yu, G.; Li, Y.; He, Q.Y. Quantitative proteomics characterization on the antitumor effects of isodeoxyelephantopin against nasopharyngeal carcinoma. Proteomics 2013, 13, 3222–3232. [Google Scholar] [CrossRef]
- Farha, A.K.; Dhanya, S.R.; Mangalam, S.N.; Geetha, B.S.; Latha, P.G.; Remani, P. Deoxyelephantopin impairs growth of cervical carcinoma SiHa cells and induces apoptosis by targeting multiple molecular signaling pathways. Cell Biol. Toxicol. 2014, 30, 331–343. [Google Scholar] [CrossRef]
- Chan, C.K.; Chan, G.; Awang, K.; Abdul Kadir, H. Deoxyelephantopin from elephantopus scaber inhibits HCT116 human colorectal carcinoma cell growth through apoptosis and cell cycle arrest. Molecules 2016, 21, 385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabeer, F.A.; Sreedevi, G.B.; Nair, M.S.; Rajalekshmi, D.S.; Gopalakrishnan, L.P.; Kunjuraman, S.; Prathapan, R. Antineoplastic effects of deoxyelephantopin, a sesquiterpene lactone from Elephantopus scaber, on lung adenocarcinoma (A549) cells. J. Integr. Med. 2013, 11, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.-H.; Nakagawa-Goto, K.; Lee, K.-H.; Shyur, L.-F. A Novel Plant Sesquiterpene Lactone Derivative, DETD-35, Suppresses BRAFV600E Mutant Melanoma Growth and Overcomes Acquired Vemurafenib Resistance in Mice. Mol. Cancer Ther. 2016, 15, 1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiau, J.-Y.; Chang, Y.-Q.; Nakagawa-Goto, K.; Lee, K.-H.; Shyur, L.-F. Phytoagent Deoxyelephantopin and Its Derivative Inhibit Triple Negative Breast Cancer Cell Activity through ROS-Mediated Exosomal Activity and Protein Functions. Front. Pharmacol. 2017, 8, 398. [Google Scholar] [CrossRef] [Green Version]
- Picman, A.K. Biological activities of sesquiterpene lactones. Biochem. Syst. Ecol. 1986, 14, 255–281. [Google Scholar] [CrossRef]
- Ichikawa, H.; Nair, M.S.; Takada, Y.; Sheeja, D.A.; Kumar, M.S.; Oommen, O.V.; Aggarwal, B.B. Isodeoxyelephantopin, a novel sesquiterpene lactone, potentiates apoptosis, inhibits invasion, and abolishes osteoclastogenesis through suppression of nuclear factor-κB (NF-κB) activation and NF-κB-regulated gene expression. Clin. Cancer. Res. 2006, 12, 5910–5918. [Google Scholar] [CrossRef] [Green Version]
- Zou, G.; Gao, Z.; Wang, J.; Zhang, Y.; Ding, H.; Huang, J.; Chen, L.; Guo, Y.; Jiang, H.; Shen, X. Deoxyelephantopin inhibits cancer cell proliferation and functions as a selective partial agonist against PPARγ. Biochem. Pharmacol. 2008, 75, 1381–1392. [Google Scholar] [CrossRef]
- Choi, S.-S.; Park, J.; Choi, J.H. Revisiting PPARγ as a target for the treatment of metabolic disorders. BMB Rep. 2014, 47, 599. [Google Scholar] [CrossRef] [Green Version]
- Staels, B.; Fruchart, J.-C. Therapeutic roles of peroxisome proliferator–activated receptor agonists. Diabetes 2005, 54, 2460–2470. [Google Scholar] [CrossRef] [Green Version]
- Lagoutte, R.; Serba, C.; Abegg, D.; Hoch, D.G.; Adibekian, A.; Winssinger, N. Divergent synthesis and identification of the cellular targets of deoxyelephantopins. Nat. Comm. 2016, 7, 1–11. [Google Scholar] [CrossRef]
- Hudis, C.A.; Gianni, L. Triple-negative breast cancer: An unmet medical need. Oncologist 2011, 16, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa-Goto, K.; Chen, J.-Y.; Cheng, Y.-T.; Lee, W.-L.; Takeya, M.; Saito, Y.; Lee, K.-H.; Shyur, L.-F. Novel sesquiterpene lactone analogues as potent anti-breast cancer agents. Mol. Oncol. 2016, 10, 921–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Wang, X.; Sun, L.; Xie, L.; Xu, X. Zinc or indium-mediated Barbier-type allylation of aldehydes with 3-bromomethyl-5H-furan-2-one in aqueous media: An efficient synthesis method for α-methylene-γ-butyrolactone. Org. Biomol. Chem. 2012, 10, 3991–3998. [Google Scholar] [CrossRef]
- Chany, A.C.; Casarotto, V.; Schmitt, M.; Tarnus, C.; Guenin-Macé, L.; Demangel, C.; Mirguet, O.; Eustache, J.; Blanchard, N. A diverted total synthesis of mycolactone analogues: An insight into Buruli ulcer toxins. Chem. Eur. J. 2011, 17, 14413–14419. [Google Scholar] [CrossRef]
- Nicolaou, K.; Bulger, P.G.; Sarlah, D. Metathesis reactions in total synthesis. Angew. Chem. Int. Ed. 2005, 44, 4490–4527. [Google Scholar] [CrossRef]
- Bruno, M.; Rosselli, S.; Maggio, A.; Raccuglia, R.A.; Napolitano, F.; Senatore, F. Antibacterial evaluation of cnicin and some natural and semisynthetic analogues. Planta Med. 2003, 69, 277–281. [Google Scholar] [CrossRef]
- Rao, A.S.; Kelkar, G.; Bhattacharyya, S. Terpenoids—XXI: The structure of costunolide, a new sesquiterpene lactone from costus root oil. Tetrahedron 1960, 9, 275–283. [Google Scholar] [CrossRef]
- Rasul, A.; Parveen, S.; Ma, T. Costunolide: A novel anti-cancer sesquiterpene lactone. Bangladesh J. Pharmacol. 2012, 7, 6–13. [Google Scholar] [CrossRef] [Green Version]
- Van Beek, T.A.; Maas, P.; King, B.M.; Leclercq, E.; Voragen, A.G.; De Groot, A. Bitter sesquiterpene lactones from chicory roots. J. Agric. Food Chem. 1990, 38, 1035–1038. [Google Scholar] [CrossRef]
- Fischer, N.H. Sesquiterpene Lactones: Biogenesis and Biomimetic Transformations. In Biochemistry of the Mevalonic Acid Pathway to Terpenoids; Springer: Baton Rouge, LA, USA, 1990; Volume 24, pp. 161–201. [Google Scholar]
