
Pro-Inflammatory Effects of Indoxyl Sulfate in Mice: Impairment of Intestinal Homeostasis and Immune Response

 , , , ,
, , , , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. IS Serum Levels
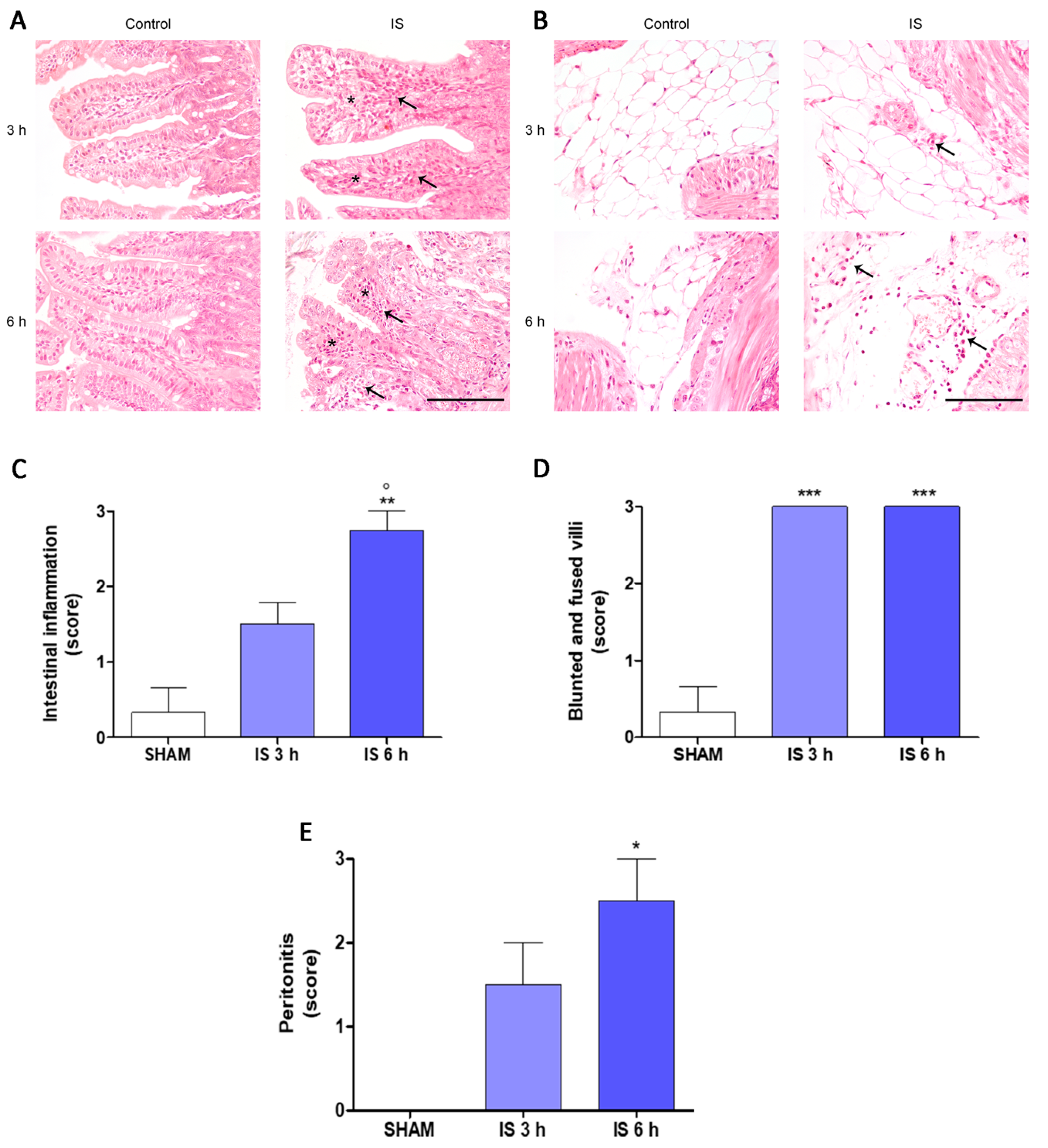
2.2. Histopathological Examination
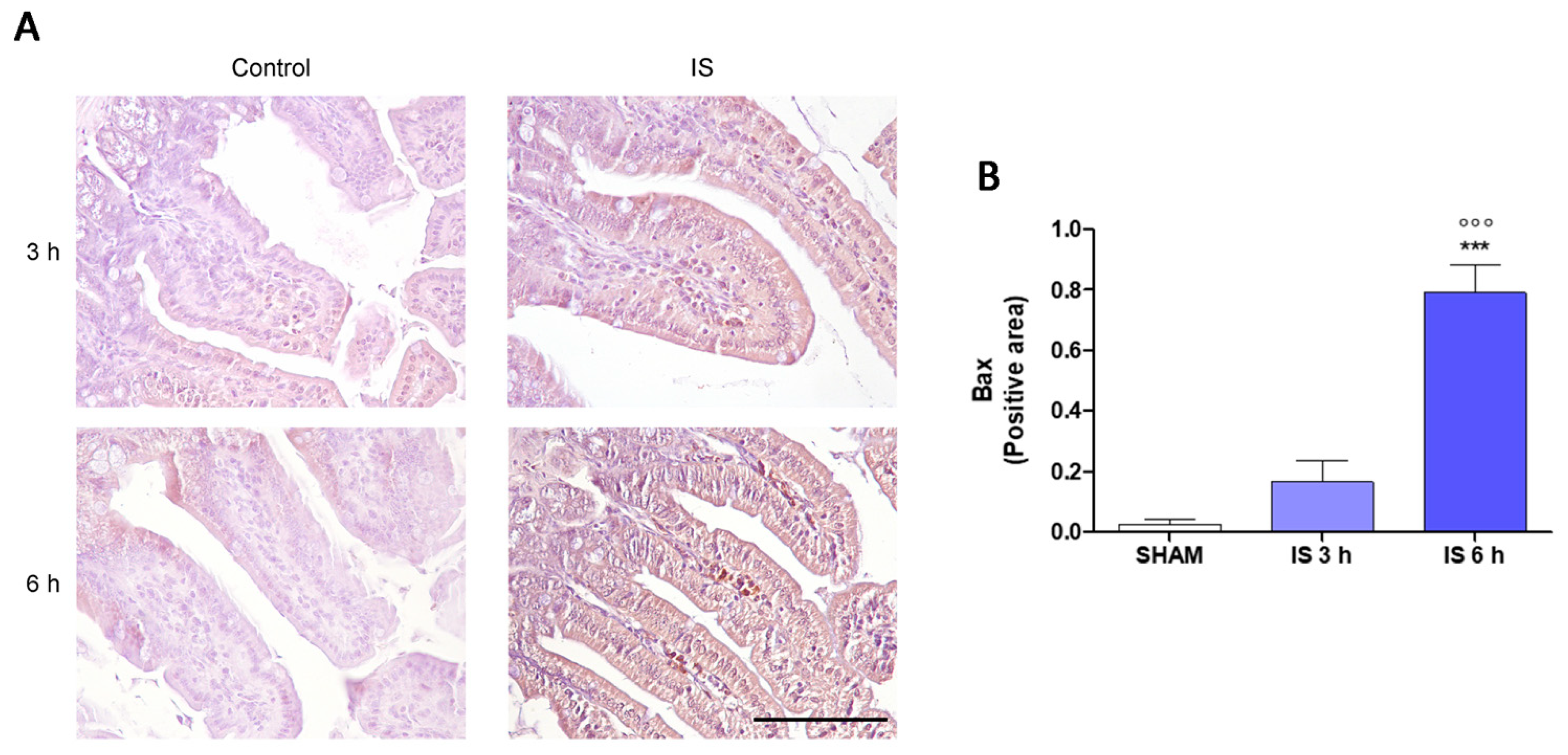
2.3. Immunohistochemical Localization of COX-2, Nitrotyrosine and BAX
2.4. IS Enhanced Pro-Inflammatory Parameters, Especially in Inflammatory Conditions
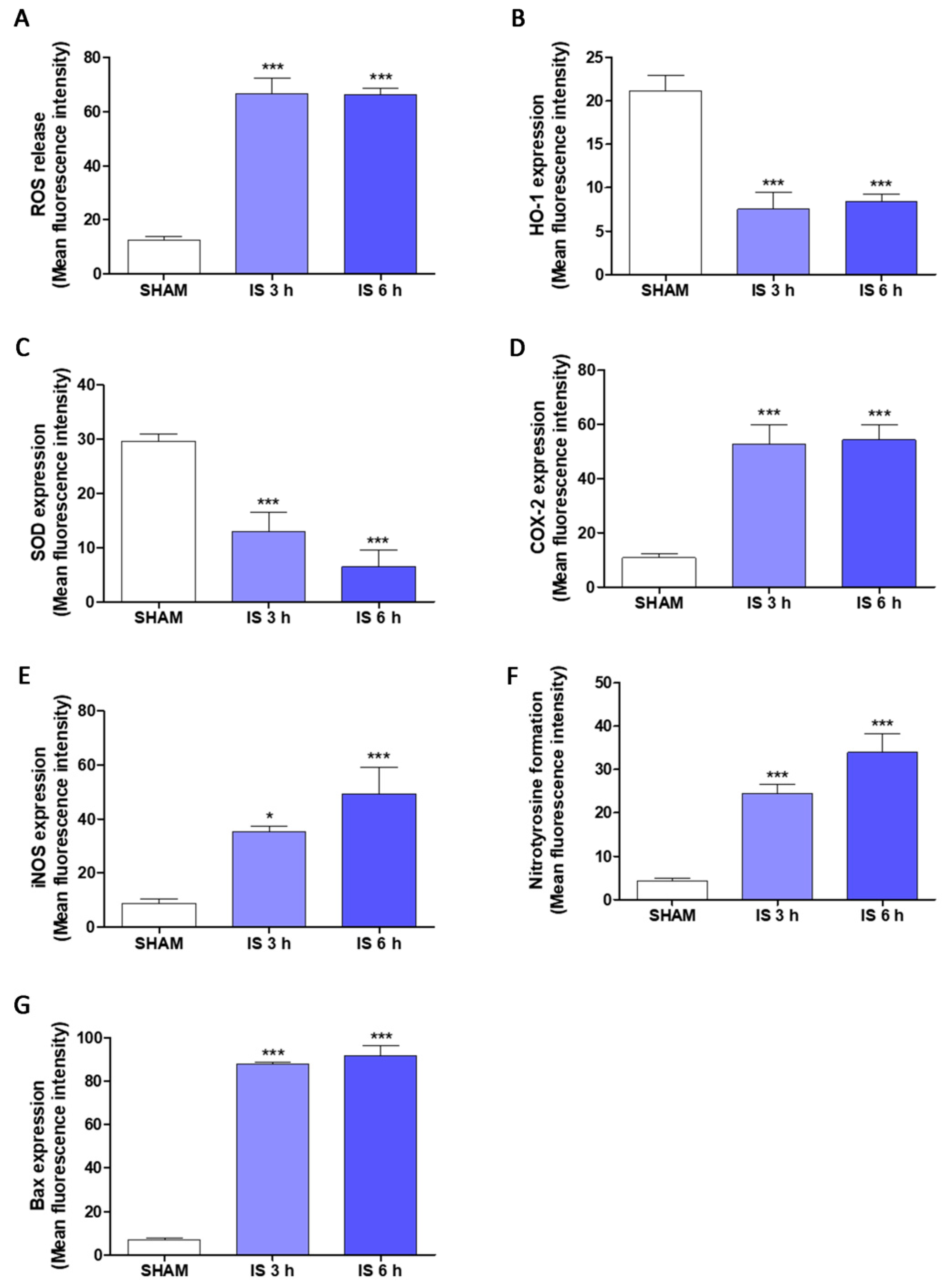
2.5. IS Increased Pro-Inflammatory, Pro-Oxidant and Pro-Apoptotic Parameters in Primary Murine Peritoneal Macrophages