- Kanno, S.-i.; Kitajima, Y.; Kakuta, M.; Osanai, Y.; Kurauchi, K.; Ujibe, M.; Ishikawa, M. Costunolide-induced apoptosis is caused by receptor-mediated pathway and inhibition of telomerase activity in NALM-6 cells. Biol. Pharm. Bull. 2008, 31, 1024–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kassuya, C.A.L.; Cremoneze, A.; Barros, L.F.L.; Simas, A.S.; da Rocha Lapa, F.; Mello-Silva, R.; Stefanello, M.É.A.; Zampronio, A.R. Antipyretic and anti-inflammatory properties of the ethanolic extract, dichloromethane fraction and costunolide from Magnolia ovata (Magnoliaceae). J. Ethnopharmacol. 2009, 124, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Eliza, J.; Daisy, P.; Ignacimuthu, S. Antioxidant activity of costunolide and eremanthin isolated from Costus speciosus (Koen ex. Retz) Sm. Chem.-Biol. Interact. 2010, 188, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Chen, Y.; Zhang, J.; Wang, L.; Jin, Z.; Huang, H.; Man, S.; Gao, W. Evaluation of protective effects of costunolide and dehydrocostuslactone on ethanol-induced gastric ulcer in mice based on multi-pathway regulation. Chem.-Biol. Interact. 2016, 250, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; He, X.; Yang, C. Costunolide promotes imatinib-induced apoptosis in chronic myeloid leukemia cells via the Bcr/Abl–Stat5 pathway. Phytother. Res. 2018, 32, 1764–1769. [Google Scholar] [CrossRef]
- Hu, M.; Liu, L.; Yao, W. Activation of p53 by costunolide blocks glutaminolysis and inhibits proliferation in human colorectal cancer cells. Gene 2018, 678, 261–269. [Google Scholar] [CrossRef]
- Li, Q.; Wang, Z.; Xie, Y.; Hu, H. Antitumor activity and mechanism of costunolide and dehydrocostus lactone: Two natural sesquiterpene lactones from the Asteraceae family. Biomed. Pharmacother. 2020, 125, 109955. [Google Scholar] [CrossRef]
- Liu, C.-Y.; Chang, H.-S.; Chen, I.-S.; Chen, C.-J.; Hsu, M.-L.; Fu, S.-L.; Chen, Y.-J. Costunolide causes mitotic arrest and enhances radiosensitivity in human hepatocellular carcinoma cells. Radiat. Oncol. 2011, 6, 56. [Google Scholar] [CrossRef] [Green Version]
- Rasul, A.; Yu, B.; Yang, L.-F.; Arshad, M.; Khan, M.; Ma, T.; Yang, H. Costunolide, a sesquiterpene lactone induces G2/M phase arrest and mitochondria-mediated apoptosis in human gastric adenocarcinoma SGC-7901 cells. J. Med. Plant Res. 2012, 6, 1191–1200. [Google Scholar]
- Yang, Y.-I.; Kim, J.-H.; Lee, K.-T.; Choi, J.-H. Costunolide induces apoptosis in platinum-resistant human ovarian cancer cells by generating reactive oxygen species. Gynecol. Oncol. 2011, 123, 588–596. [Google Scholar] [CrossRef]
- Choi, Y.K.; Seo, H.S.; Choi, H.S.; Choi, H.S.; Kim, S.R.; Shin, Y.C.; Ko, S.-G. Induction of Fas-mediated extrinsic apoptosis, p21WAF1-related G2/M cell cycle arrest and ROS generation by costunolide in estrogen receptor-negative breast cancer cells, MDA-MB-231. Mol. Cell. Biochem. 2012, 363, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Lu, T.; Wang, G.-D.; Ma, C.; Zhou, Y.-F. Costunolide, an active sesquiterpene lactone, induced apoptosis via ROS-mediated ER stress and JNK pathway in human U2OS cells. Biomed. Pharmacother. 2016, 80, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhao, X.; Gong, X. Costunolide induces lung adenocarcinoma cell line A549 cells apoptosis through ROS (reactive oxygen species)—Mediated endoplasmic reticulum stress. Cell Biol. Int. 2016, 40, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.J.; Itokawa, T.; Shibuya, M.; Kuwano, M.; Ono, M.; Higuchi, R.; Miyamoto, T. Costunolide, a sesquiterpene lactone from Saussurea lappa, inhibits the VEGFR KDR/Flk-1 signaling pathway. Cancer Lett. 2002, 187, 129–133. [Google Scholar] [CrossRef]
- Mahfouz, N.; Tahtouh, R.; Alaaeddine, N.; El Hajj, J.; Sarkis, R.; Hachem, R.; Raad, I.; Hilal, G. Gastrointestinal cancer cells treatment with bevacizumab activates a VEGF autoregulatory mechanism involving telomerase catalytic subunit hTERT via PI3K-AKT, HIF-1α and VEGF receptors. PLoS ONE 2017, 12, e0179202. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, T.N.; Huysentruyt, L.C. On the origin of cancer metastasis. Crit. Rev. Oncog. 2013, 18, 43. [Google Scholar] [CrossRef] [Green Version]
- Jeong, D.; Watari, K.; Shirouzu, T.; Ono, M.; Koizumi, K.; Saiki, I.; Kim, Y.-C.; Tanaka, C.; Higuchi, R.; Miyamoto, T. Studies on lymphangiogenesis inhibitors from Korean and Japanese crude drugs. Biol. Pharm. Bull. 2013, 36, 152–157. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, S.K.; Abraham, A.; Bhat, B.; Jaggi, M.; Singh, A.T.; Sanna, V.K.; Singh, G.; Agarwal, S.K.; Mukherjee, R.; Burman, A.C. Synthesis of 13-amino costunolide derivatives as anticancer agents. Bioorg. Med. Chem. Lett. 2006, 16, 4195–4199. [Google Scholar] [CrossRef]
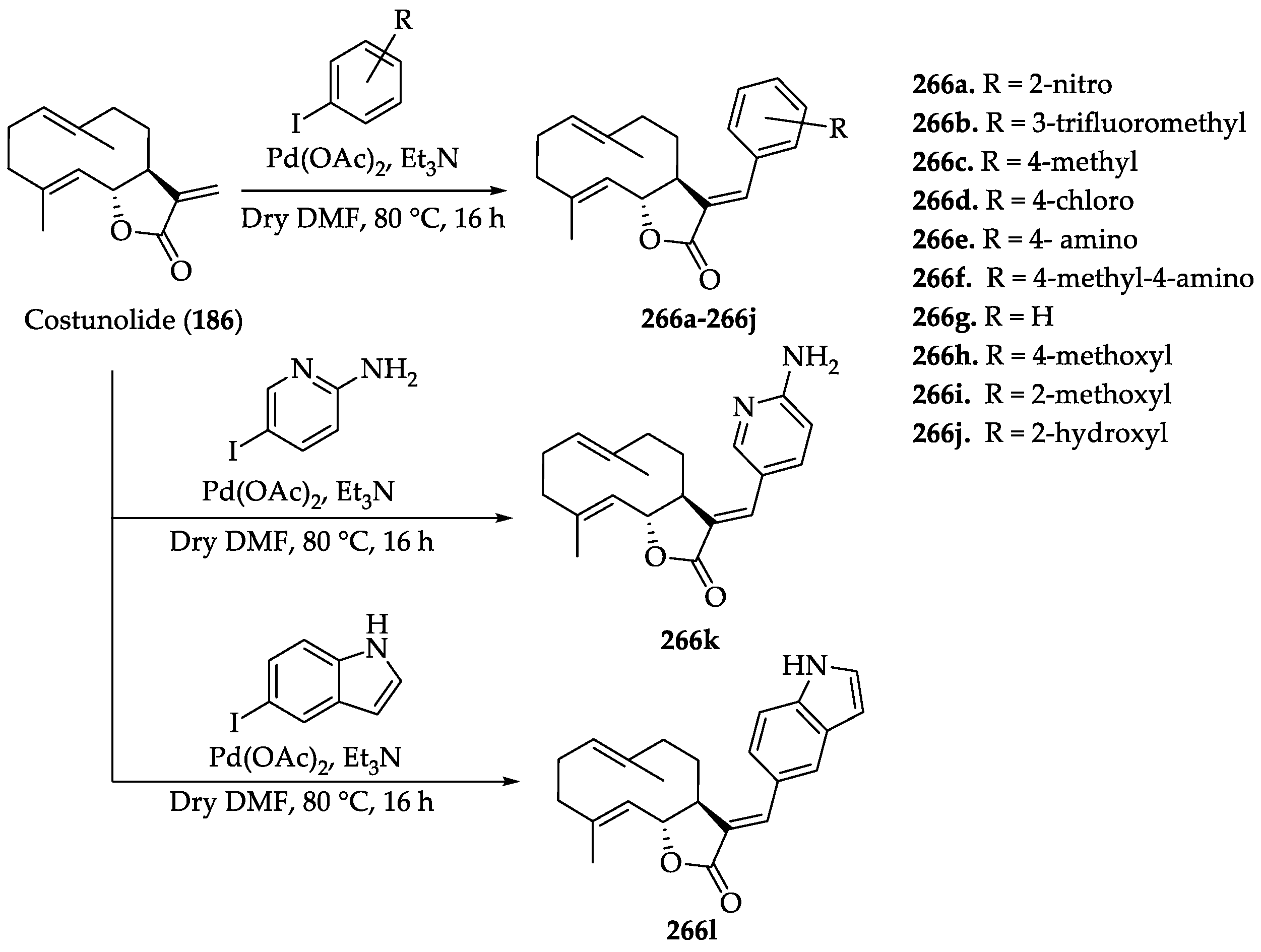
- Vadaparthi, P.R.; Kumar, C.P.; Kumar, K.; Venkanna, A.; Nayak, V.L.; Ramakrishna, S.; Babu, K.S. Synthesis of costunolide derivatives by Pd-catalyzed Heck arylation and evaluation of their cytotoxic activities. Med. Chem. Res. 2015, 24, 2871–2878. [Google Scholar] [CrossRef]
- Grieco, P.A.; Nishizawa, M. Total synthesis of (+)-costunolide. J. Org. Chem. 1977, 42, 1717–1720. [Google Scholar] [CrossRef]
- Shibuya, H.; Ohashi, K.; Kawashima, K.; Hori, K.; Murakami, N.; Kitagawa, I. Synthesis of (±)-Costunolide, an Antitumor Germacranolide, From E, E-Farnesol by Use of a Low-Valent Chromium Reagent. Chem. Lett. 1986, 15, 85–86. [Google Scholar] [CrossRef]
- Majdi, M.; Liu, Q.; Karimzadeh, G.; Malboobi, M.A.; Beekwilder, J.; Cankar, K.; de Vos, R.; Todorović, S.; Simonović, A.; Bouwmeester, H. Biosynthesis and localization of parthenolide in glandular trichomes of feverfew (Tanacetum parthenium L. Schulz Bip.). Phytochemistry 2011, 72, 1739–1750. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, H.; Kageura, T.; Inoue, Y.; Morikawa, T.; Yoshikawa, M. Absolute stereostructures and syntheses of saussureamines A, B, C, D and E, amino acid–sesquiterpene conjugates with gastroprotective effect, from the roots of Saussurea lappa. Tetrahedron 2000, 56, 7763–7777. [Google Scholar] [CrossRef]
- Geethangili, M.; Tzeng, Y.-M. Review of pharmacological effects of Antrodia camphorata and its bioactive compounds. Evid. Based Complementaryy Altern. Med. 2011, 2011, 212641. [Google Scholar]
- Ao, Z.-H.; Xu, Z.-H.; Lu, Z.-M.; Xu, H.-Y.; Zhang, X.-M.; Dou, W.-F. Niuchangchih (Antrodia camphorata) and its potential in treating liver diseases. J. Ethnopharmacol. 2009, 121, 194–212. [Google Scholar] [CrossRef]
- Rao, Y.K.; Wu, A.T.; Geethangili, M.; Huang, M.-T.; Chao, W.-J.; Wu, C.-H.; Deng, W.-P.; Yeh, C.-T.; Tzeng, Y.-M. Identification of antrocin from Antrodia camphorata as a selective and novel class of small molecule inhibitor of Akt/mTOR signaling in metastatic breast cancer MDA-MB-231 cells. Chem. Res. Toxicol. 2011, 24, 238–245. [Google Scholar] [CrossRef]
- Yeh, C.-T.; Huang, W.-C.; Rao, Y.K.; Ye, M.; Lee, W.-H.; Wang, L.-S.; Tzeng, D.T.; Wu, C.-H.; Shieh, Y.-S.; Huang, C.-Y.F. A sesquiterpene lactone antrocin from Antrodia camphorata negatively modulates JAK2/STAT3 signaling via microRNA let-7c and induces apoptosis in lung cancer cells. Carcinogenesis 2013, 34, 2918–2928. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.-H.; Wu, A.T.; Tzeng, D.T.; Huang, C.-C.; Tzeng, Y.-M.; Chao, T.-Y. Antrocin, a bioactive component from Antrodia cinnamomea, suppresses breast carcinogenesis and stemness via downregulation of β-catenin/Notch1/Akt signaling. Phytomedicine 2019, 52, 70–78. [Google Scholar] [CrossRef]
- Chen, Y.-A.; Tzeng, D.T.; Huang, Y.-P.; Lin, C.-J.; Lo, U.; Wu, C.-L.; Lin, H.; Hsieh, J.-T.; Tang, C.-H.; Lai, C.-H. Antrocin sensitizes prostate cancer cells to radiotherapy through inhibiting PI3K/AKT and MAPK signaling pathways. Cancers 2019, 11, 34. [Google Scholar] [CrossRef]
- Li, F.-Z.; Li, S.; Zhang, P.-P.; Huang, Z.-H.; Zhang, W.-B.; Gong, J.; Yang, Z. A chiral pool approach for asymmetric syntheses of (−)-antrocin,(+)-asperolide C, and (−)-trans-ozic acid. Chem. Comm. 2016, 52, 12426–12429. [Google Scholar] [CrossRef]
- Angamuthu, V.; Tai, D.-F. Synthesis of Natural (−)-Antrocin and Its Enantiomer via Stereoselective Aldol Reaction. Molecules 2020, 25, 831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz, S.; Cuesta, J.; González, A.; Bonjoch, J. Synthesis of (−)-nakamurol A and assignment of absolute configuration of diterpenoid (+)-Nakamurol A. J. Org. Chem. 2003, 68, 7400–7406. [Google Scholar] [CrossRef] [PubMed]
- Skepper, C.K.; Quach, T.; Molinski, T.F. Total synthesis of enigmazole A from Cinachyrella enigmatica. Bidirectional bond constructions with an ambident 2, 4-disubstituted oxazole synthon. J. Am. Chem. Soc. 2010, 132, 10286–10292. [Google Scholar] [CrossRef] [PubMed]
- Shao, F.-Y.; Wang, S.; Li, H.-Y.; Chen, W.-B.; Wang, G.-C.; Ma, D.-L.; Wong, N.S.; Xiao, H.; Liu, Q.-Y.; Zhou, G.-X. EM23, a natural sesquiterpene lactone, targets thioredoxin reductase to activate JNK and cell death pathways in human cervical cancer cells. Oncotarget 2016, 7, 6790. [Google Scholar] [CrossRef] [Green Version]
- Tonissen, K.F.; Di Trapani, G. Thioredoxin system inhibitors as mediators of apoptosis for cancer therapy. Mol. Nutr. Food Res. 2009, 53, 87–103. [Google Scholar] [CrossRef]
- Lincoln, D.T.; Ali, E.E.; Tonissen, K.F.; Clarke, F.M. The thioredoxin-thioredoxin reductase system: Over-expression in human cancer. Anticancer Res. 2003, 23, 2425. [Google Scholar]
- Powis, G.; Mustacich, D.; Coon, A. The role of the redox protein thioredoxin in cell growth and cancer. Free Radic. Biol. Med. 2000, 29, 312–322. [Google Scholar] [CrossRef]
- Chan, C.-O.; Jin, D.-P.; Dong, N.-P.; Chen, S.-B.; Mok, D.K.W. Qualitative and quantitative analysis of chemical constituents of Centipeda minima by HPLC-QTOF-MS & HPLC-DAD. J. Pharm. Biomed. Anal. 2016, 125, 400–407. [Google Scholar]
- ChangLong, L.; HeZhen, W.; YongPing, H.; YanFang, Y.; YanWen, L.; JianWen, L. 6-O-Angeloylenolin induces apoptosis through a mitochondrial/caspase and NF-κB pathway in human leukemia HL60 cells. Biomed. Pharmacother. 2008, 62, 401–409. [Google Scholar] [CrossRef]
- Li, C.; Wu, H.; Yang, Y.; Liu, J.; Chen, Z. Sesquiterpene lactone 6-O-angeloylplenolin reverses vincristine resistance by inhibiting YB-1 nuclear translocation in colon carcinoma cells. Oncol. Lett. 2018, 15, 9673–9680. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Du, Y.; Nan, J.; Zhang, X.; Qin, X.; Wang, Y.; Hou, J.; Wang, Q.; Yang, J. Brevilin A, a novel natural product, inhibits janus kinase activity and blocks STAT3 signaling in cancer cells. PLoS ONE 2013, 8, e63697. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.M.-L.; Chan, B.D.; Wong, W.-Y.; Leung, T.-W.; Qu, Z.; Huang, J.; Zhu, L.; Lee, C.-S.; Chen, S.; Tai, W.C.-S. Synthesis and Evaluation of Novel Anticancer Compounds Derived from the Natural Product Brevilin A. ACS Omega 2020, 5, 14586–14596. [Google Scholar] [CrossRef] [PubMed]
- Cooke, M.; Zhou, X.; Casado-Medrano, V.; Lopez-Haber, C.; Baker, M.J.; Garg, R.; Ann, J.; Lee, J.; Blumberg, P.M.; Kazanietz, M.G. Characterization of AJH-836, a diacylglycerol-lactone with selectivity for novel PKC isozymes. J. Biol. Chem. 2018, 293, 8330–8341. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.-H.; Kim, S.Y.; Lee, J.; Marquez, V.E.; Lewin, N.E.; Pearce, L.V.; Blumberg, P.M. Macrocyclic diacylglycerol-bis-lactones as conformationally constrained analogues of diacylglycerol-lactones. Interactions with protein kinase C. J. Med. Chem. 2004, 47, 4000–4007. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.; Benedetti, L.G.; Abera, M.B.; Wang, H.; Abba, M.; Kazanietz, M.G. Protein kinase C and cancer: What we know and what we do not. Oncogene 2014, 33, 5225–5237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mochly-Rosen, D.; Das, K.; Grimes, K.V. Protein kinase C, an elusive therapeutic target? Nat. Rev. Drug Discov. 2012, 11, 937–957. [Google Scholar] [CrossRef] [Green Version]