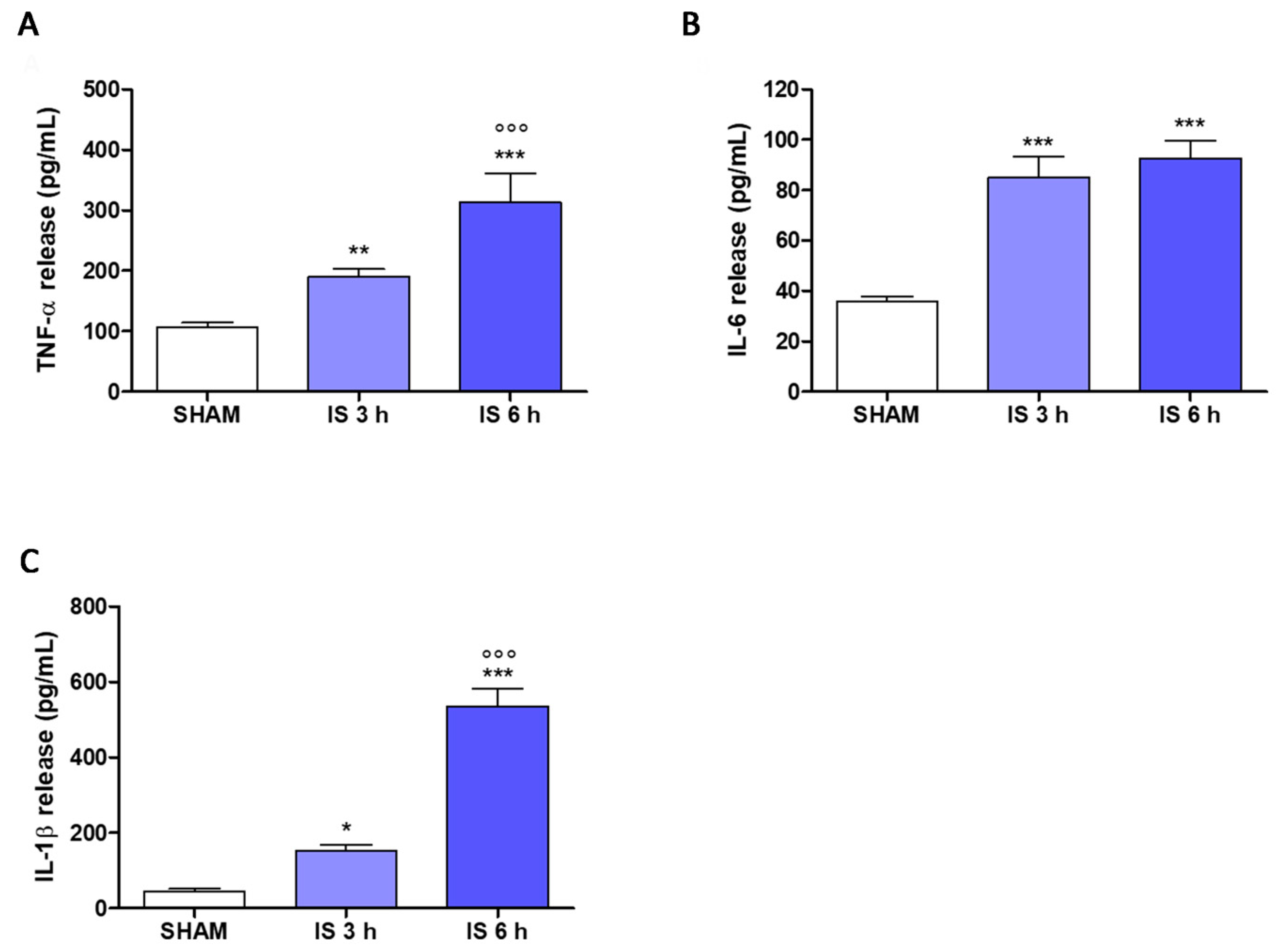
2.6. Cytokines Levels in Mice Serum
3. Discussion
4. Materials and Methods
4.1. In Vivo Studies
4.1.1. Reagents
4.1.2. Animals
4.1.3. Experimental Groups
- SHAM + vehicle group: vehicle solution was given by i.p.