- Bosco, R.; Melloni, E.; Celeghini, C.; Rimondi, E.; Vaccarezza, M.; Zauli, G. Fine tuning of protein kinase C (PKC) isoforms in cancer: Shortening the distance from the laboratory to the bedside. Mini-Rev. Med. Chem. 2011, 11, 185–199. [Google Scholar] [CrossRef]
- Lee, J.; Lewin, N.E.; Acs, P.; Blumberg, P.M.; Marquez, V.E. Conformationally Constrained Analogues of Diacylglycerol. 13.1 Protein Kinase C Ligands Based on Templates Derived from 2, 3-Dideoxy-l-erythro (threo)-hexono-1, 4-lactone and 2-Deoxyapiolactone. J. Med. Chem. 1997, 40, 1560. [Google Scholar] [CrossRef] [Green Version]
- Ono, Y.; Fujii, T.; Igarashi, K.; Kuno, T.; Tanaka, C.; Kikkawa, U.; Nishizuka, Y. Phorbol ester binding to protein kinase C requires a cysteine-rich zinc-finger-like sequence. Proc. Natl. Acad. Sci. USA 1989, 86, 4868–4871. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.-H.; Kim, Y.; Won, S.-H.; Park, S.-K.; Lee, C.W.; Kim, H.-M.; Lewin, N.E.; Perry, N.A.; Pearce, L.V.; Lundberg, D.J. Polar 3-alkylidene-5-pivaloyloxymethyl-5′-hydroxymethyl-γ-lactones as protein kinase C ligands and antitumor agents. Bioorg. Med. Chem. Lett. 2010, 20, 1008–1012. [Google Scholar] [CrossRef] [Green Version]
- Duan, D.; Sigano, D.M.; Kelley, J.A.; Lai, C.C.; Lewin, N.E.; Kedei, N.; Peach, M.L.; Lee, J.; Abeyweera, T.P.; Rotenberg, S.A. Conformationally constrained analogues of diacylglycerol. 29. Cells sort diacylglycerol-lactone chemical zip codes to produce diverse and selective biological activities. J. Med. Chem. 2008, 51, 5198–5220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishizuka, Y. Protein kinase C and lipid signaling for sustained cellular responses. FASEB J. 1995, 9, 484–496. [Google Scholar] [CrossRef] [PubMed]
- Ann, J.; Yoon, S.; Baek, J.; Kim, D.H.; Lewin, N.E.; Hill, C.S.; Blumberg, P.M.; Lee, J. Design and synthesis of protein kinase C epsilon selective diacylglycerol lactones (DAG-lactones). Eur. J. Med. Chem. 2015, 90, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Cooke, M.; Casado-Medrano, V.; Ann, J.; Lee, J.; Blumberg, P.M.; Abba, M.C.; Kazanietz, M.G. Differential Regulation of Gene Expression in Lung Cancer Cells by Diacyglycerol-Lactones and a Phorbol Ester Via Selective Activation of Protein Kinase C Isozymes. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Boden, E.P.; Keck, G.E. Proton-transfer steps in Steglich esterification: A very practical new method for macrolactonization. J. Org. Chem. 1985, 50, 2394–2395. [Google Scholar] [CrossRef]
- Seigler, D. Plant Secondary Metabolites; Springer: Berlin/Heidelberg, Germany, 1998. [Google Scholar]
- Farhat, F.; Tariq, A.; Zikrea, A.; Fatima, R.N. Diterpenes from Different Fungal Sources and Their 13C-NMR Data. In Terpenes and Terpenoids; IntechOpen: London, UK, 2018; p. 111. [Google Scholar]
- Thisoda, P.; Rangkadilok, N.; Pholphana, N.; Worasuttayangkurn, L.; Ruchirawat, S.; Satayavivad, J. Inhibitory effect of Andrographis paniculata extract and its active diterpenoids on platelet aggregation. Eur. J. Pharmacol. 2006, 553, 39–45. [Google Scholar] [CrossRef]
- Chakravarti, R.; Chakravarti, M.D. Andrographolide, the Active Constituent of Andrographis Paniculata Nees. A Preliminary Communication. Ind. Med. Gaz. 1951, 86, 96. [Google Scholar]
- Aromdee, C. Modifications of andrographolide to increase some biological activities: A patent review (2006–2011). Expert Opin. Ther. Pat. 2012, 22, 169–180. [Google Scholar] [CrossRef]
- Mishra, S.K.; Tripathi, S.; Shukla, A.; Oh, S.H.; Kim, H.M. Andrographolide and analogues in cancer prevention. Front. Biosci. Elite Ed. 2015, 7, 255. [Google Scholar] [CrossRef]
- Dai, L.; Wang, G.; Pan, W. Andrographolide inhibits proliferation and metastasis of SGC7901 gastric cancer cells. Biomed Res. Int. 2017, 2017. [Google Scholar] [CrossRef]
- Monger, A.; Boonmuen, N.; Suksen, K.; Saeeng, R.; Kasemsuk, T.; Piyachaturawat, P.; Saengsawang, W.; Chairoungdua, A. Inhibition of topoisomerase IIα and induction of apoptosis in gastric cancer cells by 19-triisopropyl andrographolide. Asian Pac. J. Cancer Rev. 2017, 18, 2845. [Google Scholar]
- Mir, H.; Kapur, N.; Singh, R.; Sonpavde, G.; Lillard, J.W., Jr.; Singh, S. Andrographolide inhibits prostate cancer by targeting cell cycle regulators, CXCR3 and CXCR7 chemokine receptors. Cell Cycle 2016, 15, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Chun, J.Y.; Tummala, R.; Nadiminty, N.; Lou, W.; Liu, C.; Yang, J.; Evans, C.P.; Zhou, Q.; Gao, A.C. Andrographolide, an herbal medicine, inhibits interleukin-6 expression and suppresses prostate cancer cell growth. Genes Cance 2010, 1, 868–876. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.C.; Wong, C.C.; Sagineedu, S.R.; Loke, S.C.; Lajis, N.H.; Stanslas, J. SRJ23, a new semisynthetic andrographolide derivative: In vitro growth inhibition and mechanisms of cell cycle arrest and apoptosis in prostate cancer cells. Cell Biol. Toxicol. 2014, 30, 269–288. [Google Scholar] [CrossRef]
- Wang, W.; Guo, W.; Li, L.; Fu, Z.; Liu, W.; Gao, J.; Shu, Y.; Xu, Q.; Sun, Y.; Gu, Y. Andrographolide reversed 5-FU resistance in human colorectal cancer by elevating BAX expression. Biochem. Pharmacol. 2016, 121, 8–17. [Google Scholar] [CrossRef]