- IS 3 h (800 mg/kg; i.p.) group: IS was administered i.p. for 3 h
- IS 6 h (800 mg/kg; i.p.) group: IS was administered i.p. for 6 h;
4.1.4. IS Serum Evaluation by HPLC
4.1.5. Histopathological Analysis
4.1.6. Immunohistochemistry
4.2. In Vitro Studies
4.2.1. Cell Culture
4.2.2. Cellular Treatment
4.2.3. TNF-α Determination
4.2.4. Evaluation of COX-2 and iNOS Expression and Nitrotyrosine Formation by Flow Cytometry Analysis
4.3. Ex Vivo Studies
4.3.1. Primary Murine Peritoneal Macrophages
4.3.2. Measurement of Intracellular Reactive Oxygen Species (ROS) Release
4.3.3. Detection of HO-1, SOD-2, COX-2, iNOS and Bax Expression and Nitrotyrosine Formation by Cytofluorimetry
4.3.4. TNF-α, IL-1 and IL-1β Serum Levels
4.4. Data Analysis and Statistical Evaluation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cachofeiro, V.; Goicochea, M.; de Vinuesa, S.G.; Oubiña, P.; Lahera, V.; Luño, J. Oxidative stress and inflammation, a link between chronic kidney disease and cardiovascular disease. Kidney Int. Suppl. 2008, 111, S4–S9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenvinkel, P. Inflammation in end-stage renal disease: The hidden enemy. Nephrology (Carlton) 2006, 11, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, S.; Pergola, P.E.; Zager, R.A.; Vaziri, N.D. Targeting Nrf2 activation to ameliorate oxidative stress and inflammation in Chronic Kidney Disease. Kidney Int. 2013, 83, 1029–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrero, J.J.; Stenvinkel, P. Persistent inflammation as a catalyst for other risk factors in chronic kidney disease: A hypothesis proposal. Clin. J. Am. Soc. Nephrol. 2009, 4 (Suppl. 1), S49–S55. [Google Scholar] [CrossRef] [Green Version]
- Lau, W.L.; Ix, J.H. Clinical detection, risk factors, and cardiovascular consequences of medial arterial calcification: A pattern of vascular injury associated with aberrant mineral metabolism. Semin. Nephrol. 2013, 33, 93–105. [Google Scholar] [CrossRef]
- Carrero, J.J.; Stenvinkel, P. Inflammation in end-stage renal disease–what have we learned in 10 years? Semin. Dial. 2010, 23, 498–509. [Google Scholar] [CrossRef]
- Mihai, S.; Codrici, E.; Popescu, I.D.; Enciu, A.M.; Necula, L.G.; Anton, G.; Tanase, C. Inflammation and Chronic Kidney Disease: Current Approaches and Recent Advances. Kidney Dis. Pathophysiol. Clin. Improv. 2018, 2018, 2180373. [Google Scholar]
- Lau, W.L.; Kalantar-Zadeh, K.; Vaziri, N.D. The gut as a source of inflammation in chronic kidney disease. Nephron 2015, 130, 92–98. [Google Scholar] [CrossRef] [Green Version]
- Shi, K.; Wang, F.; Jiang, H.; Liu, H.; Wei, M.; Wang, Z.; Xie, L. Gut bacterial translocation may aggravate microinflammation in hemodialysis patients. Dig. Dis. Sci. 2014, 59, 2109–2117. [Google Scholar] [CrossRef]
- Feroze, U.; Kalantar-Zadeh, K.; Sterling, K.A.; Molnar, M.Z.; Noori, N.; Benner, D.; Shah, V.; Dwivedi, R.; Becker, K.; Kovesdy, C.P.; et al. Examining associations of circulating endotoxin with nutritional status, inflammation, and mortality in hemodialysis patients. J. Ren. Nutr. 2012, 22, 317–326. [Google Scholar] [CrossRef] [Green Version]
- Szeto, C.C.; Kwan, B.C.; Chow, K.M.; Lai, K.B.; Chung, K.Y.; Leung, C.B.; Li, P.K.T. Endotoxemia is related to systemic inflammation and atherosclerosis in peritoneal dialysis patients. Clin. J. Am. Soc. Nephrol. 2008, 3, 431–436. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, C.W.; Harrison, L.E.; Eldehni, M.T.; Jefferies, H.J.; Szeto, C.C.; John, S.G.; Sigrist, M.K.; Burton, J.O.; Hothi, D.; Korsheed, S.; et al. Circulating endotoxemia: A novel factor in systemic inflammation and cardiovascular disease in chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2011, 6, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Anders, H.J.; Andersen, K.; Stecher, B. The intestinal microbiota, a leaky gut, and abnormal immunity in kidney disease. Kidney Int. 2013, 83, 1010–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, G.; Hörl, W.H. Immune Dysfunction in Uremia—An Update. Toxins 2012, 4, 962–990. [Google Scholar] [CrossRef] [Green Version]
- Autore, G.; Marzocco, S.; Sorrentino, R.; Mirone, V.; Baydoun, A.; Pinto, A. In vitro and in vivo TNFα synthesis by methylguanidine, an uremic catabolyte. Life Sci. 1999, 65, 121–127. [Google Scholar] [CrossRef] [Green Version]