- Lin, H.-H.; Shi, M.-D.; Tseng, H.-C.; Chen, J.-H. Andrographolide sensitizes the cytotoxicity of human colorectal carcinoma cells toward cisplatin via enhancing apoptosis pathways in vitro and in vivo. Toxicol. Sci. 2014, 139, 108–120. [Google Scholar] [CrossRef] [Green Version]
- Shu, Y.; Sun, J.; Cai, P.; Wang, W.; Han, X.; Gu, Y. An open-label, randomized, controlled clinical trial to explore the curative effects between the treatment of capecitabine and andrographolide and the single capecitabine in the patients with pathological and/or histologic diagnosed unresectable, advanced, recurrent, and metastatic colorectal cancer. Am. J. Clin. Oncol. 2017, 35, TPS819. [Google Scholar]
- Mi, S.; Xiang, G.; Yuwen, D.; Gao, J.; Guo, W.; Wu, X.; Wu, X.; Sun, Y.; Su, Y.; Shen, Y. Inhibition of autophagy by andrographolide resensitizes cisplatin-resistant non-small cell lung carcinoma cells via activation of the Akt/mTOR pathway. Toxicol. Appl. Pharm. 2016, 310, 78–86. [Google Scholar] [CrossRef]
- Yuan, H.; Sun, B.; Gao, F.; Lan, M. Synergistic anticancer effects of andrographolide and paclitaxel against A549 NSCLC cells. Pharm. Biol. 2016, 54, 2629–2635. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.C.W.; Jeyaraj, E.J.; Sagineedu, S.R.; Wong, W.S.F.; Stanslas, J. SRS06, a new semisynthetic andrographolide derivative with improved anticancer potency and selectivity, inhibits nuclear factor-κB nuclear binding in the A549 non-small cell lung cancer cell line. Pharmacology 2015, 95, 70–77. [Google Scholar] [CrossRef]
- Gao, H.-T.; Wang, B.-L.; Li, W.-D.Z. Synthetic applications of homoiodo allylsilane II. Total syntheses of (−)-andrographolide and (+)-rostratone. Tetrahedron 2014, 70, 9436–9448. [Google Scholar] [CrossRef]
- Li, W.-D.Z.; Yang, J.-H. A novel synthesis of functionalized allylsilanes. Organic Lett. 2004, 6, 1849–1852. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Fujitani, R.; Takeda, Y.; Takaishi, Y.; Yamada, T.; Kido, M.; Miura, I. On the diterpenoids of Andrographis paniculata: X-ray crystallographic analysis of andrographolide and structure determination of new minor diterpenoids. Chem. Pharm. Bull. 1984, 32, 2117–2125. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Wurm, T.; Sharma Poudel, B.; Krische, M.J. Enantioselective Total Synthesis of Andrographolide and 14-Hydroxy-Colladonin: Carbonyl Reductive Coupling and trans-Decalin Formation by Hydrogen Transfer. Angew. Chem. 2020, 59, 23169–23173. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, K.; Wan, K.K.; Oppedisano, A.; Crossley, S.W.; Shenvi, R.A. Simple, chemoselective hydrogenation with thermodynamic stereocontrol. J. Am. Chem. Soc. 2014, 136, 1300–1303. [Google Scholar] [CrossRef]
- Hayashi, Y.; Takahashi, S.; Ona, H.; Sakan, T. Structures of nagilactone a, b, c, and d, novel nor-and bisnorditerpenoids. Tetrahedron Lett. 1968, 9, 2071–2076. [Google Scholar] [CrossRef]
- Qi, Y.Y.; Su, J.; Zhang, Z.J.; Li, L.W.; Fan, M.; Zhu, Y.; Wu, X.D.; Zhao, Q.S. Two New Anti-Proliferative C18-Norditerpenes from the Roots of Podocarpus macrophyllus. Chem. Biodiversity 2018, 15, e1800043. [Google Scholar] [CrossRef]
- Sato, K.; Inaba, Y.; Park, H.-S.; Akiyama, T.; Koyama, T.; Fukaya, H.; Aoyagi, Y.; Takeya, K. Cytotoxic Bisnor-and Norditerpene Dilactones Having 7α, 8α-Epoxy-9, 11-enolide Substructure from Podocarpus macrophyllus D. D ON. Chem. Pharm. Bull. 2009, 57, 668–679. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.-L.; Feng, Z.-L.; Su, M.-X.; Jiang, X.-M.; Chen, X.; Wang, Y.; Li, A.; Lin, L.-G.; Lu, J.-J. Downregulation of Cyclin B1 mediates nagilactone E-induced G2 phase cell cycle arrest in non-small cell lung cancer cells. Eur. J. Pharmacol. 2018, 830, 17–25. [Google Scholar] [CrossRef]
- Guo, J.; Jiang, X.-M.; Chen, X.-P.; Wang, Y.-T.; Li, A.; Lin, L.-G.; Li, H.; Lu, J.-J. Identification of nagilactone E as a protein synthesis inhibitor with anticancer activity. Acta Pharmacol. Sin. 2020, 41, 698–705. [Google Scholar]
- Liu, K.; Chen, H.-L.; Wang, S.; Gu, M.-M.; Chen, X.-M.; Zhang, S.-L.; Yu, K.-J.; You, Q.-S. High expression of RIOK2 and NOB1 predict human non-small cell lung cancer outcomes. Sci. Rep. 2016, 6, 28666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, Y.; Matsumoto, T.; Nishizawa, M.; Togami, M.; Hyono, T.; Nishikawa, N.; Uemura, M.; Sakan, T. Total synthesis of nagilactone F, a biologically active norditerpenoid dilactone isolated from Podocarpus nagi. J. Org. Chem. 1982, 47, 3428–3433. [Google Scholar] [CrossRef]
- Hanessian, S.; Boyer, N.; Reddy, G.J.; Deschênes-Simard, B. Total synthesis of oidiodendrolides and related norditerpene dilactones from a common precursor: Metabolites CJ-14,445, LL-Z1271γ, oidiolactones A, B, C, and D, and nagilactone F. Org. Lett. 2009, 11, 4640–4643. [Google Scholar] [CrossRef] [PubMed]
- Ling, T.; Chowdhury, C.; Kramer, B.A.; Vong, B.G.; Palladino, M.A.; Theodorakis, E.A. Enantioselective synthesis of the antiinflammatory agent (−)-acanthoic acid. J. Org. Chem. 2001, 66, 8843–8853. [Google Scholar] [CrossRef] [PubMed]
- Waters, S.P.; Tian, Y.; Li, Y.-M.; Danishefsky, S.J. Total synthesis of (−)-scabronine G, an inducer of neurotrophic factor production. J. Am. Chem. Soc. 2005, 127, 13514–13515. [Google Scholar] [CrossRef]
- Welch, S.; Hagan, C.; White, D.; Fleming, W.; Trotter, J. A stereoselective total synthesis of the antifungal mold metabolite 7.alpha.-methoxy-3a,10b-dimethyl-1,2,3,3a.alpha.,5a.alpha.,7,10b.beta.,10c.alpha.-octahydro-4H,9H-furo[2′,3′,4′:4,5]naphtho[2,1-c]pyran-4,10-dione. J. Am. Chem. Soc. 1977, 99, 549–556. [Google Scholar] [CrossRef]