- Marzocco, S.; Di Paola, R.; Serraino, I.; Sorrentino, R.; Meli, R.; Mattaceraso, G.; Cuzzocrea, S.; Pinto, A.; Autore, G. Effect of methylguanidine in carrageenan-induced acute inflammation in the rats. Eur. J. Pharmacol. 2004, 484, 341–350. [Google Scholar] [CrossRef]
- Marzocco, S.; Di Paola, R.; Genovese, T.; Sorrentino, R.; Britti, D.; Scollo, G.; Pinto, A.; Cuzzocrea, S.; Autore, G. Methylguanidine reduces the development of non septic shock induced by zymosan in mice. Life Sci. 2004, 75, 1417–1433. [Google Scholar] [CrossRef]
- Marzocco, S.; Di Paola, R.; Ribecco, M.T.; Sorrentino, R.; Domenico, B.; Genesio, M.; Pinto, A.; Autore, G.; Cuzzocrea, S. Effect of methylguanidine in a model of septic shock induced by LPS. Free Radic. Res. 2004, 38, 1143–1153. [Google Scholar] [CrossRef]
- Marzocco, S.; Popolo, A.; Bianco, G.; Pinto, A.; Autore, G. Pro-apoptotic effect of methylguanidine on hydrogen peroxide-treated rat glioma cell line. Neurochem. Int. 2010, 57, 518–524. [Google Scholar] [CrossRef]
- Barreto, F.C.; Barreto, D.V.; Liabeuf, S.; Meert, N.; Glorieux, G.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A.; European Uremic Toxin Work Group (EUTox). Serum indoxyl sulfate is associated with vascular disease and mortality in chronic kidney disease patients. Clin. J. Am. Soc. Nephrol. 2009, 4, 1551–1558. [Google Scholar] [CrossRef] [Green Version]
- Niwa, T. Indoxyl sulfate is a nephro-vascular toxin. J. Ren. Nutr. 2010, 20, S2–S6. [Google Scholar] [CrossRef] [PubMed]
- Niwa, T.; Ise, M. Indoxyl sulfate, a circulating uremic toxin, stimulates the progression of glomerular sclerosis. J. Lab. Clin. Med. 1994, 124, 96–104. [Google Scholar] [PubMed]
- Bolati, D.; Shimizu, H.; Higashiyama, Y.; Nishijima, F.; Niwa, T. Indoxyl sulfate induces epithelial-to-mesenchymal transition in rat kidneys and human proximal tubular cells. Am. J. Nephrol. 2011, 34, 318–323. [Google Scholar] [CrossRef] [PubMed]
- Adesso, S.; Magnus, T.; Cuzzocrea, S.; Campolo, M.; Rissiek, B.; Paciello, O.; Autore, G.; Pinto, A.; Marzocco, S. Indoxyl Sulfate Affects Glial Function Increasing Oxidative Stress and Neuroinflammation in Chronic Kidney Disease: Interaction between Astrocytes and Microglia. Front. Pharmacol. 2017, 12, 370. [Google Scholar] [CrossRef] [PubMed]
- Adesso, S.; Paterniti, I.; Cuzzocrea, S.; Fujioka, M.; Autore, G.; Magnus, T.; Pinto, A.; Marzocco, S. AST-120 Reduces Neuroinflammation Induced by Indoxyl Sulfate in Glial Cells. J. Clin. Med. 2018, 7, 365. [Google Scholar] [CrossRef] [Green Version]
- Adesso, S.; Ruocco, M.; Rapa, S.F.; Piaz, F.D.; Di Iorio, B.R.; Popolo, A.; Autore, G.; Nishijima, F.; Pinto, A.; Marzocco, S. Effect of Indoxyl Sulfate on the Repair and Intactness of Intestinal Epithelial Cells: Role of Reactive Oxygen Species’ Release. Int. J. Mol. Sci. 2019, 20, 2280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adesso, S.; Popolo, A.; Bianco, G.; Sorrentino, R.; Pinto, A.; Autore, G.; Marzocco, S. The uremic toxin indoxyl sulphate enhances macrophage response to LPS. PLoS ONE 2013, 8, e76778. [Google Scholar] [CrossRef] [Green Version]
- Mihai, S.; Codrici, E.; Popescu, I.D.; Enciu, A.M.; Albulescu, L.; Necula, L.G.; Mambet, C.; Anton, G.; Tanase, C. Inflammation-Related Mechanisms in Chronic Kidney Disease Prediction, Progression, and Outcome. J. Immunol. Res. 2018, 2018, 2180373. [Google Scholar] [CrossRef]
- Marzocco, S.; Dal Piaz, F.; Di Micco, L.; Torraca, S.; Sirico, M.L.; Tartaglia, D.; Autore, G.; Di Iorio, B. Very low protein diet reduces indoxyl sulfate levels in chronic kidney disease. Blood Purif. 2013, 35, 196–201. [Google Scholar] [CrossRef]
- Rapa, S.F.; Di Iorio, B.R.; Campiglia, P.; Heidland, A.; Marzocco, S. Inflammation and Oxidative Stress in Chronic Kidney Disease-Potential Therapeutic Role of Minerals, Vitamins and Plant-Derived Metabolites. Int. J. Mol. Sci. 2020, 21, 263. [Google Scholar] [CrossRef] [Green Version]
- Chauveau, P.; Koppe, L.; Combe, C.; Lasseur, C.; Trolonge, S.; Aparicio, M. Vegetarian diets and chronic kidney disease. Nephrol. Dial. Transplant. 2019, 34, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Di Iorio, B.R.; Rocchetti, M.T.; De Angelis, M.; Cosola, C.; Marzocco, S.; Di Micco, L.; di Bari, I.; Accetturo, M.; Vacca, M.; Gobbetti, M.; et al. Nutritional Therapy Modulates Intestinal Microbiota and Reduces Serum Levels of Total and Free Indoxyl Sulfate and P-Cresyl Sulfate in Chronic Kidney Disease (Medika Study). J. Clin. Med. 2019, 8, 1424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marzocco, S.; Fazeli, G.; Di Micco, L.; Autore, G.; Adesso, S.; Dal Piaz, F.; Heidland, A.; Di Iorio, B. Supplementation of Short-Chain Fatty Acid, Sodium Propionate, in Patients on Maintenance hemodialysis-"Beneficial Effects on Inflammatory Parameters and Gut-Derived Uremic Toxins"-A Pilot Study (PLAN Study). J. Clin. Med. 2018, 7, 315. [Google Scholar] [CrossRef] [Green Version]
- Ichii, O.; Otsuka-Kanazawa, S.; Nakamura, T.; Ueno, M.; Kon, Y.; Chen, W.; Rosenberg, A.Z.; Kopp, J.B. Podocyte injury caused by indoxyl sulfate, a uremic toxin and aryl-hydrocarbon receptor ligand. PLoS ONE 2014, 9, e108448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaziri, N.D.; Dure-Smith, B.; Miller, R.; Mirahmadi, M.K. Pathology of gastrointestinal tract in chronic hemodialysis patients: An autopsy study of 78 cases. Am. J. Gastroenterol. 1985, 80, 608–611. [Google Scholar]
- Yu, C.; Wang, Z.; Tan, S.; Wang, Q.; Zhou, C.; Kang, X.; Zhao, S.; Liu, S.; Fu, H.; Yu, Z.; et al. Chronic Kidney Disease Induced Intestinal Mucosal Barrier Damage Associated with Intestinal Oxidative Stress Injury. Gastroenterol. Res. Pract. 2016, 2016, 6720575. [Google Scholar] [CrossRef] [Green Version]
- Lau, W.L.; Liu, S.M.; Pahlevan, S.; Yuan, J.; Khazaeli, M.; Ni, Z.; Chan, J.Y.; Vaziri, N.D. Role of Nrf2 dysfunction in uremia-associated intestinal inflammation and epithelial barrier disruption. Dig. Dis. Sci. 2015, 60, 1215–1222. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, A.; Krieg, R.; Massey, H.D.; Carl, D.; Ghosh, S.; Gehr, T.W.B.; Ghosh, S.S. Sodium butyrate ameliorates insulin resistance and renal failure in CKD rats by modulating intestinal permeability and mucin expression. Nephrol. Dial. Transplant. 2019, 34, 783–794. [Google Scholar] [CrossRef]
- Caggiano, G.; Cosola, C.; Di Leo, V.; Gesualdo, M.; Gesualdo, L. Microbiome modulation to correct uremic toxins and to preserve kidney functions. Curr. Opin. Nephrol. Hypertens. 2020, 29, 49–56. [Google Scholar] [CrossRef]
- Chugh, S.; Chaudhry, S.; Ryan, T.; Margetts, P.J. Peritoneal Membrane Injury and Peritoneal Dialysis. Adv. Nephrol. 2014, 2014, 573685. [Google Scholar] [CrossRef] [Green Version]
- Contreras-Velázquez, J.C.; Soto, V.; Jaramillo-Rodríguez, Y.; Samaniego-Ríos, L.I.; Quiñones-Pérez, V.; Avila, M.; Amato, D.; Paniagua, R. Clinical outcomes and peritoneal histology in patients starting peritoneal dialysis are related to diabetic status and serum albumin levels. Kidney Int. 2008, 108, S34–S41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cianchi, F.; Cuzzocrea, S.; Vinci, M.C.; Messerini, L.; Comin, C.E.; Navarra, G.; Perigli, G.; Centorrino, T.; Marzocco, S.; Lenzi, E.; et al. Heterogeneous expression of cyclooxygenase-2 and inducible nitric oxide synthase within colorectal tumors: Correlation with tumor angiogenesis. Dig. Liver Dis. 2010, 42, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; DuBois, R.N. The Role of COX-2 in Intestinal Inflammation and Colorectal Cancer. Oncogene 2010, 29, 781–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, C.; Tan, S.; Wang, Z.; Deng, B.; Li, J.; Wang, Q.; Zhou, C.; Kang, X.; Yu, Z.; Zhuang, S. Chronic Kidney Disease Elicits an Intestinal Inflammation Resulting in Intestinal Dysmotility Associated with the Activation of Inducible Nitric Oxide Synthesis in Rat. Digestion 2018, 97, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Blander, J.M. On cell death in the intestinal epithelium and its impact on gut homeostasis. Curr. Opin. Gastroenterol. 2018, 34, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Parker, A.; Vaux, L.; Patterson, A.M.; Modasia, A.; Muraro, D.; Fletcher, A.G.; Byrne, H.M.; Maini, P.K.; Watson, A.J.M.; Pin, C. Elevated apoptosis impairs epithelial cell turnover and shortens villi in TNF-driven intestinal inflammation. Cell Death Dis. 2019, 10, 108. [Google Scholar] [CrossRef]