- Barrero, A.F.; Sánchez, J.F.; Elmerabet, J.; Jiménez-González, D.; Macías, F.A.; Simonet, A.M. Enantiospecific syntheses of the potent bioactives nagilactone F and the mould metabolite LL-Z1271α an evaluation of their allelopathic potential. Tetrahedron 1999, 55, 7289–7304. [Google Scholar] [CrossRef]
- Feng, L.; Hwang, D. Method of Use of Radicicol for Treatment of Inflammation and Endotoxemia. International Patent Application WO9625928A1, 29 August 1996. [Google Scholar]
- Winssinger, N.; Barluenga, S.; Karplus, M. Synthesis of Resorcylic Acid Lactones Useful as Therapeutic Agents. International Patent Application WO2009091921A1, 23 July 2009. [Google Scholar]
- Chen, R.; Rubenstein, A.E.; Shen, X.; Yu, J.-C.; Giovannini, M. Preparation of Radicicol and Related Macrocyclic Compounds Which Inhibit HSP90 for Therapeutic Use in the Treatment of Neurofibromatosis. International Patent Application WO2008150302A1, 11 December 2008. [Google Scholar]
- Der, S.S. Reagents, Compositions Based on Heat Shock Response Activation and/or Antioxidant Response, and Methods for Improving Viability and Function of Cells, Tissues and Organs. International Patent Application WO2017214709A1, 21 December 2017. [Google Scholar]
- Danishefsky, S.J.; Garbaccio, R.M.; Baeschlin, D.K.; Stachel, S.J.; Solit, D.; Shtil, A.; Rosen, N. Preparation of therapeutic macrocyclic natural product derivatives. International Patent Application WO2002016369A2, 28 February 2002. [Google Scholar]
- Botchkareva, N.; Ahluwalia, G.S.; Shander, D. Use of Heat Shock Protein Inhibitors for the Reduction of Hair Growth. International Patent Application WO2005105023A1, 10 November 2005. [Google Scholar]
- Santi, D.V.; Reid, R.C.; Hutchinson, R.C.; Sundermann, K.F.; Lau, J. Resorcylic Acid Lactone Kinase Inhibitors, and Their Therapeutic Use for the Treatment of Cancers and Other Conditions. International Patent Application WO2006036941A2, 6 April 2006. [Google Scholar]
- Tremble, P. Methods and Compositions Comprising an Ubiquitin Activator for Inhibiting Narrowing in Mammalian Vascular Pathways. U.S. Patent Application US20040243224A1, 2 December 2004. [Google Scholar]
- Winssinger, N.; Barluenga, S. Preparation of Macrolides as Irreversible Inhibitors Useful for the Treatment of Kinase-Related Pathologies. International Patent Application WO2011036299A1, 31 March 2011. [Google Scholar]
- Alici, E.; Duru, A.; Sutlu, T. Enhanced Gene Delivery to Natural Killer Cells, Hematopoietic Stem Cells and Macrophages. International Patent Application WO2017059177A2, 6 April 2017. [Google Scholar]
- Pollack, A.; Dvashi, Z. Treatments for Fibrotic Diseases Using Transforming Growth Factor β Activated Kinase 1 (TAK1) inhibitors. U.S. Patent Application US20160206591A1, 21 July 2016. [Google Scholar]
- Pearce, C.; Croatt, M.P.; Fakhouri, L.; Oberlies, N.H. Preparation of Difluororesorcylic acid Lactone Derivatives for Use as TAK-1 Inhibitors. International Patent Application WO2016196256A2, 8 December 2016. [Google Scholar]
- Boivin, R.; Chiba, K.; Davis, H.A.; Diepitro, L.; Du, H.; Eguchi, Y.; Fujita, M.; Gilbert, S.; Goto, M.; Harmange, J.C. Preparation of Macrocyclic Compounds for Use in Pharmaceutical and Cosmetic Compositions Which Regulate Various Genes Involved in Immune and Inflammatory Responses. International Patent Application WO2003076424A1, 18 September 2003. [Google Scholar]
- Chiba, K.; Du, H.; Eguchi, Y.; Fujita, M.; Goto, M.; Gusovsky, F.; Harmange, J.-C.; Inoue, A.; Kawada, M.; Kawai, T. Preparation of Macrocyclic Compounds for the Treatment of Inflammation and Autoimmune Disorders. U.S. Patent Application US20040224936A1, 11 November 2004. [Google Scholar]
- Neel, B.G.; Mohi, G. Combination of mTOR Inhibitor and a Tyrosine Kinase Inhibitor for the Treatment of Neoplasms. International Patent Application WO2004004644A2, 15 January 2004. [Google Scholar]
- Litvin, O.; Rosen, N.; Pe’er, D. Methods of Treating Cancer Using Combinations of Interferon and MAPK Pathway Inhibitors. U.S. Patent Application US20150086509A120150326, 26 March 2015. [Google Scholar]
- Sim, T.; Yoon, H.; Kim, J. Preparation of Resorcyclic Acid Lactone Compound as Anticancer Agent. Korea Patent Application KR1387400B1, 21 April 2014. [Google Scholar]
- Fusan, R.; Jordan, C.T.; Kolev, J.N. Parthenolide Derivatives, Methods for Their Preparation and Their Use as Anticancer Agents. U.S. Patent Application US20160115508A1, 28 April 2016. [Google Scholar]
- Al-Fatlawi, A.A.; Al-Fatlawi, A.A.; Rahisuddin, I.; Ahmad, A. Effect of parthenolide on growth and apoptosis regulatory genes of human cancer cell lines. Pharm. Biol. 2015, 53, 104–109. [Google Scholar] [CrossRef] [Green Version]
- Crooks, P.; Jordan, C.T.; Wei, X. Use of Parthenolide Derivatives as Antileukemic and Cytotoxic Agents. U.S. Patent Application US008716329B2, 5 June 2014. [Google Scholar]
- Shanmugam, R.; Kusumanchi, P.; Cheng, L.; Crooks, P.; Neelakantan, S.; Matthews, W.; Nakshatri, H.; Sweeney, C.J. A water-soluble parthenolide analogue suppresses in vivo prostate cancer growth by targeting NFκB and generating reactive oxygen species. Prostate 2010, 70, 1074–1086. [Google Scholar] [CrossRef]
- Pei, S.; Guzman, M.L.; Nasim, S.; Shi, L.; Crooks, P.A.; Jordan, C.T. Analysis of the Anti-Leukemia Mechanism of Parthenolide; American Society of Hematology: Washington, DC, USA, 2009. [Google Scholar]
- Kim, Y.; Chun, J.; Kim, M. Pharmaceutical Composition for Preventing and Treating Breast Cancer Containing Inula Helenium Hexane Fraction Having STAT3 Inhibitory Activity or Compound Isolated Therefrom as Active Ingredient. International Patent Application WO2015130081, 9 March 2015. [Google Scholar]