- Han, K.H.; Park, J.M.; Jeong, M.; Han, Y.M.; Go, E.J.; Park, J.; Kim, H.; Han, J.G.; Kwon, O.; Hahm, K.B. Heme oxygenase-1 induction and anti-inflammatory actions of Atractylodes macrocephala and Taraxacum herba extracts prevented colitis and was more effective than sulfasalazine in preventing relapse. Gut Liv. 2017, 11, 655–666. [Google Scholar] [CrossRef] [Green Version]
- Yasukawa, K.; Tokuda, H.; Tun, X.; Utsumi, H.; Yamada, K. The detrimental effect of nitric oxide on tissue is associated with inflammatory events in the vascular endothelium and neutrophils in mice with dextran sodium sulfate-induced colitis. Free Radic. Res. 2012, 46, 1427–1436. [Google Scholar] [CrossRef]
- Ishigami, J.; Matsushita, K. Clinical epidemiology of infectious disease among patients with chronic kidney disease. Clin. Exp. Nephrol. 2019, 23, 437–447. [Google Scholar] [CrossRef] [Green Version]
- Maloy, K.J.; Powrie, F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature 2011, 474, 298–306. [Google Scholar] [CrossRef]
- Tumur, Z.; Shimizu, H.; Enomoto, A.; Miyazaki, H.; Niwa, T. Indoxyl sulfate upregulates expression of ICAM-1 and MCP-1 by oxidative stress-induced NF-kappaB activation. Am. J. Nephrol. 2010, 31, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Bolati, D.; Shimizu, H.; Yisireyili, M.; Nishijima, F.; Niwa, T. Indoxyl sulfate, a uremic toxin, downregulates renal expression of Nrf2 through activation of NF-κB. BMC Nephrol. 2013, 14, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.J.; Vaziri, N.D. Contribution of impaired Nrf2-Keap1 pathway to oxidative stress and inflammation in chronic renal failure. Am. J. Physiol. Ren. Physiol. 2010, 298, F662–F671. [Google Scholar] [CrossRef] [Green Version]
- Yamawaki, K.; Kanda, H.; Shimazaki, R. Nrf2 activator for the treatment of kidney diseases. Toxicol. Appl. Pharmacol. 2018, 360, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.T.; Wu, P.H.; Tsai, Y.C.; Hsu, Y.L.; Wang, H.Y.; Kuo, M.C.; Kuo, P.L.; Hwang, S.J. Indoxyl Sulfate Induces Apoptosis Through Oxidative Stress and Mitogen-Activated Protein Kinase Signaling Pathway Inhibition in Human Astrocytes. J. Clin. Med. 2019, 8, 191. [Google Scholar] [CrossRef] [Green Version]
- Hénaut, L.; Mary, A.; Chillon, J.M.; Kamel, S.; Massy, Z.A. The Impact of Uremic Toxins on Vascular Smooth Muscle Cell Function. Toxins 2018, 10, 218. [Google Scholar] [CrossRef] [Green Version]
- Malaponte, G.; Bevelacqua, V.; Fatuzzo, P.; Rapisarda, F.; Emmanuele, G.; Travali, S.; Mazzarino, M.C. IL-1beta, TNF-alpha and IL-6 release from monocytes in haemodialysis patients in relation to dialytic age. Nephrol. Dial. Transplant. 2002, 17, 1964–1970. [Google Scholar] [CrossRef] [Green Version]
- Stenvinkel, P.; Ketteler, M.; Johnson, R.J.; Lindholm, B.; Pecoits-Filho, R.; Riella, M.; Heimbürger, O.; Cederholm, T.; Girndt, M. IL-10, IL-6, and TNF-alpha: Central factors in the altered cytokine network of uremia--the good, the bad, and the ugly. Kidney Int. 2005, 67, 1216–1233. [Google Scholar] [CrossRef] [Green Version]
- Duranton, F.; Cohen, G.; De Smet, R.; Rodriguez, M.; Jankowski, J.; Vanholder, R.; Argiles, A.; Abou Deif, O.; Drueke, T.; Baurmeister, U.; et al. Normal and Pathologic Concentrations of Uremic Toxins. J. Am. Soc. Nephrol. 2012, 23, 1258–1270. [Google Scholar] [CrossRef] [Green Version]
- Tbahriti, H.F.; Meknassi, D.; Moussaoui, R.; Messaoudi, A.; Zemour, L.; Kaddous, A.; Bouchenak, M.; Mekki, K. Inflammatory status in chronic renal failure: The role of homocysteinemia and pro-inflammatory cytokines. World J. Nephrol. 2013, 2, 31–37. [Google Scholar] [CrossRef]
- Zhu, W.; Stevens, A.P.; Dettmer, K.; Gottfried, E.; Hoves, S.; Kreutz, M.; Holler, E.; Canelas, A.B.; Kema, I.; Oefner, P.J. Quantitative profiling of tryptophan metabolites in serum, urine, and cell culture supernatants by liquid chromatography-tandem mass spectrometry. Anal. Bioanal. Chem. 2011, 401, 3249–3261. [Google Scholar] [CrossRef] [PubMed]
- Erben, U.; Loddenkemper, C.; Doerfel, K.; Spieckermann, S.; Haller, D.; Heimesaat, M.M.; Zeitz, M.; Siegmund, B.; Kühl, A.A. A guide to histomorphological evaluation of intestinal inflammation in mouse models. Int. J. Clin. Exp. Pathol. 2014, 7, 4557–4576. [Google Scholar] [PubMed]