- Shyur, L.; Chao, W.; Cheng, Y. Use of Deoxyelephantopin (DET) and Analogues Thereof for Reducing Side Effects of an Anti-cancer Agent. U.S. Patent Application US9173868B2, 3 November 2015. [Google Scholar]
- Shyur, L.; Chao, W.; Cheng, Y. Use of Deoxyelephantopin (DET) and Analogues Thereof for Treatment of Melanoma. U.S. Patent Application US8754121B2, 17 June 2014. [Google Scholar]
- Kabeer, F.A.; Rajalekshmi, D.S.; Nair, M.S.; Prathapan, R. Molecular mechanisms of anticancer activity of deoxyelephantopin in cancer cells. Integr. Med. Res. 2017, 6, 190–206. [Google Scholar] [CrossRef]
- Su, M.; Wu, X.; Chung, H.Y.; Li, Y.; Ye, W. Antiproliferative activities of five Chinese medicinal herbs and active compounds in Elephantopus scaber. Nat. Prod. Commun. 2009, 4, 1934578X0900400802. [Google Scholar] [CrossRef] [Green Version]
- Shyur, L.; Lee, K.; Nakagawa, K.; Feng, J.; Chen, J.; Lee, W.; Cheng, Y.; Huang, J. Sesquiterpene Derivatives and Their Use in Inflammation or Cancer Treatment. U.S. Patent Application US10238631B2, 26 April 2019. [Google Scholar]
- Su, W.; Jia, H.; Zhang, W.; Yan, X.; Duan, J.; Wang, T.; Cai, Y. Costunolide Derivatives. U.S. Patent Application US7488836B2, 10 February 2009. [Google Scholar]
- Yan, Z.; Xu, T.; An, Z.; Hu, Y.; Chen, W.; Ma, J.; Shao, C.; Zhu, F. Costunolide induces mitochondria-mediated apoptosis in human gastric adenocarcinoma BGC-823 cells. BMC Complement. Altern. Med. 2019, 19, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheu, C.; Hattori, M. Compounds Isolated from Antrodia Cinnamomea and Use Thereof. U.S. Patent Application US20100210865A1, 19 August 2010. [Google Scholar]
- Yang, Z.; Tzeng, Y.; Li, C.; Luo, T.; Shi, H.; Yeh, C. Method for Chemical Synthesis of Antrocin and Use Thereof for Suppressing Non-Small Cell Lung Cancer. U.S. Patent Application US9045450B2, 2 June 2015. [Google Scholar]
- Tzeng, Y.; Yang, Z.; Yeh, C. Antrocin Containing Pharmaceutical Compositions for Inhibiting Cancer Cells. U.S. Patent Application US20120100175A1, 26 April 2012. [Google Scholar]
- Zhao, C.; Chen, X.; Du, Y.; Yang, J.; Wang, Q. Application of Brevilin A When Serving as JAK-STATs Signal Target Inhibitor. CN102836151, 26 December 2012. [Google Scholar]
- Wang, Y.; Jiang, X.; Jiang, J.; Zhang, Z.; Yang, Z.; Yu, P. Andrographolide Derivatives and Use Thereof in Manufacture of Medicaments. International Patent Application WO2009018780A1, 12 February 2009. [Google Scholar]
- Murphy, J.; Heptinstall, S.; Mitchell, J. Randomised double-blind placebo-controlled trial of feverfew in migraine prevention. Lancet 1988, 332, 189–192. [Google Scholar] [CrossRef]
- Hewamana, S.; Lin, T.T.; Jenkins, C.; Burnett, A.K.; Jordan, C.T.; Fegan, C.; Brennan, P.; Rowntree, C.; Pepper, C. The novel nuclear factor-κB inhibitor LC-1 is equipotent in poor prognostic subsets of chronic lymphocytic leukemia and shows strong synergy with fludarabine. Clin. Cancer Res. 2008, 14, 8102–8111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peese, K. New agents for the treatment of leukemia: Discovery of DMAPT (LC-1). Drug Discov. Today 2010, 7, 322. [Google Scholar] [CrossRef]
- Yanhong, G. Study of Andrographolides with or Without Capecitabine to Treat Colorectal Cancer. NCT01993472. Available online: https://clinicaltrials.gov/ct2/show/NCT01993472 (accessed on 29 November 2020).



































































































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
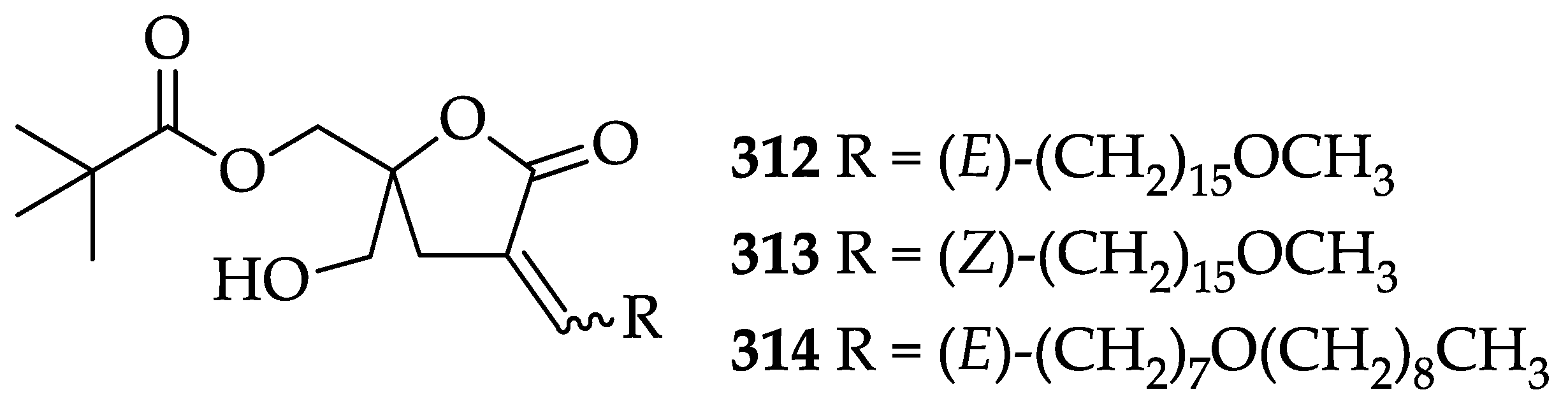{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
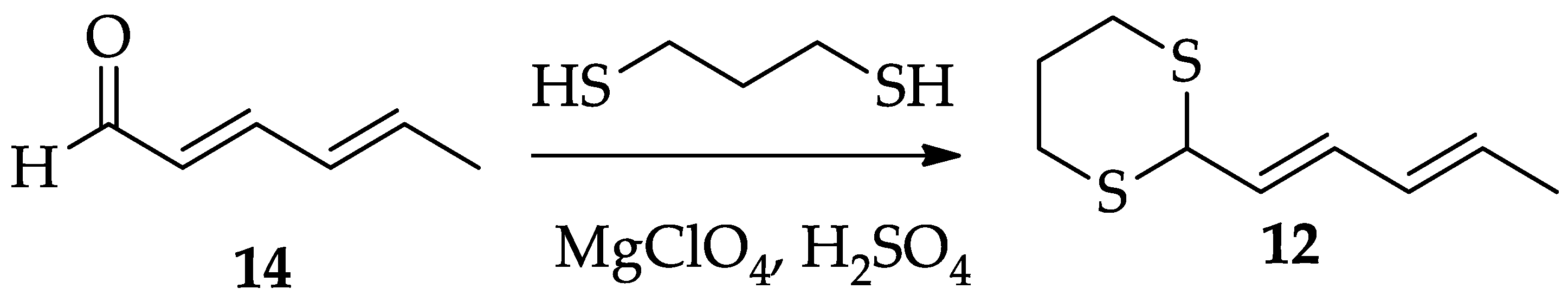{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
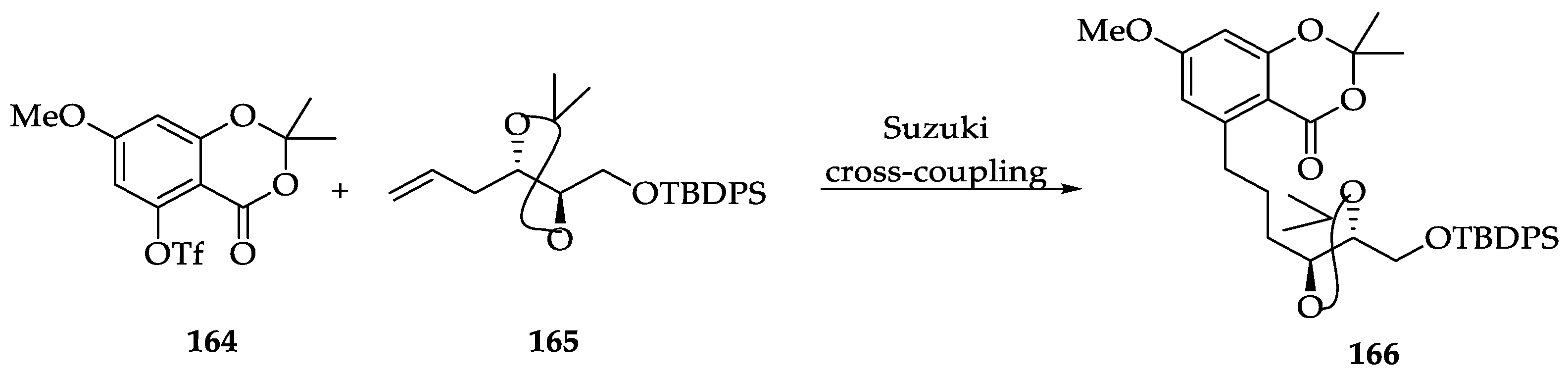{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
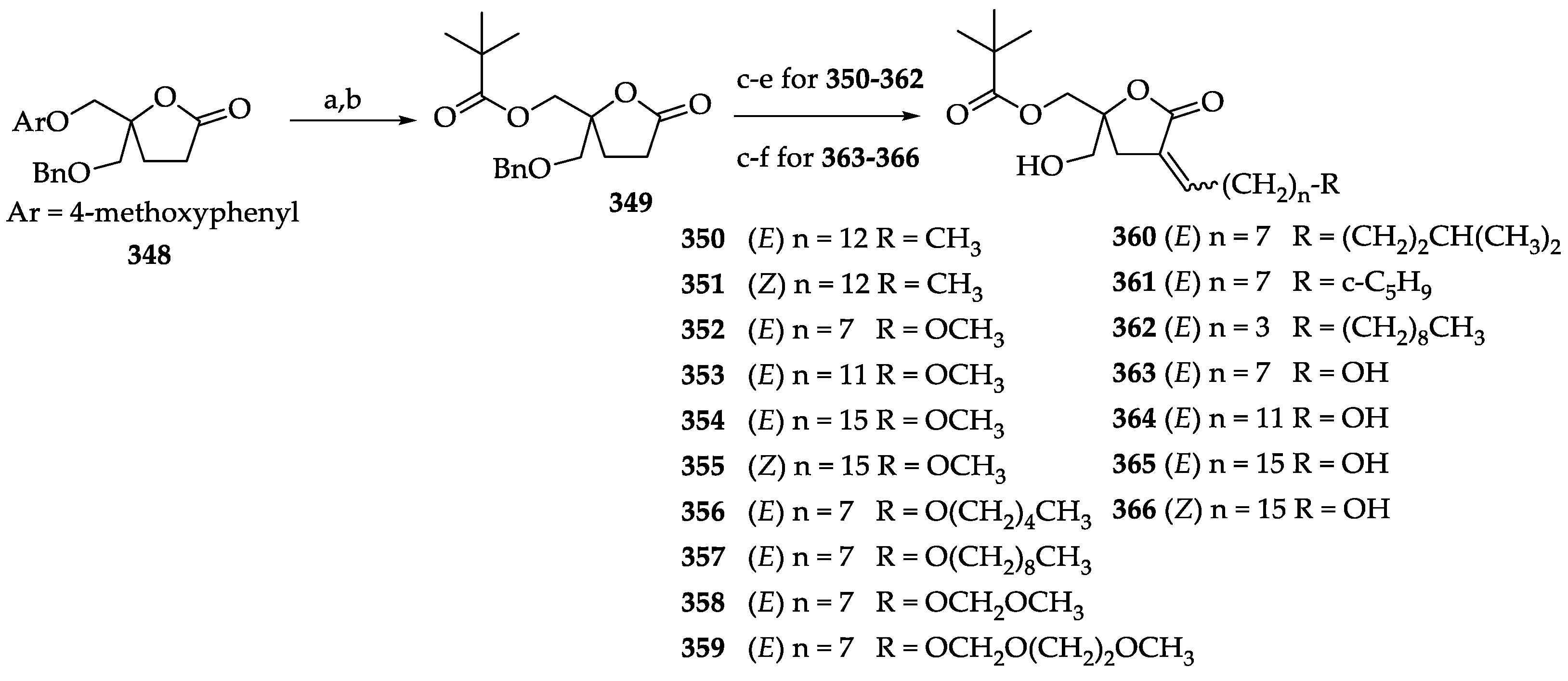{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class | Compound | Summary of Biological Activities | Patents | References |
|---|---|---|---|---|
| RALs | Radicicol (1) | It selectively inhibits HSP90 function (IC50 = 20–23 nM). Poor in vivo activity, probably due to chemical instability in serum and its rapid conversion into inactive metabolites. | [235,236,237,238] | [24] |
| RALs | 32 | It selectively inhibits HSP90 function (IC50 = 160 nM). | [239] | [29] |
| RALs | KF25706 (39) | Significant growth-inhibitory activity against human breast carcinoma MX-1 cells transplanted into nude mice at a dose of 100 mg/kg twice daily for five consecutive iv injections. | [240] | [31] |
| RALs | Hypothemycin (2) | RAS-signaling pathway inhibitor. It also inhibits the production of several cytokines such as IL2, IL6, IFNγ, and TNFα. It inhibited the growth of HT29 and HCT116 cells in serum-free defined medium, IC50 = 0.078 mM and 0.90 mM, respectively. | [241,242,243] | [35] |
| RALs | LL-Z1640-2 (3) | It selectively inhibits (TGF)-β-activated kinase 1 (TAK1) with high potency TAK1, (IC50 = 8.1 nM). Strongly inhibition JNK/p38 pathway. It also inhibits MEK1 (IC50 = 411 nM) and three other MAPKKKs (IC50 ≥ 268 nM against MEKK1, ASK1, and MEKK4). It can be applied clinically to CNS autoimmune disorders. | [244,245,246] | [38] |
| RALs | 74a-d | Its activity is comparable to 3, but with improved solubility and pharmacokinetic properties. | [46] | |
| RALs | ER803064 (79) | Increased metabolic stability and reduced potency than 3, active in vivo, but the ED50 value (13.2 mg/kg, iv) was fairly high in regard to TNF-α suppression | [246,247] | [48] |
| RALs | 83 and 84 | A MEK1 and MEKK1 inhibitor. Similar in vitro potency to natural product 3 and improved in vivo potency by iv administration. TNFα-PLAP IC50s: 32 nM for 83, 15 nM for 84. ED50: 6.5 mg/kg for 84. | [247,248] | [49] |