- De Biase, D.; Esposito, F.; De Martino, M.; Pirozzi, C.; Luciano, A.; Palma, G.; Raso, G.M.; Iovane, V.; Marzocco, S.; Fusco, A.; et al. Characterization of inflammatory infiltrate of ulcerative dermatitis in C57BL/6NCrl-Tg(HMGA1P6)1Pg mice. Lab. Anim. 2019, 53, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Cimmino, I.; Margheri, F.; Prisco, F.; Perruolo, G.; D’Esposito, V.; Laurenzana, A.; Fibbi, G.; Paciello, O.; Doti, N.; Ruvo, M.; et al. Prep1 regulates angiogenesis through a PGC-1α–mediated mechanism. FASEB J. 2019, 33, 13893–13904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raehtz, K.D.; Barrenas, F.; Xu, C.; Busman-Sahay, K.; Valentine, A.; Law, L.; Ma, D.; Policicchio, B.B.; Wijewardana, V.; Brocca-Cofano, E.; et al. African green monkeys avoid SIV disease progression by preventing intestinal dysfunction and maintaining mucosal barrier integrity. PLoS Pathog. 2020, 16, e1008333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Law, A.M.K.; Yin, J.X.M.; Castillo, L.; Young, A.I.J.; Piggin, C.; Rogers, S.; Caldon, C.E.; Burgess, A.; Millar, E.K.A.; O’Toole, S.A.; et al. Andy’s Algorithms: New automated digital image analysis pipelines for FIJI. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Rapa, S.F.; Di Paola, R.; Cordaro, M.; Siracusa, R.; D’Amico, R.; Fusco, R.; Autore, G.; Cuzzocrea, S.; Stuppner, H.; Marzocco, S. Plumericin Protects against Experimental Inflammatory Bowel Disease by Restoring Intestinal Barrier Function and Reducing Apoptosis. Biomedicines 2021, 12, 67. [Google Scholar] [CrossRef]
- Adesso, S.; Russo, R.; Quaroni, A.; Autore, G.; Marzocco, S. Astragalus membranaceus Extract Attenuates Inflammation and Oxidative Stress in Intestinal Epithelial Cells via NF-κB Activation and Nrf2 Response. Int. J. Mol. Sci. 2018, 19, 800. [Google Scholar] [CrossRef] [Green Version]
- Rapa, S.F.; Waltenberger, B.; Di Paola, R.; Adesso, S.; Siracusa, R.; Peritore, A.F.; D’Amico, R.; Autore, G.; Cuzzocrea, S.; Stuppner, H.; et al. Plumericin prevents intestinal inflammation and oxidative stress in vitro and in vivo. FASEB J. 2020, 34, 1576–1590. [Google Scholar] [CrossRef] [Green Version]
- Pepe, G.; Rapa, S.F.; Salviati, E.; Bertamino, A.; Auriemma, G.; Cascioferro, S.; Autore, G.; Quaroni, A.; Campiglia, P.; Marzocco, S. Bioactive Polyphenols from Pomegranate Juice Reduce 5-Fluorouracil-Induced Intestinal Mucositis in Intestinal Epithelial Cells. Antioxidants 2020, 9, 699. [Google Scholar] [CrossRef]
- Basilicata, M.G.; Pepe, G.; Rapa, S.F.; Merciai, F.; Ostacolo, C.; Manfra, M.; Di Sarno, V.; Autore, G.; De Vita, D.; Marzocco, S.; et al. Anti-Inflammatory and Antioxidant Properties of Dehydrated Potato-Derived Bioactive Compounds in Intestinal Cells. Int. J. Mol. Sci. 2019, 20, 6087. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rapa, S.F.; Prisco, F.; Popolo, A.; Iovane, V.; Autore, G.; Di Iorio, B.R.; Dal Piaz, F.; Paciello, O.; Nishijima, F.; Marzocco, S. Pro-Inflammatory Effects of Indoxyl Sulfate in Mice: Impairment of Intestinal Homeostasis and Immune Response. Int. J. Mol. Sci. 2021, 22, 1135. https://doi.org/10.3390/ijms22031135
Rapa SF, Prisco F, Popolo A, Iovane V, Autore G, Di Iorio BR, Dal Piaz F, Paciello O, Nishijima F, Marzocco S. Pro-Inflammatory Effects of Indoxyl Sulfate in Mice: Impairment of Intestinal Homeostasis and Immune Response. International Journal of Molecular Sciences. 2021; 22(3):1135. https://doi.org/10.3390/ijms22031135
Chicago/Turabian StyleRapa, Shara Francesca, Francesco Prisco, Ada Popolo, Valentina Iovane, Giuseppina Autore, Biagio Raffaele Di Iorio, Fabrizio Dal Piaz, Orlando Paciello, Fuyu Nishijima, and Stefania Marzocco. 2021. "Pro-Inflammatory Effects of Indoxyl Sulfate in Mice: Impairment of Intestinal Homeostasis and Immune Response" International Journal of Molecular Sciences 22, no. 3: 1135. https://doi.org/10.3390/ijms22031135
APA StyleRapa, S. F., Prisco, F., Popolo, A., Iovane, V., Autore, G., Di Iorio, B. R., Dal Piaz, F., Paciello, O., Nishijima, F., & Marzocco, S. (2021). Pro-Inflammatory Effects of Indoxyl Sulfate in Mice: Impairment of Intestinal Homeostasis and Immune Response. International Journal of Molecular Sciences, 22(3), 1135. https://doi.org/10.3390/ijms22031135