| RALs | 90 | Active against MNK2 kinase (IC50 = 7.2 μM). | [50] | |
| RALs | L-783277 (4) | Highly potent inhibitory activity against MEK (IC50 of 4 nM). Potent inhibitory activities against several kinases including VEGFR2/3, FLT1/3/4, MEK1/2, KDR, and PDGFRα but with low kinome selectivity. | [241,243,249,250] | [47,52] |
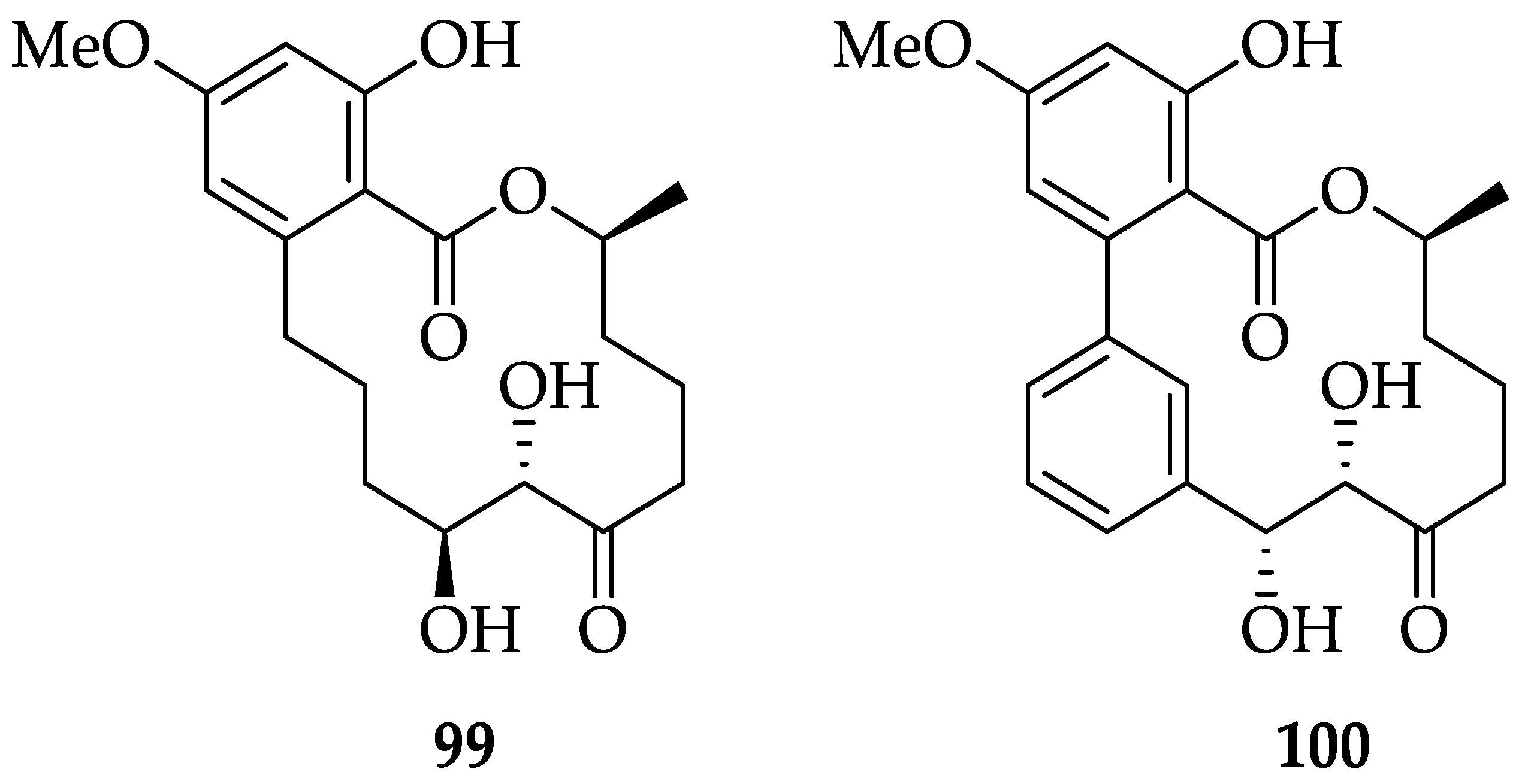
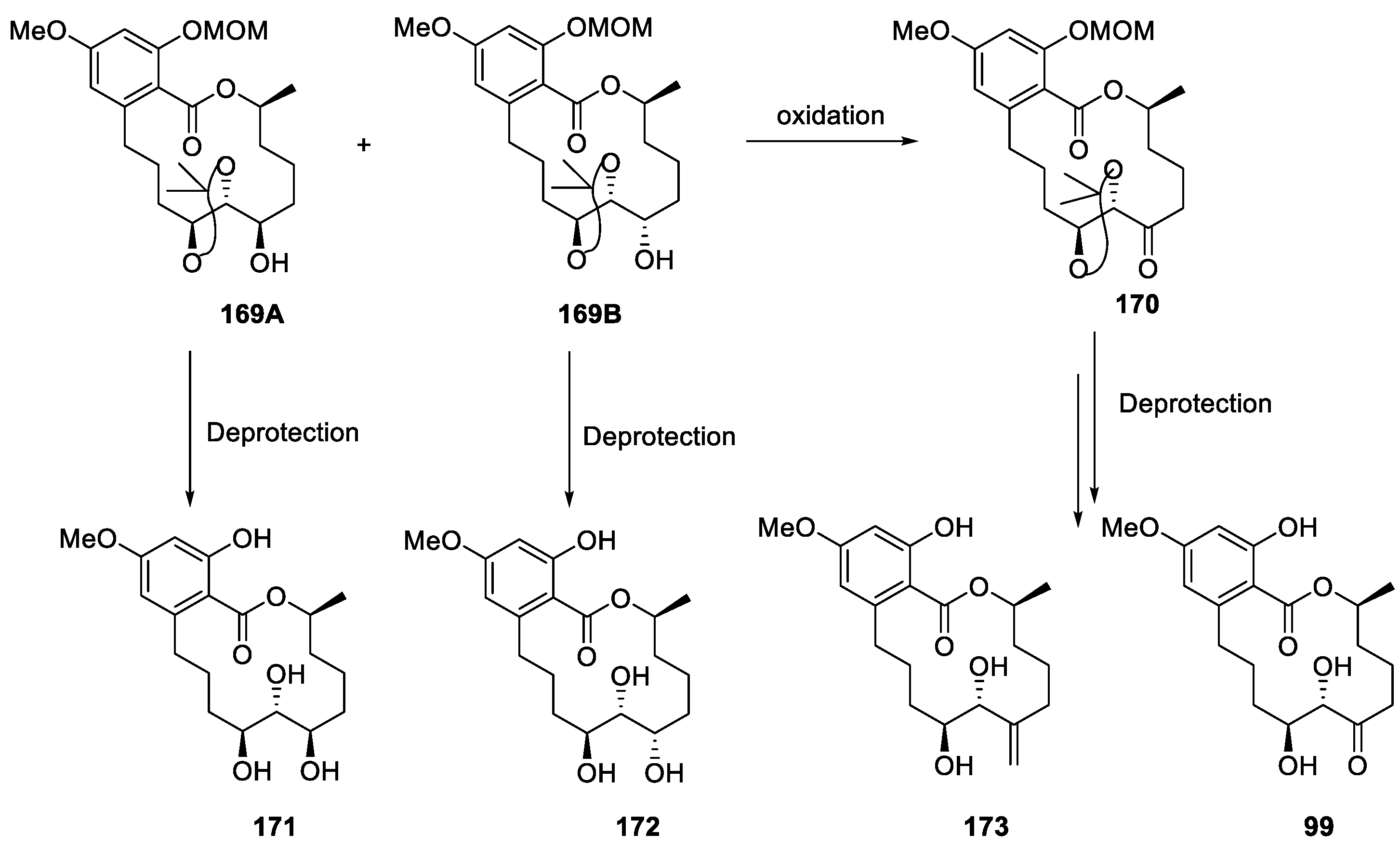
| RALs | 99 | A selective and potent ALK1 inhibitor It inhibits ALK1 with an IC50 value of 62 nM and activates Smad4 by phosphorylating Smad1/5. It acts by selectively blocking BMP9-induced ALK1 signaling in C2C12 cells. | [56,57,58] | |
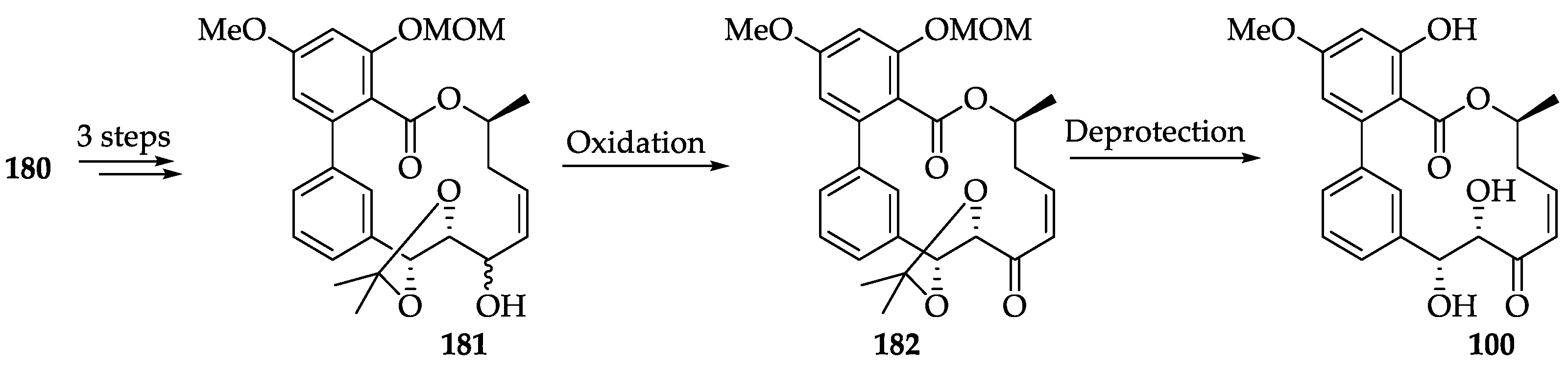
| RALs | 100 | Potent dual VEGFR3 and VEGFR2 inhibitor (VEGFR3 IC50 = 1.15 nM, VEGFR2 IC50 = 3.56 nM). It effectively suppresses both lymphangiogenesis and angiogenesis in a 3D-microfluidic tumor lymphangiogenesis assay and in vivo corneal assay. | [251] | [54] |
| SLs | Parthenolide (183) | IC50 values against SiHa and MCF-7 cells (8.42 and 9.54 μM, respectively). It prevents resistance of MDA-MB-231 to doxorubicin and mitoxantrone. Cytotoxicity in a wide variety of human cancers, targeting IKK-β, and FAK 1 inhibition In a mouse xenograft model, it decreased tumor size in combination with docetaxel. | [252] | [80,81,82,83,253] |
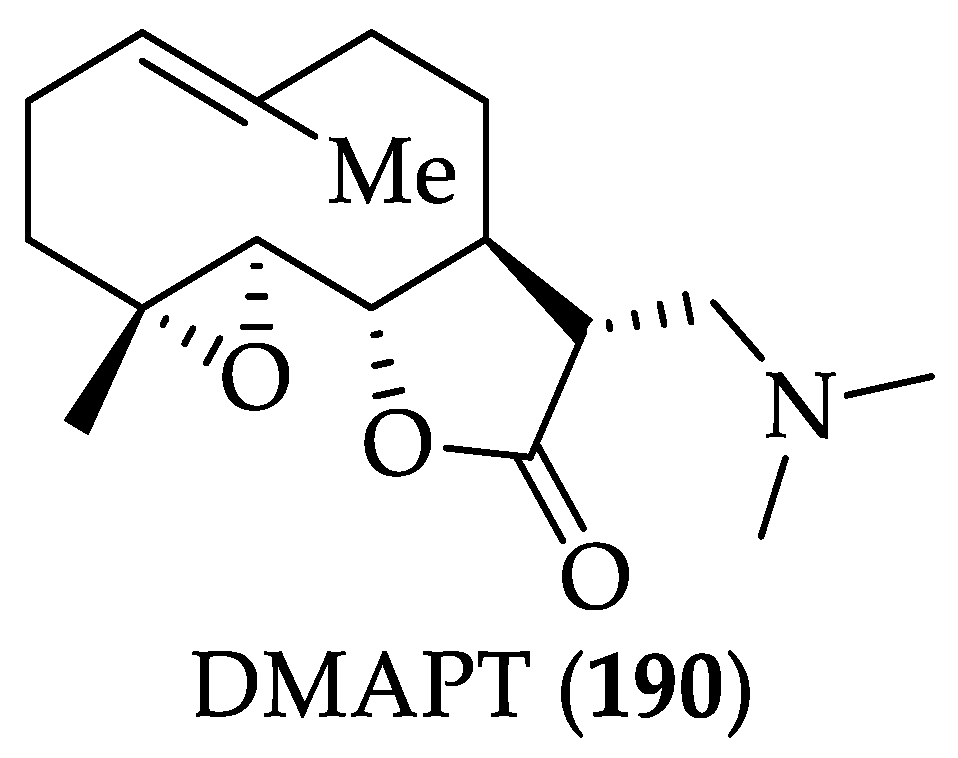
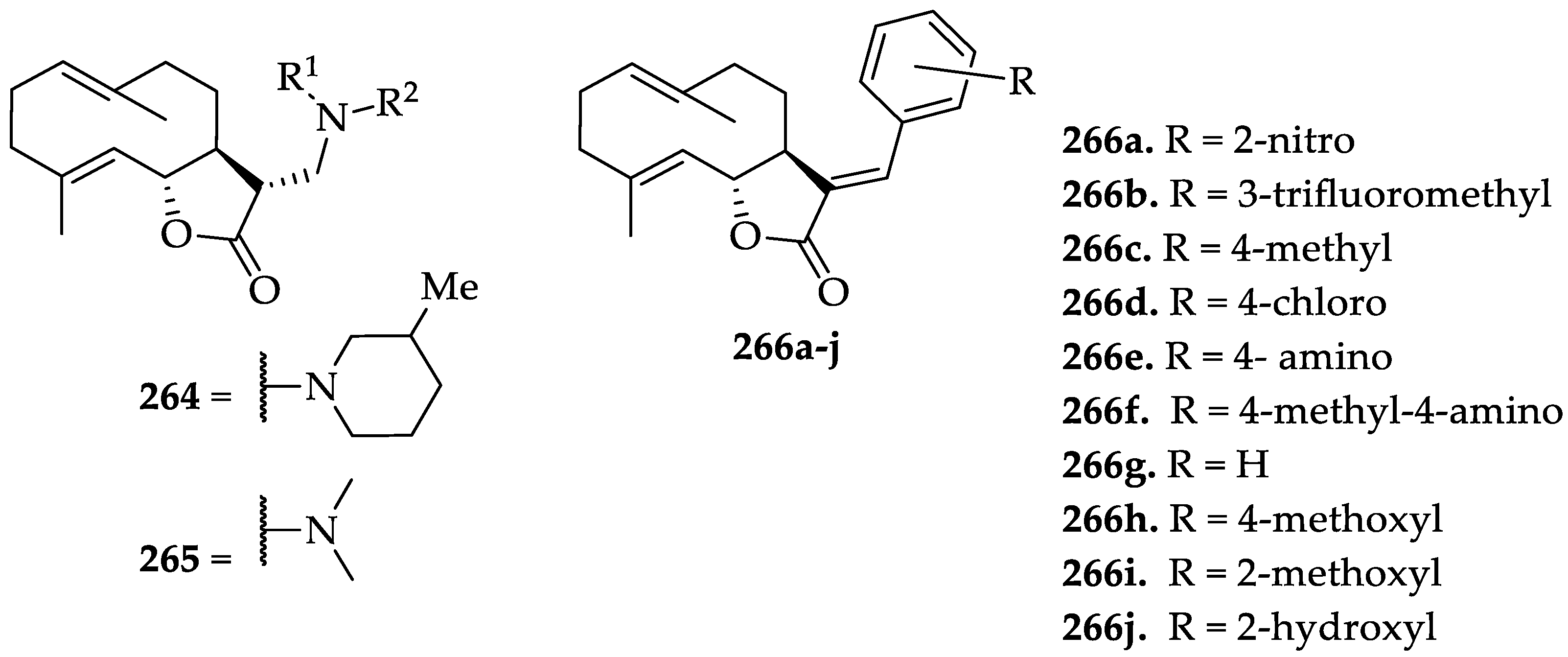
| SLs | DMAPT (190) | It selectively eliminates AML stem cells. DMAPT (190) significantly suppressed PC-3 tumor growth until day 95 compared with control (P = 0.0007) and resulted in greater tumor control than that observed with docetaxel (P = 0.007). In A549 subcutaneous xenograft, it reduced tumor growth by 54% (100 mg/kg/day, po). In UMUC-3 (transitional carcinoma) xenograft, it suppressed tumor growth by 63% (100 mg/kg oral twice/day). It has an increased PK profile compared to 183 | [252,254] | [89,255,256] |
| SLs | Alantolactone (184) | IC50 values against MDA-MB 231 and HUVEC cells (40 μM and 14.2 μM, respectively). | [257] | [104] |
| SLs | Deoxyelephantopin (185) | IC50 values against HCT116 (colorectal), K562 (CML), KB (oral), and T47D (breast) cancer cell lines are 7.46, 4.02, 3.35, 1.86 μg/mL, respectively. IC50 values against PC-3, CNE, and HL-60 cells are 4.6, 2.6, and 0.9 μM, respectively. It is an inhibitor of NF-κB and targets PPAR-γ. | [258,259] | [130,131,260,261] |
| SLs | DETD-35 (232) | IC50 value against MDA-MB-231 is 3.5 μM. In combination with paclitaxel, it shows synergistic effects on MDA-MB 231 cells. It also synergistic effects with vemurafenib to overcome BRAFV600E mutant melanoma in a mouse model | [262] | [127,137] |
| SLs | Costunolide (186) | It shows inhibitory activities on TR-LE cells. IC50 value against SW-620 cells is 7.8 μM. IC50 value against BGC-823 cells at 24 and 48 h is 32.80 and 23.12 μM, respectively. | [263] | [162,163,264] |
| SLs | 264 | IC50 value against SW-620 cells is 3.3 μM. | [163] | |
| SLs | Antrocin (187) | IC50 value against MDA-MB-231 cells is 0.6 μM. IC50 values against H441 (wt-EGFR) and H1975 (EGFRT790M) are 0.75 μM and 0.83 μM, respectively. It suppressed tumorigenesis in lung cancer mouse xenograft in vivo and enhanced tumor inhibitory response in treatment with JAK2 inhibitor. It showed no apparent systematic toxicity in a 28-day rat study (at 37.5 mg/kg). | [246,265,266,267] | [171,172,173] |
| SLs | EM23 (188) | IC50 values against Caski and SiHa cell lines are 5.8 and 6.6 μM, respectively. | [179] | |
| SLs | Brevilin A (189) | It selectively inhibits growth of DU145 and MDA-MB-468. It inhibits JAK-STAT signaling pathway by attenuating JAKs activity and blocking STAT3 signaling (IC50 = 10.6 μM) in cancer cells. | [268] | [186] |
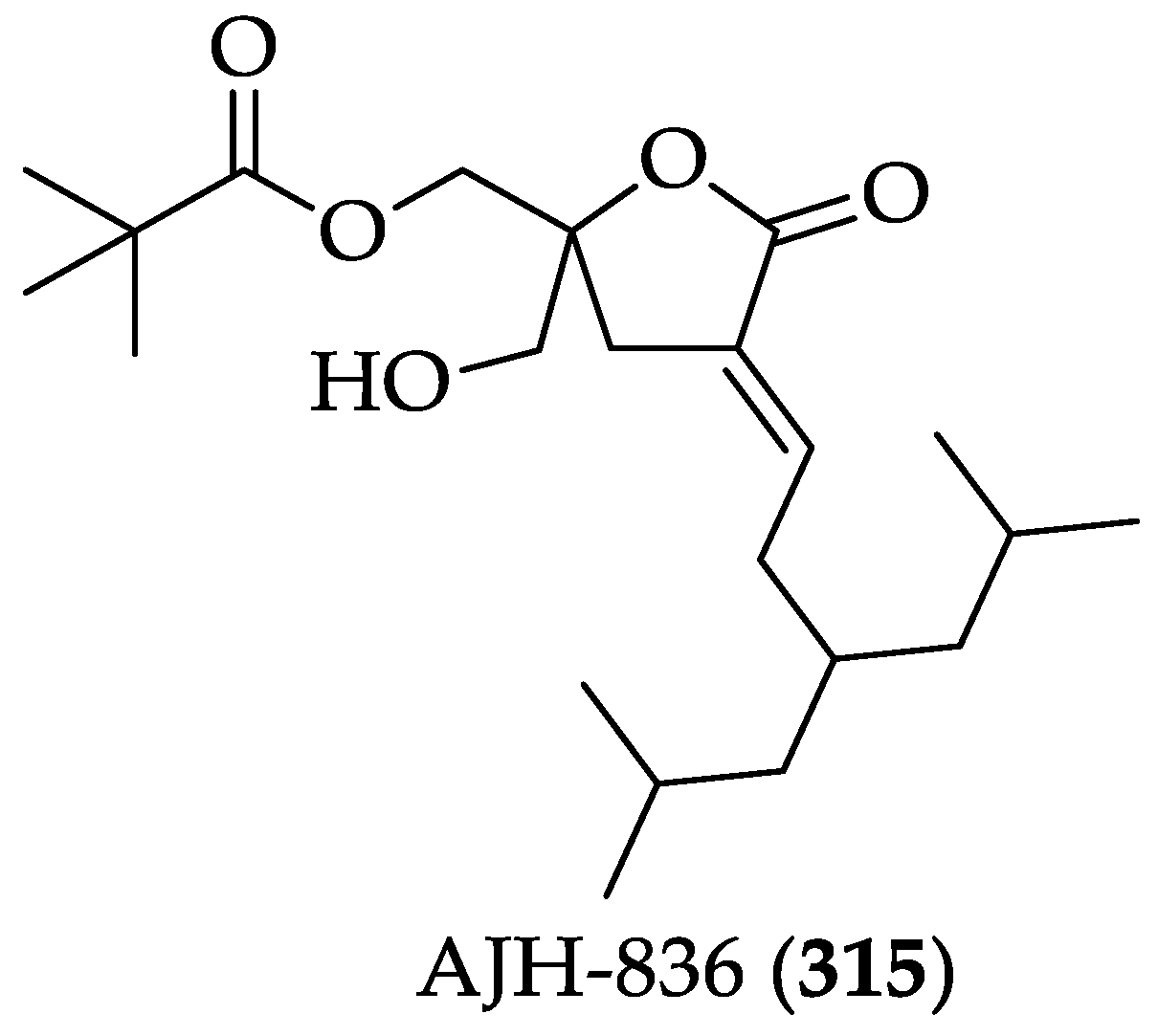
| DAGLs | AJH-836 (315) | It is a selective ligand for PKCε. (Ki of PKCα = 46 nM, Ki of PKCε = 1.43 nM) | [14,199] | |
| DLs | Andrographolide (373) | IC50 value against PCa cells is 19.95 μM. IC50 value against A549 cells of PTX + 30 μM 373 is 0.5 nM, showing significant synergy. | [205,269] | [207,211,212,216] |
| DLs | 19-triisopropyl andrographolide (343) | IC50 = 6.3 μM and 1.6 μM for MKN-45 and AGS cell lines, respectively. | [208] | |
| DLs | SRJ23 (375) | 50-fold improved IC50 for PCa cells (0.4 μM) than 373. | [211] | |
| DLs | Nagilactone C (397) | IC50 = 2–5 mM against MDA-MB-231, AGS, and Hela cell lines. | [224] | |
| DLs | Nagilactone E (398) | IC50 = 5.2 and 3.6 μM against A549 and NIC-H1975, respectively. In an A549 xenograft mouse model (10 mg/kg/d, ip), it suppressed tumor growth by 62% and inhibited tumor metastasis without apparent toxicity | [226,227] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.; Sengupta, S.; Sim, T. Natural and Synthetic Lactones Possessing Antitumor Activities. Int. J. Mol. Sci. 2021, 22, 1052. https://doi.org/10.3390/ijms22031052
Kim Y, Sengupta S, Sim T. Natural and Synthetic Lactones Possessing Antitumor Activities. International Journal of Molecular Sciences. 2021; 22(3):1052. https://doi.org/10.3390/ijms22031052
Chicago/Turabian StyleKim, Younghoon, Sandip Sengupta, and Taebo Sim. 2021. "Natural and Synthetic Lactones Possessing Antitumor Activities" International Journal of Molecular Sciences 22, no. 3: 1052. https://doi.org/10.3390/ijms22031052
APA StyleKim, Y., Sengupta, S., & Sim, T. (2021). Natural and Synthetic Lactones Possessing Antitumor Activities. International Journal of Molecular Sciences, 22(3), 1052. https://doi.org/10.3390/ijms22031052